

ABSTRACT
Herpesviruses are highly successful pathogens that persist for the lifetime of their hosts primarily because of their ability to establish and maintain latent infections from which the virus is capable of productively reactivating. Human cytomegalovirus (HCMV), a betaherpesvirus, establishes latency in CD34+ hematopoietic progenitor cells during natural infections in the body. Experimental infection of CD34+ cells ex vivo has demonstrated that expression of the viral gene products that drive productive infection is silenced by an intrinsic immune defense mediated by Daxx and histone deacetylases through heterochromatinization of the viral genome during the establishment of latency. Additional mechanistic details about the establishment, let alone maintenance and reactivation, of HCMV latency remain scarce. This is partly due to the technical challenges of CD34+ cell culture, most notably, the difficulty in preventing spontaneous differentiation that drives reactivation and renders them permissive for productive infection. Here we demonstrate that HCMV can establish, maintain, and reactivate in vitro from experimental latency in cultures of human embryonic stem cells (ESCs), for which spurious differentiation can be prevented or controlled. Furthermore, we show that known molecular aspects of HCMV latency are faithfully recapitulated in these cells. In total, we present ESCs as a novel, tractable model for studies of HCMV latency.
IMPORTANCE
Human cytomegalovirus (HCMV) is a significant human pathogen that is known for causing birth defects, blindness in AIDS patients, and organ transplant rejection. The ability of HCMV to cause disease is dependent upon its capacity to establish and maintain latent infections. Very few of the molecular mechanisms of latency have been elucidated, due in part to the lack of a tractable cell culture model. Here we present embryonic stem cells (ESCs) as a model for HCMV latency, one in which genome maintenance and reactivation could be closely monitored. HCMV establishes latency in ESCs in the same fashion as it does in CD34+ cells, the currently favored in vitro model. Hence, ESCs represent a novel model with unique properties, such as the ability to be genetically manipulated and cultured indefinitely in an undifferentiated state, that will facilitate the mechanistic examination of certain aspects of HCMV latency that have proven technically challenging in other model systems.
Introduction
Human cytomegalovirus (HCMV) is a betaherpesvirus whose virions contain an approximately 235-kb double-stranded DNA genome, which is enclosed within a protein capsid that is in turn surrounded by a proteinaceous tegument and ultimately a lipid envelope (1). HCMV infects a majority of the world’s population, causing severe disease in immunocompromised individuals and birth defects in neonates. Currently, there is no vaccine to prevent HCMV infection. Antiviral drugs against HCMV exist, including ganciclovir, cidofovir, and foscarnet, but toxicities are high, and resistant strains develop (2, 3). These drugs inhibit only productive (lytic) viral replication. Like all herpesviruses, HCMV can also achieve a latent state where it is immune to these antivirals yet poised to productively reactivate and cause disease at a later time (4, 5). Developing a better biological understanding of the latent virus is an initial step toward targeting it with antivirals for the improved treatment of HCMV infections.
Lytic infection is divided into three phases (immediate early [IE], early, and late) characterized by the expression of select viral genes and for which many molecular details are known. Latency is also divided into three phases (establishment, maintenance, and reactivation) for which little molecular details are known. During latency, most lytic-phase gene expression is absent, although certain transcripts, including LUNA, UL138, US28, UL111A (also known as viral interleukin 10 [vIL-10]) and the CLTs (cytomegalovirus [CMV] latency transcripts), accumulate during both lytic replication and latency (6). It is thought that expression of the lytic-phase-promoting IE1 and IE2 proteins must be suppressed in order to establish and maintain latency and that expression of the proteins must be activated, as it is at the start of a de novo lytic infection, to initiate reactivation.
Whether these IE genes are expressed or not is controlled by the intersection of the tegument transactivator pp71 and a cellular intrinsic immune defense mediated in part by the transcriptional corepressor Daxx (7). Capsids deposited into the cytoplasm during the entry process travel along microtubules to nuclear pores through which they release their DNA into the nucleus. These viral genomes then colocalize with cellular proteins that constitute promyelocytic leukemia nuclear bodies (PML-NBs), including Daxx, ATRX, Sp100, PML, and histone deacetylases (HDACs) (8). This results in transcriptional silencing by the assembly of heterochromatin on the viral genome (9, 10). In differentiated cells, such as fibroblasts, macrophages, or dendritic cells where lytic replication is initiated upon infection, tegument-delivered pp71 prevents this defense from silencing the viral genome by entering the nucleus, displacing ATRX from Daxx (11) and inducing Daxx degradation (12). This results in the production of the IE1 and IE2 proteins that counteract the repressive effects of the other PML-NB components and accelerate viral gene expression, respectively, thus promoting productive, lytic infection (13). In incompletely differentiated cell types where latency is established or modeled, tegument-delivered pp71 localizes to the cytoplasm, Daxx remains stable, the viral genome is heterochromatinized, and viral IE gene expression is repressed (14–17). If the intrinsic defense is artificially inactivated by Daxx knockdown or HDAC inhibition, latency is not established, IE genes are expressed, and the lytic program is initiated.
While genetic requirements for latency are emerging, few molecular details other than those described above for the establishment phase are known. This lack of mechanistic detail can be traced largely to the intractability of the systems used to study latency. Naturally latent cells are rare and cannot be enriched for or selected; thus, they have been used mostly to molecularly phenotype cell surface markers and to confirm that latent transcripts detected during experimental infections in vitro are also found in cells naturally infected in vivo. Primary CD34+ hematopoietic progenitor cells have been most effectively used to study HCMV latency (14, 17, 18). However, these preparations from donated umbilical cord blood or bone marrow represent heterogeneous mixtures of cells that are expensive to acquire and difficult to maintain in the desired differentiation state over extended periods of time. Recently, primary CD14+ monocytes have also been examined as a model for HCMV latency (19), although the technical limitations listed above apply to these cells as well.
Cells that can be stably maintained at the proper differentiation state and that faithfully recapitulate all known parameters of HCMV latency identified in primary CD34+ cells would represent an ideal model system for studying this mode of viral infection. For almost 30 years, various cells have been examined in the hopes of finding such a system (20). Unfortunately, all tested models mimic some, but not all, facets of true natural or in vitro experimental latent infections. For example, immortalized NTera2 (21) and THP-1 cell lines (22) and their differentiated derivatives model the differentiation-dependent IE gene expression observed during HCMV latency, but these cells reactivate poorly or not at all, and thus are considered models of quiescence, but not true latency. More recently, the immortalized CD34+ cell line Kasumi-3 has been used to model HCMV latency (23), but the transformed nature of these cells must be considered when comparing these infections to natural or experimental latency in primary CD34+ cells. Thus, the search for a convenient and reliable in vitro model for HCMV latency continues.
Here we present embryonic stem cells (ESCs) as a novel HCMV latency model. Unlike primary CD34+ cells, ESCs can be cultured indefinitely in an undifferentiated state, are economical for use in large-scale experiments, and have established protocols for genetic manipulation. Importantly, ESCs can be differentiated down the myeloid lineage into CD34+ cells and ultimately into dendritic cells (24), which are thought to be a site for HCMV latency reactivation in vivo (15). In this article, we show that when either clinical (FIX) or laboratory-adapted (AD169) strains of HCMV enter ESCs, tegument-delivered pp71 localizes to the cytoplasm, Daxx remains stable, IE gene expression is not detected, and latency is established. Inactivation of the intrinsic immune defense with the HDAC inhibitor valproic acid (VPA) prevents latency establishment and allows lytic replication to initiate for AD169, but not FIX. We further demonstrate that established latent infections are maintained more efficiently for FIX than for AD169, mirroring work in primary CD34+ cells (18), and that differentiation of latently infected ESCs stimulates reactivation and the release of infectious viral progeny. Finally, we capitalize on the strengths of this system, namely, the stability with which the differentiation status of the cells is maintained, to monitor genome maintenance over time and show that FIX and AD169 genomes are rapidly lost and show no evidence of replicative amplification over the time frame monitored. ESCs represent a new, tractable tissue culture model for latency studies with HCMV that will allow both genetic and molecular analyses to probe mechanistic features of this critical modality for viral persistence.
RESULTS
HCMV laboratory strain AD169 and clinical strain FIX establish latency in ESCs with similar efficiencies.
To determine whether human ESCs represent a viable model for HCMV latency, we tested known parameters of establishment, maintenance, and reactivation. When experimental latency is established in primary CD34+ cells, tegument-delivered pp71 localizes to the cytoplasm and the IE1 protein is not expressed (17). We found tegument-delivered pp71 in the cytoplasm of ESCs after infection with the laboratory-adapted AD169 strain (Fig. 1A) or with the clinical virus isolate FIX (Fig. 1D). The Oct4 protein is visualized as a marker of the undifferentiated state of these cells. In addition to pp71, tegument-delivered pp65 (Fig. 1B) also localized to the cytoplasm, as it does in other undifferentiated cells (25). Importantly, the IE1 protein was not expressed (Fig. 1C and E), indicating that HCMV establishes a latent infection within these undifferentiated cells.
FIG 1 .

HCMV enters embryonic stem cells (ESCs) but does not initiate lytic infection. ESCs grown on coverslips were infected with HCMV strain AD169 (A and C), FIX (D and E), or AD169 pp65-GFP (B) at an MOI of 3. The cells growing on coverslips were harvested 24 h postinfection, and the indicated viral (pp71 and IE1) or cellular (Oct4) proteins were visualized by indirect immunofluorescence microscopy. Viral pp65 was detected as a GFP signal by fluorescence microscopy. The nuclei were counterstained with Hoechst stain.
Treating ESCs with the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA) induces differentiation into cell types expressing markers of parietal endoderm (26), which gives rise to respiratory and gastrointestinal tissues during development and thus likely includes HCMV-permissive cell types, such as fibroblasts, endothelial cells, and epithelial cells. TPA-differentiated ESCs no longer express Oct4, but the cells are now capable of expressing the viral IE2 protein upon subsequent infection with HCMV (Fig. 2A). Tegument-delivered pp71 entered the nuclei of differentiated ESCs after infection with UV-inactivated (Fig. 2B) viral stocks. TPA differentiated ESCs into cells competent not only for the initiation of lytic infection but also the productive completion of this process, as in addition to IE2, the early protein UL44 (Fig. 2C), and the late protein pp28 (Fig. 2D) were expressed, and infectious progeny virions were released (Fig. 3). Thus, ESCs display similar differentiation-dependent viral gene expression and productive replication parameters as observed in the standard primary CD34+ cell model for HCMV latency.
FIG 2 .
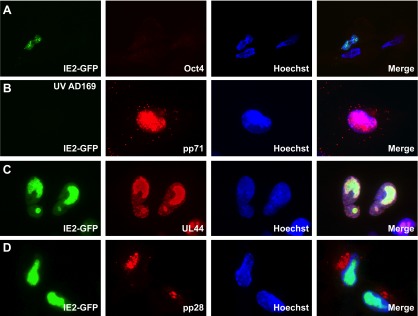
Differentiated ESCs support lytic-phase gene expression. ESCs grown on coverslips were treated with TPA for 3 days and subsequently infected with HCMV strain AD169 IE2-GFP (A, C, and D) or UV-inactivated AD169 IE2-GFP (B) at an MOI of 1. The cells growing on coverslips were harvested at 4 h (A and B), 3 days (C), or 5 days (D) postinfection. The indicated viral (pp71, UL44, and pp28) or cellular (Oct4) proteins were visualized by indirect immunofluorescence microscopy. Viral IE2 was detected as a GFP signal by fluorescence microscopy. The nuclei were counterstained with Hoechst stain.
FIG 3 .

Differentiated ESCs support productive infection. ESCs treated with TPA for 3 days were subsequently infected with HCMV strain AD169 at an MOI of 1. Infectious virions accumulated in the medium at 2 or 8 days postinfection (dpi) were quantitated by plaque assay.
ESCs express PML-NB proteins but do not assemble PML-NBs.
The cellular Daxx protein and an unidentified HDAC modulate HCMV latency through a defined mechanism. They silence viral IE gene expression when latency is established (17). Daxx localizes to PML-NBs, where other proteins that restrict viral lytic infection, such as ATRX, PML, and Sp100 are also found (11, 27, 28). Whether these other PML-NB proteins play a role during latency has not been examined; however, they are expressed in primary CD34+ cells (17).
We found that the PML-NB proteins ATRX, Daxx, PML, and Sp100 are all expressed in uninfected ESCs, though the expression pattern differs substantially from primary human fibroblasts (Fig. 4A). Compared to the levels in fibroblasts, the levels of ATRX and Daxx in ESCs are elevated, while the levels in PML and Sp100 are substantially decreased. Though these proteins are present, they do not form classic PML-NBs in ESCs (Fig. 4B). Daxx and ATRX show punctae clearly visible over a diffuse staining pattern within the nucleus. However, these proteins fail to colocalize with PML, which was found exclusively in punctae that localized to both the cytoplasm and nucleus (Fig. 4B). This unique PML staining pattern was observed with two independent antibodies (data not shown). Sp100 could not be reliably imaged, likely due to the low level of protein found in these cells. While ESCs do not appear to assemble classic PML-NBs, Daxx, the one component of these structures known to promote viral genome silencing during the establishment of latency (17), is present within the nuclei of these cells.
FIG 4 .
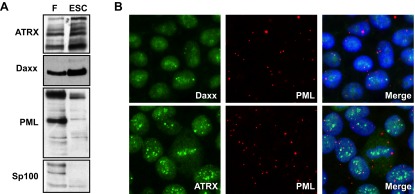
ESCs express PML-NB proteins but do not assemble canonical PML-NBs. (A) Equal amounts of protein lysates generated from uninfected fibroblasts (F) or uninfected ESCs were analyzed by Western blotting with the indicated antibodies. (B) Uninfected ESCs grown on coverslips were analyzed by indirect immunofluorescence microscopy for the indicated proteins. The merge panels show nuclei counterstained with Hoechst stain.
Viral IE gene expression is silenced by cellular and viral functions in ESCs.
To determine whether viral IE mRNA accumulation is suppressed in ESCs through the same mechanism that it is in primary CD34+ cells (17), we asked whether the cellular intrinsic defense, as well as the clinical strain-specific restriction, was active in these cells. Artificial inactivation of the cellular intrinsic defense with the HDAC inhibitor VPA allowed for IE1 transcription in ESCs infected with the laboratory-adapted AD169 strain, but not with the clinical FIX strain (Fig. 5A). VPA did not induce ESC differentiation, as tegument-delivered pp71 remained cytoplasmic and Oct4 was present in the drug-treated cells (Fig. 5B). Thus, both cellular and viral mechanisms silence viral IE gene expression upon the establishment of HCMV latency in ESCs, as they do in primary CD34+ cells. LUNA, an HCMV transcript known to be expressed during natural and experimental infection of primary CD34+ cells (29), was expressed by both AD169 and FIX strains in ESCs in the absence of VPA (Fig. 5A). In total, these results indicate that, for every parameter analyzed, HCMV establishes latency in ESCs in a manner indistinguishable from primary CD34+ cells.
FIG 5 .

HCMV gene expression in ESCs is regulated in a manner indistinguishable from experimental latency in primary CD34+ cells. (A) ESCs not treated with VPA (−) or pretreated with VPA (+) for 1 h were infected with HCMV strain AD169 (A) or HCMV strain FIX (F) at an MOI of 3. RNA extracted at 24 hpi was subjected to RT-PCR to monitor the expression of the indicated viral (IE1 and LUNA) or cellular (GAPDH) gene. (B) ESCs grown on coverslips and pretreated with VPA for 1 h were infected with AD169 at an MOI of 3. The indicated proteins were imaged by indirect immunofluorescence microscopy.
FIX maintains latency in ESCs more efficiently than AD169 does.
For HCMV to maintain latency, viral genomes must be maintained while reactivation is suppressed. Only a single study has quantitatively monitored genome maintenance in experimentally infected primary CD34+ cells. In two biological replicates, one population of cells showed evidence of genome amplification over 10 days, while a second population did not (30). We monitored total viral DNA levels in ESCs at 1 day postinfection (dpi) with either HCMV strain AD169 or FIX as an indicator of input viral genomes and then again at 3 and 10 days postinfection by Li-Cor analysis of standard PCR reactions (Fig. 6A and B) or by real-time PCR (Fig. 6C). At equivalent multiplicities, FIX appeared to deliver more viral genomes to ESCs (Fig. 6A). By day 3, viral genomes of either strain were less readily amplified (Fig. 6A) and present at less than 20% of the level for each specific strain found just 2 days prior (Fig. 6B and C). At 10 days postinfection, viral genome levels were similar to day 3 values. From this, we conclude that amplifying viral DNA replication, such as occurs during lytic infection, does not seem to occur in ESCs, and that while viral genomes are lost over time, detectable levels remain for at least 10 days.
FIG 6 .

Viral genomes are maintained in ESCs for at least 10 days. (A) ESCs were mock infected (M) or infected with HCMV strain AD169 (AD) or FIX at an MOI of 3. Total DNA isolated at the indicated day postinfection (dpi) was analyzed by PCR for the presence of viral (UL123) or cellular (GAPDH) DNA. (B and C) Viral genomes at the indicated dpi were quantitated by Li-Cor imaging for three independent biological replicates (B) or by real-time PCR for two biological replicates (C), normalized to cellular genome levels and are expressed as a percentage of the viral genome level detected on day 1 for each individual viral strain.
In primary CD34+ cells, AD169 spontaneously reactivates more frequently than FIX does (18). We acquired similar data from ESCs (Fig. 7B and C). After 20 days, populations of AD169-infected ESCs were approximately 30-fold more likely to produce infectious centers upon coincubation with fibroblasts than FIX-infected ESCs were (Fig. 7C). As both FIX and AD169 lytic-phase gene expression is silenced upon ESC infection (Fig. 1 and 5A), both viruses appear to establish latency with similar efficiencies. Combined with the hyperreactive phenotype of AD169, we interpret these results to indicate that AD169 is less able to maintain latency in ESCs than FIX is, a result in congruence with previous findings in primary CD34+ cells (18).
FIG 7 .

HCMV reactivates from latently infected ESCs upon their differentiation. (A) Reactivation assay flow chart. ESCs infected with HCMV strain AD169 (AD) or HCMV strain FIX for 10 days (10d) were treated with TPA (+) or not treated with TPA (−) for 3 days, cultured for an additional 7 days, and then separated into fractions consisting of either attached cells or clarified medium. Cells (top right) were coplated with fibroblasts (FIBROS) for the indicated number of weeks (1 week [1W] or 3 weeks [3W]), and then infectious centers were quantitated by counting GFP foci. Data from this portion of the assay are presented in panels B to F. The titers of virus in the medium fraction (bottom right) were determined directly by a plaque assay. Data from this portion of the assay are presented in panel G. (B) Representative images used to quantify GFP foci detected in untreated or TPA-treated cells infected with AD169 or FIX. (C) Quantitation of GFP foci detected in single wells of untreated, HCMV-infected ESCs. (D) Quantitation of GFP foci detected in single wells of untreated or TPA-treated ESCs infected with AD169. (E) Quantitation of GFP foci detected in single wells of untreated or TPA-treated ESCs infected with FIX. (F) Calculated fold increase for TPA-treated versus untreated cells infected with the indicated virus. (G) Infectious virions accumulated in the medium rescued from AD169-infected ESCs untreated or treated with TPA were quantitated by plaque assay. In all graphs, error bars represent standard deviations.
FIX reactivates from latency in ESCs more efficiently than AD169 does.
In order to be considered true latency, reactivation must occur in response to an appropriate stimulus, such as differentiation. ESCs infected with HCMV for 10 days were induced to differentiate with the addition of TPA for 3 days, and then 7 days later (at 20 days postinfection), clarified medium or attached cells were collected (Fig. 7A). Cells infected with AD169 (Fig. 7D) and with FIX (Fig. 7E) treated with TPA were much more likely to generate infectious centers after cocultivation with permissive fibroblasts than non-TPA-treated cells were. However, FIX was much more responsive to TPA than AD169 was (Fig. 7F).
Interestingly, while ESCs infected with either HCMV AD169 or FIX and subsequently differentiated were able to transfer infectious virus to permissive fibroblasts during coculture, only AD169-derived supernatants contained infectious progeny virions (Fig. 7G). Thus, it appears that reactivated FIX virus remained solely cell associated, while AD169 was clearly also released from cells. This is not surprising, as strong cell association is a characteristic of HCMV clinical strains (31). These experiments show that FIX virus reactivates more efficiently from ESCs than AD169 does. In total, our results indicate that ESCs represent a viable model for HCMV latency that faithfully recapitulates known latency parameters established in primary CD34+ cells.
DISCUSSION
ESCs are pluripotent cells isolated from human blastocysts that display unlimited self-renewal in an undifferentiated state. They maintain the potential to form cell types from all three embryonic germ layers and can specifically be differentiated into various myeloid lineage cells. These properties made them attractive candidates for a tractable tissue culture model for experimental HCMV latency. Here we show that ESCs establish, maintain, and reactivate experimental latency with characteristics indistinguishable from those described for primary human CD34+ cells, the most utilized and accepted current in vitro model system for studying HCMV latency. Interestingly, varicella-zoster virus, but not other alphaherpesviruses, also failed to productively replicate in ESCs, although whether this result was due to intrinsic defense mechanisms or the establishment of latency (or both) was not delineated (32).
We define the establishment of HCMV latency as delivery of the viral genome to the nucleus without the initiation of lytic-phase gene expression. Specifically, the products of the major IE locus, IE1 and IE2, are not expressed. These genes are silenced by an intrinsic defense mediated by the PML-NB resident protein Daxx and histone deacetylases. Other PML-NB proteins that suppress IE gene expression at the start of lytic infections, such as ATRX, PML, and Sp100, also likely contribute to viral gene silencing during latency, but their roles in this process have yet to be tested. Interestingly, ESCs do not have canonical PML-NBs (Fig. 4), yet the HDAC component of the intrinsic defense still silences the genome, likely indicating, as has been previously proposed, that PML-NB proteins themselves, as opposed to the structure, are the critical mediators of this defense. As in primary CD34+ cells, this defense is not inactivated in ESCs, as it is at the start of a lytic infection in fibroblasts, because tegument-delivered pp71 remains in the cytoplasm.
Maintaining latency requires that lytic-phase gene expression remain silenced and that the viral genome be retained in the nucleus. As in primary CD34+ cells, the clinical strain FIX utilizes at least one additional means of silencing lytic-phase gene expression during ESC experimental latency that is not found in the laboratory strain AD169 (Fig. 5). Thus, as in primary CD34+ cells, AD169 in ESCs is less efficient at maintaining latency, indicated by a higher incidence of spontaneous reactivation events (Fig. 7). This as yet unidentified clinical strain-specific suppressor of IE gene expression during latency appears to work independently of the cellular intrinsic defense to silence lytic-phase gene expression in an HDAC-independent manner.
During the maintenance of latency, the HCMV genome presumably resides in the nucleus to prevent the activation of cytoplasmic DNA sensors and to allow for the synthesis of latent transcripts and, upon reactivation, the expression of lytic-phase genes. How this maintenance is achieved is entirely unknown. Alphaherpesviruses maintain latency in nondividing neurons where the means to replicate viral genomes and permit them to reaccess the nucleus after cell division should not be required. Gammaherpesviruses have clearly demonstrated mechanisms for viral genome replication and partitioning in the dividing cells in which they maintain latency (33). As the in vivo latent reservoir (or reservoirs) for HCMV has not been definitively assigned (discussed below), whether or not the virus must deal with genome issues inherent in establishing latency in a dividing cell type is not appreciated.
We detected no evidence of HCMV genome replication in latent ESCs (Fig. 6). Previous work by others in primary CD34+ cells also failed to detect genome replication during latency (30, 34). However, in one of those studies (30), replication was observed in one lot of primary CD34+ cells, and recently, immortalized CD34+ cells have yielded evidence for genome replication during latency (23). Thus, the replicative fate of viral genomes while latency is maintained remains controversial. This is an important issue to resolve, as the CD34+ cells used to model HCMV latency represent mixtures of hematopoietic stem cells (HSCs) and hematopoietic progenitor cells (HPCs). HSCs, sometimes operationally divided into long-term and short-term stem cells, are multipotent and able to self-renew. However, HSCs represent only a small fraction of cell populations selected for CD34 cell surface expression. HPCs are oligopotent, do not self-renew, and represent a much larger fraction of CD34+ cell populations than HSCs do. Because some CD34+ cells retain the ability to self-renew (HSCs) while others do not (HPCs), determining in which cell type HCMV establishes and maintains latency along with determining latent genome replicative capacity will have mechanistic implications for natural latent reservoirs.
An important, confounding issue with monitoring viral DNA replication during the maintenance of HCMV latency is the inability of the population-based assays currently in use to differentiate between modest replication of latent genomes and high levels of replication (as would occur during a lytic infection) in a minor subset of differentiated cells likely present in these heterogeneous populations. Single-cell assays for viral replication during HCMV latency are likely to be much more informative but have yet to be conducted. ESCs are cultured as monolayers and can be maintained indefinitely in an undifferentiated state that supports latency. Their propensity for cell division can be manipulated by culturing density and frequency of passage. Thus, they represent an ideal model system for the single-cell experiments required to answer questions about HCMV genome replication during the maintenance of latency.
We were able to efficiently reactivate latent HCMV by treating ESCs with TPA (Fig. 7). Though we have not specifically characterized the cell types generated upon ESC differentiation with TPA, ESCs can be differentiated down the myeloid lineage into dendritic cell types (24) where HCMV reactivation is naturally triggered. The ability to differentiate ESCs in a stepwise fashion through the myeloid hierarchy, including HPCs, and ultimately into dendritic cells makes them an intriguing and attractive model with which to study mechanistic features of how viral genomes are maintained during latency and how cellular differentiation triggers HCMV reactivation.
Finally, the ability of ESCs to support all facets of experimental HCMV latency likely means that induced pluripotent stem cells (iPSCs) will also be viable models of latency. Therefore, genetically manipulated iPSCs could conceivably be generated from various knockdown, knockout, or complementing fibroblast cell lines to further expand the utility of this general model for mechanistic studies of HCMV latency.
MATERIALS AND METHODS
Cells and viruses.
Normal human dermal fibroblasts (NHDFs) were cultured as previously described (12). The Wisconsin H1 (WA01) and H9 (WA09) embryonic stem cell (ESC) lines were obtained from WiCell. Both cell lines were used for examination of IE protein expression and tegument-delivered pp71 localization with identical results. Studies analyzing transcription, genome maintenance, and differentiation prior to infection were performed in WA01 cells. Reactivation assays were performed in WA09 cells. All cells were cultured independent of a feeder layer on Matrigel (BD Biosciences) in TeSR1 medium (WiCell) as described previously (35). Briefly, cells were propagated in 6-well plates and passaged at a ratio of 1:4 to 1:6 every 3 to 5 days, using dispase (2 mg/ml from Gibco) and mechanical disruption to detach colonies for passaging. Prior to passaging, differentiated cells detected visually by changes in cell morphology were removed by suction. Between passages, the medium was changed daily on cells. During the course of experiments, differentiation status was monitored by indirect immunofluorescence for Oct4 expression. ESCs were differentiated by adding 100 ng/ml 12-O-tetradecanoylphorbol-13-acetate (TPA) for 3 days. ESCs used in experiments were typically infected at approximately 50 to 75% confluence. Viruses used were AD169 (36), AD169 pp65-GFP (37), AD169 IE2-GFP (38), and FIX-GFP (39). Cells were infected in minimal volume for 60 min, followed by the addition of medium to normal culture volumes. Note that the titers used represent “fibroblast infectious units” and do not necessarily represent the efficiency with which the virus enters or infects other cell types, including ESCs. To quantitate infection rates, single-cell suspensions of ESCs infected with FIX-GFP were generated by trypsinization and analyzed by flow cytometry (FACSCalibur; BD Biosciences) for green fluorescent protein (GFP) expression. For 5 biological replicates, approximately 7.8% ± 1.5% of ESCs expressed GFP.
Inhibitors, antibodies, and Western blots.
Valproic acid (VPA) (1 mM) (Sigma) dissolved in water was added 1 h before infection. The following antibodies were from commercial sources: Daxx (D7810; Sigma), PML (H-238 and sc-966; Santa Cruz), ATRX (sc-15408; Santa Cruz), Sp100 (AB1380; Chemicon), and UL44 (CA006-100; Virusys). Antibodies against pp71 (IE-233), IE1 (1B12), and pp28 (CMV157) and secondary antibodies have been previously described (12). For Western blot analysis, equivalent protein concentrations from cell lysates prepared in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors were analyzed as previously described (12).
Indirect immunofluorescence.
ESCs were plated and cultured on coverslips, fixed with 1% paraformaldehyde in phosphate-buffered saline (PBS) and processed as previously described (12). Individual cells were imaged using a Zeiss Axiovert 200M deconvolution fluorescence microscope with a 63× objective. Plaques were imaged with a Zeiss Axiovert 200 microscope with a 20× objective.
PCR and RT-PCR.
Infected cells were washed once, treated with trypsin for 5 min at 37°C, collected in medium, centrifuged to pellet cells, washed once with PBS, and pelleted again by low-speed centrifugation. DNA was isolated from cells using the Genome DNA minikit (catalog no. IB47202; IBI Scientific). Subsequently, DNA was analyzed by real-time PCR (quantitative PCR [qPCR]) or standard PCR followed by quantitation using the Li-Cor Odyssey Fc imaging system. qPCR was performed on an ABI 7900HT instrument, and data were analyzed using SDS 2.2.1 software. Standard PCR was performed using GoTaq polymerase (M300; Promega) and primer pairs listed below (30 cycles). Total RNA was isolated using the RNeasy minikit (catalog no. 74104; Qiagen) and quantified using a UV spectrophotometer. Equivalent amounts were treated with RNase-free DNase (catalog no. M6101; Promega) following the manufacturer’s protocol and subsequently used in a reverse transcription-PCR (RT-PCR) (40 cycles) using SuperScript III one-step RT-PCR system (catalog no. 12574-026; Invitrogen). Primer pairs included primers for the following: for IE1, sense (5′-CGTCCTTGACACGATGGAGT) and antisense (5′-ATCTGTTTGACGAGTTCTGCC) primers (that span the intron between exon 2 and exon 3); for LUNA, sense (5′-ATGACCTCTCCTCCACAC) and antisense (5′-GGAAAAACACGCGGGGGA) primers; and primers to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as previously described (12). PCR products were separated on 1.5% agarose gels and visualized by ethidium bromide staining.
Reactivation assays.
ESCs were infected at a multiplicity of infection (MOI) of 3 with either AD169-GFP or FIX-GFP in a 6-well dish. Twenty-four hours after infection, the medium was changed. Infection was allowed to proceed for 10 days, with daily medium changes, at which point cells were either left untreated or treated with 100 ng/ml TPA for 3 days. Medium was changed daily during TPA treatment and for 2 days after treatment, at which point (15 dpi) medium was left on cells for an additional 5 days. At 10 days posttreatment (20 dpi), medium and cells were collected separately. The titers of virus in medium from cells were determined in a standard plaque assay. Cells were coplated with fibroblasts and GFP centers (defined as a group of at least 4 cells) were counted after either 1 week (AD169) or 3 weeks (FIX). Data represent the number of GFP-positive centers detected per well of a 6-well dish. A summary of the assay is shown in Fig. 7.
ACKNOWLEDGMENTS
We thank Phil Balandyk for expert technical assistance and Mitch Probasco for assistance with stem cells.
This work was supported by National Institutes of Health grant AI074984 (to R.F.K.). R.F.K. is a Vilas Fellow and a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease.
Footnotes
Citation Penkert RR, Kalejta RF. 2013. Human embryonic stem cell lines model experimental human cytomegalovirus latency. mBio 4(3):e00298-13. doi:10.1128/mBio.00298-13.
REFERENCES
- 1. Mocarski E, Shenk T, Pass R. 2007. Cytomegaloviruses, p 2701–2772 In Knipe D, Howley P, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, Fields virology. Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 2. Marschall M, Stamminger T. 2009. Molecular targets for antiviral therapy of cytomegalovirus infections. Future Microbiol. 4:731–742 [DOI] [PubMed] [Google Scholar]
- 3. McGregor A, Choi KY. 2011. Cytomegalovirus antivirals and development of improved animal models. Expert Opin. Drug Metab. Toxicol. 7:1245–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goodrum F, Caviness K, Zagallo P. 2012. Human cytomegalovirus persistence. Cell. Microbiol. 14:644–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reeves M, Sinclair J. 2008. Aspects of human cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 325:297–313 [DOI] [PubMed] [Google Scholar]
- 6. Slobedman B, Cao JZ, Avdic S, Webster B, McAllery S, Cheung AK, Tan JC, Abendroth A. 2010. Human cytomegalovirus latent infection and associated viral gene expression. Future Microbiol. 5:883–900 [DOI] [PubMed] [Google Scholar]
- 7. Penkert RR, Kalejta RF. 2012. Tale of a tegument transactivator: the past, present and future of human CMV pp71. Future Virol. 7:855–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tavalai N, Stamminger T. 2011. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res. 157:128–133 [DOI] [PubMed] [Google Scholar]
- 9. Sinclair J. 2010. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys. Acta 1799:286–295 [DOI] [PubMed] [Google Scholar]
- 10. Woodhall DL, Groves IJ, Reeves MB, Wilkinson G, Sinclair JH. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 281:37652–37660 [DOI] [PubMed] [Google Scholar]
- 11. Lukashchuk V, McFarlane S, Everett RD, Preston CM. 2008. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 82:12543–12554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80:3863–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahn JH, Hayward GS. 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 71:4599–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reeves MB, Lehner PJ, Sissons JG, Sinclair JH. 2005. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 86:2949–2954 [DOI] [PubMed] [Google Scholar]
- 15. Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. U. S. A. 102:4140–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 81:9109–9120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saffert RT, Penkert RR, Kalejta RF. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J. Virol. 84:5594–5604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goodrum F, Reeves M, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hargett D, Shenk TE. 2010. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. U. S. A. 107:20039–20044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gönczöl E, Andrews PW, Plotkin SA. 1984. Cytomegalovirus replicates in differentiated but not in undifferentiated human embryonal carcinoma cells. Science 224:159–161 [DOI] [PubMed] [Google Scholar]
- 21. Dósa R, Burián K, Gönczöl E. 2005. Human cytomegalovirus latency is associated with the state of differentiation of the host cells: an in vitro model in teratocarcinoma cells. Acta Microbiol. Immunol. Hung. 52:397–406 [DOI] [PubMed] [Google Scholar]
- 22. Turtinen LW, Seufzer BJ. 1994. Selective permissiveness of TPA differentiated THP-1 myelomonocytic cells for human cytomegalovirus strains AD169 and Towne. Microb. Pathog. 16:373–378 [DOI] [PubMed] [Google Scholar]
- 23. O’Connor CM, Murphy EA. 2012. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J. Virol. 86:9854–9865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choi KD, Vodyanik M, Slukvin II. 2011. Hematopoietic differentiation and production of mature myeloid cells from human pluripotent stem cells. Nat. Protoc. 6:296–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Penkert RR, Kalejta RF. 2010. Nuclear localization of tegument-delivered pp71 in human cytomegalovirus-infected cells is facilitated by one or more factors present in terminally differentiated fibroblasts. J. Virol. 84:9853–9863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng X, Zhang J, Smuga-Otto K, Tian S, Yu J, Stewart R, Thomson JA. 2012. Protein kinase C mediated extraembryonic endoderm differentiation of human embryonic stem cells. Stem Cells 30:461–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tavalai N, Adler M, Scherer M, Riedl Y, Stamminger T. 2011. Evidence for a dual antiviral role of the major nuclear domain 10 component Sp100 during the immediate-early and late phases of the human cytomegalovirus replication cycle. J. Virol. 85:9447–9458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tavalai N, Papior P, Rechter S, Leis M, Stamminger T. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 80:8006–8018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bego M, Maciejewski J, Khaiboullina S, Pari G, St Jeor S. 2005. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J. Virol. 79:11022–11034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goodrum F, Jordan CT, Terhune SS, High K, Shenk T. 2004. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104:687–695 [DOI] [PubMed] [Google Scholar]
- 31. Sinzger C, Hahn G, Digel M, Katona R, Sampaio KL, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 89:359–368 [DOI] [PubMed] [Google Scholar]
- 32. Dukhovny A, Sloutskin A, Markus A, Yee MB, Kinchington PR, Goldstein RS. 2012. Varicella-zoster virus infects human embryonic stem cell-derived neurons and neurospheres but not pluripotent embryonic stem cells or early progenitors. J. Virol. 86:3211–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nanbo A, Sugden A, Sugden B. 2007. The coupling of synthesis and partitioning of EBV’s plasmid replicon is revealed in live cells. EMBO J. 26:4252–4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Poole E, McGregor Dallas SR, Colston J, Joseph RS, Sinclair J. 2011. Virally induced changes in cellular microRNAs maintain latency of human cytomegalovirus in CD34+ progenitors. J. Gen. Virol. 92:1539–1549 [DOI] [PubMed] [Google Scholar]
- 35. Ludwig TE, Bergendahl V, Levenstein ME, Yu J, Probasco MD, Thomson JA. 2006. Feeder-independent culture of human embryonic stem cells. Nat. Methods 3:637–646 [DOI] [PubMed] [Google Scholar]
- 36. Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 76:2316–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sanchez V, Clark CL, Yen JY, Dwarakanath R, Spector DH. 2002. Viable human cytomegalovirus recombinant virus with an internal deletion of the IE2 86 gene affects late stages of viral replication. J. Virol. 76:2973–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ibig-Rehm Y, Götte M, Gabriel D, Woodhall D, Shea A, Brown NE, Compton T, Feire AL. 2011. High-content screening to distinguish between attachment and post-attachment steps of human cytomegalovirus entry into fibroblasts and epithelial cells. Antiviral Res. 89:246–256 [DOI] [PubMed] [Google Scholar]
- 39. Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 100:14976–14981 [DOI] [PMC free article] [PubMed] [Google Scholar]