

Abstract
Acute lung injury (ALI) is a clinical syndrome characterized by hypoxia which is caused by the breakdown of the alveolar capillary barrier. IL-1β a cytokine released within the airspace in ALI, down-regulates αENaC transcription and protein expression via p38 MAP kinase-dependent signaling. While induction of the heat shock response can restore alveolar fluid clearance compromised by IL-1β following the onset of severe hemorrhagic shock in rats, the mechanisms are not fully understood. In this study, we report that the induction of the heat shock response prevents IL-1β-dependent inhibition of αENaC mRNA expression and subsequent channel function. Heat shock results in IRAK1 detergent insolubility and a disruption of Hsp90 binding to IRAK1. Likewise, TAK1, another client protein of Hsp90 and signaling component of the IL-1β pathway is also detergent insoluble after heat shock. Twenty-four hours post-heat shock, both IRAK1 and TAK1 are again detergent soluble, which correlates with the IL-1β-dependent p38 activation. Remarkably, IL-1β-dependent p38 activation 24-hour post-heat shock did not result in an inhibition of αENaC mRNA expression and channel function. Further analysis demonstrates prolonged preservation of αENaC expression by the activation of the heat shock response that involves inducible Hsp70. Inhibition of Hsp70 at 24 hours post-heat shock results in p38-dependent IL-1β inhibition of αENaC mRNA expression while over-expression of Hsp70 attenuates the p38-dependent IL-1β inhibition of αENaC mRNA expression. These studies demonstrate new mechanisms by which the induction of the heat shock response protects the barrier function of the alveolar epithelium in acute lung injury.
Keywords: Lung, Stress Protein Response, αENaC, p38 MAP Kinase, IRAK-1, TAK-1
INTRODUCTION
Acute lung injury (ALI) is a clinical syndrome characterized by alveolar flooding of a protein rich edema due to the breakdown of the alveolar capillary barrier (1). In severe lung injury, the alveolar epithelial vectorial fluid transport is inhibited by the development of the inflammatory response within the airspace of the lung (2). IL-1β, one of the most biologically active cytokines in the airspace of patients with acute lung injury (3–5), contributes to ALI development by downregulating expression of the apical epithelial sodium channel (ENaC) α-subunit through a p38 MAP kinase-dependent signaling thereby impairing vectorial sodium transport and removal of edema fluid from the alveolar space (6).
The heat shock or stress protein response (SPR) has been classically defined as a highly conserved cellular defense mechanism characterized by increased expression of stress proteins (7). This allows the cell to withstand a subsequent insult or “second hit” that would otherwise be lethal, a phenomenon referred as "thermotolerance" or "preconditioning" (8). The activation of SPR is characterized by an early phase defined by the inhibition of proinflammatory cell signaling pathways within minutes after onset of SPR. The inhibition of these intracellular pathways early after onset of SPR is due in a large part to the dissociation of Hsp90 from its client proteins, as we have published previously (9–11). Indeed, Hsp90 functions as a positive regulator of multiple cell signaling pathways by modifying the conformation of its client proteins into active signaling proteins (12). Blocking the ATPase site of Hsp90 by geldanamycin inhibits the function of these client proteins making them detergent insoluble and targeting them for degradation via the proteasome (13).
SPR is also associated with the expression of inducible heat shock proteins such as Hsp70. Several studies that include our own work have shown that the lung inflammatory response is inhibited in animals or cells that have been preconditioned by stress (14–19). For example, prior SPR induction with either heat or geldanamycin inhibits the development of ALI due to ischemia reperfusion, hemorrhagic shock or sepsis (2,20,21). In addition, the inhibitory effect of SPR can be reproduced in part by the adenoviral gene transfer of Hsp70 into the distal airspace of the lung, suggesting that the expression of intracellular Hsp70 may participate to the anti-inflammatory effect induced by SPR (22).
Previous work from our laboratory has shown that some patients with ALI undergo SPR activation that is associated with the maintenance of the alveolar epithelial fluid transport (23). We have also found that prior SPR activation prevents the IL-1β-mediated decrease in transepithelial current by maintaining the expression of αENaC in alveolar epithelial type II cells, although the mechanism by which SPR activation prevents the inhibition of alveolar fluid clearance by IL-1β is still unknown. In the present study, we show that the attenuation of IL-1β signaling by SPR activation is due to the detergent insolubility of IRAK-1 and TAK1 as well as disruption of Hsp90 from IRAK1. However, the IL-1β signaling pathway leading to p38 MAP kinase activation is reconstituted within 24 hours post-heat shock and yet αENaC transcription is still preserved. Further analysis showed the prolonged attenuation of IL-1β signaling by SPR activation is due to up-regulation of inducible Hsp70.
MATERIAL AND METHODS
Isolation and culture of rat ATII cells
Rat alveolar type II (ATII) cells were isolated as described previously (24) after obtaining approval from the UCSF Committee on Animal Research or the UAB IACUC committee. Male Sprague-Dawley rats (150–200g) were used for the isolation of ATII cells.
Isolation and culture of human ATII cells
Human alveolar ATII cells were isolated by a modification of methods previously described (24) after obtaining approval from the UCSF Committee on Human Research.
SPR activation using heat (heat shock)
The incubation time for cells placed in a 43°C CO2 incubator was determined based on the absence of cell death, using the Alamar blue assay, and the efficiency of SPR activation by measuring the intracellular increase in inducible Hsp70 protein by western blotting (data not shown). Type II alveolar epithelial cells were heat shocked for 45 minutes and returned to 37°C, as previously published (23).
Short circuit measurements
Freshly isolated rat or human ATII cells (0.75 × 106) were seeded on polycarbonate Snapwell membranes (pore size 0.4 µm, surface area 1.13 cm2). At 120–144 h, the Snapwell inserts were mounted in an Ussing chamber system (Physiologic Instruments Inc., San Diego, CA) as we have done before (24).
ELISA
p38 phosphorylation was measured using ELISA (eBioscience, San Diego, CA) following the manufacturer's instructions.
Western Blotting
Cells were washed three times with phosphate buffered saline (PBS) on ice, and either lysed with 25 mM Tris buffer (pH 7.5) containing 100 mM NaCl, 5mM MgCl2, 1 mM EDTA, 1% Triton, 1 mM sodium orthovanadate, 1 mM NaF, and protease inhibitors to determine solubility/insolubility of IRAK1 and TAK1 or lysed in 1x Laemmli sample buffer (LSB). Nuclei were pelleted at 14,000 × g for 5 minutes when cells were lysed in the Tris/Triton buffer. When cells were lysed in LSB, the DNA was sheared using an 18G needle. The protein concentrations of the samples were determined in triplicate using an adaptation of the Bradford protein assay (BioRAD, Hercules, CA). The O.D.595 was determined using a Wallac Victor2 counter (Perkin Elmer, San Jose, CA) and the protein concentrations determined using a standard curve. Equal amounts of protein were separated by gel electrophoresis using SDS-polyacrylamide for all experiments including both detergent soluble and insoluble fractions, as we have done before (11). Proteins were visualized using the Odyssey Infrared Imaging System from LI-COR®. Antibodies: inducible Hsp70 (StressGen clone 92, Plymouth Meeting, PA), Hsp90 (Assay Designs #SPA-830F, Plymouth Meeting, PA), IRAK1 (Santa Cruz #SC5288, Santa Cruz, CA), TAK1 (Cell Signaling #4505, Danvers, MA), phosphop38 (Cell Signaling #9211S), p38 (Cell Signaling #9212) and actin (Cell Signaling #4967). IRAK1 1:500, TAK1 1:500, Hsp70 1:1,000, Hsp90 1:500, phosphorylated p38 1:500, p38 1:1,000, actin 1:1,000. LICOR Goat anti-rabbit and anti-mouse secondary antibodies were used at a dilution of 1:10,000.
Isolation of membrane enriched fraction
Type II cells were scraped into a hypotonic buffer (10 mM HEPES, 10 mM NaCl, 5 mM MgCl2, 1 mM DTT, protease inhibitors, phosphatase inhibitors). Cells were allowed to swell by rotation for 30 minutes at 4°C. Cells were broken by dounce using a loose fitting pestle and 18 strokes. Nuclei were removed by centrifugation 1,000 rpm for 3 minutes at 4°C. The supernatant was subjected to centrifugation at 15,000 × g for 15 minutes at 4°C to pellet an enriched membrane fraction (9). This pelleted fraction was either resuspended in LSB and further analyzed by SDS-PAGE and Western blotting or resuspended in 50 mM Hepes, 150 mM NaCl, 1%NP40, 1mM EDTA, 10% glycerol and 1mM DTT for immunoprecipitation prior to SDS-PAGE and Western blotting analysis.
Immunoprecipitations
Rat primary type II alveolar epithelial cells were lysed as described above for isolation of membrane enriched fraction with the addition of 40mM sodium molybdate for the immunoprecipitation of IRAK1/Hsp90 complexes. The membrane-enriched fraction was incubated with primary antibodies to Hsp90 for 1 hour at 4°C. Antigen-antibody (Ag-Ab) complexes are captured using protein G sepharose. The protein G sepharose-Ag-Ab complex was pelleted at 3,000 rpms for 4 seconds. The Ag-Ab complexes were washed three times and subjected to SDS-PAGE and Western blotted for the presence of the IRAK1 protein prior to stripping and re-probing for Hsp90.
Adenovirus infection
Rat type II alveolar epithelial cells grown on transwell permeable supports were incubated in PBS containing 5.3 mM EDTA on both the basolateral and apical compartments for 60 minutes. The PBS/EDTA solution was removed and cells were placed in 1ml of PBS in the basolateral compartment and 0.3ml of PBS in the apical compartment containing recombinant adenovirus expressing Hsp70 subcloned downstream from the CMV promoter (Clifford S. Deutschman, MD, University of Pennsylvania); MOI 50. Plates were incubated at 37°C for 1 hour with rocking to evenly distribute the virus over the cells every 10 minutes. Viral inoculum was removed after 1 hour and cells were placed in media containing heat-inactivated serum. Cells were lysed 48 hours post-infection and analyzed for protein and mRNA expression.
siRNA transfections
Primary rat type II alveolar epithelial cells were transfected with siRNA using X-tremeGENE transfection reagent (Roche, Indianapolis, IN) according to manufacturers’ instructions. The ratio of lipid to siRNA was 10 µl to 150 pmoles. Cells seeded on transwells were heat shocked for 45 minutes at 43°C and allowed to recover for 1 hour prior to the addition of the lipid/siRNA transfection complex on the cells. Twenty-four hours post-transfection, cells were lysed and inducible Hsp70 expression determined by SDS-PAGE and Western blotting. For analysis of ENaC mRNA, cells were stimulated with IL-1β from 21–24 hours post-transfection prior to RNA isolation. siRNA sequence to inducible Hsp70 (Ambion, Austin, TX): sense 5’-AACAAGAUCACCAUCACCAACtt-3’. Silencer® Negative control siRNA (Ambion #AM4635).
Measurement of αENaC mRNA
Real-time RT-PCR primers and probes were designed using Primer Express software (PE-Applied Biosystems (PE-ABI), Warrington, UK). The real-time RT-PCR probes were labeled with a fluorophore reporter dye (6–carboxy-fluorescein, FAM) at the 5' end and a quencher dye (BHQ, Biosearch Technologies, Inc., Novato CA) at the 3' end. Quantitative real time RT-PCR was performed as previously published. RT-PCR was carriedout in a 25-µl reaction mixture containing 1x TaqMan UniversalPCR Master Mix (PE-Biosystems, Foster City, CA), 10 pmol ofprimers, 5 pmol of TaqMan probe, and an equivalent of 100 ngof total RNA for 40 cycles at 95 °C for 15 s and 60 °C for 1 minute. The number of cycles to threshold (CT) of fluorescence detection was normalized to the CT of glyceraldehydes 3-phosphate dehydrogenase (GAPDH) for each sample tested. Rat RT-PCR GAPDH primers: TAGF: CTGCCAAGTATGATGACATCAAGAA, TAGR: AGCCCAGGATGCCCTTTAGT, TAGP: TCGGCCGCCTGCTTCACCA. Rat RT-PCR primers: αENaC TAGF: TGATTGAATTCCACCGCTCC, αENaC TAGR: CCCGTGGATGGTGGTGTT, αENaC TAGP: CGGGAGCTCTTCCAGTTCTTCTGCAA.
Statistical analysis
For the statistical analysis we used Statview 5.0® (SAS Inc.) and MedCalc® 7.2.0.2 (MedCalc Software Inc., Mariakerke, Belgium). All data were tested for normality and due to non-uniform distribution the data were subsequently analyzed with nonparametric tests. Data were calculated as medians ± interquartile ranges. The data are presented in the figures as box plots with interquartile ranges and lower and upper ranges. The Kruskal-Wallis test followed by the Dunn’s test were used to compare three or more experimental groups. The Mann-Whitney test was used to compare two experimental conditions. A p value of <0.05 was considered statistically significant.
RESULTS
SPR activation prevents IL-1β-dependent inhibition of amiloride-sensitive current in rat and human alveolar epithelial type II cell monolayers
In our prior studies, we reported that IL-1β, a biologically active cytokine in patients with ALI, decreased the amiloride-sensitive transepithelial current as well as mRNA expression of the α subunit of the epithelial sodium channel (ENaC) via a p38-dependent pathway (6). Further studies also showed that SPR activation prevented the IL-1β–mediated decrease in transepithelial current across ATII cell monolayers, although the mechanism of this effect is not fully understood (23). In the current study, we first determined the time required for attenuation of IL-1β inhibition of the amiloride-sensitive ENaC current in alveolar epithelial cells following activation of the stress protein response. A time-course of recovery after heat shock shows that the preservation of the amiloride-sensitive ENaC current in the presence of IL-1β stimulation persists as long as 24 hours showing a range of 40-90% protection (Figure 1A). By 48 hours after heat shock, the preservation of the amiloride-sensitive ENaC current was reversed, as IL-1β inhibited the transepithelial current at the same extent as that seen in the non-heat shock control monolayers (Figure 1A). The same time-course of protection was also observed in human type II alveolar epithelial cells, indicating that the protection due to heat shock was not unique to rat alveolar epithelial type II cells (Figure 1B).
Figure 1. SPR activation prevents IL-1β-dependent inhibition of amiloride-sensitive current in rat and human alveolar epithelial type II cell monolayers.
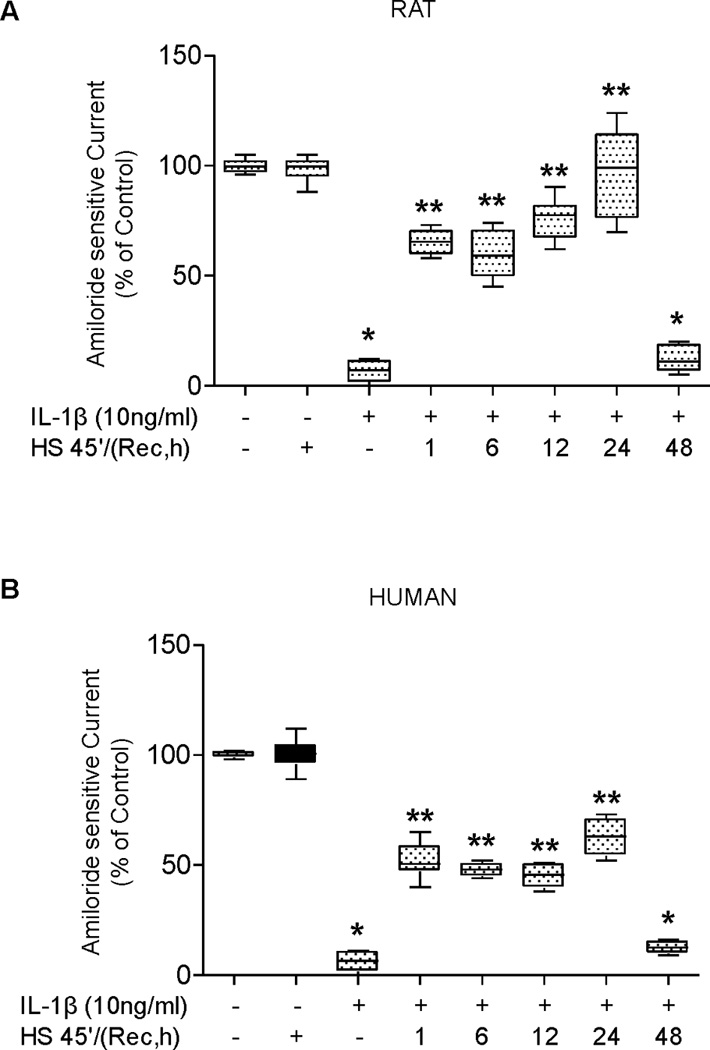
Panel A. Rat alveolar epithelial type II cell monolayers were heat shocked at 43°C and allowed to recover at 37°C for increasing times prior to the addition of IL-1β for three hours. The amiloride-sensitive fraction of the short-circuit current was measured in Ussing chambers (see Methods). Control monolayers were not subjected to SPR activation nor exposed to IL-1β stimulation. Panel B. Human alveolar epithelial type II cell monolayers were heat shocked at 43°C and allowed to recover at 37°C for increasing times prior to the addition of IL-1β for three hours. The amiloride-sensitive fraction of the short-circuit current was measured in Ussing chambers. Control monolayers were not subjected to SPR activation nor exposed to IL-1β stimulation. Panel C. Rat alveolar epithelial type II cell monolayers were treated with 10µg/ml of 17-AAG for the indicated times prior to the addition of IL-1β for three hours. The amiloride-sensitive fraction of the short-circuit current was measured in Ussing chambers. Control monolayers were not exposed to 17-AAG or to IL-1β. Panel D. Human alveolar epithelial type II cell monolayers were treated with 10µg/ml of 17-AAG for the indicated times prior to the addition of IL-1β for three hours. The amiloride-sensitive fraction of the short-circuit current was measured in Ussing chambers. Control monolayers were not exposed to 17-AAG or to IL-1β. For all panels, the results are the median ± IQRs of at least n=12 monolayers from 4 experiments; *, p < 0.05 from monolayers exposed to IL-1β vehicle; **, p < 0.05 from monolayers exposed to IL-1β.
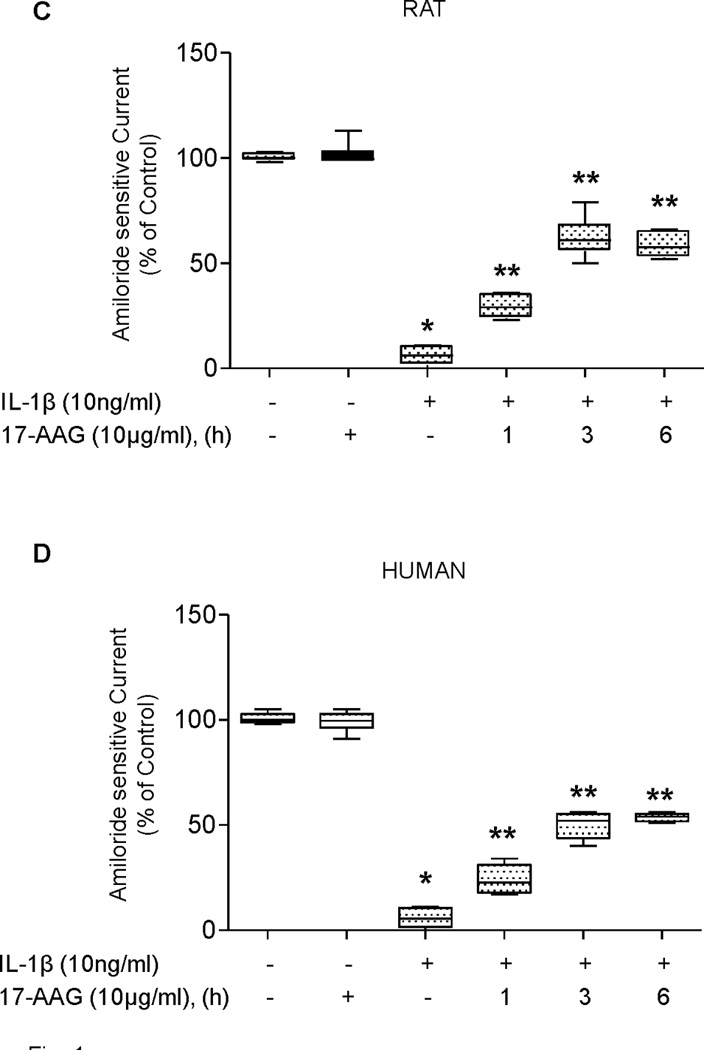
The stress protein response can also be activated in ATII cell monolayers with pharmacological agents, such as 17-allylamino-17-demothoxy-geldanamycin (17–AAG) (11). Thus, we tested whether 17-AAG would attenuate IL-1β-mediated down-regulation of the amiloride-sensitive ENaC current across ATII cell monolayers. Incubation of both rat and human type II alveolar epithelial cell monolayers with 10 µg/ml of 17-AAG showed a 25% protection as early as 1 hour pre-treatment and demonstrated a time-dependent increase in protection of ENaC channel function from IL-1β inhibition with the maximum level of protection of 50–60% of control (Figure 1C&D). These results support the hypothesis that attenuation of IL-1β inhibition of the amiloride-sensitive current is due to SPR activation and not to a non-specific effect of heat.
SPR activation with heat and 17-AAG preserves αENaC mRNA expression from IL-1β-dependent inhibition in rat alveolar epithelial type II cell monolayers
As the amiloride-sensitive fraction of the vectorial current across the alveolar epithelium is largely ENaC-dependent, we examined the effect of prior SPR activation on IL-1β-mediated inhibition of αENaC mRNA expression in ATII cell monolayers. The preservation of αENaC mRNA expression induced by SPR activation followed a similar time course to that observed for the preservation of the amiloride-sensitive ENaC current. αENaC mRNA expression after heat shock and in the presence of IL-1β averaged 75% of the mRNA expression of the non-heat shocked control for time points up to and including 24 hours. However, by 48 hours post-heat shock, this protection was reversed, as IL-1β inhibited αENaC mRNA expression at a level comparable to that seen in cells that were not heat shocked (Figure 2A). Likewise, the preservation of αENaC mRNA expression after 17-AAG treatment and IL-1β stimulation also showed a time-dependent effect. One hour incubation with 17-AAG showed a 10% increase in αENaC mRNA expression in the presence of IL-1β compared to the control (Figure 2B) that continued to increase to 40-60% after three or six hours exposure to of 17-AAG and IL-1β stimulation. Finally, there was a good correlation between the protective effect of SPR induced by heat or 17-AAG on the amiloride-sensitive current and αENaC mRNA expression (Figures 1 and 2).
Figure 2. SPR activation prevents IL-1β-dependent inhibition of αENaC mRNA expression in rat alveolar epithelial type II cell monolayers.
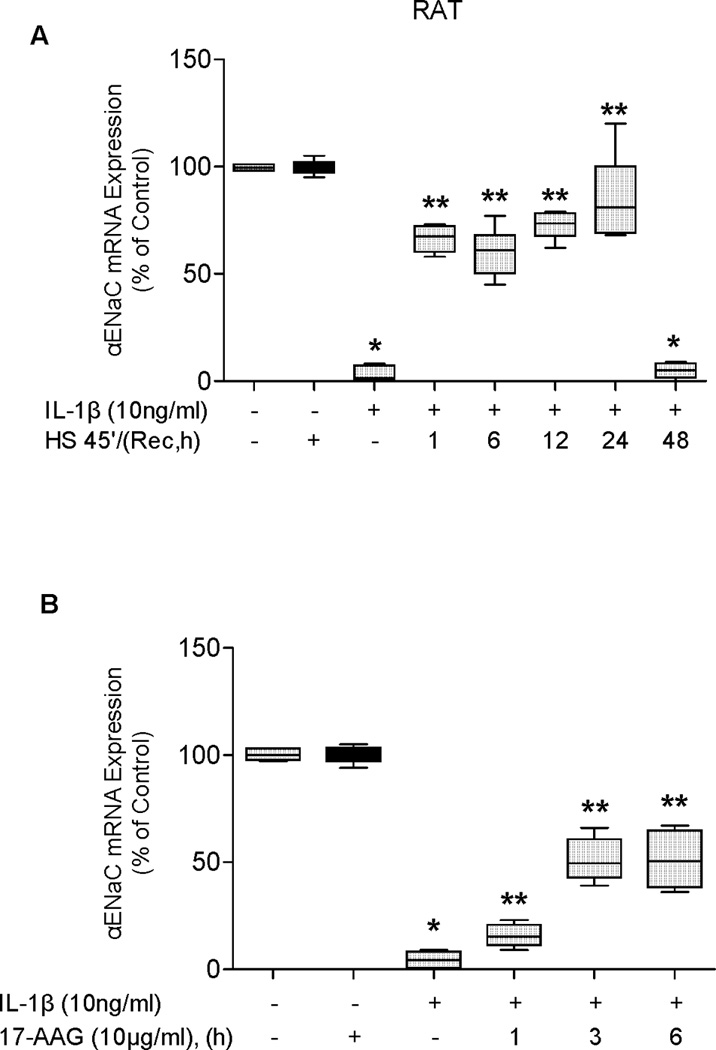
Panel A. Rat alveolar epithelial type II cell monolayers were heat shocked at 43°C and allowed to recover at 37°C for increasing times prior to the addition of IL-1β for three hours, the cells lysed, RNA isolated and and αENaC mRNA expression measured using Real-time PCR. Control monolayers were not subjected to SPR activation nor exposed to IL-1β stimulation. Panel B. Rat type II cell monolayers were treated with 10µg/ml of 17-AAG for the indicated times prior to the addition of IL-1β for three hours, the cells lysed, RNA isolated and αENaC mRNA expression measured by Real-time PCR. Control monolayers were not exposed to 17-AAG or to IL-1β. The results are the median ± IQRs of at least n=12 monolayers from 4 experiments; *, p < 0.05 from monolayers exposed to IL-1β vehicle; **, p < 0.05 from monolayers exposed to IL-1β.
SPR activation with heat and 17-AAG prevents IL-1β-dependent p38 activation in a time-dependent manner in rat alveolar epithelial type II cell monolayers
Our previous studies have shown that SPR activation causes the disruption of Hsp90 binding to its client proteins (9,11). IL-1β-dependent p38 activation is known to signal through both IRAK1 and TAK1, which are both Hsp90 client proteins (25–27). Analysis of the time course after SPR activation showed a time-dependent expression of Hsp70 (Figure 3A) and an attenuation of IL-1β-dependent activation of p38 MAPkinase, as indicated by the inhibition of the phosphorylation of this MAP kinase at 1 and 6, but not at 24 and 48 hours after SPR activation with heat (Figure 3B), but at the 24-hour time point, IL-1β-dependent inhibition of the amiloride-sensitive ENaC current and αENaC mRNA expression was still attenuated (Figure 1A). This result suggests that 24 hours after SPR activation, the IL-1β signaling pathway upstream of p38 activation has been reconstituted and that the inhibition of the effect of IL-1β on ENaC current and mRNA expression involves another cellular mechanism. Likewise, treatment of type II alveolar epithelial cells with 17-AAG shows a time-dependent effect of 17-AAG on p38 activation (Figure 3C) that correlates with a time-dependent preservation of ENaC current and mRNA expression.
Figure 3. SPR activation prevents IL-1β-dependent p38 activation in a time-dependent manner in rat alveolar epithelial type II cell monolayers.
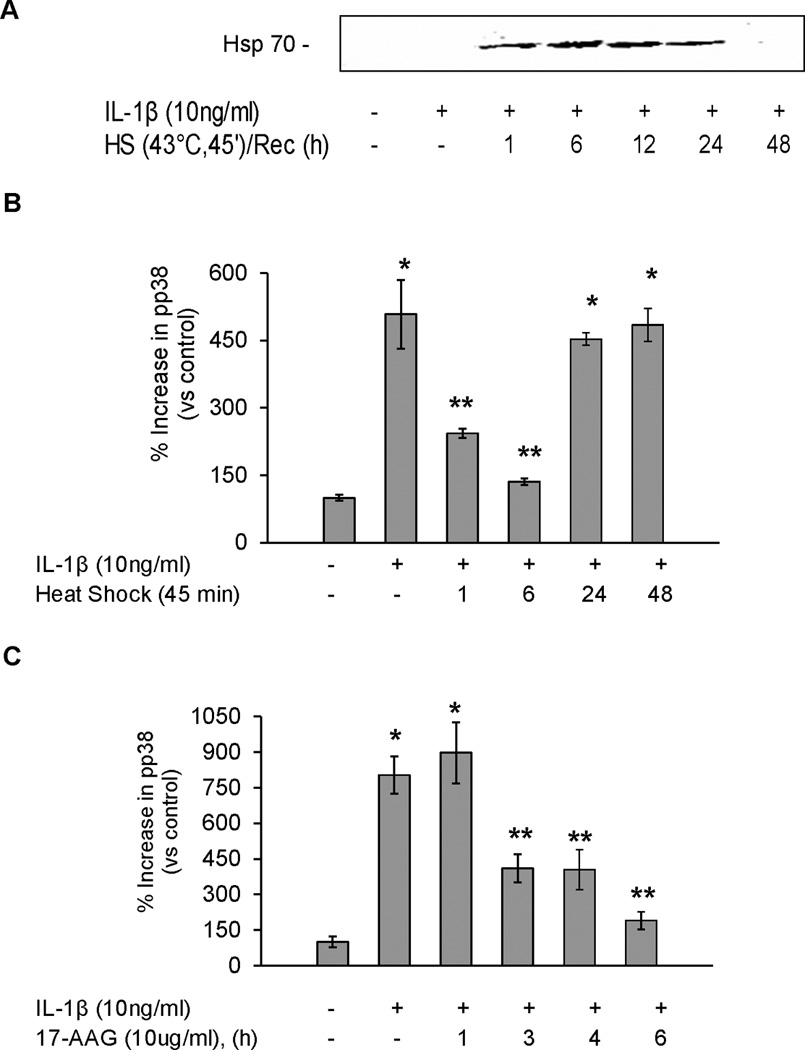
Panel A. Rat alveolar epithelial type II cell monolayers were heat shocked at 43°C and allowed to recover for increasing times at 37°C prior to the addition of IL-1β for 10 minutes, the cells lysed and inducible Hsp70 protein expression analyzed by SDS-PAGE and Western blotting.Panel B. Rat alveolar epithelial type II cell monolayers were heat shocked at 43°C and allowed to recover for increasing times at 37°C prior to the addition of IL-1β for 10 minutes, the cells lysed and changes in phosphorylated p38 MAP kinase were measured by ELISA. Panel C. Rat alveolar epithelial type II cell monolayers were treated with 10µg/ml of 17-AAG for increasing times at 37°C prior to the addition of IL-1β for 10 minutes, the cells lysed and changes in phosphorylated p38 MAP kinase protein analyzed using ELISA (*, p < 0.05 from control; **, p < 0.05 from monolayers exposed to IL-1β alone).
SPR activation with heat and 17-AAG promotes the detergent insolubility of IRAK1 and TAK1 proteins in rat alveolar epithelial type II cell monolayers
Our previous studies have shown that disruption of Hsp90 from its client protein results in detergent insolubility of the client protein (9, 11). Hsp90 binds both IRAK1 and TAK1, two proteins that are upstream of p38 activation in the IL-1β signaling pathway (25–27). Both IRAK1 and TAK1 are shown to be in a Triton X-100 soluble fraction under non-stimulated (Figure 4A&B) and IL-1β stimulated conditions (data not shown). SPR activation with heat followed by one-hour recovery resulted in the Triton X-100 insolubility of both IRAK1 and TAK1 proteins (Figure 4A&B). Treatment with MG132, a proteasome inhibitor, had no effect on the solubility of TAK1 or IRAK1. Both IRAK1 and TAK1 protein were found in a detergent soluble fraction 24 hours post-heat shock, a time point at which IL-1β dependent p38 is activation is restored. Treatment of alveolar epithelial type II cell monolayers with 17-AAG also showed a time-dependent increase in IRAK1 and TAK1 TritonX-100 insolubility (Figure 4C&D). Lastly, co-immunoprecipitation of IRAK1/Hsp90 was performed from membrane-enriched fractions. IRAK1 was found to bind Hsp90 at basal conditions (Figure 5) as previously reported (3). After SPR activation with heat, there was a disruption of the binding of Hsp90 to its client protein IRAK1 (Figure 5).
Figure 4. SPR activation promotes the insolubility of IRAK1 and TAK1 proteins in rat alveolar epithelial type II cell monolayers.
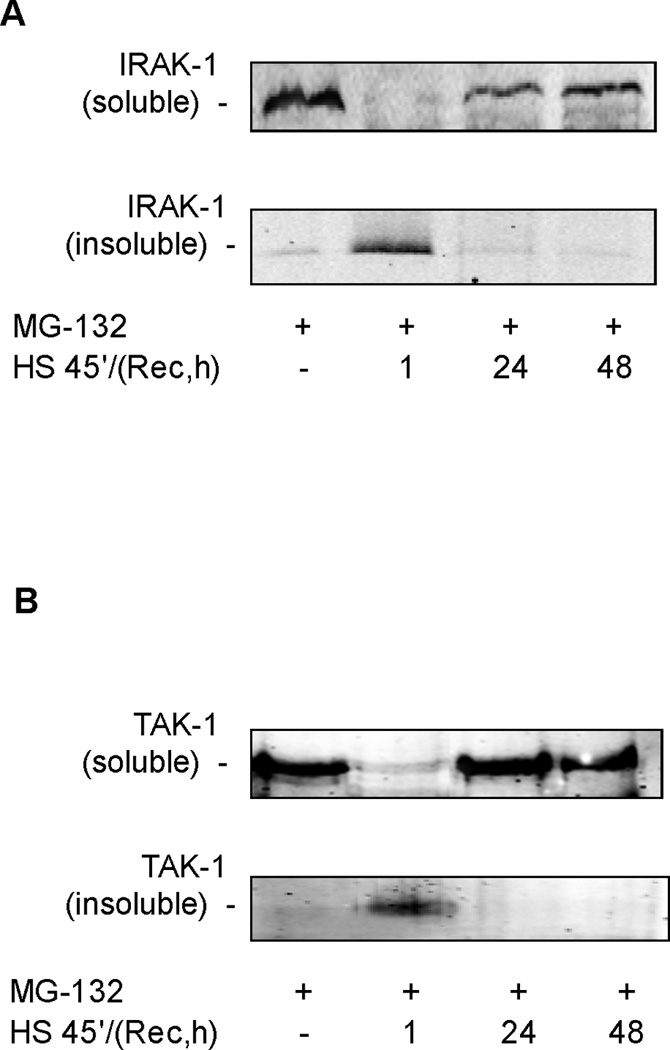
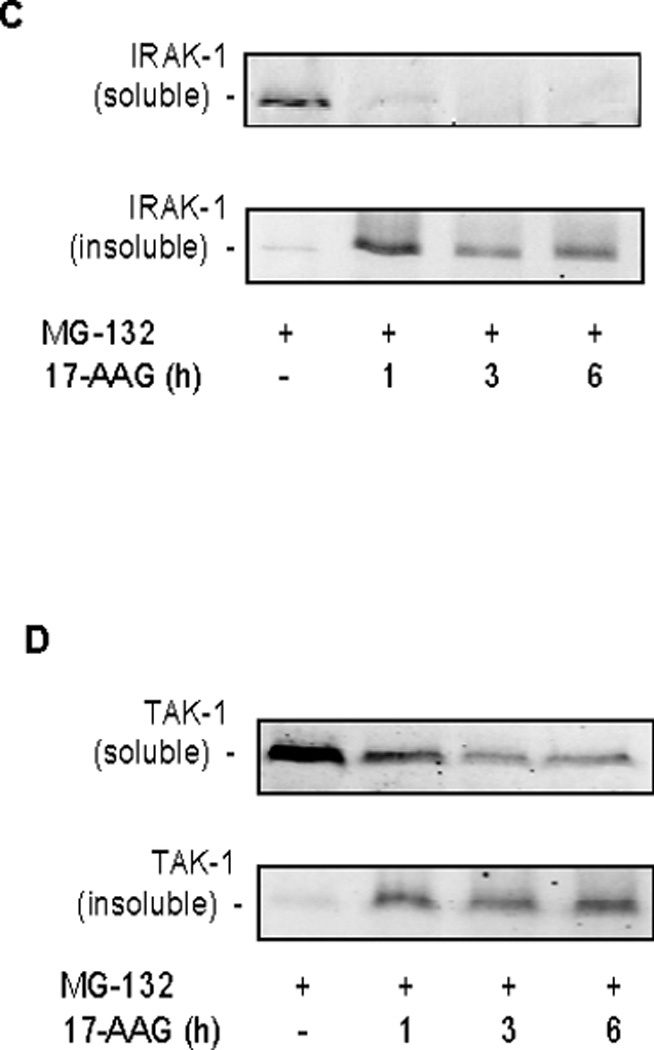
Panel A. Rat alveolar epithelial type II cell monolayers were heat shocked at 43°C and allowed to recover for 1, 24 and 48 hours at 37°C prior to differential detergent cell lysis using Triton X-100 and analysis of IRAK1 protein expression by SDS-PAGE and Western blotting. Panel B. Rat alveolar epithelial type II cell monolayers were heat shocked at 43°C and allowed to recover for 1, 24 and 48 hours at 37°C prior to differential detergent cell lysis using Triton X-100 and analysis of TAK1 protein expression by SDS-PAGE and Western blotting. Panel C. Rat alveolar epithelial type II cell monolayers were treated with 10µg/ml of 17-AAG for increasing times at 37°C prior to differential detergent cell lysis using Triton X-100 and analysis of IRAK1 protein expression by SDS-PAGE and Western blotting. Panel D. Rat alveolar epithelial type II cell monolayers were treated with 10µg/ml of 17-AAG for increasing times at 37°C prior to differential detergent cell lysis using Triton X-100 and analysis of TAK1 protein expression by SDS-PAGE and Western blotting.
Figure 5. SPR activation disrupts IRAK1/Hsp90 protein binding in rat alveolar epithelial type II cell monolayers.
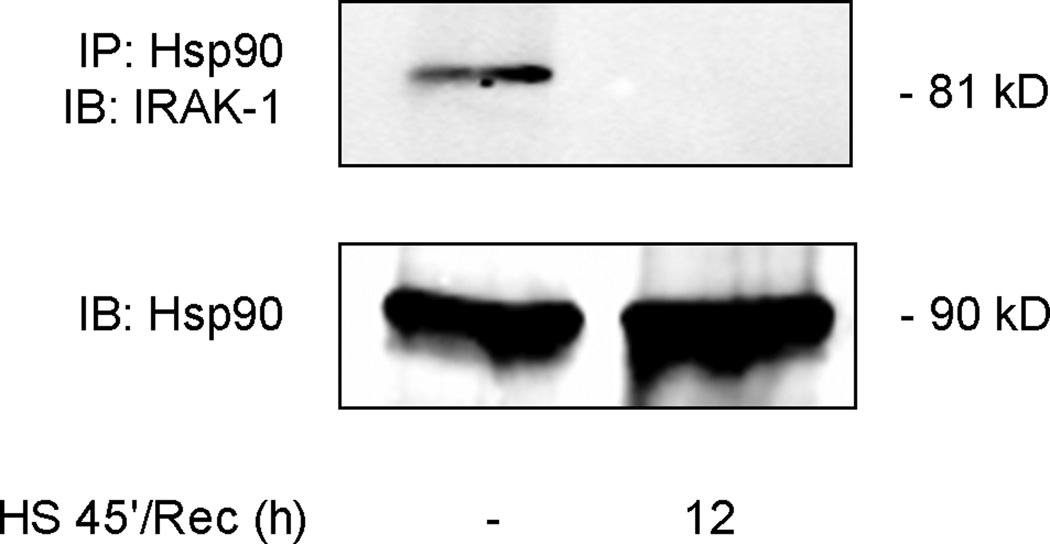
Rat alveolar epithelial type II cell monolayers were heat shocked at 43°C and allowed to recover for 12 hours at 37°C prior to hypotonic cell lysis, isolation of a membrane enriched fraction, immunoprecipitation using a polyclonal antibody to Hsp90 and analysis of IRAK1 and Hsp90 protein expression by SDS-PAGE and Western blotting.
Overexpression of Hsp70 and inhibition of SPR-induced Hsp70 expression inversely affect αENaC mRNA expression in rat alveolar epithelial type II cell monolayers
Our previous studies have shown a role for intracellular inducible Hsp70 in attenuating pro-inflammatory signaling pathways (9). Due to the fact that 24 hours after SPR activation IL-1β-dependent p38 activation did not result in a down-regulation of αENaC mRNA, the role of inducible Hsp70 expression in contributing to this effect was examined. Hsp70 was overexpressed in rat alveolar epithelial type II cell monolayers using a recombinant Hsp70 adenovirus in the absence of SPR activation (Figure 6A). In the presence of Hsp70 overexpression in ATII cell monolayers, IL-1β phosphorylated p38 MAP kinase (Figure 6A). However, overexpression of Hsp70 resulted in the preservation of αENaC mRNA expression in the presence of IL-1β stimulation (Figure 6B). Conversely, treatment of the type II alveolar epithelial cell monolayers with Hsp70 siRNA after heat shock caused the inhibition of inducible Hsp70 expression and restored the inhibitory effect of IL-1β on αENaC mRNA expression (Figure 6C&D).
Figure 6. Overexpression of Hsp70 protein and inhibition of SPR-dependent Hsp70 protein expression inversely affect αENaC mRNA expression in rat alveolar epithelial type II cell monolayers.
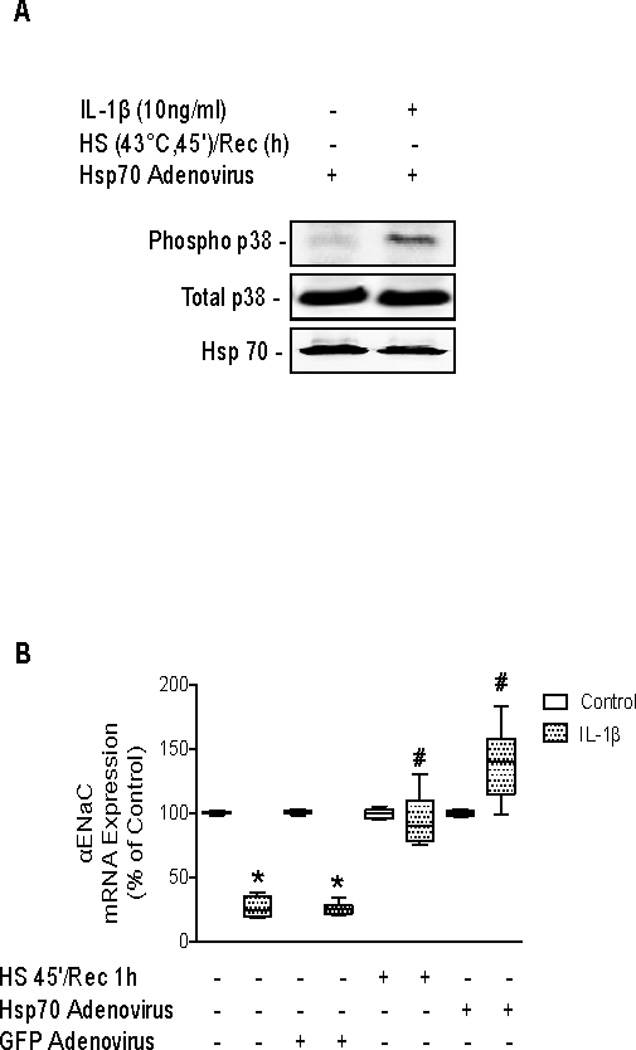
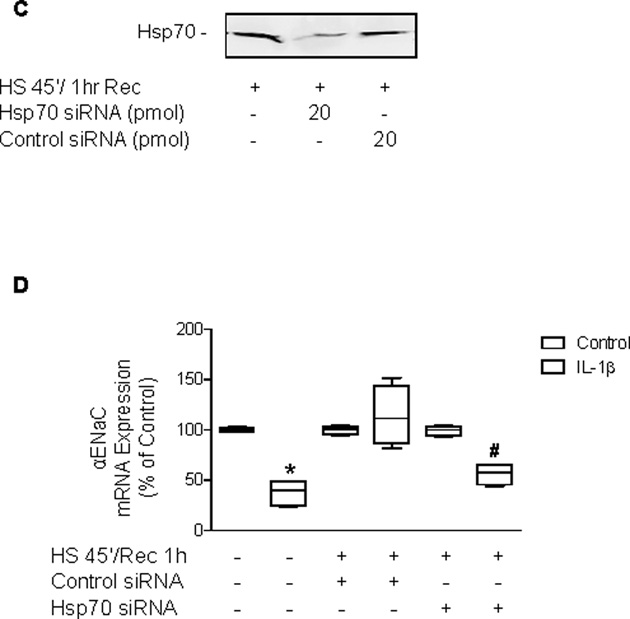
Panel A. Rat alveolar epithelial type II cell monolayers were infected with a recombinant Hsp70 expressing adenovirus for 48 hours prior to the addition of IL-1β for 10 minutes. The cells were lysed and phosphorylated and total p38 MAP kinase protein expression analyzed on the same blot at the same time by SDS-PAGE and Western blotting using the Odyssey Infrared Imaging System from LI-COR®. One representative blot is shown. Two additional experiments showed comparable results. Panel B. Rat alveolar epithelial type II cell monolayers were infected with a recombinant Hsp70 expressing adenovirus or a control GFP adenovirus for 48 hours prior to the addition of IL-1β for 3 hours. Cells were lysed, RNA isolated and Real-time PCR performed to determine αENaC mRNA expression. The results are the median ± IQRs of at least n=9 monolayers from 3 experiments; *, p < 0.05 from monolayers exposed to IL-1β vehicle; #, p < 0.05 from monolayers exposed to IL-1β. Panel C. Rat alveolar epithelial type II cell monolayers were heat shocked and recovered at 37°C for 1 hour before transfection with siRNA to inducible Hsp70 or a control siRNA for 24 hours. Cells were lysed and Hsp70 protein expression was determined by SDS-PAGE and Western blotting. Panel D. Rat alveolar epithelial type II cell monolayers were heat shocked and recovered at 37°C for 1 hour before transfection with siRNA to inducible Hsp70 or a control siRNA for 24 hours, the last three hours included the addition of IL-1β. Cells were lysed, RNA isolated and Real-time PCR performed to determine αENaC mRNA expression. The results are the median ± IQRs of at least n=9 monolayers from 3 experiments; *, p < 0.05 from monolayers exposed to IL-1β vehicle; #, p < 0.05 from monolayers heat shocked, transfected with control siRNA and exposed to IL-1β.
DISCUSSION
In this study, we demonstrate that in rat and human alveolar epithelial type II cells, (a) prior SPR activation prevents the IL-1β-mediated inhibition of the amiloride-sensitive short-circuit current and αENaC mRNA expression; (b) SPR-mediated attenuation of IL-1β-dependent p38 MAP kinase activation is associated with the reversible detergent insolubility of IRAK-1 and TAK-1 (both Hsp90 client proteins); (c) detergent solubility of IRAK1 and TAK1 24 hours post-heat stress correlates with p38 MAP kinase activation, but not with IL-1β-dependent inhibition of αENaC mRNA expression and channel function; (d) inducible Hsp70 plays an important role in SPR-mediated prolonged attenuation of IL-1β-dependent inhibition of αENaC mRNA expression.
Activation of the SPR has been shown to attenuate cell and organ response to inflammatory stimuli induced by several pathological conditions (15–19,28). For example, we previously reported that prior SPR activation restored the cAMP-mediated stimulation of the vectorial alveolar fluid transport (2) that was completely inhibited after severe hemorrhagic shock by an IL-1β-dependent mechanism (29). In further work, we also found that IL-β inhibited ENaC gene and protein expression and channel function via a p38 MAP kinase-dependent mechanism (6). However, the molecular mechanism(s) by which SPR activation prevents the inhibitory effect of IL-1β on ENaC expression and function were still unknown.
In the present study, we found that SPR activated by heat or pharmacologically by 17-AAG, an Hsp90 inhibitor that is known to activate a heat shock response (and induces up-regulation of Hps70 in ATII cells (2), prevented the IL-1β-mediated inhibition of the amiloride-sensitive short-circuit current and αENaC mRNA expression in rat and human alveolar epithelial type II cell monolayers This indicates that the SPR effect was not due to a non-specific effect of heat. We have previously reported that there is a good correlation between the IL-1β-mediated inhibition of αENaC mRNA, plasma membrane protein expression and ENaC channel function at the cell membrane of ATII cells (6). What are the mechanisms by which SPR preserves αENaC mRNA expression and function in ATII cells after exposure to IL-1β? There are several mechanisms that can explain this SPR effect. First, SPR activation has been shown to cause the dissociation of Hsp90 from its client proteins resulting in the temporary inhibition of the function of the particular client protein, its detergent insolubility and thus its degradation via the proteasomal pathway (13,25-27). We have previously reported that SPR activation inhibits several inflammatory signaling pathways via this mechanism (9–11). Importantly, once the client protein becomes detergent insoluble, the enzymatic activity of this protein is inhibited until the protein becomes detergent soluble again provided that it is not fully degraded by the proteasomal pathway. These results are consistent with a mechanism that we have previously reported in alveolar epithelial cells for the Hsp90 client protein IkappaB kinase (11).
Several proteins that are upstream of p38 MAP kinase in the IL-1β-dependent signaling pathway, such as IRAK-1 and TAK-1, have recently been shown to be client proteins of Hsp90 (17,25). We found that activation of SPR using heat, which disrupts Hsp90 binding to IRAK-1, results in the immediate detergent insolubility of both IRAK-1 and TAK1 with the concomitant inhibition of p38 MAP kinase phosphorylation and correlates with maximal preservation of αENaC mRNA expression and amiloride-sensitive current. In contrast, SPR activation using 17-AAG instead of heat, shows a slower kinetics of attenuation of IL-1β-dependent p38 MAP kinase activation as a one-hour treatment with 17-AAG prior to exposure to IL-1β inhibits p38 MAP kinase phosphorylation by 50% compared to the non heat shocked cell monolayers treated with IL-1β. This reduction in p38 MAP phosphorylation correlates with a 50% preservation of both αENaC mRNA expression and amiloride-sensitive current observed in the cell monolayers treated with 17-AAG prior to exposure to IL-1β. Interestingly, our results indicate that a one-hour treatment with 17-AAG results in the complete insolubility of IRAK-1 while only a fraction of TAK1 is insoluble under these conditions. Our data showing partial insolubility of TAK1 is consistent with the work by Liu et al. (26) that reported that Hsp90 binding to TAK1 is required for its folding and stability, but is released when TAK1 binds to TAB1. Thus, the soluble fraction of TAK1 likely represents a stable form of TAK1 that does not form a protein complex with Hsp90. By 24 hours post-heat shock, the IL-1β signaling pathway upstream of p38 MAP kinase is restored, as both IRAK-1 and TAK1 are found in the detergent soluble fraction and IL-1β phosphorylates p38 MAP kinase in ATII cell monolayers. However, IL-1β does not inhibit αENaC mRNA expression and amiloride-sensitive current in ATII cell monolayers up to 48 hours after SPR activation, indicating that other mechanism(s) are implicated in the inhibition of IL-1β cell signaling by SPR in ATII cells.
What are the mechanism(s) by which SPR preserves αENaC mRNA expression and function in the presence of p38 MAP kinase activation? It has previously been shown that heat stress attenuates IL-1β signaling by dephosphorylating p38 MAP kinase through the induction of MAP kinase phosphatase (MKP-1) expression (30). MKP-1 is indeed upregulated in response to heat shock via transcriptional and post-transcriptional mechanisms (31,32). In addition, studies by Lee and colleagues reported that after heat shock, Hsp70 binds to MKP-1, increasing the levels of phosphorylated MKP-1 and accelerating MAP kinase inactivation (33). Thus, the simplest model explaining our results at the 24-hour time point after SPR activation would be that once the p38 MAP kinase is phosphorylated and translocates to the nucleus, it is dephosphorylated by MKP-1 allowing transcription of αENaC. For example, Nimah et al. have shown in THP-1 cells, a monocyte cell line, that there is a significant increase in nuclear MKP-1 protein expression 24 hours after SPR activation that is associated with an absence of p38 phosphorylation by E. coli endotoxin in THP-1 cells (34). In contrast, our results demonstrate that IL-1β phosphorylates p38 MAP kinase 24 hours after SPR activation with the same intensity as in non-heat shocked cell monolayers. Thus, it is unlikely that the induction of nuclear expression of MKP-1 protein would fully explain the inhibition of IL-1β signaling during the late phase of SPR in alveolar epithelial type II cell monolayers. We have previously reported that the induction of Hsp70 plays an important role in the prolonged SPR-mediated inhibition of the Stat1-iNOS pathway in alveolar macrophages (9). In the present study, we found that Hsp70 was expressed up to 24 hours after SPR activation in ATII cell monolayers. Furthermore, adenoviral gene transfer of Hsp70 prevented the inhibitory effect of on ENaC mRNA expression and channel function without inhibiting p38 MAP kinase activation by IL-1β̃ These results are consistent with those from Shi et al. who reported in RAW cells that over-expression of Hsp70 had no inhibitory effect on p38 MAP kinase activation (35). Conversely, the inhibition of Hsp70 protein expression by siRNA in heat shocked ATII cells prevented the SPR-mediated effect on inhibition of αENaC gene expression by IL-1β. What mechanism(s) could then explain the effect of Hsp70 protein overexpression on the attenuation of IL-1β-dependent inhibition of αENaC gene expression in ATII cells? Published work from Rubenstein’s laboratory has shown that overexpression of Hsp70, but not Hsc70, increases mENaC channel expression and activity at the plasma membrane of oocytes (14). Other investigators have reported comparable results for the effect of HSP70 on CFTR expression and function at the plasma membrane in lung epithelial cells (36). Taken together, these published studies as well as our present results suggest that the induction of Hsp70 following SPR activation preserves αENaC mRNA levels and increases targeting of ion channels to the plasma membrane of lung epithelial cells without modulating the cell signaling pathways of inflammatory mediators, such as IL-1β. However, further studies are needed to understand the mechanism(s) by which HSP70 modulates expression and function of ion channels in lung epithelial cells.
Clinical implications
We had previously reported that the release of extracellular inducible Hsp70, a marker of an intracellular stress protein response, in the pulmonary edema fluid of patients with ALI preserves alveolar fluid clearance and is associated with survival (23). The present study provides a new potentially clinically relevant mechanism to explain the protective effect of SPR activation on the vectorial alveolar epithelial fluid transport after onset of ALI because IL-1β is one of the most biologically active cytokines in the distal airspaces of ALI patients (3–5) and causes increased lung vascular permeability and inhibits the vectorial fluid transport across the distal lung epithelium (6). However, it should be pointed out that there is a large body of evidence that IL-1 beta signaling is only one of multiple signaling pathways that can negatively affect the removal of the pulmonary edema fluid by the alveolar epithelium after onset of ALI in humans (reviewed in 37). The multiplicity of the signaling pathways involved in inhibiting the function of the alveolar epithelial ion channels may explain in part why most of the phase 3 clinical trials that included a single drug to treat ALI patients have produced negative results.
In summary, our studies demonstrate that SPR activation significantly attenuates IL-1β inhibition of αENaC mRNA expression and the amiloride-sensitive current via multiple mechanisms. It has previously been shown that heat shock increases the nuclear expression of MKP-1, a phosphatase that inhibits the nuclear activity of p38 MAP kinase. Herein, we found that SPR activation also attenuates IL-1β-dependent p38 MAP kinase activation by disrupting the binding of IRAK-1 and TAK1 to Hsp90 resulting in their detergent insolubility and targeting them for degradation via the proteasomal pathway. Furthermore, SPR activation also causes a prolonged attenuation of p38-dependent inhibition of αENaC mRNA expression via the expression of the inducible Hsp70. These results provide additional understanding about the mechanisms by which SPR activation is associated with better survival in patients with acute lung injury or severe trauma (23, 38).
Acknowledgments
Funding Support: Academic Senate Grant UCSF (MBH); American Lung Association Fellowship Award and NIH T32 Training Grant (JR); NIH RO1 GM62188 (JFP)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
MBH and JR contributed equally to this manuscript
REFERENCES
- 1.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 2.Pittet JF, Lu LN, Geiser T, Lee H, Matthay MA, Welch WJ. Stress preconditioning attenuates oxidative injury to the alveolar epithelium of the lung following haemorrhage in rats. J Physiol. 2002;538:583–597. doi: 10.1113/jphysiol.2001.013102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olman MA, White KE, Ware LB, Simmons WL, Benveniste EN, Zhu S, Pugin J, Matthay MA. Pulmonary edema fluid from patients with early lung injury stimulates fibroblast proliferation through IL-1 beta-induced IL-6 expression. J Immunol. 2004;172:2668–2677. doi: 10.4049/jimmunol.172.4.2668. [DOI] [PubMed] [Google Scholar]
- 4.Pugin J, Ricou B, Steinberg KP, Suter PM, Martin TR. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1. Am J Respir Crit Care Med. 1996;153:1850–1856. doi: 10.1164/ajrccm.153.6.8665045. [DOI] [PubMed] [Google Scholar]
- 5.Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med. 1999;27:304–312. doi: 10.1097/00003246-199902000-00036. [DOI] [PubMed] [Google Scholar]
- 6.Roux J, Kawakatsu H, Gartland B, Pespeni M, Sheppard D, Matthay MA, Canessa CM, Pittet JF. Interleukin-1 beta decreases expression of the epithelial sodium channel alpha-subunit in alveolar epithelial cells via a p38 MAPK-dependent signaling pathway. J Biol Chem. 2005;280:18579–18589. doi: 10.1074/jbc.M410561200. [DOI] [PubMed] [Google Scholar]
- 7.Welch WJ. Heat shock proteins functioning as molecular chaperones: their roles in normal and stressed cells. Philos Trans R Soc Lond B Biol Sci. 1993;339:327–333. doi: 10.1098/rstb.1993.0031. [DOI] [PubMed] [Google Scholar]
- 8.Minowada G, Welch WJ. Clinical implications of the stress response. J Clin Invest. 1995;95:3–12. doi: 10.1172/JCI117655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Howard M, Roux J, Lee H, Miyazawa B, Lee J, Gartland B, Howard AJ, Matthay MA, Carles M, Pittet JF. Activation of the stress protein response inhibits the STAT1 signaling pathway and iNOS function in alveolar macrophages: role of Hsp90 and Hsp70. Thorax. 2010;65:346–53. doi: 10.1136/thx.2008.101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pespeni MH, Hodnett M, Abayasiriwardana KS, Roux J, Howard M, Broaddus VC, Pittet JF. Sensitization of mesothelioma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by heat stress via the inhibition of the 3-phosphoinositide-dependent kinase 1/Akt pathway. Cancer Res. 2007;67:2865–2871. doi: 10.1158/0008-5472.CAN-06-3871. [DOI] [PubMed] [Google Scholar]
- 11.Pittet JF, Lee H, Pespeni M, O'Mahony A, Roux J, Welch WJ. Stress-induced inhibition of the NF-kappaB signaling pathway results from the insolubilization of the IkappaB kinase complex following its dissociation from heat shock protein 90. J Immunol. 2005;174:384–394. doi: 10.4049/jimmunol.174.1.384. [DOI] [PubMed] [Google Scholar]
- 12.Zhao R, Davey M, Hsu YC, Kaplanek P, Tong A, Parsons AB, Krogan N, Cagney G, Mai D, Greenblatt J, Boone C, Emili A, Houry WA. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120:715–727. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 13.Kamal A, Boehm MF, Burrows FJ. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med. 2004;10:283–290. doi: 10.1016/j.molmed.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldfarb SB, Kashlan OB, Watkins JN, Suaud L, Yan W, Kleyman TR, Rubenstein RC. Differential effects of Hsc70 and Hsp70 on the intracellular trafficking and functional expression of epithelial sodium channels. Proc Natl Acad Sci USA. 2006;103:5817–5822. doi: 10.1073/pnas.0507903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K, Kim W, Rhee JE, Jo YH, Lee JH, Kim KS, Kwon WY, Suh GJ, Lee CC, Singer AJ. Induced hypothermia attenuates the acute lung injury in hemorrhagic shock. J Trauma. 2010;68:373–381. doi: 10.1097/TA.0b013e3181a73eea. [DOI] [PubMed] [Google Scholar]
- 16.Koh Y, Lim CM, Kim MJ, Shim TS, Lee SD, Kim WS, Kim DS, Kim WD. Heat shock response decreases endotoxin-induced acute lung injury in rats. Respirology. 1999;4:325–330. doi: 10.1046/j.1440-1843.1999.00200.x. [DOI] [PubMed] [Google Scholar]
- 17.McCormick PH, Chen G, Tlerney S, Kelly CJ, Bouchier-Hayes DJ. Clinically relevant thermal preconditioning attenuates ischemia-reperfusion injury. J Surg Res. 2003;109:24–30. doi: 10.1016/s0022-4804(02)00035-5. [DOI] [PubMed] [Google Scholar]
- 18.Ribeiro SP, Villar J, Downey GP, Edelson JD, Slutsky AS. Sodium arsenite induces heat shock protein-72 kilodalton expression in the lungs and protects rats against sepsis. Crit Care Med. 1994;22:922–929. doi: 10.1097/00003246-199406000-00008. [DOI] [PubMed] [Google Scholar]
- 19.Villar J, Ribeiro SP, Mullen JB, Kuliszewski M, Post M, Slutsky AS. Induction of the heat shock response reduces mortality rate and organ damage in a sepsis-induced acute lung injury model. Crit Care Med. 1994;22:914–921. [PubMed] [Google Scholar]
- 20.Chatterjee A, Dimitropoulou C, Drakopanayiotakis F, Antonova G, Snead C, Cannon J, Venema RC, Catravas JD. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. Am J Respir Crit Care Med. 2007;176:667–675. doi: 10.1164/rccm.200702-291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiratsuka M, Yano M, Mora BN, Nagahiro I, Cooper JD, Patterson GA. Heat shock pretreatment protects pulmonary isografts from subsequent ischemia-reperfusion injury. J Heart Lung Transplant. 1998;17:1238–1246. [PubMed] [Google Scholar]
- 22.Weiss YG, Maloyan A, Tazelaar J, Raj N, Deutschman CS. Adenoviral transfer of HSP-70 into pulmonary epithelium ameliorates experimental acute respiratory distress syndrome. J Clin Invest. 2002;110:801–806. doi: 10.1172/JCI15888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganter MT, Ware LB, Howard M, Roux J, Gartland B, Matthay MA, Fleshner M, Pittet JF. Extracellular heat shock protein 72 is a marker of the stress protein response in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;291:L354–361. doi: 10.1152/ajplung.00405.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roux J, Carles M, Koh H, Goolaerts A, Ganter MT, Chesebro BB, Howard M, Houseman BT, Finkbeiner W, Shokat KM, Paquet AC, Matthay MA, Pittet JF. Transforming growth factor beta1 inhibits cystic fibrosis transmembrane conductance regulator-dependent cAMP-stimulated alveolar epithelial fluid transport via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem. 2010;285:4278–4290. doi: 10.1074/jbc.M109.036731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Nardo D, Masendycz P, Ho S, Cross M, Fleetwood AJ, Reynolds EC, Hamilton JA, Scholz GM. A central role for the Hsp90.Cdc37 molecular chaperone module in interleukin-1 receptor-associated-kinase-dependent signaling by toll-like receptors. J Biol Chem. 2005;280:9813–9822. doi: 10.1074/jbc.M409745200. [DOI] [PubMed] [Google Scholar]
- 26.Liu XY, Seh CC, Cheung PC. HSP90 is required for TAK1 stability but not for its activation in the pro-inflammatory signaling pathway. FEBS Lett. 2008;582:4023–4031. doi: 10.1016/j.febslet.2008.10.053. [DOI] [PubMed] [Google Scholar]
- 27.Shi L, Zhang Z, Fang S, Xu J, Liu J, Shen J, Fang F, Luo L, Yin Z. Heat shock protein 90 (Hsp90) regulates the stability of transforming growth factor beta-activated kinase 1 (TAK1) in interleukin-1 beta-induced cell signaling. Mol Immunol. 2009;46:541–550. doi: 10.1016/j.molimm.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 28.Godzich M, Hodnett M, Frank JA, Su G, Pespeni M, Angel A, Howard MB, Matthay MA, Pittet JF. Activation of the stress protein response prevents the development of pulmonary edema by inhibiting VEGF cell signaling in a model of lung ischemia-reperfusion injury in rats. FASEB J. 2006;20:1519–1521. doi: 10.1096/fj.05-4708fje. [DOI] [PubMed] [Google Scholar]
- 29.Laffon M, Lu LN, Modelska K, Matthay MA, Pittet JF. Alpha-adrenergic blockade restores normal fluid transport capacity of alveolar epithelium after hemorrhagic shock. Am J Physiol Lung. 1999;1277:L760–L768. doi: 10.1152/ajplung.1999.277.4.L760. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Liu Y. Regulation of innate immune response by MAP kinase phosphatase-1. Cell Signal. 2007;19:1372–1382. doi: 10.1016/j.cellsig.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanlorenzo L, Zhao B, Spight D, Denenberg AG, Page K, Wong HR, Shanley TP. Heat shock inhibition of lipopolysaccharide-mediated tumor necrosis factor expression is associated with nuclear induction of MKP-1 and inhibition of mitogen-activated protein kinase activation. Crit Care Med. 2004;32:2284–2292. doi: 10.1097/01.ccm.0000145580.96994.c9. [DOI] [PubMed] [Google Scholar]
- 32.Wong HR, Dunsmore KE, Page K, Shanley TP. Heat shock-mediated regulation of MKP-1. Am J Physiol Cell Physiol. 2005;289:C1152–C1158. doi: 10.1152/ajpcell.00138.2005. [DOI] [PubMed] [Google Scholar]
- 33.Lee KH, Lee CT, Kim YW, Han SK, Shim YS, Yoo CG. Preheating accelerates mitogen-activated protein (MAP) kinase inactivation post-heat shock via a heat shock protein 70-mediated increase in phosphorylated MAP kinase phosphatase-1. J Biol Chem. 2005;280:13179–13186. doi: 10.1074/jbc.M410059200. [DOI] [PubMed] [Google Scholar]
- 34.Nimah M, Zhao B, Denenberg AG, Bueno O, Molkentin J, Wong HR, Shanley TP. Contribution of MKP-1 regulation of p38 to endotoxin tolerance. Shock. 2005;23:80–87. doi: 10.1097/01.shk.0000145206.28812.60. [DOI] [PubMed] [Google Scholar]
- 35.Shi Y, Tu Z, Tang D, Zhang H, Liu M, Wang K, Calderwood SK, Xiao X. The inhibition of LPS-induced production of inflammatory cytokines by HSP70 involves inactivation of the NF-kappaB pathway but not the MAPK pathways. Shock. 2006;26:277–284. doi: 10.1097/01.shk.0000223134.17877.ad. [DOI] [PubMed] [Google Scholar]
- 36.Choo-Kang LR, Zeitlin PL. Induction of HSP70 promotes DeltaF508 CFTR trafficking. Am J Physiol Lung Cell Mol Physiol. 2001;281:L58–L68. doi: 10.1152/ajplung.2001.281.1.L58. [DOI] [PubMed] [Google Scholar]
- 37.Matthay MA, Ware LB, Zimmerman A. The acute respiratory distress syndrome. J Clin invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pittet JF, Lee H, Morabito D, Howard MB, Welch WJ, Mackersie RC. Serum levels of Hsp 72 measured early after trauma correlate with survival. J Trauma. 2002;52:611–617. doi: 10.1097/00005373-200204000-00001. [DOI] [PubMed] [Google Scholar]