

Abstract
Altered mechanical loading of the heart leads to hypertrophy, decompensated heart failure and fatal arrhythmias. However, the molecular mechanisms that link mechanical and electrical dysfunction remain poorly understood. Growing evidence suggest that ventricular electrical remodeling (VER) is a process that can be induced by altered mechanical stress, creating persistent electrophysiological changes that predispose the heart to life-threatening arrhythmias. While VER is clearly a physiological property of the human heart, as evidenced by “T wave memory”, it is also thought to occur in a variety of pathological states associated with altered ventricular activation such as bundle branch block, myocardial infarction, and cardiac pacing. Animal models that are currently being used for investigating stretch-induced VER have significant limitations. The zebrafish has recently emerged as an attractive animal model for studying cardiovascular disease and could overcome some of these limitations. Owing to its extensively sequenced genome, high conservation of gene function, and the comprehensive genetic resources that are available in this model, the zebrafish may provide new insights into the molecular mechanisms that drive detrimental electrical remodeling in response to stretch. Here, we have established a zebrafish model to study mechano-electrical feedback in the heart, which combines efficient genetic manipulation with high-precision stretch and high-resolution electrophysiology. In this model, only ninety minutes of ventricular stretch caused VER and recapitulated key features of VER found previously in the mammalian heart. Our data suggest that the zebrafish model is a powerful platform for investigating the molecular mechanisms underlying mechano-electrical feedback and VER in the heart.
Keywords: zebrafish cardiac electrophysiology, electrical remodeling, stretch-induced VER, arrhythmias, mechano-electrical feedback
1. Introduction
1.1. Stretch-induced ventricular electrical remodeling in the heart
Ventricular electrical remodeling (VER) is a persistent change in the electrophysiological properties of myocardium in response to a change in heart rate or activation sequence (Jeyaraj et al., 2010). Abnormal ventricular activation is commonly associated with a variety of cardiac pathologies including conduction system disease, myocardial infarction, hypertrophy and heart failure and is induced by ventricular pacing. The alteration of ventricular activation by pacing causes an inversion of the T wave of the electrocardiogram which was first described by Chatterjee et al. (1969). This T wave abnormality was later termed “T-wave memory” by Rosenbaum et al. (1982), because it persists long after the cessation of pacing and dissipates at a rate that depends on the history of prior pacing (Wecke et al., 2005). Importantly, T-wave memory exhibits accumulation, i.e. the duration and the degree of the T wave abnormality observed is dependent on the duration and degree of abnormal activation (del Balzo and Rosen, 1992). Because electrical excitation and mechanical contraction are tightly coupled (Bers, 2002), any alteration of electrical activation changes the temporal and spatial properties of contraction, leading to mechanical dyssynchrony (Bank et al., 2011; Gjesdal et al., 2011). Therefore, it was hypothesized that an alteration of myocardial mechanics is the underlying cause of cardiac memory. Sosunov et al. (2008) examined this possibility in a Langendorff-perfused rabbit heart. They reported that stretching just a single point on the epicardium induced a T-wave alteration which was similar to that observed in cardiac memory. This result clearly supports the hypothesis that altered mechanical activity, rather than the direction of current flow per se, is a key determinant of cardiac memory. Recently, using an in vivo dog model of pacing-induced VER, we found that periods of mechanical stretch are in fact a mechanism for triggering VER and T wave memory (Jeyaraj et al., 2007). Importantly, action potential durations were markedly increased in high-strain regions and unchanged in low-strain regions, accounting for the inversion of the T wave.
Taken together, these reports suggest that tissue stretch is a prerequisite for T-wave memory and that long-term alteration of ventricular activation causes persistent electrical remodeling via a mechano-electrical feedback mechanism (Marrus and Nerbonne, 2008). It is important to point out that in this context, VER produces persistent electrophysiological changes, such as action potential prolongation, which outlast the period of applied stretch. This should be distinguished from transient mechano-electrical effects which have been studied extensively and are only present as long as cardiac muscle is actively stretched (Iribe et al., 2009; Kohl et al., 2006; Nishimura et al., 2006; Nishimura et al., 2008).
1.2. Clinical consequences of ventricular electrical remodeling
Results from both randomized clinical trials and smaller mechanistic studies have suggested that long-term ventricular pacing has significant adverse effects, ranging from impaired mechanical function (Tantengco et al., 2001) to increased risk of hospitalization, new or worsened heart failure, and death (Sweeney et al., 2003; Thambo et al., 2004). The DAVID trial showed that patients who received continuous dual-chamber pacing had significantly worse outcomes than patients with back-up, i.e. less frequent, ventricular pacing, presented by worsening of heart failure and increased mortality (Wilkoff et al., 2002). This result was confirmed by the MADIT II trial, which demonstrated that ventricular pacing was linked to worsening of heart failure (Moss et al., 2002). Some reports suggested that there is a direct relationship between ventricular pacing, cardiac memory, mechanical dysfunction and increased susceptibility to arrhythmias (Alessandrini et al., 1997; Medina-Ravell et al., 2003), thus highlighting the need to further investigate the effects of ventricular pacing. This is particularly paramount for pacing therapy in children with congenital or acquired atrioventricular (AV) block because in these patients, pacing has been shown to be highly beneficial (van Geldorp et al., 2011).
When the normal pattern of activation is disturbed by ventricular pacing, impulse propagation occurs between ventricular muscle cells, rather than through the fast His-Purkinje network, causing slowing of conduction (Scher and Young, 1955; Scher et al., 1953; Vassallo et al., 1984) and mechanical dyssynchrony (Fang et al., 2010; Rosenbush et al., 1982). In a study of 93 patients with sinus node dysfunction who had been paced for at least 6 months, Fang et al. (2010) found that half had developed significant mechanical dyssynchrony. Those patients who developed dyssynchrony had significantly larger ventricles and lower ejection fraction than those who did not develop dyssynchrony. Furthermore, in another report, Zhang et al. (2008a) showed that, of 79 patients who were paced from the right ventricle more than 90% of the time, 26% developed systolic heart failure within the 8-year follow-up period. Right-ventricular pacing created left-ventricular mechanical dysfunction and accelerated the progression of heart failure (Tse and Lau, 1997; Zhang et al., 2008a). Conversely, clinical studies aimed at synchronizing electrical activation and mechanical contraction by bi-ventricular pacing improved survival (Abraham et al., 2002; Bristow et al., 2004; Tu et al., 2011). In a recent review, Bank et al. (2011) have summarized many of these clinical observations.
1.3. Molecular mechanisms of ventricular electrical remodeling
The aforementioned studies established clear correlations between pacing-induced mechanical dyssynchrony and electrical dysfunction. Therefore, a number of investigations have been aimed at understanding the molecular mechanisms that link abnormal mechanical stretch with adverse electrical remodeling in the heart. Recent reports have been focused on the G protein-coupled mechanoreceptor pathway as a possible mechanism (Patel et al., 2010; Sharif-Naeini et al., 2010; Storch et al., 2012). This pathway is an ideal candidate to explain stretch-induced VER because it couples a pressure sensor (for example, the angiotensin II type I receptor, AT1R) to a depolarizing, calcium-permeable ion channel such as the transient receptor potential canonical, TRPC, channel (Rowell et al., 2010) via a Gq protein-coupled signaling cascade. There is substantial evidence that Gq protein-mediated signaling is involved in the maladaptive hypertrophic response of the heart to pressure overload (Akhter et al., 1998; Wettschureck et al., 2001) that causes cellular Ca2+ influx and hypertrophy via activation of the NFAT-calcineurin pathway (Bush et al., 2006; Kuwahara et al., 2006; Nakayama et al., 2006; Onohara et al., 2006; Seth et al., 2009). Moreover, expression of the protein ‘regulator of G-protein signaling 2’ (RGS2), an endogenous inhibitor of Gqα function, was reduced in ventricular myocardium from hypertensive patients (Semplicini et al., 2006), from end-stage heart failure patients with left-ventricular assist devices (Takeishi et al., 2000), and in a dog model of dyssynchronous heart failure (Chakir et al., 2009). Consistent with this, mice lacking RGS2 responded more rapidly to pressure overload than their wild-type counterparts because of Gq hyper-activation, leading to hypertrophy, heart failure and early death (Takimoto et al., 2009). Activation of AT1R (Bkaily et al., 2003; Gusev et al., 2009), down-regulation of RGS2 (Calo et al., 2008; Klaiber et al., 2010; Semplicini et al., 2006) and membrane stretch (Malhotra et al., 1999; Onohara et al., 2006) have been shown to independently activate Gq signaling and Ca2+ influx through TRPC channels.
Other signaling pathways are known to transduce mechanical signals into electrophysiological and structural remodeling events, and these pathways include: protein kinase A, protein kinase C, protein kinase G, calmodulin-activated kinase, calcineurin, mitogen-activated protein kinase, integrin and tyrosine kinase (Heineke and Molkentin, 2006; Romer et al., 2006; Sadoshima and Izumo, 1997). They activate a number of key transcription factors (for example, MEF2, GATA4, etc.) that regulate myocyte growth and function. Some mechanosensitive ion channels (MSC) that are present in the sarcolemmal membrane and in the extracellular matrix/cytoskeleton have been shown to be directly gated by stretch, thereby affecting excitation-contraction coupling on a beat-to-beat basis (Iribe et al., 2009; Kohl et al., 2006). In the beating heart, it is likely that the final common pathway linking mechanical signals to electrophysiological and structural remodeling is not represented by a single signaling pathway, but rather a combination of these mechanisms.
1.4. Limitations of current animal models for studying mechano-electrical feedback mechanisms in the heart
Studies in intact hearts from large animals (for example dog, sheep or lamb) with physiologies that are close to human have provided some important insights into the VER phenotypes which are clinically relevant and would have otherwise been difficult to obtain from patients (Dixon and Spinale, 2009). Although these large-animal experiments at the intact-organ level are informative because of their high level of complexity, they allow only limited control of tissue parameters. For example, perfusion of whole-organ preparations with drugs affecting excitation-contraction coupling causes highly non-uniform electrophysiological effects due to significant drug concentration gradients (Lacroix et al., 1999). Moreover, tissue stretch by pacing or pressure is difficult to control in magnitude and direction in a large 3-dimensional intact heart preparation because mechanical load is distributed heterogeneously across the endocardial, epicardial and transmural walls, causing regional structural and electrical remodeling that is difficult to localize or access experimentally (Spragg et al., 2003; van Oosterhout et al., 1998). For the same reasons, gene manipulation studies at the whole-organ level aimed at investigating the influences of altered protein expression on arrhythmia susceptibility have proven difficult (Rapti et al., 2011). Simplified cell culture models have been developed that circumvent some of the complexities inherent to intact tissue models and provide in vitro preparations that can be tested under controlled conditions, including high-precision stretch (Zhang et al., 2008b). Presently available cardiac cell culture models are derived from neonatal rat, neonatal mouse or embryonic chick (Tung and Zhang, 2006); although more recently, other cell culture models have become available that are derived from human stem cell sources (Arbel et al., 2010). These models significantly contributed to our understanding of electrical remodeling in response to stretch, but they lack important structural and electrical properties of adult, in vivo, cardiac tissue, which define cardiac electrical activity and are needed for an accurate representation of arrhythmia susceptibility. Among those properties are cell shapes, cell architecture (e.g. fiber structures), and cellular composition of ion channels and junction proteins, as reviewed by Kleber et al. (2004). Recent advances in tissue engineering have allowed the reproduction of some aspects of cellular architecture (Bian et al., 2012; McCain and Parker, 2011), but while electrically functional, these artificial tissues have been re-assembled from dissociated cardiac myocytes and are therefore fundamentally different from naturally grown heart tissue.
1.5. The zebrafish as a new animal model to study mechano-electrical feedback in the heart
Over the past decade the zebrafish (danio rerio) has emerged in the field of cardiovascular research as an attractive model organism. The popularity of this animal model is largely a result of its fecundity, diploid genome, and the transparency of its embryos, making it possible to study organ development in vivo (Haffter and Nusslein-Volhard, 1996). Sequencing of the zebrafish genome by the Sanger Institute transformed this species into a major genetic model organism (Vogel, 2000). Any gene in the zebrafish genome is readily manipulated using antisense morpholinos (Nasevicius and Ekker, 2000), making high-throughput, phenotype-driven genetic and chemical screens possible. Recently, stable gene knockouts have become feasible using zinc-finger nuclease technologies (Meng et al., 2008; Urnov et al., 2010). Zebrafish grow to about 3 cm in length, but during embryonic and larval stages, they are approximately 1–2 mm long. During these stages, zebrafish can live for several days in standard well plates, surviving on nutrients that are provided by their yolk sacs. Zebrafish are inexpensive to raise, and a pair of adults can routinely lay hundreds of fertilized eggs in a single morning. Therefore, even a small fish facility can produce thousands of embryos in a day making large-scale screens feasible. The zebrafish is ideal for phenotype-driven screens because cardiac phenotypes can be observed in vivo using a simple dissection microscope (Anderson and Ingham, 2003; Grunwald and Eisen, 2002; Patton and Zon, 2001). Several of the mutants identified in these screens exhibited cardiac arrhythmias (Stainier et al., 1996). Closer examinations of the hearts from these mutants lead to the discovery of many zebrafish cardiac ion channels (Chen et al., 1996; Novak et al., 2006; Warren et al., 2001) and calcium-handling proteins (Zhang et al., 2010), which are known to play important roles in cardiac excitation-contraction coupling in the human heart. These and other reports show that there are striking similarities in ion channel expression and function between the human and the zebrafish heart and indicate that the electrophysiological mechanisms governing the generation of the human and zebrafish ventricular action potentials are similar (Baker et al., 1997; Milan and Macrae, 2008; Shin and Fishman, 2002; Warren and Fishman, 1998). Thus, for many electrophysiological phenomena the zebrafish should prove to be a suitable model.
For the electrophysiologist, one particularly interesting feature of the zebrafish is that severe cardiac defects can be studied that would be embryonic lethal in other animal models. The zebrafish embryo is small enough so that it can survive for several days by diffusion alone and in complete absence of blood circulation (Burggren and Pinder, 1991; Pelster and Burggren, 1996; Stainier et al., 1996). Compared with other animal models, this is an important advantage because mechano-electrical feedback resulting from physiological cardiac contraction can be reduced or completely eliminated. This was first demonstrated by the silent heart (sih) mutant, which was found to result from a mutation in the tnnt2 gene encoding the thin-filament contractile protein cardiac troponin T. Mutation in this gene causes complete cessation of cardiac contraction (Sehnert et al., 2002). Dosage of a morpholino targeted against this gene allowed a controlled reduction of the wild-type transcript, thereby creating morphant zebrafish with hearts that showed reduced contractility (Becker et al., 2011). These tnnt2 morphants reproduced many of the cellular and transcriptional responses seen in patients with sarcomeric protein mutations that cause contractile dysfunction. A similar noncontractile phenotype is caused by a mutation in the unc-45b gene, which encodes a chaperone required for myofibril assembly (Etard et al., 2007). Interestingly, hearts from both noncontractile tnnt2 and unc-45 morphants generated depolarizing action potentials, but APDs, conduction velocities and calcium transients were significantly disturbed (Becker et al., 2011; Panakova et al., 2010). These reports show that mechano-electrical feedback mechanisms from cardiac contraction are important for the development of normal cardiac electric activity and calcium signaling in the heart.
In summary, the zebrafish embryo possesses a number of important advantages compared with other animal models that make it attractive for studying mechano-electrical feedback mechanisms in the heart: (1) a diploid genome, with almost every gene identified to have a human ortholog with a comparable function, (2) powerful genetic tools (e.g. morpholino oligos) that allow efficient knockdown of any gene of interest, (3) a large number of identified cardiac ion channels and calcium-handling proteins that are known to play important roles in excitation-contraction coupling in the human heart, (4) heart rates and ventricular action potentials that are comparable to those in humans, and (5) a small (<0.5 mm) heart that allows precise experimental control of the experimental conditions, e.g. extracellular ion concentrations, or as described here, the magnitude and direction of mechanical load using the carbon fiber method. To test the hypothesis that the zebrafish is a suitable animal model for studying mechano-electrical feedback mechanisms in the heart, we established a zebrafish heart stretch system that combines efficient genetic manipulation with high-precision stretch and high-resolution electrophysiology.
2. Materials and methods
2.1. Heart isolation
All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and approved by the Institutional Animal Care and Use Committee at Case Western Reserve University. Hearts were isolated from zebrafish embryos 72 hours post-fertilization (h.p.f.) and placed in Tyrode’s solution which contained (in mM) Na+ (136), K+ (5.4), Mg2+ (1.0), PO43− (0.3), Ca2+ (1.8), glucose (5.0) and HEPES (10.0) at pH 7.4. For stretch experiments and fluorescence recordings, hearts were placed into an experimental perfusion chamber with integrated field stimulation electrodes (RC27NE, Warner Instruments, LLC, CT, USA) that contained Tyrode’s solution. For fluorescence measurements, the Tyrode’s solution was supplemented with the excitation-contraction uncoupler blebbistatin (10 μM, 203390, EMD Chemicals Inc., Philadelphia, PA) to eliminate contractions. The perfusion chamber was mounted onto the stage of an inverted microscope (Axiovert-100, Zeiss, Germany), as shown in Fig. 1 A. All experiments were carried out at 21 degrees Celsius.
Fig. 1. Experimental system for the application of controlled stretch to zebrafish embryo hearts.

The zebrafish heart stretch system was designed using a conventional inverted fluorescence microscope with attached micromanipulators (panel A). A high-speed CCD camera was used for measuring action potentials and conduction velocities. Two carbon fibers were attached to the ventricle of an isolated zebrafish embryo heart (panel B). The terminal 2 mm of a glass pipette was bent by 25° and a 1 mm long section of carbon fiber was glued into each pipette using a cyanoacrylate-based adhesive (panel C). Zebrafish hearts were stretched from the force-free position (panel D, left) to the stretched position (panel D, right) using the micromanipulators.
2.2. Experimental system for the application of controlled stretch to zebrafish embryo hearts
We developed a system for the precise application of controlled stretch to the ventricles of isolated zebrafish embryo hearts based on a carbon fiber method that was previously applied to isolated cardiac myocytes (Iribe et al., 2007). Briefly, carbon fibers (Tsukuba Materials, Ltd., Tsukuba, Japan), 10 μm in diameter, were cut into 1 mm long pieces and glued into glass capillaries that were pulled from 2 mm-outer diameter, 1.12 mm-inner diameter glass pipettes (1B200F-6 World Precision Instruments, FL, USA) using cyanoacrylate adhesive (Fig. 1 B, C). The final section (~ 2 mm long) of the glass capillary containing the fiber was bent by approximately 25° to allow near-parallel alignment with the bottom of the perfusion chamber (Fig. 1 C). Each pipette was mounted on a high-precision motorized micro-manipulator (MP-285, Sutter Instruments, CA, USA). Before each experiment, fibers were aligned near horizontal and near parallel to each other. They were brought into contact with the ventricle of an isolated heart and firmly attached via electrostatic forces as described by Yasuda et al. (2001). Position control was accomplished using the motorized micromanipulators. Mechanical load was applied by moving the micromanipulators in opposite directions causing the fibers to deflect and the ventricular tissue to stretch (Fig. 1 D and 2 A).
Fig. 2. Carbon fiber technique for applying controlled stretch to zebrafish embryo hearts.

Time-dependent carbon fiber deflections and tip displacements were monitored during the experiment via the attached CCD camera. Increasing the mechanical load by moving the micromanipulators apart caused an increase in fiber deflection and a near-proportional increase in % tissue stretch (panel A). Mechanical loading of the zebrafish ventricle caused an immediate, but transient, elevation of the contractile force and rate of contraction (panel B). Contraction rates were the same at the end of the stretch period (panel C). “Small” and “large” stretch amounts were defined as relative (panel D) and absolute (panel E) increases in tissue length between the fiber tips. Stretch directions were defined corresponding to the zebrafish heart anatomy (panel F): hearts were stretched along an axial (Ax) direction by attaching one fiber near the outer curvature (OC) and the other one near the outflow tract (OFT) of the ventricle. The transversal (Tr) stretch direction was defined perpendicular to the axial direction and across the atrium (panel F, G).
The positions of the micromanipulator and the carbon fiber tips were monitored in real time during the experiment to control the amount of stretch. Tip distances were recorded at a frame rate of 30 s−1 using a high-speed CCD camera (PentaMAX, Princeton Instruments, NJ, USA) that was mounted on the microscope. Fiber positions were measured using custom-designed edge detection software written in Matlab (R2011, MathWorks, MA, USA, Fig. 2 B). Slippage, i.e. sliding of the fibers relative to the heart surface, was apparent by a gradual increase of the stretch transient baseline and was used as an exclusion criterion. Hearts were left contracting in sinus rhythm against the mechanical load of the carbon fibers for 90 minutes. Then, they were separated from the fibers and stained with the transmembrane potential-sensitive dye Di-8-ANEPPS (D3167, Invitrogen, NY) for the measurements of action potential durations and conduction velocities.
2.3. High-resolution optical mapping of isolated zebrafish embryo hearts
We previously described in detail the measurement of action potentials and the estimation of conduction velocities in isolated zebrafish embryo hearts (Panakova et al., 2010). Briefly, hearts were stained for 20 minutes with 84 μM of Di-8-ANEPPS. They were then thoroughly rinsed with Tyrode’s solution to remove all extracellular dye. Subsequently, individual hearts were placed into the perfusion chamber which contained Tyrode’s solution supplemented with the excitation-contraction uncoupler blebbistatin (10 μM, 203390, EMD Chemicals Inc., PA) to ensure that all measurements were done in mechanically unloaded hearts. This drug has previously been shown to effectively uncouple the excitation-contraction process in the zebrafish embryonic heart without significantly altering the action potential morphology (Jou et al., 2010). The perfusion chamber was then re-positioned onto the stage of the inverted microscope for fluorescence analysis. All fluorescence measurements reported in the figures were carried out approximately 40 minutes after the end of stretch. This time was required for the careful detachment, staining, rinsing and re-positioning of the hearts onto the microscope stage. Fluorescence light emission was recorded using the attached CCD camera at a frame rate of 200 s−1. Using a magnification of 40 x, the pixel-to-pixel distance was 5.5 μm. Two fluorescence recordings were carried out in sequence for each heart: the first recording was done during sinus-node activation and in the absence of any field stimulation to estimate conduction velocities, and the second recording was done during steady-state field stimulation at a rate of 60 beats-per-minute (bpm) to measure action potential durations (APDs). Stimulation voltages were set at approximately 1.2 times the activation threshold causing simultaneous excitation of the entire heart independent of the field direction. At least 30 beats were counted before each APD measurement. Conduction velocity vector fields were estimated using an established algorithm (Bayly et al., 1998), which we previously applied to action potential measurements from 72-h.p.f-old embryonic (Panakova et al., 2010) and adult (Kikuchi et al., 2010; Wang et al., 2011) zebrafish. Briefly, fluorescence images were exported as 16 bit TIFF stacks and processed spatially using a 2 × 2-pixel averaging filter. Activation times were calculated for each pixel as the time from baseline to 50% depolarization. In computer simulations of two-dimensional cardiac tissue, this criterion for electrical activation was found to correlate well with the time of maximum depolarizing sodium inward current (Fast and Kleber, 1995). Isochronal maps that represent the positions of the wavefront in 5 ms time intervals were generated from the activation times using the contour plotting function provided by Matlab (Fig. 3 A). Ventricles and atria were each divided into 3 non-overlapping regions-of-interest (ROIs), 6 × 6 pixels in size (33 × 33 μm2 of tissue area) covering the inner curvature (IC), mid-ventricular myocardium (MV) and outer curvature (OC) as shown in Fig. 3 A. Taking into account the activation time and spatial coordinates of each pixel, conduction velocity vector maps were estimated in each ROI (Fig. 3 A, B). The action potential duration (APD) was defined as the time from 20 % depolarization to 80 % repolarization and measured during steady-state field pacing at 60 bpm (Fig. 3 C). All measurements of APD and conduction velocity were averaged across the three ROIs in the ventricle (and atrium) to obtain overall measures of ventricular (and atrial) APDs and conduction velocities.
Fig. 3. Electrophysiological properties of the zebrafish embryo heart.
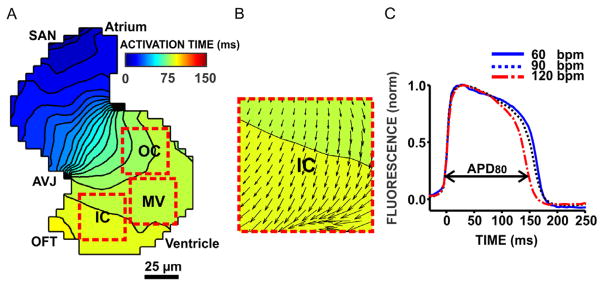
Action potentials propagated from the sino-atrial node (SAN) via the atrio-ventricular junction (AVJ) to the outflow tract (OFT, panel A). Isochronal maps show the positions of the depolarizing wavefront in 5 ms time intervals. Colors represent the direction of activation (from blue to red). Conduction velocity vector maps (panel B) were estimated in 35 × 35 μm2 large, non-overlapping ROIs covering the outer curvature (OC), mid-ventricular myocardium (MV) and inner curvature (IC) using an established algorithm described by Bayly et al. (1998). The action potential duration (APD80) was defined as the time from 20 % depolarization to 80 % repolarization (panel C). Increasing the stimulation frequency from 60 beats-per-minute (bpm) to 120 bpm shortened APD as expected from the mammalian heart.
3. Results
3.1. Electrophysiological properties of the zebrafish embryo heart
Ventricular action potentials were characterized by a fast upstroke and a pronounced plateau phase, similar in shape and duration to the human ventricular action potential. Ventricular action potentials shortened with increasing heart rates (Fig. 3 C), an observation that is well known from the mammalian heart as APD restitution (Pastore et al., 2006). The maximum APD differences within a broad range of spontaneous heart rates (60 bpm to 120 bpm) were smaller than 20 ms. Importantly, this is far less than the APD changes that were associated with stretch-induced VER, reassuring that any spontaneous variations in APD due to heart rate fluctuations do not obscure the reliable detection of VER responses in this model. However, to completely avoid any rate dependencies, all APD measurements were carried out during field stimulation at 60 bpm. Mean conduction velocities during sinus activation were higher in the ventricle than in the atrium, with slow conduction zones emerging at the sino-atrial node (SAN), the atrio-ventricular junction (AVJ) and the outflow tract (Fig. 3 A), as we have previously described (Panakova et al., 2010).
3.2. Time course study to determine the optimal time point for stretch experiments in the developing embryo heart
An important consideration for using embryonic, rather than adult, zebrafish was the possibility of combining ventricular stretch with efficient genetic manipulation using morpholino antisense oligos. These compounds are most effective in embryos that are less than 90 h.p.f. old (Nasevicius and Ekker, 2000). Therefore, our goal was to test whether there was a time interval during zebrafish heart development that was late enough so that developmental influences on the stretch phenotype were small but early enough so that morpholinos could still be used effectively. We measured APDs and conduction velocities in the ventricles from developing zebrafish embryo hearts in 24-hour intervals, i.e. from 24 h.p.f. to 120 h.p.f. (Fig. 4). We found that APDs shortened and ventricular conduction velocities increased markedly within the first 48 hours of development, but became very stable after 72 h.p.f. In fact, mean APDs and conduction velocities were not significantly different between 72 h.p.f. and 120 h.p.f., suggesting that during this time electrophysiological changes caused by cardiac development are negligible. Given the relative stability of these properties at 72 h.p.f., this was an optimal time point of development to apply mechanical stretch interventions.
Fig. 4. Time course study to determine the optimal time point for stretch experiments in the developing zebrafish embryo heart.
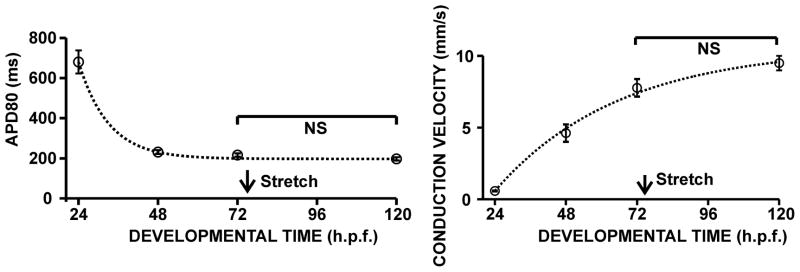
Ventricular action potential durations (APD80, left) and ventricular conduction velocities (right) were calculated from hearts isolated in 24 hour intervals, between 24 and 120 h.p.f. to evaluate the influence of development on electrical activity during the time of stretch. The time point of 72 h.p.f. was chosen for all subsequent stretch experiments and is indicated by the arrow.
3.3. Validation of the carbon fiber technique for applying controlled stretch to zebrafish embryo hearts
The carbon fibers attached reliably to the ventricles of isolated 72 h.p.f zebrafish embryo hearts upon touch as previously described for isolated cardiac myocytes (Iribe et al., 2007; Yasuda et al., 2001). The magnitude of stretch could be controlled via the attached micromanipulators. Moving the fiber tips apart increased the ventricular mechanical load as evident from the changes in the fiber deflection (Fig. 2 A) which is in first approximation proportional to the restoring force (Iribe et al., 2007; Nishimura et al., 2004). The fiber deflection was calculated as the difference between the manipulator and the fiber tip displacements. Because hearts contracted in sinus rhythm, changes in contractile force and frequency could readily be observed. The zebrafish ventricle responded to the increase in mechanical load with an immediate increase in the amplitude and rate of contraction (Fig. 2 B). This effect has been described previously for larger mammalian heart preparations (Bollensdorff et al., 2011). However, contraction rates decreased shortly after the stretch was applied and eventually became independent of the stretch amount at the end of the stretch period (Fig. 2 C). It was possible to stretch ventricles by up to 300 % of their unloaded length, before detachment occurred (Fig. 2 D). Despite these large stretch amounts, contractions remained periodic and stable throughout the stretch experiment. “Small” and “large” stretch, respectively, were defined as relative increases in tissue length of (25 ± 3) % and (230 ± 50) %, and measured as the relative increase in fiber-tip distance (Fig. 2 D, E). Small stretch was comparable in magnitude with relative stretch amounts that were previously reported in isolated rat, guinea pig and frog cardiac myocytes (Iribe and Kohl, 2008; Iribe et al., 2009; Riemer and Tung, 2003). Large stretch was chosen to be on the same order of magnitude as the relative increase in circumferential strain that we previously measured in paced dog hearts using tagged MRI imaging (Jeyaraj et al., 2007). This was done to assure that the magnitudes of stretch that we tested fell within physiologically reasonable ranges. To study the influence of stretch direction on the stretch response, we stretched ventricles across an outer curvature-outflow tract axis (axial stretch, Ax), and along a transversal axis (transversal stretch, Tr), which was perpendicular to the axial direction (Fig. 2 F, G).
3.4. Demonstration of stretch-induced ventricular electrical remodeling in the zebrafish embryo heart
In the zebrafish ventricle, 90 minutes of axial ventricular stretch caused a profound, more than two-fold increase in the ventricular action potential duration (APD) compared with unstretched control hearts (Fig. 5 A, C). The APD was unchanged in the atrium of the post-stretch hearts (Fig. 5 B, C), demonstrating that, as expected, the stretch response was localized to the cardiac chamber that was subjected to stretch, i.e. the ventricle. This electrophysiological stretch response was maintained 40 minutes after the end of stretch and in mechanically unloaded hearts. To test if the remodeled phenotype persisted even longer than 40 minutes, we measured APDs in four hearts 90 minutes after the end of stretch. We found that in these hearts, APD prolongation was still significant, i.e. 357 ± 30 ms in post-stretch hearts vs. 216 ± 8 ms in unstretched control hearts, demonstrating that the electrophysiological change outlasted the period of applied stretch. We then examined the dependence of APD prolongation on the magnitude and direction of stretch. We found that small axial stretch caused the same increase in APD as large axial stretch, indicating that over a large range of stretch amounts, the APD phenotype was independent of the mechanical load. However, changing the direction of stretch, as defined in Fig. 2, from axial stretch to transversal stretch, attenuated the stretch response (Fig. 5 D): Large transversal stretch resulted in a smaller, not significant increase of the mean APD compared with unstretched control hearts, indicating that the stretch response was dependent on the direction of applied stretch. Within the ventricles of the unstretched control hearts, APDs were distributed homogeneously (Fig. 5 E). However, it is possible that the carbon fiber method caused heterogeneous ventricular stretch, with the largest mechanical loads occurring between the fibers, resulting in a heterogeneous response. We therefore tested if stretch increased the spatial heterogeneity of APD, i.e. the dispersion of repolarization, as this may have occurred in the heart shown in Fig. 5 F. We calculated the dispersion of repolarization as the maximum absolute differences in APD between the three ROIs that are shown for a representative unstretched heart in Fig. 3 A. Interestingly, ventricular stretch did not significantly increase the dispersion of repolarization in the ventricle, suggesting that the stretch response was fairly uniform throughout the ventricle (Fig. 5 G).
Fig. 5. Demonstration of stretch-induced ventricular electrical remodeling in the zebrafish embryo heart.

Action potentials were prolonged in the ventricle (panel A) but not in the atrium (panel B) of a representative post-stretch heart (red trace) compared with an unstretched control heart (black trace). APDs were significantly increased in the ventricles (n=6, panel C) from post-stretch hearts but not in the atria (n=9, panel C). Small axial Ax stretch (n=5, panel D) caused the same APD prolongation as large axial Ax stretch (n=6, panel D). However, large transversal Tr stretch (n=5) significantly attenuated the stretch response because the resulting increase in the mean APD was not significant compared to the mean APD of unstretched control hearts (n=6, panel D). Color maps from an unstretched control heart (panel E) and a post-stretch heart (panel F) show that the increase in APD was restricted to the area where stretch was applied, i.e. the ventricle. Stretch did not significantly increase the APD dispersion, as calculated from the maximum absolute differences in APD between the OC, MV and IC ROIs (panel G). NS=not significant.
We then compared the conduction velocities in stretched and unstretched hearts (Fig. 6). Large ventricular axial stretch significantly slowed the conduction velocities in the ventricle but not in the atrium (Fig. 6 A–C). This decrease in the conduction velocity was not significant for small axial and large transversal stretch (Fig. 6 D), suggesting that conduction slowing was less sensitive to stretch than APD prolongation. Taken together, our data demonstrate that the response of the zebrafish ventricle to stretch is dependent on the amount and the direction of stretch and that large axial stretch causes the most severe electrophysiological phenotype.
Fig. 6. Demonstration of persistent conduction slowing in stretched zebrafish ventricles.

Ventricular stretch decreased the ventricular conduction velocities as evident from representative 5 ms isochronal maps of an unstretched control heart (panel A) and a post-stretch heart (panel B). Ventricular conduction velocities were significantly decreased in post-stretch hearts (n=6, panel C) compared with unstretched control hearts (n=5, panel C). Stretch did not influence the atrial conduction velocities. Only large axial Ax stretch (n=6) significantly reduced the ventricular conduction velocities compared with unstretched control hearts (n=5, panel D). Neither small axial Ax stretch (n=5) nor large transversal Tr stretch (n=4) caused significant conduction slowing compared with the unstretched control hearts (panel D). NS=not significant.
4. Discussion
The goal of this study was to examine the possibility of using the zebrafish model to investigate mechano-electrical feedback mechanisms in the heart. The power of this approach resides in the possibility to rapidly test the overlapping functions of genes that are involved in the response of the intact vertebrate heart to stretch. The similarities that exist between the zebrafish and the human heart, in particular the documented high conservation of gene expression and function, should facilitate translation at every level to other model systems or to human (Shin and Fishman, 2002).
4.1. Electrical stability of the developing zebrafish heart during assessment of ventricular electrical remodeling
One of the anticipated challenges in assessing stretch responses in a model that is undergoing cardiac development is that it might be difficult to determine if any given electrophysiological change was attributable to cardiac development or to the stretch perturbation itself. We found that 72 h.p.f. was a suitable time point for our stretch experiments, because in the vicinity of that time, developmental changes in APD and conduction velocity are small (Fig. 4) and morpholino antisense strategies are still very effective (Ekker, 2000). In a recent time course study, Leong et al. (2010) demonstrated that expression levels of the zebrafish kcnh2 gene encoding the α-subunit of the rapid delayed rectifier current (IKr) were stable after 72 h.p.f. These results suggest that developmental influences on the electrophysiological stretch response are small – yet they may still exist and contribute to the stretch phenotype, especially for longer stretch durations. As in every embryonic model, these developmental changes may not be completely eliminated but could possibly be controlled by carefully characterizing the model and keeping the experiment durations short.
4.2. Ventricular stretch caused ventricular electrical remodeling in the isolated zebrafish embryo heart
Zebrafish ventricles responded with profound and persistent electrophysiological changes to stretch. Only 90 minutes of ventricular stretch caused a significant prolongation of the ventricular action potential duration (Fig. 5) and a decrease of the ventricular conduction velocity (Fig. 6). When stretch was applied, the zebrafish heart responded with an immediate increase in the contraction rate (Fig. 2 B), suggesting that there was some acute influence of ventricular stretch on the sino-atrial node. However, this effect was transient because contraction rates decreased within the first minute of stretch and eventually became independent of the stretch amount (Fig. 2 C). Moreover, there was no effect of ventricular stretch on APDs and conduction velocities in the atria of post-stretch hearts, reaffirming that it was mechanical stretch that induced the persistent electrophysiological response. This result is consistent with previously published data from mammalian VER models. The APD was prolonged in regions of high circumferential strain that were induced by ventricular pacing in our dog model (Jeyaraj et al., 2007). In a similar model, Patel et al. (2001) reported reduced expression of the gap junction protein connexin 43 and conduction slowing after induction of T-wave memory. However, as in most of the published electrophysiological studies about cardiac memory to date, strain patterns were not recorded.
In our zebrafish model, the electrophysiological stretch response was maintained for at least 90 minutes after the end of stretch and in the absence of any mechanical load. In contrast to this, activation of mechanosensitive ion channels that mediate electrophysiological changes on a beat-to-beat basis are fully reversible when the mechanical load is removed (Bollensdorff et al., 2011; Iribe et al., 2009). Because the response outlasted the duration of stretch, we define the zebrafish stretch response here as VER in accordance with our previous definition (Jeyaraj et al., 2010; Jeyaraj et al., 2007). These observations raise an important question: how could only 90 minutes of stretch result in a subsequent maintained effect? A possible mechanism may be the post-transcriptional modification of ion channels, pumps and exchangers, for example via phosphorylation. Phosphorylation of contractile proteins and activation of the Na+/H+ exchanger were found in acutely stretched rabbit (Monasky et al., 2010) and rat (Alvarez et al., 1999; Calaghan and White, 2004; Perez et al., 2011) heart muscle preparations, respectively, and have been suggested to be important contributors to the instantaneous slow-force response to a change in muscle length (Luers et al., 2005). Another recent study identified several post-translational modifications of focal adhesion proteins in uniaxially stretched fibroblasts (Hoffman et al., 2012), demonstrating that stretch-induced protein modifications can occur in a variety of cell types independently from transcription or translation. It is not known, however, how long such protein changes would persist in the zebrafish heart.
It is further conceivable that transcriptional changes are responsible for the zebrafish stretch response. Studies in cultured neonatal rat myocytes demonstrated that brief periods of stretch, lasting less than 60 minutes, induced features of the hypertrophic response, including changes in contractile protein expression and activation of signal transduction pathways (Komuro et al., 1990; Komuro et al., 1991; Komuro et al., 1996; Sadoshima and Izumo, 1993). One study concluded that just 5 minutes of stretch were sufficient to induce rapid secretion of VEGF and increased expression of both VEGF and VEGF receptor mRNA in cultured cardiac myocytes (Seko et al., 1999). More recent data in a cell culture model indicated that 60 minutes of pulsatile stretch were sufficient to cause substatial upregulation of intercellular gap junction proteins likely leading to the significant conduction velocity increase that was observed (Zhuang et al., 2000). These and other experimental data on the effects of mechanical forces on the remodeling of gap junctions were reviewed in detail by Jeffrey Saffitz and André Kléber (2004). Taken together, the results of these studies demonstrate that short periods of stretch, lasting from several minutes to hours, can produce electrical remodeling. Clearly, additional expression studies and direct exploration of candidate pathways are necessary to understand the molecular mechanism of stretch-induced VER in the zebrafish.
It is possible that tissue damage contributed to the conduction slowing observed in post-stretch hearts. To test this hypothesis, we used a significantly smaller stretch amount (Fig. 2 D, E) corresponding to a smaller loading force (Fig. 2 A). A similar stretch amount had previously been applied using the same method to isolated rat, guinea pig and frog cardiac myocytes without causing damage (Iribe and Kohl, 2008; Iribe et al., 2009; Riemer and Tung, 2003). We found that in the zebrafish ventricle, the much smaller axial stretch amount did not cause significant conduction slowing (Fig. 6 D), suggesting that it preserved normal cell-to-cell coupling. However, small axial stretch caused the same APD prolongation as large axial stretch (Fig. 5 D). This result indicates that the stretch-induced APD prolongation was independent of any mechanical uncoupling or tissue damage that might have contributed to the conduction slowing. Additionally, we obtained consistently high signal-to-noise ratios (>10) from stretched ventricular areas, even from those areas that were located right between the fibers and subjected to the largest stretch amounts. This indicates that the stretched ventricular myocytes were viable and generated action potentials. Our observations are supported by previous studies showing that fish are able to increase stroke volumes by up to 300 % to increase cardiac output, unlike mammals who accomplish this by accelerating heart rate (Patrick et al., 2011; Shiels et al., 2006). Although we cannot rule out structural changes, our optical mapping data indicate that stretch-induced APD prolongation occurs independent from electrical uncoupling.
We also found that the stretch-induced VER phenotype was dependent on the direction of stretch. Axial stretch increased the APD (Fig. 5 D) and slowed ventricular conduction (Fig. 6 D), while transversal stretch resulted in a significantly attenuated response. Previous reports showed in rat neonatal cell cultures that the electrophysiological stretch response depended on how stretch was applied. Heterogeneous shear stress increased the APD and decreased the conduction velocity, while uniform hydrostatic pressure did not cause any statistically significant changes (Zhang et al., 2008b). Cyclic linear axial stretch in a similar model increased the conduction velocity, likely due to an upregulation of gap junction proteins (Zhuang et al., 2000). Static stretch produced significantly smaller changes than pulsatile stretch. Anisotropic biaxial stretch of micropatterned cells grown as linear strands showed upregulation of connexin 43 and N-cadherin with transversal stretch, but not with axial stretch (Gopalan et al., 2003).
Last, some components of the heart stretch response may be species-dependent. For example, short-term (seconds to minutes) stretch by pressure overload caused APD prolongation in the intact rabbit heart (Sung et al., 2003), while APD shortening occurred in an open-chest lamb model (Chen et al., 2004). Decreased mechanical load by intravenous infusion of sodium nitroprusside, a potent vasodilator, increased left ventricular APD in the pig heart linearly with decreased systolic ventricular pressure (Dean and Lab, 1989). This variability in the stretch response might be due to subtle electrophysiological differences in ion channel expression and function that exist between species leading to very different cellular behaviors, as illustrated in a recent simulation study by O’Hara and Rudy (2011), thus leaving the possibility that the molecular mechanisms underlying the electrophysiological stretch response are universal.
5. Conclusions
VER represents a serious clinical problem that affects a large number of adults and children with cardiac conduction system defects or ventricular pacing. Recent data from patients and animal models suggest that VER is triggered by a mechano-electrical feedback mechanism, but the molecular pathways that link mechanical stretch and electrical remodeling are not known. Our results indicate that the zebrafish is a suitable animal model for studying the molecular mechanisms that cause stretch-induced VER because it reproduces the key electrophysiological phenotypes that have been described in mammalian VER models, and yet, allows efficient genetic manipulation and precise experimental control. In the future, the zebrafish VER model could provide important insights into the complex molecular processes that drive detrimental electrical remodeling in the heart in response to stretch.
Acknowledgments
This project was supported by a grant from the National Institutes of Health (R01-HL0054807, David S Rosenbaum) and a postdoctoral fellowship award from the American Heart Association (10POST4150110, Andreas A Werdich). We thank the members of the McDermott laboratory in the Department of Otolaryngology-Head and Neck Surgery at Case Western Reserve University for expert help with zebrafish.
Footnotes
7. Editors’ Note
Please see also related communications in this issue by AUTHOR-1 et al. (2012) and AUTHOR-2 et al. (2012).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham WT, Fisher WG, Smith AL, Delurgio DB, Leon AR, Loh E, Kocovic DZ, Packer M, Clavell AL, Hayes DL, Ellestad M, Trupp RJ, Underwood J, Pickering F, Truex C, McAtee P, Messenger J Evaluation MSGMIRC. Cardiac resynchronization in chronic heart failure. N Engl J Med. 2002;346:1845–53. doi: 10.1056/NEJMoa013168. [DOI] [PubMed] [Google Scholar]
- Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998;280:574–577. doi: 10.1126/science.280.5363.574. [DOI] [PubMed] [Google Scholar]
- Alessandrini RS, McPherson DD, Kadish AH, Kane BJ, Goldberger JJ. Cardiac memory: a mechanical and electrical phenomenon. Am J Physiol. 1997;272:H1952–9. doi: 10.1152/ajpheart.1997.272.4.H1952. [DOI] [PubMed] [Google Scholar]
- Alvarez BV, Perez NG, Ennis IL, Camilion de Hurtado MC, Cingolani HE. Mechanisms underlying the increase in force and Ca(2+) transient that follow stretch of cardiac muscle: a possible explanation of the Anrep effect. Circ Res. 1999;85:716–22. doi: 10.1161/01.res.85.8.716. [DOI] [PubMed] [Google Scholar]
- Anderson KV, Ingham PW. The transformation of the model organism: a decade of developmental genetics. Nat Genet. 2003;33(Suppl):285–93. doi: 10.1038/ng1105. [DOI] [PubMed] [Google Scholar]
- Arbel G, Caspi O, Huber I, Gepstein A, Weiler-Sagie M, Gepstein L. Methods for human embryonic stem cells derived cardiomyocytes cultivation, genetic manipulation, and transplantation. Methods Mol Biol. 2010;660:85–95. doi: 10.1007/978-1-60761-705-1_6. [DOI] [PubMed] [Google Scholar]
- Baker K, Warren KS, Yellen G, Fishman MC. Defective “pacemaker” current (Ih) in a zebrafish mutant with a slow heart rate. Proc Natl Acad Sci USA. 1997;94:4554–4559. doi: 10.1073/pnas.94.9.4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank AJ, Gage RM, Burns KV. Right Ventricular Pacing, Mechanical Dyssynchrony, and Heart Failure. J Cardiovasc Transl Res. 2011 doi: 10.1007/s12265-011-9341-8. [DOI] [PubMed] [Google Scholar]
- Bayly PV, KenKnight BH, Rogers JM, Hillsley RE, Ideker RE, Smith WM. Estimation of conduction velocity vector fields from epicardial mapping data. IEEE Trans Biomed Eng. 1998;45:563–571. doi: 10.1109/10.668746. [DOI] [PubMed] [Google Scholar]
- Becker JR, Deo RC, Werdich AA, Panakova D, Coy S, MacRae CA. Human cardiomyopathy mutations induce myocyte hyperplasia and activate hypertrophic pathways during cardiogenesis in zebrafish. Disease models & mechanisms. 2011;4:400–10. doi: 10.1242/dmm.006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bian W, Juhas M, Pfeiler TW, Bursac N. Local Tissue Geometry Determines Contractile Force Generation of Engineered Muscle Networks. Tissue Eng Part A. 2012 doi: 10.1089/ten.tea.2011.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bkaily G, Sleiman S, Stephan J, Asselin C, Choufani S, Kamal M, Jacques D, Gobeil F, Jr, D’Orleans-Juste P. Angiotensin II AT1 receptor internalization, translocation and de novo synthesis modulate cytosolic and nuclear calcium in human vascular smooth muscle cells. Can J Physiol Pharmacol. 2003;81:274–287. doi: 10.1139/y03-007. [DOI] [PubMed] [Google Scholar]
- Bollensdorff C, Lookin O, Kohl P. Assessment of contractility in intact ventricular cardiomyocytes using the dimensionless ‘Frank-Starling Gain’ index. Pflugers Arch. 2011;462:39–48. doi: 10.1007/s00424-011-0964-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, DeMets D, White BG, DeVries DW, Feldman AM Comparison of Medical Therapy P, Defibrillation in Heart Failure I. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med. 2004;350:2140–50. doi: 10.1056/NEJMoa032423. [DOI] [PubMed] [Google Scholar]
- Burggren WW, Pinder AW. Ontogeny of cardiovascular and respiratory physiology in lower vertebrates. Annu Rev Physiol. 1991;53:107–35. doi: 10.1146/annurev.ph.53.030191.000543. [DOI] [PubMed] [Google Scholar]
- Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR, Olson EN, McKinsey TA. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem. 2006;281:33487–96. doi: 10.1074/jbc.M605536200. [DOI] [PubMed] [Google Scholar]
- Calaghan S, White E. Activation of Na+-H+ exchange and stretch-activated channels underlies the slow inotropic response to stretch in myocytes and muscle from the rat heart. J Physiol. 2004;559:205–14. doi: 10.1113/jphysiol.2004.069021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo LA, Pagnin E, Ceolotto G, Davis PA, Schiavo S, Papparella I, Semplicini A, Pessina AC. Silencing regulator of G protein signaling-2 (RGS-2) increases angiotensin II signaling: insights into hypertension from findings in Bartter’s/Gitelman’s syndromes. JHypertens. 2008;26:938–945. doi: 10.1097/HJH.0b013e3282f60d98. [DOI] [PubMed] [Google Scholar]
- Chakir K, Daya SK, Aiba T, Tunin RS, Dimaano VL, Abraham TP, Jaques-Robinson KM, Lai EW, Pacak K, Zhu WZ, Xiao RP, Tomaselli GF, Kass DA. Mechanisms of enhanced beta-adrenergic reserve from cardiac resynchronization therapy. Circulation. 2009;119:1231–1240. doi: 10.1161/CIRCULATIONAHA.108.774752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee K, Harris AM, Davies JG, Leatham A. T-wave changes after artificial pacing. Lancet. 1969;1:759–760. doi: 10.1016/s0140-6736(69)91758-9. [DOI] [PubMed] [Google Scholar]
- Chen JN, Haffter P, Odenthal J, Vogelsang E, Brand M, van Eeden FJ, Furutani-Seiki M, Granato M, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Kelsh RN, Mullins MC, Nusslein-Volhard C. Mutations affecting the cardiovascular system and other internal organs in zebrafish. Development. 1996;123:293–302. doi: 10.1242/dev.123.1.293. [DOI] [PubMed] [Google Scholar]
- Chen RL, Penny DJ, Greve G, Lab MJ. Stretch-induced regional mechanoelectric dispersion and arrhythmia in the right ventricle of anesthetized lambs. Am J Physiol Heart Circ Physiol. 2004;286:H1008–14. doi: 10.1152/ajpheart.00724.2003. [DOI] [PubMed] [Google Scholar]
- Dean JW, Lab MJ. Effect of changes in load on monophasic action potential and segment length of pig heart in situ. Cardiovasc Res. 1989;23:887–96. doi: 10.1093/cvr/23.10.887. [DOI] [PubMed] [Google Scholar]
- del Balzo U, Rosen MR. T wave changes persisting after ventricular pacing in canine heart are altered by 4-aminopyridine but not by lidocaine. Implications with respect to phenomenon of cardiac ‘memory’. Circulation. 1992;85:1464–1472. doi: 10.1161/01.cir.85.4.1464. [DOI] [PubMed] [Google Scholar]
- Dixon JA, Spinale FG. Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail. 2009;2:262–71. doi: 10.1161/CIRCHEARTFAILURE.108.814459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekker SC. Morphants: a new systematic vertebrate functional genomics approach. Yeast. 2000;17:302–306. doi: 10.1002/1097-0061(200012)17:4<302::AID-YEA53>3.0.CO;2-#. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etard C, Behra M, Fischer N, Hutcheson D, Geisler R, Strahle U. The UCS factor Steif/Unc-45b interacts with the heat shock protein Hsp90a during myofibrillogenesis. Developmental biology. 2007;308:133–43. doi: 10.1016/j.ydbio.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Fang F, Chan JY, Yip GW, Xie JM, Zhang Q, Fung JW, Lam YY, Yu CM. Prevalence and determinants of left ventricular systolic dyssynchrony in patients with normal ejection fraction received right ventricular apical pacing: a real-time three-dimensional echocardiographic study. Eur J Echocardiogr. 2010;11:109–18. doi: 10.1093/ejechocard/jep171. [DOI] [PubMed] [Google Scholar]
- Fast VG, Kleber AG. Cardiac Tissue Geometry As A Determinant of Unidirectional Conduction Block - Assessment of Microscopic Excitation Spread by Optical Mapping in Patterned Cell-Cultures and in A Computer-Model. Cardiovascular Research. 1995;29:697–707. [PubMed] [Google Scholar]
- Gjesdal O, Remme EW, Opdahl A, Skulstad H, Russell K, Kongsgaard E, Edvardsen T, Smiseth OA. Mechanisms of abnormal systolic motion of the interventricular septum during left bundle-branch block. Circ Cardiovasc Imaging. 2011;4:264–73. doi: 10.1161/CIRCIMAGING.110.961417. [DOI] [PubMed] [Google Scholar]
- Gopalan SM, Flaim C, Bhatia SN, Hoshijima M, Knoell R, Chien KR, Omens JH, McCulloch AD. Anisotropic stretch-induced hypertrophy in neonatal ventricular myocytes micropatterned on deformable elastomers. Biotechnology and Bioengineering. 2003;81:578–587. doi: 10.1002/bit.10506. [DOI] [PubMed] [Google Scholar]
- Grunwald DJ, Eisen JS. Headwaters of the zebrafish -- emergence of a new model vertebrate. Nat Rev Genet. 2002;3:717–24. doi: 10.1038/nrg892. [DOI] [PubMed] [Google Scholar]
- Gusev K, Domenighetti AA, Delbridge LM, Pedrazzini T, Niggli E, Egger M. Angiotensin II-mediated adaptive and maladaptive remodeling of cardiomyocyte excitation-contraction coupling. Circ Res. 2009;105:42–50. doi: 10.1161/CIRCRESAHA.108.189779. [DOI] [PubMed] [Google Scholar]
- Haffter P, Nusslein-Volhard C. Large scale genetics in a small vertebrate, the zebrafish. Int J Dev Biol. 1996;40:221–227. [PubMed] [Google Scholar]
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Hoffman LM, Jensen CC, Chaturvedi A, Yoshigi M, Beckerle MC. Stretch-induced actin remodeling requires targeting of zyxin to stress fibers and recruitment of actin regulators. Mol Biol Cell. 2012 doi: 10.1091/mbc.E11-12-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iribe G, Helmes M, Kohl P. Force-length relations in isolated intact cardiomyocytes subjected to dynamic changes in mechanical load. Am J Physiol Heart Circ Physiol. 2007;292:H1487–H1497. doi: 10.1152/ajpheart.00909.2006. [DOI] [PubMed] [Google Scholar]
- Iribe G, Kohl P. Axial stretch enhances sarcoplasmic reticulum Ca2+ leak and cellular Ca2+ reuptake in guinea pig ventricular myocytes: experiments and models. Prog Biophys Mol Biol. 2008;97:298–311. doi: 10.1016/j.pbiomolbio.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Iribe G, Ward CW, Camelliti P, Bollensdorff C, Mason F, Burton RA, Garny A, Morphew MK, Hoenger A, Lederer WJ, Kohl P. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res. 2009;104:787–95. doi: 10.1161/CIRCRESAHA.108.193334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyaraj D, Ashwath M, Rosenbaum DS. Pathophysiology and clinical implications of cardiac memory. Pacing Clin Electrophysiol. 2010;33:346–52. doi: 10.1111/j.1540-8159.2009.02630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyaraj D, Wilson LD, Zhong J, Flask C, Saffitz JE, Deschenes I, Yu X, Rosenbaum DS. Mechanoelectrical feedback as novel mechanism of cardiac electrical remodeling. Circulation. 2007;115:3145–3155. doi: 10.1161/CIRCULATIONAHA.107.688317. [DOI] [PubMed] [Google Scholar]
- Jou CJ, Spitzer KW, Tristani-Firouzi M. Blebbistatin effectively uncouples the excitation-contraction process in zebrafish embryonic heart. Cell Physiol Biochem. 2010;25:419–24. doi: 10.1159/000303046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DY, Poss KD. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 2010;464:601–5. doi: 10.1038/nature08804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaiber M, Kruse M, Volker K, Schroter J, Feil R, Freichel M, Gerling A, Feil S, Dietrich A, Londono JE, Baba HA, Abramowitz J, Birnbaumer L, Penninger JM, Pongs O, Kuhn M. Novel insights into the mechanisms mediating the local antihypertrophic effects of cardiac atrial natriuretic peptide: role of cGMP-dependent protein kinase and RGS2. Basic Res Cardiol. 2010;105:583–95. doi: 10.1007/s00395-010-0098-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiological Reviews. 2004;84:431–488. doi: 10.1152/physrev.00025.2003. [DOI] [PubMed] [Google Scholar]
- Kohl P, Bollensdorff C, Garny A. Effects of mechanosensitive ion channels on ventricular electrophysiology: experimental and theoretical models. Exp Physiol. 2006;91:307–21. doi: 10.1113/expphysiol.2005.031062. [DOI] [PubMed] [Google Scholar]
- Komuro I, Kaida T, Shibazaki Y, Kurabayashi M, Katoh Y, Hoh E, Takaku F, Yazaki Y. Stretching cardiac myocytes stimulates protooncogene expression. J Biol Chem. 1990;265:3595–8. [PubMed] [Google Scholar]
- Komuro I, Katoh Y, Kaida T, Shibazaki Y, Kurabayashi M, Hoh E, Takaku F, Yazaki Y. Mechanical loading stimulates cell hypertrophy and specific gene expression in cultured rat cardiac myocytes. Possible role of protein kinase C activation. J Biol Chem. 1991;266:1265–8. [PubMed] [Google Scholar]
- Komuro I, Kudo S, Yamazaki T, Zou Y, Shiojima I, Yazaki Y. Mechanical stretch activates the stress-activated protein kinases in cardiac myocytes. FASEB J. 1996;10:631–6. doi: 10.1096/fasebj.10.5.8621062. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–26. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix D, Extramiana F, Delfaut P, Adamantidis M, Grandmougin D, Klug D, Kacet S, Dupuis B. Factors affecting epicardial dispersion of repolarization: a mapping study in the isolated porcine heart. Cardiovasc Res. 1999;41:563–74. doi: 10.1016/s0008-6363(98)00269-7. [DOI] [PubMed] [Google Scholar]
- Leong IU, Skinner JR, Shelling AN, Love DR. Identification and expression analysis of kcnh2 genes in the zebrafish. Biochem Biophys Res Commun. 2010;396:817–24. doi: 10.1016/j.bbrc.2010.04.157. [DOI] [PubMed] [Google Scholar]
- Luers C, Fialka F, Elgner A, Zhu D, Kockskamper J, von Lewinski D, Pieske B. Stretch-dependent modulation of [Na+]i, [Ca2+]i, and pHi in rabbit myocardium--a mechanism for the slow force response. Cardiovasc Res. 2005;68:454–63. doi: 10.1016/j.cardiores.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Malhotra R, Sadoshima J, Brosius FC, III, Izumo S. Mechanical stretch and angiotensin II differentially upregulate the renin-angiotensin system in cardiac myocytes In vitro. Circ Res. 1999;85:137–146. doi: 10.1161/01.res.85.2.137. [DOI] [PubMed] [Google Scholar]
- Marrus SB, Nerbonne JM. Mechanisms linking short- and long-term electrical remodeling in the heart...is it a stretch? Channels (Austin) 2008:2. doi: 10.4161/chan.2.2.6104. [DOI] [PubMed] [Google Scholar]
- McCain ML, Parker KK. Mechanotransduction: the role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Pflugers Archiv: European journal of physiology. 2011;462:89–104. doi: 10.1007/s00424-011-0951-4. [DOI] [PubMed] [Google Scholar]
- Medina-Ravell VA, Lankipalli RS, Yan GX, Antzelevitch C, Medina-Malpica NA, Medina-Malpica OA, Droogan C, Kowey PR. Effect of epicardial or biventricular pacing to prolong QT interval and increase transmural dispersion of repolarization: does resynchronization therapy pose a risk for patients predisposed to long QT or torsade de pointes? Circulation. 2003;107:740–746. doi: 10.1161/01.cir.0000048126.07819.37. [DOI] [PubMed] [Google Scholar]
- Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol. 2008;26:695–701. doi: 10.1038/nbt1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan DJ, Macrae CA. Zebrafish genetic models for arrhythmia. Prog Biophys Mol Biol. 2008;98:301–8. doi: 10.1016/j.pbiomolbio.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monasky MM, Biesiadecki BJ, Janssen PM. Increased phosphorylation of tropomyosin, troponin I, and myosin light chain-2 after stretch in rabbit ventricular myocardium under physiological conditions. J Mol Cell Cardiol. 2010;48:1023–8. doi: 10.1016/j.yjmcc.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss AJ, Zareba W, Hall WJ, Klein H, Wilber DJ, Cannom DS, Daubert JP, Higgins SL, Brown MW, Andrews ML. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346:877–883. doi: 10.1056/NEJMoa013474. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Wilkin BJ, Bodi I, Molkentin JD. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 2006;20:1660–70. doi: 10.1096/fj.05-5560com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–20. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Kawai Y, Nakajima T, Hosoya Y, Fujita H, Katoh M, Yamashita H, Nagai R, Sugiura S. Membrane potential of rat ventricular myocytes responds to axial stretch in phase, amplitude and speed-dependent manners. Cardiovasc Res. 2006;72:403–11. doi: 10.1016/j.cardiores.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Seo K, Nagasaki M, Hosoya Y, Yamashita H, Fujita H, Nagai R, Sugiura S. Responses of single-ventricular myocytes to dynamic axial stretching. Prog Biophys Mol Biol. 2008;97:282–97. doi: 10.1016/j.pbiomolbio.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Yasuda S, Katoh M, Yamada KP, Yamashita H, Saeki Y, Sunagawa K, Nagai R, Hisada T, Sugiura S. Single cell mechanics of rat cardiomyocytes under isometric, unloaded, and physiologically loaded conditions. Am J Physiol Heart Circ Physiol. 2004;287:H196–H202. doi: 10.1152/ajpheart.00948.2003. [DOI] [PubMed] [Google Scholar]
- Novak AE, Taylor AD, Pineda RH, Lasda EL, Wright MA, Ribera AB. Embryonic and larval expression of zebrafish voltage-gated sodium channel alpha-subunit genes. Dev Dyn. 2006;235:1962–73. doi: 10.1002/dvdy.20811. [DOI] [PubMed] [Google Scholar]
- O’Hara T, Rudy Y. Quantitative Comparison of Cardiac Ventricular Myocyte Electrophysiology and Response to Drugs in Human and Non-Human Species. Am J Physiol Heart Circ Physiol. 2011 doi: 10.1152/ajpheart.00785.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, Mori Y, Nagao T, Kurose H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006;25:5305–5316. doi: 10.1038/sj.emboj.7601417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panakova D, Werdich AA, Macrae CA. Wnt11 patterns a myocardial electrical gradient through regulation of the L-type Ca(2+) channel. Nature. 2010;466:874–8. doi: 10.1038/nature09249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore JM, Laurita KR, Rosenbaum DS. Importance of spatiotemporal heterogeneity of cellular restitution in mechanism of arrhythmogenic discordant alternans. Heart Rhythm. 2006;3:711–9. doi: 10.1016/j.hrthm.2006.02.1034. [DOI] [PubMed] [Google Scholar]
- Patel A, Sharif-Naeini R, Folgering JR, Bichet D, Duprat F, Honore E. Canonical TRP channels and mechanotransduction: from physiology to disease states. Pflugers Arch. 2010;460:571–81. doi: 10.1007/s00424-010-0847-8. [DOI] [PubMed] [Google Scholar]
- Patel PM, Plotnikov A, Kanagaratnam P, Shvilkin A, Sheehan CT, Xiong W, Danilo P, Jr, Rosen MR, Peters NS. Altering ventricular activation remodels gap junction distribution in canine heart. J Cardiovasc Electrophysiol. 2001;12:570–7. doi: 10.1046/j.1540-8167.2001.00570.x. [DOI] [PubMed] [Google Scholar]
- Patrick SM, White E, Shiels HA. Rainbow trout myocardium does not exhibit a slow inotropic response to stretch. J Exp Biol. 2011;214:1118–22. doi: 10.1242/jeb.048546. [DOI] [PubMed] [Google Scholar]
- Patton EE, Zon LI. The art and design of genetic screens: zebrafish. Nat Rev Genet. 2001;2:956–66. doi: 10.1038/35103567. [DOI] [PubMed] [Google Scholar]
- Pelster B, Burggren WW. Disruption of hemoglobin oxygen transport does not impact oxygen-dependent physiological processes in developing embryos of zebra fish (Danio rerio) Circ Res. 1996;79:358–62. doi: 10.1161/01.res.79.2.358. [DOI] [PubMed] [Google Scholar]
- Perez NG, Nolly MB, Roldan MC, Villa-Abrille MC, Cingolani E, Portiansky EL, Alvarez BV, Ennis IL, Cingolani HE. Silencing of NHE-1 blunts the slow force response to myocardial stretch. Journal of applied physiology. 2011;111:874–80. doi: 10.1152/japplphysiol.01344.2010. [DOI] [PubMed] [Google Scholar]
- Rapti K, Chaanine AH, Hajjar RJ. Targeted gene therapy for the treatment of heart failure. Can J Cardiol. 2011;27:265–83. doi: 10.1016/j.cjca.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer TL, Tung L. Stretch-induced excitation and action potential changes of single cardiac cells. Prog Biophys Mol Biol. 2003;82:97–110. doi: 10.1016/s0079-6107(03)00008-7. [DOI] [PubMed] [Google Scholar]
- Romer LH, Birukov KG, Garcia JG. Focal adhesions: paradigm for a signaling nexus. Circ Res. 2006;98:606–16. doi: 10.1161/01.RES.0000207408.31270.db. [DOI] [PubMed] [Google Scholar]
- Rosenbaum MB, Blanco HH, Elizari MV, Lazzari JO, Davidenko JM. Electrotonic modulation of the T wave and cardiac memory. Am J Cardiol. 1982;50:213–222. doi: 10.1016/0002-9149(82)90169-2. [DOI] [PubMed] [Google Scholar]
- Rosenbush SW, Ruggie N, Turner DA, Von Behren PL, Denes P, Fordham EW, Groch MW, Messer JV. Sequence and timing of ventricular wall motion in patients with bundle branch block. Assessment by radionuclide cineangiography. Circulation. 1982;66:1113–9. doi: 10.1161/01.cir.66.5.1113. [DOI] [PubMed] [Google Scholar]
- Rowell J, Koitabashi N, Kass DA. TRP-ing up heart and vessels: canonical transient receptor potential channels and cardiovascular disease. J Cardiovasc Transl Res. 2010;3:516–24. doi: 10.1007/s12265-010-9208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes: potential involvement of an autocrine/paracrine mechanism. EMBO J. 1993;12:1681–92. doi: 10.1002/j.1460-2075.1993.tb05813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–571. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- Saffitz JE, Kleber AG. Effects of mechanical forces and mediators of hypertrophy on remodeling of gap junctions in the heart. Circ Res. 2004;94:585–91. doi: 10.1161/01.RES.0000121575.34653.50. [DOI] [PubMed] [Google Scholar]
- Scher AM, Young AC. Spread of excitation during premature ventricular systoles. Circ Res. 1955;3:535–42. doi: 10.1161/01.res.3.5.535. [DOI] [PubMed] [Google Scholar]
- Scher AM, Young AC, Malmgren AL, Paton RR. Spread of electrical activity through the wall of the ventricle. Circ Res. 1953;1:539–47. doi: 10.1161/01.res.1.6.539. [DOI] [PubMed] [Google Scholar]
- Sehnert AJ, Huq A, Weinstein BM, Walker C, Fishman M, Stainier DY. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. NatGenet. 2002;31:106–110. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- Seko Y, Seko Y, Takahashi N, Shibuya M, Yazaki Y. Pulsatile stretch stimulates vascular endothelial growth factor (VEGF) secretion by cultured rat cardiac myocytes. Biochem Biophys Res Commun. 1999;254:462–5. doi: 10.1006/bbrc.1998.9969. [DOI] [PubMed] [Google Scholar]
- Semplicini A, Lenzini L, Sartori M, Papparella I, Calo LA, Pagnin E, Strapazzon G, Benna C, Costa R, Avogaro A, Ceolotto G, Pessina AC. Reduced expression of regulator of G-protein signaling 2 (RGS2) in hypertensive patients increases calcium mobilization and ERK1/2 phosphorylation induced by angiotensin II. J Hypertens. 2006;24:1115–24. doi: 10.1097/01.hjh.0000226202.80689.8f. [DOI] [PubMed] [Google Scholar]
- Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J, Tsiokas L, Winn M, Abramowitz J, Rockman HA, Birnbaumer L, Rosenberg P. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res. 2009;105:1023–1030. doi: 10.1161/CIRCRESAHA.109.206581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif-Naeini R, Folgering JH, Bichet D, Duprat F, Delmas P, Patel A, Honore E. Sensing pressure in the cardiovascular system: Gq-coupled mechanoreceptors and TRP channels. J Mol Cell Cardiol. 2010;48:83–9. doi: 10.1016/j.yjmcc.2009.03.020. [DOI] [PubMed] [Google Scholar]
- Shiels HA, Calaghan SC, White E. The cellular basis for enhanced volume-modulated cardiac output in fish hearts. J Gen Physiol. 2006;128:37–44. doi: 10.1085/jgp.200609543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JT, Fishman MC. From Zebrafish to human: modular medical models. AnnuRev Genomics Hum Genet. 2002;3:311–340. doi: 10.1146/annurev.genom.3.031402.131506. [DOI] [PubMed] [Google Scholar]
- Sosunov EA, Anyukhovsky EP, Rosen MR. Altered ventricular stretch contributes to initiation of cardiac memory. Heart Rhythm. 2008;5:106–113. doi: 10.1016/j.hrthm.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Spragg DD, Leclercq C, Loghmani M, Faris OP, Tunin RS, DiSilvestre D, McVeigh ER, Tomaselli GF, Kass DA. Regional alterations in protein expression in the dyssynchronous failing heart. Circulation. 2003;108:929–932. doi: 10.1161/01.CIR.0000088782.99568.CA. [DOI] [PubMed] [Google Scholar]
- Stainier DY, Fouquet B, Chen JN, Warren KS, Weinstein BM, Meiler SE, Mohideen MA, Neuhauss SC, Solnica-Krezel L, Schier AF, Zwartkruis F, Stemple DL, Malicki J, Driever W, Fishman MC. Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development. 1996;123:285–292. doi: 10.1242/dev.123.1.285. [DOI] [PubMed] [Google Scholar]
- Storch U, Mederos YSM, Gudermann T. G Protein Mediated Stretch Reception. Am J Physiol Heart Circ Physiol. 2012 doi: 10.1152/ajpheart.00818.2011. [DOI] [PubMed] [Google Scholar]
- Sung D, Mills RW, Schettler J, Narayan SM, Omens JH, McCulloch AD. Ventricular filling slows epicardial conduction and increases action potential duration in an optical mapping study of the isolated rabbit heart. J Cardiovasc Electrophysiol. 2003;14:739–49. doi: 10.1046/j.1540-8167.2003.03072.x. [DOI] [PubMed] [Google Scholar]
- Sweeney MO, Hellkamp AS, Ellenbogen KA, Greenspon AJ, Freedman RA, Lee KL, Lamas GA. Adverse effect of ventricular pacing on heart failure and atrial fibrillation among patients with normal baseline QRS duration in a clinical trial of pacemaker therapy for sinus node dysfunction. Circulation. 2003;107:2932–2937. doi: 10.1161/01.CIR.0000072769.17295.B1. [DOI] [PubMed] [Google Scholar]
- Takeishi Y, Jalili T, Hoit BD, Kirkpatrick DL, Wagoner LE, Abraham WT, Walsh RA. Alterations in Ca2+ cycling proteins and G alpha q signaling after left ventricular assist device support in failing human hearts. Cardiovasc Res. 2000;45:883–888. doi: 10.1016/s0008-6363(99)00415-0. [DOI] [PubMed] [Google Scholar]
- Takimoto E, Koitabashi N, Hsu S, Ketner EA, Zhang M, Nagayama T, Bedja D, Gabrielson KL, Blanton R, Siderovski DP, Mendelsohn ME, Kass DA. Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J Clin Invest. 2009;119:408–420. doi: 10.1172/JCI35620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantengco MV, Thomas RL, Karpawich PP. Left ventricular dysfunction after long-term right ventricular apical pacing in the young. J Am Coll Cardiol. 2001;37:2093–100. doi: 10.1016/s0735-1097(01)01302-x. [DOI] [PubMed] [Google Scholar]
- Thambo JB, Bordachar P, Garrigue S, Lafitte S, Sanders P, Reuter S, Girardot R, Crepin D, Reant P, Roudaut R, Jais P, Haissaguerre M, Clementy J, Jimenez M. Detrimental ventricular remodeling in patients with congenital complete heart block and chronic right ventricular apical pacing. Circulation. 2004;110:3766–72. doi: 10.1161/01.CIR.0000150336.86033.8D. [DOI] [PubMed] [Google Scholar]
- Tse HF, Lau CP. Long-term effect of right ventricular pacing on myocardial perfusion and function. J Am Coll Cardiol. 1997;29:744–9. doi: 10.1016/s0735-1097(96)00586-4. [DOI] [PubMed] [Google Scholar]
- Tu R, Zhong G, Zeng Z, Wu W, Wu H, Cao X, Aung LH. Cardiac resynchronization therapy in patients with mild heart failure: a systematic review and meta-analysis of randomized controlled trials. Cardiovasc Drugs Ther. 2011;25:331–40. doi: 10.1007/s10557-011-6313-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung L, Zhang Y. Optical imaging of arrhythmias in tissue culture. J Electrocardiol. 2006;39:S2–6. doi: 10.1016/j.jelectrocard.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–46. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- van Geldorp IE, Vanagt WY, Prinzen FW, Delhaas T. Chronic ventricular pacing in children: toward prevention of pacing-induced heart disease. Heart Fail Rev. 2011;16:305–14. doi: 10.1007/s10741-010-9207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oosterhout MF, Prinzen FW, Arts T, Schreuder JJ, Vanagt WY, Cleutjens JP, Reneman RS. Asynchronous electrical activation induces asymmetrical hypertrophy of the left ventricular wall. Circulation. 1998;98:588–595. doi: 10.1161/01.cir.98.6.588. [DOI] [PubMed] [Google Scholar]
- Vassallo JA, Cassidy DM, Marchlinski FE, Buxton AE, Waxman HL, Doherty JU, Josephson ME. Endocardial activation of left bundle branch block. Circulation. 1984;69:914–23. doi: 10.1161/01.cir.69.5.914. [DOI] [PubMed] [Google Scholar]
- Vogel G. Genomics. Sanger will sequence zebrafish genome. Science. 2000;290:1671. [PubMed] [Google Scholar]
- Wang J, Panakova D, Kikuchi K, Holdway JE, Gemberling M, Burris JS, Singh SP, Dickson AL, Lin YF, Sabeh MK, Werdich AA, Yelon D, Macrae CA, Poss KD. The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development. 2011;138:3421–30. doi: 10.1242/dev.068601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren KS, Baker K, Fishman MC. The slow mo mutation reduces pacemaker current and heart rate in adult zebrafish. Am J Physiol Heart Circ Physiol. 2001;281:H1711–H1719. doi: 10.1152/ajpheart.2001.281.4.H1711. [DOI] [PubMed] [Google Scholar]
- Warren KS, Fishman MC. “Physiological genomics”: mutant screens in zebrafish. Am J Physiol. 1998;275:H1–H7. doi: 10.1152/ajpheart.1998.275.1.H1. [DOI] [PubMed] [Google Scholar]
- Wecke L, Gadler F, Linde C, Lundahl G, Rosen MR, Bergfeldt L. Temporal characteristics of cardiac memory in humans: vectorcardiographic quantification in a model of cardiac pacing. Heart Rhythm. 2005;2:28–34. doi: 10.1016/j.hrthm.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Wettschureck N, Rutten H, Zywietz A, Gehring D, Wilkie TM, Chen J, Chien KR, Offermanns S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat Med. 2001;7:1236–1240. doi: 10.1038/nm1101-1236. [DOI] [PubMed] [Google Scholar]
- Wilkoff BL, Cook JR, Epstein AE, Greene HL, Hallstrom AP, Hsia H, Kutalek SP, Sharma A. Dual-chamber pacing or ventricular backup pacing in patients with an implantable defibrillator: the Dual Chamber and VVI Implantable Defibrillator (DAVID) Trial. JAMA. 2002;288:3115–3123. doi: 10.1001/jama.288.24.3115. [DOI] [PubMed] [Google Scholar]
- Yasuda SI, Sugiura S, Kobayakawa N, Fujita H, Yamashita H, Katoh K, Saeki Y, Kaneko H, Suda Y, Nagai R, Sugi H. A novel method to study contraction characteristics of a single cardiac myocyte using carbon fibers. American Journal of Physiology-Heart and Circulatory Physiology. 2001;281:H1442–H1446. doi: 10.1152/ajpheart.2001.281.3.H1442. [DOI] [PubMed] [Google Scholar]
- Zhang PC, Llach A, Sheng XY, Hove-Madsen L, Tibbits GF. Calcium handling in zebrafish ventricular myocytes. Am J Physiol Regul Integr Comp Physiol. 2010 doi: 10.1152/ajpregu.00377.2010. [DOI] [PubMed] [Google Scholar]
- Zhang XH, Chen H, Siu CW, Yiu KH, Chan WS, Lee KL, Chan HW, Lee SW, Fu GS, Lau CP, Tse HF. New-onset heart failure after permanent right ventricular apical pacing in patients with acquired high-grade atrioventricular block and normal left ventricular function. J Cardiovasc Electrophysiol. 2008a;19:136–41. doi: 10.1111/j.1540-8167.2007.01014.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sekar RB, McCulloch AD, Tung L. Cell cultures as models of cardiac mechanoelectric feedback. Prog Biophys Mol Biol. 2008b;97:367–82. doi: 10.1016/j.pbiomolbio.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Yamada KA, Saffitz JE, Kleber AG. Pulsatile stretch remodels cell-to-cell communication in cultured myocytes. Circ Res. 2000;87:316–22. doi: 10.1161/01.res.87.4.316. [DOI] [PubMed] [Google Scholar]