

Summary
Mammalian Sin1 plays key roles in the regulation of mitogen activated protein kinase (MAPK) and mammalian target of rapamycin (mTOR) signaling. Sin1 is an essential component of mTOR complex (mTORC) 2. The function of Sin1 and mTORC2 remains largely unknown in T cells. Here we investigate Sin1 function in T cells using mice which lack Sin1 in the hematopoietic system. Sin1 deficiency blocks the mTORC2 dependent Akt phosphorylation in T cells during development and activation. Sin1 deficient T cells exhibit normal thymic cellularity and percentages of double negative, double positive and single positive CD4 and CD8 thymocytes. Sin1 deficiency does not impair T cell receptor (TCR) induced growth and proliferation, and normal CD4+ helper cell differentiation. However Sin1 deficiency results in an increased proportion of Foxp3+ natural T regulatory (nTreg) cells in the thymus. We show that the TGF-β dependent differentiation of CD4+ T cells in vitro is enhanced by the inhibition of mTOR but not loss of Sin1 function. Our results reveal that Sin1 and mTORC2 are dispensable for the development and activation of T cells but play a role in natural Treg cell differentiation.
Introduction
Mammalian target of rapamycin (mTOR) is a conserved serine/threonine protein kinase that regulates cell growth and metabolism [1]. Mammalian TOR is inhibited by rapamycin which is potent suppressor of T cell-mediated immune responses [2]. Rapamycin inhibits IL-2 dependent T cell proliferation, promotes the expansion of regulatory T (Treg) cells and has recently been shown to promote the development of memory CD8+ T cells [3–5]. Mammalian TOR function is mediated by at least two distinct multi-protein complexes called mTOR complex 1 (mTORC1), containing mTOR, raptor, mLST8 (GβL) and PRAS40, and mTORC2, containing Rictor, Sin1, and mLST8 in addition to mTOR. Nutrients, growth factors, hormones, and energy signals, activate mTORC1 to phosphorylate the translational regulators S6K and 4EBP1 leading to increased cellular protein synthesis and ribosome biogenesis [1]. Mammalian TORC2 regulates actin polymerization and cytoskeleton function [1], controls Akt activation and specificity in a PI3K-dependent manner by phosphorylating the Akt hydrophobic motif (S473 on Akt1), and regulates the stability of Akt and conventional PKC in a PI3K-independent manner by phosphorylating the turn motif (T450 on Akt1) [6–8]. Mammalian TORC2 is less sensitive to rapamycin inhibition than mTORC1, however chronic rapamycin treatment may inhibit mTORC2. Therefore, previous studies utilizing rapamycin to study mTOR were unable to properly evaluate the contribution of mTORC2 to T cell immunity. In addition, mTOR also posses a rapamycin independent mTORC1 function [9]. Therefore, it is unclear how mTORC1 and mTORC2 each specifically contribute to T cell function.
Recent genetic studies have begun to elucidate the mechanism of mTOR function and regulation in T cells. Delgoffe et al recently reported that CD4-Cre mediated T cell specific mTOR deletion impairs T cell proliferation and inhibits TH1, TH2, and TH17 differentiation without blocking early T cell activation [10]. Mammalian TOR deficiency also greatly enhanced Treg differentiation in vitro, while T cells lacking Rheb, a small GTPase which positively regulates mTORC1 function, failed to spontaneously differentiate into Treg cells upon activation suggesting that mTORC2 may play a prominent role in regulating Treg differentiation [10]. Two recent studies from independent labs have explored the function of mTORC2 in T cells using mice that specifically lack Rictor expression in T cells [11, 12]. In the first study, Lee et al show that rictor−/− T cells lack functional mTORC2 and exhibit defects in Akt and PKCθ phosphorylation as well as decreased NF-κB activity, reduced proliferation, impaired T helper cell differentiation and increased CD4+FoxP3+ Treg differentiation [12]. While in the second study, Delgoffe et al show that rictor−/− T cells exhibit defects in proliferation and TH2 differentiation, they do not observe deficiencies in TH1, TH17 or Treg differentiation [11].
In this study, we reconstituted lethally irradiated wild type mice with Sin1−/− fetal liver hematopoietic stem cells (HSC) and examined the T cell development, growth, proliferation, and CD4+ effector cell differentiation in cells obtained from these mice. We show that the loss of Sin1 in T cells disrupts mTORC2 function and blocks Akt phosphorylation at the HM and TM sites. Although mTORC2 function is abolished in Sin1−/− T cells, we find that Sin1 is not required for thymic T cell development. These data reveal that Akt HM and TM phosphorylation are not required for thymic T cell development even though Akt plays an essential role in maintaining the metabolism and viability of thymocytes undergoing TCRβ selection. Furthermore, mature T cell growth, proliferation or CD4+ helper T cell differentiation is unaffected by Sin1 deficiency. However, we observe that Sin1−/− thymic T cells give rise to a greater proportion of natural Treg cells than wild type thymocytes. These data support a role for mTORC2 in the regulation of Treg differentiation. We also provide evidence that Akt1 and Akt2 are not required for mTORC2 mediated regulation of thymic Treg development.
Materials and Methods
Mice
Sin1−/− mice and Akt1−/−, Akt2−/− and Akt1−/− Akt2−/− mice were described previously [6, 13]. CD45.1+ congenic mice were purchased from The Jackson Laboratory and used as recipients for the fetal liver hematopoietic cell transfers. Mice receiving fetal liver cell transplants were irradiated with 700–900 cGy prior to cell transfer. 0.5–1×106 total fetal liver cells were suspended in sterile 1xPBS and injected via the tail vein. Successful donor cell engraftment was verified by the presence of CD45.2+ peripheral blood mononuclear cells. All mice were housed in the animal facilities at Yale University and all animal procedures were approved by the Yale IACU Committee.
OP9-DL1/progenitor T cell cultures
Mouse fetal liver hematopoietic cells were obtained from embryonic day 11.5–12.5 Sin1+/+ and Sin1−/− littermate embryos. Fetal liver cells were cultured on confluent OP9-DL1 bone marrow stromal cells in RPMI1640 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 5 μg/ml gentamicin, 50 μM β-mercaptoethanol and 10 ng/ml mouse IL-7 (Constem, CT). Stable T cell lines were grown at 37°C in an atmosphere containing 5% CO2.
Lymphocyte staining and flow cytometry
Cells were washed with FACS buffer (1% FBS in 1x phosphate buffered saline (PBS) with 0.1% NaN3), incubated with indicated antibodies on ice for 30 min, then washed two more times with FACS buffer, and fixed in 1% paraformaldehyde in PBS before being analyzed with a LSRII flow cytometer (BD Biosciences). For intracellular cytokine staining, cells were stimulated with phorbol 12-myristate 13-acetate (PMA, Sigma) (50 ng/ml) + ionomycin (Sigma) (500 ng/ml) for 6 hours in the presence of Golgi-stop (BD Bioscience) for the last four hours. Cells were first surface stained, fixed/permeablized with a Cytofix/Cytoperm kit (BD Bioscience), and stained with antibodies against indicated cytokines. Intracellular FoxP3 and T-bet staining were carried out according to manufacturer’s instruction (EBioscience). For co-staining FoxP3 with GFP, cells were fixed by cytofix buffer (BD Bioscience), permeablized by ice-cold methanol and stained with indicated antibodies in the 1x Perm/Wash buffer (BD Bioscience).
Cell Purification and Culture
Splenocytes and lymph node cells were first stained with anti-CD4 biotin and CD4+ cells were magnetically purified using a Biotin-selection kit (Stem cell). Purified CD4 T cells were stimulated with plate bound anti-CD3 (3 μg/ml; 2C11) and soluble anti-CD28 (2 μg/ml; 37N) in T cell medium (RPMI 1640, 10% FBS, 1x antibiotics, 1x non-essential amino acid and 50 μM β-mercaptoethanol). When indicated, recombinant (r) cytokines were added into the culture: TH1: anti-IL-4 (5 μg/ml; 11B11) and IL-12 (10 ng/ml; PeproTech); iTreg: anti-IL-4 (5 μg/ml, 11B11), anti-IFN γ (5 μg/ml; R46A2), rhIL-2 (100 U/ml, Peprotech) and indicated concentration of rhTGF-β (Peprotech); TH17: anti-IL4 (5 μg/ml), anti-IFN γ (5 μg/ml), IL-6 (20 ng/ml, Peprotech), and indicated concentration of rhTGF-β (Peprotech). When indicated, the following inhibitors were used in this study: Rapamycin (LC laboratories); pp242 [14].
T Cell Stimulation and Immunoblotting
Naïve CD4 T cells were activated with anti-CD3/anti-CD28 antibodies in the presence of IL-2 (50 U/ml) for four days. Activated cells were then split into fresh culture medium with IL-2 (100 U/ml) and expanded for an additional four days. Cultured T cells were rested in T cell medium without IL-2 overnight and stimulated with either plate bound anti-CD3 (5 μg/ml)+anti-CD28 (2 μg/ml) for various time points. Stimulated T cells were washed with ice-cold PBS and lysed with RIPA buffer plus freshly added protease inhibitors and phosphatase inhibitors. Total cell lysates were used for immunoblot analysis. To detect S6 and Akt S473 phosphorylation following TCR stimulation, CD4 T cells were first stained with anti-CD3 (5 μg/ml) for 30 min on ice. After wash, T cells were cross-linked with anti-Hamster IgG for 3min, fixed with Phosflow fix buffer I (BD Bioscience), and stained with anti-pS6 S235/236 or anti-pAkt S473 (Cell Signaling) in Phosflow perm/wash buffer (BD Bioscience) for 30 minutes at room temperature followed by Alex-fluor 647 conjugated anti-Rabbit IgG (Cell signaling) in Phosflow perm/wash buffer for 15 minutes at room temperature.
T cell proliferation
Purified CD4 T cells were labeled with CFSE (3 nM) at 37°C for 10 min. CFSE labeled cells were stimulated with plate bound anti-CD3 and anti-CD28 as described[15, 16].
Results
Sin1 is not required for the development of major thymic T cell subsets
We generated chimeric mice by transplanting E12.5 fetal liver cells from Sin1+/+ or Sin1−/− embryos into lethally irradiated wild type (WT) CD45.1 congenic mice [13]. Analysis of thymic T cell populations in these chimeric mice revealed that Sin1 deficient hematopoietic stem cells gave rise to equivalent proportions of CD4/CD8 double negative (DN), CD4/CD8 double positive (DP), CD4+ single positive (SP) and CD8+ SP T cells as Sin1+/+ cells (Figure 1a). We also derived progenitor T cells from Sin1+/+ and Sin1−/− fetal liver hematopoietic cells to further characterize the role of Sin1 in early T cell development. Sin1+/+ or Sin1−/− fetal liver HSCs were cultured on OP9-DL1 stromal cells with IL-7 to generate stable T cell lines which resemble CD4/CD8 double negative thymocytes [17]. Phenotypic analysis of the in vitro derived Sin1+/+ and Sin1−/− T cells revealed that Sin1 is not required for the development of DN1, DN2, DN3 or DN4 T cells (Figure 1b, upper panels). Furthermore, analysis of these progenitor T cells revealed that Sin1 is not required for TCR beta chain expression (Figure 1b, lower panels).
Figure 1.

Sin1 is not required for T cell development.
a) CD4+ and CD8+ cells in the thymus of Sin1+/+ (WT) or Sin1−/− (KO) chimeric mice were assessed by flow cytometry.
b) Sin1 WT or KO fetal liver cells were cultured on OP9-DL1 stromal cells with IL-7 to derive progenitor T cells. Cell surface expression of CD44, CD25 (upper panels) and TCRβ chain (lower panels) was measured by flow cytometry. Representative FACS plots of 1 WT and 2 independent Sin1 KO T cell cultures are shown.
c) Sin1 WT and KO T cells were cultured on OP9-DL1 stromal cells with IL-7 and then analyzed by immunoblotting for the indicated proteins. ERK2 serves as a loading control. Pan-PKC p-HM antibody (PKCβII p-S660) detects phosphorylated hydrophobic motif (HM) of PKC.
d) Total FoxO1 expression in WT and Sin1 KO T cells cultured on OP9-DL1 stromal cells with IL-7 was measured by immunoblotting. ERK2 serves as a loading control. FoxO1 phosphorylation at T24 in WT and Sin1 KO T cellswas measured by immunoblotting and normalized to total FoxO1. The data in c and d are representative of 1 WT and 2 KO T cell cultures from two independent experiments.
To assess the effect of Sin1 on mTORC2 dependent signaling we examined Akt S473 phosphorylation in Sin1+/+ and Sin1−/− OP9-DL1 T cells. As expected, Akt S473 phosphorylation was abolished in the Sin1 deficient T cells (Figure 1c). We also observed that PKC hydrophobic motif phosphorylation was impaired in the Sin1−/− T cells (Figure 1c). We have previously shown that FoxO1 expression is increased in Sin1−/− pro-B cells and FoxO phosphorylation is impaired in Sin1−/− fibroblasts and pro-B cells [6, 13]. Consistently, FoxO1 expression was also increased in the Sin1−/− T cells relative to the Sin+/+ T cells (Figure 1d). FoxO1 phosphorylation was also decreased in Sin1−/− T cells relative to Sin1+/+ T cells (Figure 1d). These data show that Sin1 deficiency impairs mTORC2 dependent signaling in developing T cells. However Sin1 deficiency does not significantly alter thymic T cell development.
Next we examined if Sin1 deficiency has any effect on peripheral T cell populations. We observed equivalent proportions of splenic CD4+ and CD8+ T cells in Sin1+/+ and Sin1−/− chimeric mice (Figure 2a). We also measured the proportion of cytokine producing CD4+ effector T cells in the periphery of unimmunized chimeric mice. We found that the proportion of IFN-γ, IL-4 or IL-17A expressing CD4+ T cells in the spleen of unimmunized Sin1−/− chimeric mice was comparable to that of Sin1+/+ mice (Figure 2b). These data indicate that Sin1 may not be required for peripheral T cell differentiation.
Figure 2.

Sin1 deficiency increases the number of thymic natural regulatory T cells
a) Splenocytes from Sin1+/+ (WT) or Sin1−/− (KO) chimeric mice were analyzed for percentages of CD4 and CD8 T cells. The FACS plots shown are pre-gated on CD45.2+ donor cells.
b) Splenocytes from Sin1+/+ (WT) or Sin1−/− (KO) chimeric mice were stimulated with PMA and ionomycin for 4 hours in the presence of Golgi-Stop and assayed for IFN-γ, IL-17A, and IL-4 expression by flow cytometry. The FACS plots shown are pre-gated on donor CD4+CD8− CD45.2+ cells.
c) The expression of CD62L and CD44 was measured on splenic CD4+ T cells from Sin1+/+ or Sin1−/− chimeric mice by flow cytometry. The mean fluorescent intensity (MFI) of CD62L in the CD44lowCD62Lhi gate are as follows: Sin1+/+ MFI=8520 and Sin1−/− MFI=17400. The FACS plots shown are pre-gated on donor CD4+CD8− CD45.2+ cells.
d) The expression of IL-7R (CD127) on splenic donor CD4+CD45.2+ T cells from Sin1+/+ or Sin1−/− chimeric mice was determined by flow cytometry.
e) The thymic (upper panel) and splenic (lower panel) donor CD4+Foxp3+CD45.2+ T cells in Sin1+/+ or Sin1−/− chimeric mice were determined by flow cytometry.
f) Chimeric mice containing host WT and Sin1−/− (KO) T cells were generated by transferring Sin1−/− fetal liver cells into sub-lethally irradiated WT (CD45.1+) hosts. The percent of thymic WT CD4+Foxp3+ T cells (CD45.1+) and donor Sin1−/− CD4+Foxp3+ T cells (CD45.2+) in a chimeric mouse was determined by flow cytometry.
Numbers in the plots indicate percentages of the gated populations. All flow cytometry plots shown are representative of Sin1+/+ (n=3) and Sin1−/− (n=4) fetal liver chimeric mice.
We have previously shown that suppression of FoxO1 and FoxO3a transcriptional activity by Akt is dependent on Sin1 and mTORC2 in MEFs and in B cells [6, 13]. FoxO1 is a positive regulator of L-selectin (CD62L), CD127 (IL-7 receptor alpha chain, IL7r) and FoxP3 gene expression in T cells [18, 19]. Therefore we asked if Sin1−/− T cells exhibit increased expression of these FoxO1 dependent genes. CD62L expression was increased on the splenic CD4+CD44lowCD62L+ Sin1−/− T cells relative to Sin1+/+ T cells (Sin1+/+, MFI=8520 vs. Sin1−/− MFI=17400 (Figure 2c) but CD127 expression was equivalent on Sin1+/+ and Sin1−/− peripheral T cells (Figure 2d).
The transcription factor Foxp3 is the master regulator of Treg development. To assess the possible role of Sin1 in Treg development we first determined the proportion of thymic Tregs in Sin1+/+ and Sin1−/− chimeric mice. We observed that Sin1−/− thymocytes gave rise to 2 fold more CD25+Foxp3+ Treg cells when compared to Sin1+/+ thymocytes (4% Sin1+/+ CD4+CD25+FoxP3+ vs. 10% Sin1−/− CD4+CD25+Foxp3+) (Figure 2e), indicating that Sin1 may be a suppressor of thymic Treg differentiation. The proportion of CD25+Foxp3+ T cells in the spleens of Sin1+/+ and Sin1−/− chimeric mice was not significantly different (9% Sin1+/+ CD4+CD25+Foxp3+ vs. 10% Sin1−/− CD4+CD25+Foxp3+) (Figure 2e).
To determine if the Sin1 mediated suppression of thymic Treg development is cell intrinsic, we generated Sin1−/− chimeric mice containing an equivalent ratio of Sin1−/− fetal liver cells (CD45.2+) and WT cells (CD45.1+). There were two times more Sin1−/− CD25+Foxp3+Tregs that WT Tregs (7% Sin1+/+ CD4+CD25+Foxp3+ vs. 15% Sin1−/− CD4+CD25+Foxp3+) in the same host (Figure 2f). These data indicate that Sin1 inhibits the development of thymic Treg development in a cell intrinsic manner.
Akt is a negative regulator of Treg development [20] and Akt activity is directly regulated by mTORC2 [6, 13]. Since Sin1−/− cells lack mTORC2 function and exhibit deficiencies in Akt phosphorylation and function, we hypothesized that Akt may mediate mTORC2 dependent signals to suppress thymic Treg development. To test this hypothesis we measured the proportion of thymic Treg cells in Akt deficient mice. We determined the proportion of CD4+FoxP3+ Treg cells in the thymus of wild type, Akt1−/− or Akt2−/− mice. We found that Akt1−/− and Akt2−/− mice had an equivalent proportion of CD4+Foxp3+ T cells when compared to WT mice (Figure 3a). In addition, we also analyzed thymic Treg development in Akt1−/− Akt2−/− fetal liver cell chimeric mice (these mice die at late embryonic stage E18–19). Consistent with the previous reports [21], we observed that thymocyte development was blocked at the DN to DP transition in Akt1−/− Akt2−/− chimeric mice (data not shown). However a small number of Akt1−/− Akt2−/− thymocytes were capable of developing to the CD4+ SP stage. We measured the proportion of Foxp3+CD4+ T cells within this population of Akt1−/− Akt2−/− CD4+ SP cells and found that the proportion of Treg cells was similar to that observed in mice reconstituted with wild type fetal liver cells (Figure 3b).
Figure 3.
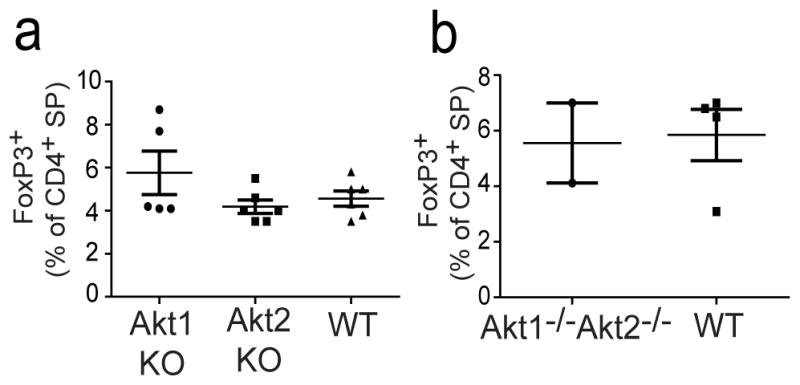
Loss of Akt1 and Akt2 does not alter thymic Treg development.
a) The thymic CD4+Foxp3+ T cells in wild type (WT), Akt1−/−, and Akt2−/− mice were assessed by flow cytometry. Graphs show the mean proportion of thymic CD4+CD8− Foxp3+ T cells from WT (n=5), Akt1−/− (n=6), and Akt2−/− (n=6) mice analyzed. The data shown are combined from three independent experiments.
b) Fetal livers from either Akt1−/− Akt2−/− (dKO) mice (n=2) or wild type (WT) mice (n=4) were used to reconstitute lethally irradiated B6.SJL mice. Ten weeks after reconstitution, donor thymic CD4+CD8− Foxp3+ T cells in WT or Akt1−/− Akt2−/− mice were measured by flow cytometry. The data are from two independent experiments. Each dot in the graph represents one individual mouse, and error bars are standard deviation.
Sin1 is not required for T cell growth or proliferation
Mammalian TOR is a master regulator of cellular growth. Therefore we asked if Sin1/mTORC2 was involved in regulating T cell growth and proliferation. We found that the size of resting CD4+ and CD8+ T cells from lymph nodes or spleen of Sin1+/+ and Sin1−/− fetal liver chimeric mice was similar (Figure 4a, data not shown). Next, we stimulated Sin1+/+ and Sin1−/− T cells with anti-CD3 plus anti-CD28 and assessed T cell size change and proliferation. Sin1 deficiency did not impair the blast cell growth (size increase) following T cell activation (Figure 4b and 4c). CD4+ T cells from Sin1+/+ and Sin1−/− chimeric mice also exhibited a similar activation induced proliferative capacity as determined by a CFSE dilution assay (Figure 4d). Finally, we examined the proliferation and survival of Sin1+/+ and Sin1−/− CD4+ T cells activated in the presence of TGF-β. We observed that Sin1 deficiency did not impair the proliferation of in vitro differentiated CD4+Foxp3+ T cells (Figure 4e). No difference in the proportion of live cells in the cultures of Sin1+/+ and Sin1−/− T cells was observed (Figure 4f). These data suggest that Sin1 is not required for T cell volume (size) growth of either resting or activated T cells and that Sin1 is not required for the proliferation and survival of activated T cells.
Figure 4.

Sin1 is not required for T cell growth and proliferation.
a) The relative cell size of resting lymph node CD4+ and CD8+ T cells from Sin1−/− (KO) chimeric mice was measured by forward light scatter (FSC). Each chimeric mouse also contained host WT T cells, which serve as individual internal controls. WT and KO cells were distinguished by differential CD45.1/CD45.2 expression. Error bars indicate standard error and p-values were derived by two-tailed paired t-test analysis.
b) Lymph node CD4+ or CD8+ T cells from three Sin1−/− (KO) chimeric mice were stimulated in vitro with anti-CD3 plus anti-CD28 for 48 hours and relative cell size was determined by measuring FSC. Representative histograms of resting (shaded) and stimulated host WT (black line) or Sin1−/− (dotted line) T cells from one of the three mice analyzed are shown.
c) The relative cell size of activated WT or Sin1−/− CD4+ and CD8+ T cells. The data shown are the average of three mice described in b. Error bars indicate standard error and p-values were calculated using two-tailed paired t-test analysis.
d) Purified Sin1+/+ or Sin1−/− CD4+ T cells were stimulated in vitro with anti-CD3 plus anti-CD28 for 4 days, and cell proliferation was measured by CFSE dilution. Histograms shown are pre-gated on CD45.2+ donor derived cells and are representative of two independent experiments.
e). Purified Sin1+/+ (WT) or Sin1−/− (KO) CD4+ T cells were stimulated in vitro with anti-CD3 plus anti-CD28 with 1 ng/ml TGF-β for 4 days, and proliferation of Foxp3+ and Foxp3− cells was determined by CFSE dilution.
f) Purified Sin1+/+ (WT) or Sin1−/− (KO) CD4+ T cells were stimulated in vitro with anti-CD3 plus anti-CD28 with 1 ng/ml TGF-β for 4 days. CD45.2+ cells were gated and the proportion of live cells was determined by forward and side light scatter gating.
Sin1 is not required for TH1, TH2, and TH17 effector T cell differentiation in vitro
To test the function of Sin1 in effector T cell differentiation, we purified CD4+ T cells from Sin1+/+ or Sin1−/− chimeric mice, activated these cells in vitro and differentiated these cells under TH1, TH2 or TH17 polarizing conditions. Sin1+/+ and Sin1−/− T cells cultured under TH1, TH2, or TH17 polarizing conditions gave rise to equivalent proportions of IFN-γ (30% Sin1+/+ vs. 35% Sin1−/−), IL-4 (6% Sin1+/+ vs. 5% Sin1−/−), or IL-17 (15% Sin1+/+ vs. 14% Sin1−/−) expressing cells, respectively (Figure 5a). We obtained the same results when we co-cultured Sin1−/− T cells with wild type congenic T cells under the same TH polarizing conditions (data not shown) indicating that Sin1 is not required for effector T cell differentiation into the TH1, TH2, or TH17 lineages.
Figure 5.

Sin1 is not required for CD4+ TH effector cell differentiation.
a) Purified CD4+ T cells from chimeric mice were stimulated with anti-CD3 plus anti-CD28 under indicated TH1 TH17, andTH2 polarizing conditions as indicated and then analyzed by flow cytometry. Plots are gated on donor T cells from WT or Sin1−/− (KO) chimeric mice. Plots shown are representative of two independent experiments.
b) Sin1+/+ (WT) or Sin1−/− (KO) CD4+ T cells were stimulated with anti-CD3 for 3 minutes and Akt S473 phosphorylation was measured by flow cytometry. Histograms shown are pre-gated on CD45.2+ donor cells and are representative of two independent experiments.
c) Sin1−/− (KO) or Sin1+/+ (WT) CD4+ T cells were stimulated with anti-CD3 for 0, 15 or 30 minutes and Akt T450 phosphorylation and total Akt was measured by immunoblotting. Total ERK2 serves as a loading control.
Sin1 is required for mTORC2 dependent phosphorylation of Akt in T cells
To examine if Akt phosphorylation at the mTORC2 target sites S473 and T450 was defective in Sin1−/− T cells, resting Sin1+/+ or Sin1−/− CD4+ T cells were stimulated with anti-CD3 antibody and Akt S473 phosphorylation was measured. As expected, compared to unstimulated T cells, anti-CD3 stimulation induced Akt S473 phosphorylation in Sin1+/+ T but failed to induce this phosphorylation in Sin1−/− T cells (Figure 5b). Consistent with our previous observations in Sin1−/− fibroblasts and B cells, Akt T450 phosphorylation in Sin1−/− T cells was also deficient (Figure 5c). These data show that Sin1 deficient T cells lack mTORC2 function and show defective Akt phosphorylation at the HM and TM sites.
Sin1 and mTOR differentially regulate TGF-β dependent Treg differentiation
Our observation that Sin1 deficiency promotes thymic Treg development is consistent with a current model in which mTORC2-Akt signal inhibits FoxO1 activity, which is required for Treg differentiation [10, 12]. To test if Sin1 may also inhibit the TGF-β dependent Treg differentiation of peripheral CD4+ T cells, purified Sin1+/+ or Sin1−/− CD4+ T cells were differentiated in the presence or absence of TGF-β. Without TGF-β Sin1+/+ and Sin1−/− CD4+ T gave rise to very few numbers of Foxp3+ cells (1.4% vs. 1.6%) (Figure 6a). In the presence of TGF-β, Sin1−/− CD4+ T cells consistently gave rise to fewer Foxp3+ Treg cells when compared to Sin1+/+ CD4+ T cells (28% vs. 38%, respectively) (Figure 6a). These data are surprising since we predicted that loss of mTORC2 function would enhance Treg differentiation similar to that of Sin1−/− thymocytes. Our results raise the possibility that Sin1 may have mTORC2 independent functions that may influence TGF-β dependent Treg differentiation in the periphery.
Figure 6.

Sin1 and mTOR differentially regulate TGF-β dependent Treg differentiation.
a) Sin1+/+ (WT) or Sin1−/− (KO) CD4+ T cells were stimulated in vitro with anti-CD3 plus anti-CD28 antibodies in the presence or absence of 1 ng/ml TGF-β. After four days Foxp3 expressing cells were enumerated by flow cytometry. Data shown are representative of two independent experiments.
b) Sin1+/+ CD4+ T cells were stimulated in vitro with anti-CD3 plus anti-CD28 antibodies plus 1 ng/ml TGF-β in the presence of rapamycin (Rapa. 30 nM), pp242 (100 nM) or carrier (ctrl.). The proportion of Foxp3+ cells generated after four days was measured by flow cytometry. Data are representative of two independent experiments.
c) Sin1+/+ CD4+ T cells were activated with anti-CD3 in the presence of indicated inhibitors and assayed for induction of S6 p-S235/S236 and Akt p-S473 by flow cytometry. Data are representative of two independent experiments.
To directly test the function of mTOR during Treg differentiation, we induced Treg differentiation of WT naïve CD4+ T cells with TGF-β in vitro in the presence or absence of mTOR inhibitors rapamycin or pp242 [14]. Rapamycin specifically inhibits mTORC1 while pp242, a specific mTOR kinase inhibitor, targets both mTORC1 and mTORC2 [14]. We observed that rapamycin (30 nM) did not significantly change the proportion of Treg cells generated in the presence of TGF-β (untreated = 53% vs. rapamycin treated = 50%). However, pp242 treatment (100 nM) consistently resulted in an increase in the proportion of Treg cells generated in response to TGF-β (untreated = 53% vs. pp242 treated = 68%) (Figure 6b). Both rapamycin and pp242 blocked mTORC1 dependent phosphorylation of ribosomal protein S6 while only pp242 blocked mTORC2 dependent HM site phosphorylation of Akt (Figure 6c). Overall our data support a model in which inhibition of both mTORC1 and mTORC2 is necessary to promote TGF-β induced Treg differentiation.
Discussion
In this study, we provide the first evidence examining the function of Sin1 in T cells. Our analysis of Sin1−/− fetal liver chimeric mice reveals that Sin1 is largely dispensable for the development of thymic T cells and peripheral CD4+ and CD8+ T cell populations. Since Sin1 is essential for mTORC2 function, our data also indicates that mTORC2 is not required for T cell development. Akt is the best characterized mTORC2 target and is required for T cell development [6, 7, 22]. Akt1−/− Akt2−/− T cells show a profound block in thymic development at the DN to DP transition due to a dramatic increase in the rate of thymocyte cell death [22]. Sin1−/− T cells develop normally despite having a partial loss of Akt function due to impaired HM and TM phosphorylation. The dramatic difference in T cell developmental phenotypes observed in Akt1−/− Akt2−/− and Sin1−/− chimeric mice indicates that the functional outcomes of Akt signaling can be subdivided into HM phosphorylation independent signals and HM phosphorylation dependent signals. Our data show that T cell development is not dependent on Akt HM phosphorylation. These findings are consistent with our previously proposed model in which mTORC2 dependent Akt HM phosphorylation is required to confer Akt specificity toward a limited subset of Akt substrates [6]. Our data also suggest that Akt, when activated via phosphorylation of activation loop, plays a central role for DN-DP transition, most likely by controlling the survival of thymic T cells. Furthermore, our data suggests that phosphorylation of Akt at the activation loop may be sufficient to support TCR/CD3 mediated peripheral T cell proliferation and survival.
Since mTOR is an evolutionarily conserved regulator of cellular growth and metabolism, we investigated if Sin1 deletion may affect the size of resting peripheral T cells or activated T cells and proliferation. Sin1 deficiency had little effect on resting T cell growth and activation induced blast cell growth. Furthermore, Sin1 deficiency did not impair antigen receptor/co-receptor dependent T cell proliferation in vitro. These results contrast with those reported in mice bearing a T cell specific rictor deletion which show a modest defect in activation induced T cell proliferation [12, 23]. It is possible that the differences in the in vitro T cell stimulation conditions between our assays may account for the difference in experimental results since we stimulated our T cells in the presence of plate-bound anti-CD3 antibody plus soluble anti-CD28 in the presence of exogenous IL-2.
FoxO1 is an mTORC2 dependent Akt substrate which has been shown to play a key role in regulating T cell development, homeostasis and effector cell differentiation [19, 24]. FoxO1 is required for proper expression of the genes that encode L-selectin (CD62L), interleukin 7 receptor alpha chain (CD127) and FoxP3 [18, 19, 24]. We have previously shown that Sin1 deficiency results in decreased FoxO1 phosphorylation at the Akt target sites, leading to increased FoxO1 transcriptional activity [6, 13]. Consistently, we observed an increased proportion of FoxP3 expressing nTregs in the thymus and an increased expression of CD62L expression on naive peripheral CD4+ T cells in Sin1−/− chimeric mice. Surprisingly, Sin1 deficiency did not affect IL-7R expression on resting peripheral T cells. We have previously shown that in developing progenitor B cells, the mTORC2-Akt-FoxO1 signaling negatively regulates IL-7R expression [13]. IL-7R expression is suppressed in antigen activated T cells. It is possible that the loss of mTORC2 function has no effect on IL-7R expression in resting T cells because these cells normally have a very low level of Akt signaling. Mammalian TORC2 may play a more important role in suppressing IL-7R expression after T cell activation since TCR signaling strongly induces the Akt signaling pathway. Alternatively, it is possible that another kinase may phosphorylate and regulate FoxO1 activity in place of Akt in Sin1−/− T cells. The serum & glucocorticoid dependent kinases (SGK)s may also phosphorylate FoxO proteins and negatively regulate FoxO transcriptional activity [25]. This may explain why we did not observe a complete loss of FoxO1 phosphorylation in Sin1−/− T cells. SGK1 has been shown to be positively regulated by both mTORC1 and mTORC2 dependent mechanisms [26, 27]. Since mTORC1 activity is not inhibited by Sin1 deficiency it is possible that SGK1 may play an important role in the regulation of FoxO1 in Sin1−/− T cells. Interestingly, like our previous observation in pro-B cells [13], we observed a significant increase in FoxO1 expression in Sin1−/− T cells. These data raise the possibility that Sin1 may regulate FoxO1 expression although the exact mechanism through which this regulation occurs is currently unclear.
We have also determined if Akt mediates the Sin1-mTORC2 signals to regulate the development of thymic nTreg cells by examining the nTreg development in Akt1−/−, Akt2−/− and Akt1−/−Akt2−/− mice. We had previously used a similar experimental approach to identity Akt2 as the specific mediator of mTORC2 dependent FoxO1 regulation in B cells [13]. Disruption of Akt1, Akt2 or both Akt1 and Akt2 did not alter the proportion of CD4+ thymic nTreg cells when compared to wild type mice. Therefore, it is possible that either Akt3 is the principle mediator of mTORC2 dependent FoxO1 regulation or, alternatively, FoxO1 may be inhibited by the serum & glucocorticoid dependent kinases.
We also explored the function of Sin1 in CD4+ T helper cell differentiation. We did not observe any deficiency in the ability of Sin1−/− CD4+ T cells to differentiate into TH1, TH2 or TH17 effector cells. These data also differ from the results reported in rictor−/− T cells from two different groups [12, 23]. Lee and colleagues reported that Rictor deficient CD4+ T cells show impaired TH1 and TH2 differentiation while Delgoffe and colleagues only observed a deficiency in TH2 differentiation in rictor−/− T cells. Lee et al also report that PKC phosphorylation is deficient in rictor−/− T cells and that ectopic expression of PKCθ rescues TH2 differentiation in rictor−/− T cells. Interestingly, we observe that PKC-HM phosphorylation is deficient in Sin1−/− T cells however we failed to observe a deficiency in TH2 differentiation in Sin1−/− T cells. It is possible that the disparity between our data and those observed in rictor−/− T cells could be partially due to differences in the in vitro experimental conditions used to induce TH cell differentiation in the three studies. Alternatively it is possible that Rictor may also influence TH cell differentiation through a mechanism which is independent of mTORC2. Analysis of the roles Rictor and Sin1 in the context of a physiologic T cell immune response should resolve these issues.
Our observation that Sin1 deficiency in T cells results in an increased proportion of thymic Treg cells is consistent with previous studies linking mTOR and FoxO transcription factors to regulatory T cell differentiation. Surprisingly however, we observed that peripheral Sin1−/− CD4+ T cells gave rise to fewer FoxP3+ cells when stimulated in the presence of TGF-β. The unexpected finding that Sin1−/− T cells had slightly decreased TGF-β dependent Treg differentiation suggests that Sin1 may regulate Treg development independent of mTORC2 function. It is possible that Sin1 may regulate TGF-β dependent Treg differentiation through the MAPK signaling pathway [28]. In this regard, we have recently shown that deletion of MEKK2/3, which bind to and are negatively regulated by Sin1, augments TGF-β dependent Treg differentiation [29]. Future investigations into the role of Sin1-MAPK signaling in T cells will help elucidate the mechanism underlying this phenotype.
Acknowledgments
We thank Drs. A Lorenzo and W Sessa (Yale University) for the Akt1 and Akt2 knockout mice, KM Shokat (UCSF) for providing the pp242. This work is supported in part by grant AI063348 (NIH) (to BS). ASL is a Leukemia and Lymphoma Society fellow, and XC was a recipient of Gershon and Trudeau Fellowship from Yale University.
Footnotes
Conflict of Interest Statement
The authors claim no conflict of interest involved in this study.
Human Studies
No human research was conducted in this study.
References
- 1.Wullschleger S, Loewith R, Hall MN. TOR Signaling in Growth and Metabolism. Cell. 2006;124:471. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 2.Mondinoa A, Mueller DA. mTOR at the crossroads of T cell proliferation and tolerance . Molecular Mechanisms Supporting Peripheral T cell Tolerance: Potential Therapeutic Approaches to Autoimmunity and Allograft. Rejection. 2007;19:162–172. doi: 10.1016/j.smim.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuo CJ, Chung J, Fiorentino DF, Flanagan WM, Blenis J, Crabtree GR. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature. 1992;358:70–73. doi: 10.1038/358070a0. [DOI] [PubMed] [Google Scholar]
- 4.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–4748. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 5.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 7.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 8.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C, Lowry C, Newton AC, Mao Y, Miao RQ, Sessa WC, Qin J, Zhang P, Su B, Jacinto E. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. Embo J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, Sabatini DM. mTOR Complex 1 Regulates Lipin 1 Localization to Control the SREBP Pathway. Cell. 146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32:743–753. doi: 10.1016/j.immuni.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lazorchak AS, Liu D, Facchinetti V, Di Lorenzo A, Sessa WC, Schatz DG, Su B. Sin1-mTORC2 suppresses rag and il7r gene expression through Akt2 in B cells. Mol Cell. 2010;39:433–443. doi: 10.1016/j.molcel.2010.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Zhang F, Chen F, Liu D, Zheng Y, Zhang Y, Dong C, Su B. MEKK3 regulates IFN-gamma production in T cells through the Rac1/2-dependent MAPK cascades. J Immunol. 2011;186:5791–5800. doi: 10.4049/jimmunol.1002127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Chang X, Facchinetti V, Zhuang Y, Su B. MEKK3 is essential for lymphopenia-induced T cell proliferation and survival. J Immunol. 2009;182:3597–3608. doi: 10.4049/jimmunol.0803738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmes R, Zuniga-Pflucker JC. The OP9-DL1 system: generation of T-lymphocytes from embryonic or hematopoietic stem cells in vitro. Cold Spring Harb Protoc. 2009 doi: 10.1101/pdb.prot5156. pdb prot5156. [DOI] [PubMed] [Google Scholar]
- 18.Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol. 2010;11:618–627. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- 19.Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, DePinho RA, Hedrick SM. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O’Connor E, Shokat KM, Fisher AG, Merkenschlager M. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juntilla MM, Wofford JA, Birnbaum MJ, Rathmell JC, Koretzky GA. Akt1 and Akt2 are required for alphabeta thymocyte survival and differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12105–12110. doi: 10.1073/pnas.0705285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Juntilla MM, Wofford JA, Birnbaum MJ, Rathmell JC, Koretzky GA. Akt1 and Akt2 are required for alphabeta thymocyte survival and differentiation. Proc Natl Acad Sci U S A. 2007;104:12105–12110. doi: 10.1073/pnas.0705285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ouyang W, Beckett O, Flavell RA, Li MO. An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity. 2009;30:358–371. doi: 10.1016/j.immuni.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 27.Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell. 2008;30:701–711. doi: 10.1016/j.molcel.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 28.Cheng J, Zhang D, Kim K, Zhao Y, Su B. Mip1, an MEKK2-interacting protein, controls MEKK2 dimerization and activation. Mol Cell Biol. 2005;25:5955–5964. doi: 10.1128/MCB.25.14.5955-5964.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang X, Liu D, Wang X, Lin A, Zhao H, Su B. The Kinases MEKK2 and MEKK3 Regulate Transforming Growth Factor-[beta]-Mediated Helper T. Cell Differentiation. 2011;34:201–212. doi: 10.1016/j.immuni.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]