

Abstract
Prevention of HIV-1 transmission at mucosal surfaces will likely require durable pre-existing mucosal anti-HIV-1 antibodies (Abs). Defining the ontogeny, specificities and potentially protective nature of the initial mucosal virus-specific B-cell response will be critical for understanding how to induce protective Ab responses by vaccination. Genital fluids from patients within the earliest stages of acute HIV-1 infection (Fiebig I–VI) were examined for multiple anti-HIV specificities. Gp41 (but not gp120) Env immunoglobulin (Ig)A Abs were frequently elicited in both plasma and mucosal fluids within the first weeks of transmission. However, shortly after induction, these initial mucosal gp41 Env IgA Abs rapidly declined with a t1/2 of ∼2.7 days. B-cell-activating factor belonging to the TNF family (BAFF) was elevated immediately preceding the appearance of gp41 Abs, likely contributing to an initial T-independent Ab response. HIV-1 transmission frequently elicits mucosal HIV-1 envelope-specific IgA responses targeted to gp41 that have a short half-life.
Introduction
Immunoglobulin (Ig)A antibodies (Abs) are a first line of immune protection for many viral infections at mucosal surfaces. The mechanisms by which mucosal IgA is induced and how innate immune recognition of pathogenic microorganisms have a role in Ig class switch recombination are an area of intensive study that ultimately will inform HIV-1 vaccine design1. There is a resurgence of interest in the potential role of pre-existing mucosal IgA Abs in protection against HIV-1. Recent studies in non-human primates indicate that Abs to gp41 (ref. 2) and in particular IgA Abs to gp41 (ref. 3), if pre-existing, may have important functional roles in protection from infection. In acute HIV-1 infection (AHI), humoral responses arise too late to be effective in the control of HIV-1 replication,4 and there is significant impairment of the B-cell response (reviewed in refs 5 and 6). HIV-1 transmission results in rapid turnover of CD4+ T lymphocytes7 and specific depletion of CCR5+ CD4+ T cells in the gut.8, 9, 10 Moreover, HIV-specific CD4+ T cells are preferentially infected and lost during infection.11, 12 Thus, T cell help for maturation of the B-cell response is likely limited during the early stages of HIV-1 infection. HIV-1 also specifically causes deficiencies in B-cell function,13, 14, 15 and viral cofactors such as the viral envelope glycoprotein gp120 (refs 16 and 17) and Nef18, 19 have been proposed to mediate B-cell dysfunction in HIV-1 infection. A cytokine storm early in AHI20 may also account for some aspects of B-cell dysfunction.
There is a narrow window of vulnerability after virus exposure that could allow Abs with antiviral function to inhibit HIV-1 at mucosal surfaces.21, 22 Pre-existing HIV-1-specific mucosal Abs present at the time of transmission could block HIV-1 acquisition. The mechanisms by which Abs can inhibit HIV-1 movement across the mucosal barrier include direct virus neutralization, viral aggregation, inhibition of transcytosis, intra-epithelial neutralization, phagocytosis, inhibition through mucus, and Fc receptor-mediated neutralization (Ab-dependent cellular cytotoxicity) (reviewed in refs 23 and 24). Analyses of the initial mucosal Ab responses to HIV-1 infection will enable a more complete understanding of the effects of HIV-1 on the B-cell arm of the immune response. Moreover, mucosal Abs that are easily elicited by HIV-1 and target the HIV-1 Env may be a potential target for vaccine design if the durability of the response can be improved. Antibodies that are easy to elicit and do not require the same extensive level of somatic hypermutation thought to be required for broadly neutralizing Abs24 would, if protective, be ideal as targets for HIV-1 vaccine immunogen design. Thus, understanding the properties of the initial IgA response in the mucosa following HIV-1 transmission should enable a better understanding of how to best elicit durable humoral immunity for preventing HIV-1 acquisition and/or virus replication.
Here we report that IgA Abs to gp41 Env arise initially in acute infection and are frequently detected in genital fluids. Mucosal IgA Abs to gp120 Env and to other parts of HIV-1 arise later during acute infection. Moreover, we examined potential regulators of the initial Ab response and found that B-cell-activating factor belonging to the TNF family (B-cell-activating factor (BAFF)) was elevated preceding the spike in anti-HIV-1 Abs. This early BAFF elevation coupled with the lack of a robust and sustained mucosal HIV-1-specific IgA response is consistent with an initial HIV-1-specific Ab response that is T independent. Thus, the initial mucosal Env Ab response is limited in specificity to gp41 during the early stages of infection and has a short half-life.
Results
Frequency, specificity and timing of anti-HIV-1 mucosal Abs during AHI
To determine if mucosal Abs to the HIV-1 envelope arise within the first few months of AHI and whether mucosal Abs to other regions of HIV-1 are elicited during AHI, we examined the frequency of detection of IgA and IgG responses to the HIV-1 envelope proteins, gp41 and gp120 (Figure 1a and c); and to HIV-1 antigens Gag, Tat, Nef, Integrase (p31), and reverse transcriptase (RT) (Figure 1b and d) in up to 30 patients (N=170 samples) for plasma and up to 23 patients (N=119 samples) for mucosal samples (Supplementary Table S1 online). HIV-1 gp41-specific IgA was detected in the genital secretions of 20/23 (87%) acutely infected individuals and plasma gp41-specific IgA was present in 29/30 (97%) of AHI patients, all of whom were positive for HIV-1 gp41 IgG in both plasma and genital secretions. Plasma HIV-1 gp41-specific IgM was frequently detected in 25/28 (89%) patients; however, only about half of acute patients had detectable mucosal gp41-specific IgM (11/21, 52%). Additionally, the vast majority of patients had gp41 IgG and IgA Abs that were specific for the immunodominant epitope (Figure 1c); whereas, there were no detectable membrane proximal external region (MPER) epitope-specific responses above 1 ng/ml of mucosal fluid. Anti-gp120 IgA Abs were detected at later stages of acute infection in plasma and mucosal samples and they were less frequently detected than gp41 Env-specific Abs. Mucosal gp120 IgA was present in 27% of AHI patients, and mucosal gp120 IgG was present in 40% of AHI patients.
Figure 1.

Frequent detection of anti-gp41 mucosal immunoglobulin (Ig)A, but infrequent detection of mucosal anti-gp120 IgA within 133 days from enrollment. Frequency of HIV-specific antibodies in (a, b) plasma in 14–30 CHAVI 001 subjects and (c, d) mucosal sites in 13–23 CHAVI 001 subjects within 133 days of enrollment. IgG and IgA specific for the immunodominant epitope of gp41 were also tested in mucosal samples from 12 individuals. nt, not tested.
To determine whether other HIV-1 proteins, in addition to the HIV-1 Env, stimulated a humoral response in acute infection, we assessed Ab responses to Gag, p66 RT, p31 Integrase, Tat, and Nef. The majority of patients developed systemic and mucosal IgG and IgA responses to Gag and p66 RT, whereas IgG and IgA Ab responses to p31 Integrase, Tat, and Nef were less commonly detected by ∼133 days post enrollment (Figure 1b and d).
To evaluate the timing of the appearance of HIV-specific IgA during AHI, we determined the frequency of plasma (Table 1a) and mucosal (Table 1b) IgA specificities by Fiebig stage.25 We evaluated plasma samples from 30 Center for HIV/AIDS Vaccine Immunology (CHAVI) 001 subjects (most were enrolled during stage 3 or later) and 25 plasma donors (Fiebig stages I/II onward until sample collection stopped). In plasma, gp41 IgA and Gag IgA were present in 26.1% and 8.7% of subjects during Fiebig stages I/II, respectively. RT- and Nef-specific IgA were not detected in samples obtained during Fiebig stages I/II and were more commonly detected in samples from stages IV–VI. HIV gp120- and p31-specific IgA were less frequent even at later stages V/VI. In mucosal samples from 23 CHAVI 001 subjects, we observed that gp41, Gag-, and gp120-specific IgA were first detected during Fiebig stage IV and were more frequently detected during Fiebig stages V/VI. RT-, Nef-, and p31-specific IgA were only detected in samples obtained during Fiebig stages V/VI. In the HIV-1 seroconversion plasma donor cohort, we also examined the time since T0 (plasma viral RNA=100 copies/ml)4, 20, 26 for the appearance of IgA Abs. During Fiebig stages I–VI, in those plasma donors with detectable HIV-1-specific plasma IgA, the median time to appearance of gp41-specific IgA was 13.5 days post T0 (range of 9–18 days), whereas the median time for the appearance of Gag-specific IgA was 25.5 days post T0 (range of 14–40 days) (Table 1c). Gp120 IgA Ab responses were not detected in these samples due to the early Fiebig staging in this cohort, consistent with the lack of early IgG gp120 responses we previously reported.4 Taken together, these data show that HIV-specific IgA is frequently present in the genital tract, as well as, present systemically during AHI. The initial Ab specificities include predominantly the immunodominant epitope in gp41, whereas Abs to gp120 and other HIV-1 proteins appear later in some individuals.
Table 1. Timing of systemic and mucosal gp41-specific IgA antibodies.
|
gp41 |
Gag |
RT |
Nef |
gp120 |
P31 |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fiebig stage | No./totala | % | No./total | % | No./total | % | No./total | % | No./total | % | No./total | % |
| (a) Plasma IgA | ||||||||||||
| I/II | 6/23 | 26.1 | 2/23 | 8.7 | NT | — | NT | — | NT | — | NT | — |
| III | 7/14 | 50.0 | 3/14 | 21.4 | NT | — | NT | — | NT | — | NT | — |
| IV | 15/17 | 88.2 | 10/17 | 58.8 | 3/7 | 42.9 | 3/7 | 42.9 | 2/8 | 25.0 | 0/7 | 0.0 |
| V/VI | 39/40 | 97.5 | 35/40 | 87.5 | 13/15 | 86.7 | 8/14 | 57.1 | 11/29 | 37.9 | 2/14 | 14.3 |
| (b) Mucosal IgA | ||||||||||||
| I/II | 0/1 | 0 | 0/1 | 0 | NT | — | NT | — | NT | — | NT | — |
| III | 0/2 | 0 | 0/2 | 0 | NT | — | NT | — | NT | — | NT | — |
| IV | 7/21 | 33.3 | 4/8 | 50.0 | 0/7 | 0 | 0/7 | 0 | 1/7 | 14.3 | 0/7 | 0 |
| V/VI | 18/23 | 78.3 | 17/23 | 73.9 | 5/14 | 35.7 | 1/14 | 7.1 | 4/14 | 28.6 | 1/14 | 7.1 |
|
(c) | ||
|---|---|---|
| Antigen | No. of subjectsb | Median time to antibody response (range (days))c |
| gp41 | 14 | 13.5 (9–18) |
| p55 | 6 | 25.5 (14–40) |
Abbreviations: Ig, immunoglobulin; NT, not tested.
(a) Frequency of HIV-1-specific plasma IgA responses per Fiebig25 stage in up to 30 CHAVI and 25 plasma donor patients is shown. Frequency was calculated by the number of plasma samples positive per each Fiebig stage over the total number plasma samples measured in that stage. (b) Frequency of HIV-specific mucosal IgA responses were measured in mucosal samples per Fiebig stage from up to 23 acute HIV-1 infection (AHI) patients. (c) Systemic HIV-1 gp41-specific IgA appears before anti-Gag IgA antibodies. HIV-specific IgA responses were measured in 25 HIV+ seroconversion plasma donor panels by standard enzyme-linked immunosorbent assay and aligned to T0 (the time at which viral load is first detectable), as previously described.
Number of subjects having an antigen-specific IgA positive sample in the designated Fiebig stage divided by the total number of subjects in that designated Fiebig stage.
Number of plasma donor subjects with positive IgA response.
Median time from T0 (the first day the viral load reaches 100 copies/ml).
HIV-1-specific mucosal Ab concentrations
To examine the concentrations of HIV-1-specific IgA responses in both plasma and mucosal samples following HIV-1 transmission, we evaluated HIV-1-specific Ab responses in the CHAVI 001 AHI cohort that were enrolled during AHI and followed for up to 133 days after enrollment.6, 27 Mucosal Ab concentrations were measured in 64 samples from 12 patients (seminal plasma from nine males and cervicovaginal lavage from three females) to quantify HIV-1-specific Ab concentrations relative to total (non-specific) Ab concentrations. During AHI, HIV-1-specific IgG and IgA were both frequently detected in systemic and mucosal compartments, and the observed peak concentrations of anti-gp41 Abs (IgG, IgA, and IgM) (Table 2) were measured during Fiebig stage III through V/VI. On an average, there were over two logs more HIV-1 gp41-specific IgG in the plasma compared with IgA at the peak of the response during AHI. This was partly reflective of the difference in the total non-HIV-specific levels of IgG/IgA that is present in plasma (mean 43.8 mg/ml of IgG vs. 4.9±mg/ml IgA, an 8.9-fold difference). In the genital secretions, gp41-specific IgG was on an average 11-fold higher than gp41-specific IgA (Table 2). However, total IgG and IgA levels within the mucosal samples that we examined were more similar to one another (168±STDEV 209.5 μg/ml of total IgG vs. 133.2±STDEV 278.2 μg/ml of total IgA); consistent with normal Ab ranges28 and consistent with previous reports that demonstrated that, unlike intestinal fluid containing high levels of SIgA, semen and cervicovaginal fluid contain more IgG than IgA.29 As expected, in mucosal secretions total IgM levels were lower than total IgG and IgA. In those samples with HIV-1-specific IgM, the specific activity levels are higher than the IgG and IgA measurements, in part, due to the lower concentrations of total IgM (mean 10.2 μg/ml±STDEV 11.7 μg/ml) that were present. In addition, there were no significant differences in total or gp41-specific IgG and IgA in plasma or genital fluid collections between males and females from this study (Supplementary Figure S1 online). Taken together, the levels of HIV-1 Env-specific IgA in genital fluids during acute infection were low compared with HIV-1 Env-specific IgG.
Table 2. Peak concentration of HIV-1 gp41-specific antibodies in plasma and mucosal samples observed during Fiebig stage III through V/VI of AHI.
|
Plasma gp41-specific Ab (μg/ml) |
Mucosal gp41-specific Ab (μg/mg) |
|||||
|---|---|---|---|---|---|---|
| Number positive (n) | Median (range) | Mean±s.d. | Number positive (n) | Median (range) | Mean±s.d. | |
| IgG | ||||||
| All | 23 | 546.3 (5.4–5194.8) | 1242.4±1485.8 | 12 | 35.6 (2.4–444.9) | 102.7±133.7 |
| Male | 16 | 1300.1 (5.4–5194.8) | 1618.8±1632.8 | 9 | 32.5 (2.4–444.9) | 111.2±151.8 |
| Female | 7 | 353.5 (128.8–1761.1) | 555.4±554.8 | 3 | 72.1 (10.6–148.1 ) | 76.0±68.9 |
| IgA | ||||||
| All | 23 | 12.5 (0.7–41.2) | 12.5±9.4 | 12 | 4.4 (0.4–52.4) | 9.2±14.2 |
| Male | 16 | 13.0 (0.7–41.2) | 14.4± 10.3 | 9 | 9.0 (0.7–52.4) | 11.6±15.7 |
| Female | 7 | 4.8 (1.2–13.0) | 7.0±4.6 | 3 | 0.8 (0.4–3.2) | 1.4±1.5 |
| IgM | ||||||
| All | 12 | 7.9 (0.3–26.7) | 9.8±8.0 | 5 | 26,939 (15,110–112,989) | 40,719±40,780 |
| Male | 10 | 6.7 (0.3–13.7) | 6.9±4.2 | 5 | 26,939 (15,110–112,989) | 40,719±40,780 |
| Female | 2 | 24.8 (22.9–26.7) | 24.8±2.7 | 0 | — | — |
Abbreviations: Ab, antibody; AHI, acute HIV-1 infection; Ig, immunoglobulin.
Peak plasma and mucosal HIV-1 gp41-specific antibody levels were determined in CHAVI 001 patients during the first 133 days post enrollment. Concentration is expressed in μg/ml equivalents determined with a standard curve using 2F5 IgG, IgA, or IgM. Mucosal antibody levels are expressed as specific activity (μg of gp41-specific antibody/mg of total antibody).
Differences in systemic and mucosal IgA and IgG concentrations and specificities
To examine whether the HIV-1-specific mucosal Ab response was likely derived locally or transudated from plasma, we compared the levels of HIV-specific Abs in plasma and genital tract compartments across all time points within the first 133 days post enrollment. We first compared the antigen-specific Ab responses in plasma to those in mucosal samples (Figure 2a and b). Of all comparisons, systemic and mucosal gp41-specific IgG most strongly correlated to one other (Figure 2a), in contrast to gp41-specific IgA (Figure 2b) that correlated weakly (R=0.322, P=0.040), suggesting that some IgA may be locally produced in the mucosal compartment. We then examined IgG vs. IgA in plasma and mucosal samples (Figure 2c and d) to determine whether any correlations existed between IgG and IgA levels in either compartment. Plasma HIV-1 gp41 Env IgG and IgA correlated (R=0.532, P<0.0001) (Figure 2c). However, mucosal gp41 Env-specific IgG and IgA responses correlate weakly (Figure 2d) (R=0.366 and R=0.017). As noted by the circles, Ab concentrations in numerous samples lined one axis or the other (not on an x=y line). These data indicate that some HIV-1-specific Ab responses present at mucosal sites represent transudate. However, the discordance between mucosal and systemic HIV-specific Abs suggests that some fraction of the measured HIV-1-specific mucosal response may be locally produced. Thus, measurement of plasma HIV-1 Env IgA does not entirely reflect the level or specificity of mucosal HIV-1 Env IgA.
Figure 2.

Difference in systemic and mucosal immunoglobulin (Ig)A and IgG concentrations and specificities. Correlations of gp41 Env-specific (a) IgG and (b) IgA in genital secretions vs. plasma during acute HIV-1 infection (AHI). Correlations of (c) plasma and (d) mucosal levels of HIV-1 Env-specific IgG vs. IgA during AHI. Specific activity in mucosal samples is calculated as μg of HIV-specific antibody (Ab) per mg total Ab. Concentrations of anti-gp41 Ab calculated per 2F5 monoclonal Ab equivalents and concentrations of anti-gp120 Abs are calculated as the binding units in the linear range multiplied by the dilution factor divided by total Ab concentration.
Short half-life of the mucosal anti-gp41 Env IgA in AHI
To address the question of whether the initial mucosal Ab response to HIV-1 infection is transient and therefore may have been difficult to detect previously30, we examined 12 CHAVI 001 patients longitudinally (out to 133 days post enrollment) (Figure 3) to determine the kinetics of the HIV-1-specific IgA and IgG responses in both plasma (Figure 3a and c) and mucosal compartments (Figure 3b and d). To normalize for changes in total Ab concentrations, specific activity (HIV-1 gp41-specific Ab/total Ab) was determined for each mucosal sample. Although mucosal HIV-1-specific IgA responses were detected frequently in AHI, there was an early peak and subsequent decline during the later phase of acute infection in 11 out of 12 patients (91.7%). This was in contrast to the predominantly increasing or steady mucosal gp41 IgG response. Likewise, of the 12 patients that we studied with matching longitudinal plasma and mucosal samples, 10 (83.3%) of these patients had declining gp41-specific IgA in the plasma (Figure 3e).
Figure 3.

Rapid decline in mucosal HIV-specific immunoglobulin (Ig)A in acute HIV-1 infection subjects. HIV-1-specific IgG and IgA antibody concentrations are shown for two representative subjects with (a, c) paired plasma and (b, d) genital fluid (seminal plasma and cervicovaginal lavage (CVL). (a) Plasma and (b) mucosal HIV-specific IgM is shown for one subject. (e) Mucosal HIV gp41-specific IgA kinetics in 11 patients aligned to the peak response.
We applied an exponential decay model31 to determine the Ab half-life of gp41-specific IgA in the plasma and mucosal compartments during AHI among individuals with at least a 2-fold decrease in Ab response (Table 3). The model that fit best for the mucosal samples assumes a lower asymptote greater than zero (Ab responses plateaus at a non-zero level), whereas the model for the plasma samples assumes that the lower asymptote is zero (Ab response eventually declines all the way to zero). Although the half-life of plasma gp41-specific IgA was much longer (48.19 days (95% confidence interval (CI)=34.57–61.81)) than the half-life of mucosal IgA (2.71 days (95% CI=2.06–3.36)), the fold decline (the delta from peak to nadir) of HIV-1-specific IgA was similar in mucosal (6.20-fold (95% CI=−0.51, 12.92) and plasma (8.65-fold (95% CI=3.38–13.93) samples.
Table 3. Half-life estimates for initial gp41 IgA decline in AHI (exponential decay model) for individuals with at least a 2-fold decrease in antibody response.
|
HIV-1 gp41 IgA |
||||
|---|---|---|---|---|
|
Half-life (days) |
Fold decreasea |
|||
| Estimate | 95% CI | Estimate | 95% CI | |
| Plasma (N=10) | 48.19 | 34.57, 61.81 | 8.65 | 3.38, 13.93 |
| Mucosal (N=11) | 2.71 | 2.06, 3.36 | 6.20 | −0.51, 12.92 |
AHI, acute HIV-1 infection; CI, confidence interval; Ig, immunoglobulin.
The model that fit best for the mucosal samples assumes a lower asymptote greater than zero (antibody response plateaus at a non-zero level), whereas the model for the plasma samples assumes that the lower asymptote is zero (antibody response eventually declines all the way to zero).
At lower asymptote for mucosal and 150 days post peak for plasma.
Confirmation of short half-life of anti-gp41 Env IgA in plasma in additional AHI cohorts
To determine whether the overall systemic gp41-specific IgA decline in the CHAVI 001 patients was common to other acute infection cohorts, we examined additional patients for whom we had longitudinal samples available for study in the CHAVI 001 cohort and, as well, compared Ab kinetics to that observed in two other cohorts: the plasma donor cohort4 and Trinidad cohort32, 33. In sequential plasma samples from 44 patients, we found that 28 patients demonstrated discordant gp41-specific IgA and IgG responses (Table 4), in that, gp41-specific IgA responses declined whereas gp41-specific IgG responses either remained stable or increased over time. The kinetics of HIV-1-specific IgG and IgA in the plasma donor cohort are shown in Figure 4, where the HIV-1-specific IgG response appears at a median time of 13 days post T0 (ref. 4) and continues to rise whereas gp41-specific IgA declined. These data demonstrate that the HIV-1 gp41-specific IgA decline that we observed in the CHAVI 001 cohort could be confirmed in additional AHI cohorts.
Table 4. Decline in systemic gp41-specific IgA in multiple cohorts.
| No. of subjects | |||
|---|---|---|---|
|
Cohort |
No. Samples per subjecta |
IgG+/IgA+b |
IgA↓c |
| Plasma donor | 4–29 | 14 | 6 |
| Trinidad | 8–17 | 7 | 6 |
| CHAVI 001 | 4–10 | 23 | 16 |
| Total | 44 | 28 | |
Abbreviation: Ig, immunoglobulin.
The number of longitudinal samples per subject analyzed over the course of acute infection are indicated.
The number of subjects with at least four time points that were tested that had detectable anti-gp41 IgG and IgA to measure decline are indicated.
Number of subjects with increasing/stable IgG and ≥2.5-fold decline in IgA from peak level.
Figure 4.

Initial decline in anti-gp41 immunoglobulin (Ig)A, but not anti-gp41 IgG during acute HIV-1 infection. Differential kinetics of systemic HIV gp41-specific IgA and IgG antibodies following HIV-1 transmission in plasma donor subjects aligned by T0 (plasma viral RNA=100 copies/ml) in acute infection. OD, optical density.
Short half-life of IgA Abs in AHI is specific to HIV-1 IgA
The decline in gp41-specific IgA that we observed during AHI suggested that HIV-1 does not stimulate long-lived HIV-1-specific IgA+ B cells. To determine whether this was a generalized downregulation of all IgA responses, we also examined memory IgA responses specific to tetanus toxoid and influenza antigens from vaccination (Figure 5a–c). Tetanus toxoid and influenza IgA Ab responses could not be fit to the exponential decay model and did not decline during AHI during the time of gp41 IgA Ab decline. Moreover, the average levels of total plasma IgA and IgG within this cohort did not decline during acute infection. These data imply that the decline in the IgA response is specific for HIV-1 and is not due to a general downregulation of all memory IgA responses; rather, the short half-life of HIV-1-specific IgA in AHI is more likely due to Env stimulation of a short-lived B-cell response to HIV-1.
Figure 5.

Immunoglobulin (Ig)A decline is specific for HIV-1 infection. During acute infection HIV-1 gp41-specific antibodies (a) decline over time, in contrast to IgA antibodies specific for (b) tetanus toxoid and (c) influenza (Fluzone). All antibody responses are plotted as a percentage of levels measured at the peak of the gp41-specific IgA response.
Specific elevations in BAFF before initial HIV-1 Env Ab
To investigate potential mechanisms of HIV-1 antigen-specific IgA decline, we measured the plasma and mucosal levels of two cytokines, BAFF and APRIL (a proliferation inducing ligand), known to regulate B-cell survival and activation. We first measured longitudinal samples from seronegative donors and found that BAFF levels typically remained stable under 1 ng/ml over time (N=7 donors, 10–12 time points each, from up to 60–120 days). We then examined plasma samples from acute hepatitis B virus (HBV) (N=10) and acute hepatitis C virus (HCV) (N=10) infection and found that levels of BAFF were also stable during the acute phase of these infections, remaining at levels similar to those found in uninfected patients whereas viremia increased (Figure 6a). We then examined levels of plasma BAFF in AHI (N=20) and found that there was a peak in the levels of BAFF, above 1 ng/ml, soon after HIV-1 transmission that preceded the peak in the HIV-1-specific Ab response (Figure 6b). Elevations in BAFF were also observed in mucosal fluid near the time of the initial HIV-1-specific Ab response (N=5 cervicovaginal lavage and N=4 longitudinal seminal plasma samples) (Figure 6c). Levels of APRIL in these same patients also showed an increase at similar times to that of BAFF, although the levels of APRIL were generally higher (peaking at 2–12 ng/ml). The transient upregulation of APRIL during this period was specific to AHI, as there were not similar elevations in APRIL in acute HBV and HCV plasma donor panels at similar time points. To determine whether higher transient levels of BAFF could be a consequence of higher T-cell activation and IFNγ production due to viral replication during AHI, we examined whether plasma BAFF and APRIL correlated with viral load (Figure 6d and e). BAFF levels, but not APRIL, were weakly correlated with viral load. HIV-1 gp41 Env IgA Ab levels did not correlate with viral load (R=−0.140, P=0.174). These data suggest that the levels of BAFF, APRIL and HIV-1-specific IgA do not directly reflect increased virus replication and immune activation in these patients.
Figure 6.

B-cell-activating factor (BAFF) is elevated before antibody production in acute HIV-1 infection, but is not elevated during acute hepatitis B virus (HBV) and hepatitis C virus (HCV) infections. (a) Plasma panels from subjects with acute HBV infection (N=10 donors, 10–12 time points each, up to 50 days)) and acute HCV infection (N=10 donors, 7–15 time points each, up to 50 days) were measured for levels of BAFF. Data from one representative subject infected with each virus are shown. Sample time courses are aligned relative to the time when virus first reached detectable levels in plasma (200 DNA copies/ml for HBV (T200), 600 RNA copies/ml for HCV (T600)). (b) An elevation in plasma BAFF levels precedes the rise in anti-HIV immunoglobulin (Ig)G and IgA antibody levels during acute HIV-1 infection. (N=21 donors, 8–22 time points each up to 40 days post T0 were tested for BAFF; antibody levels were also detected in 9 of these; data from two representative subjects is shown). (c) Elevation in BAFF levels during the time of HIV-specific antibody elevations in mucosal samples. Longitudinal cervicovaginal lavage samples (N=5 patients) and seminal plasma (N=4 patients) were tested (data from two representative individuals is shown). Correlation between plasma (d) BAFF and (e) APRIL (a proliferation inducing ligand) levels with viral load (VL). Data from patient (outlier) are circled and when these points are removed, R=0.27, P=0.0032. OD, optical density.
Discussion
In mucosal samples collected within weeks of HIV-1 transmission, we show that mucosal IgA HIV-1-specific Abs were frequently detected (87%) in acute infection, albeit at relatively low levels compared with anti-Env IgG. Similar to the initial plasma IgG response to gp41,4 and the initial plasma B-cell response to gp41 (ref. 34), we demonstrate here that the initial mucosal Ab response is also directed to gp41 Env. The reason for the focus of the initial Ab response to gp41, rather than to gp120, is not fully understood. However, we have previously reported34 that the initial gp41 IgG Ab response can be derived from pre-existing cross-reactive memory B cells that are further driven by HIV-1 infection. Previous reports indicated that there was a paucity of HIV-1-specific IgA responses in HIV-1 infection;30 however, the HIV-1 antigens used and the later timing during acute infection of the samples likely accounted for the low detection of Ab responses in prior studies. Moreover, previous studies have reported the detection of high frequencies of HIV-1-specific IgA responses in seminal plasma from chronically infected patients.35, 36 In agreement with this, we found that in the majority of patients, gp41-specific IgA responses peaked and then declined in concentration in both the mucosal and systemic compartments during AHI, whereas gp120 IgA appeared later. The ratio of HIV-1-specific IgA: IgG was relatively low in genital fluids; although there is a higher natural ratio of IgA: IgG in mucosal fluids compared with plasma. The mechanism for the relatively low HIV-1-specific IgA, compared with HIV-1-specific IgG, in mucosal fluids during HIV-1 infection is not well understood. The ratio could be reflective of contributions from plasma transudate or a result of mechanisms that cause defects in mucosal class switching, such as HIV-1 Nef-mediated inhibition of class switching to IgA37 or potentially destruction of T-regulatory cells important in the development of IgA+ mucosal B cells.38
A long-lived protective Ab response is highly desired for any vaccine and has been particularly problematic for HIV-1 vaccines; thus, measuring the Ab half-life of HIV-1-specific responses systemically and at mucosal sites is important for understanding the mechanisms to induce long-lived effective Ab responses. Here, we found that in acute infection, gp41 Env-specific IgA responses had a very short half-life both systemically and mucosally: ∼48 days in the blood and 2.7 days in the mucosal secretions during the acute phase of HIV-1 infection. The half-life of total IgA in the blood is ∼4–9 days,39 whereas the predominant IgG subclasses (IgG1, IgG2) have a longer half-life (21 days)40 due to the ability of IgG Abs to bind to FcRn, preventing lysosomal degradation.41
We previously demonstrated that in an HIV-1 vaccine trial and anti-retroviral-treated chronic HIV-1 infection, the half-life of IgG Env gp120 Abs is short compared with tetanus and influenza Abs, is dependent on specific antigen drive, and has the hallmark of being maintained by short-lived memory B cells rather than long-lived plasma cells.42 This finding that gp41-specific mucosal IgA responses have a remarkably shorter half-life (2.7 days) than gp41-specific IgG is extraordinary and suggests isotype-specific rapid catabolism of mucosal IgA. Also, these data suggest that long-lived plasma cells secreting HIV-1-specific IgA are not generated immediately following HIV-1 transmission. However, it is important to note that the HIV-1-specific IgA response does not decline to negative. In chronic HIV-1 infection, Env-specific IgA responses are present in both plasma and at mucosal sites such as breastmilk;43 thus, likely ongoing virus replication can stimulate HIV-1-specific IgA responses. Further studies are needed to quantify and compare the levels of HIV-1-specific secretory, monomeric, and dimeric IgA in mucosal secretions throughout infection and in response to HIV-1 vaccination.
T-cell-independent class switching can occur by activation of B cells by BAFF and APRIL secretion from dendritic cells.44, 45 We found that the cytokines BAFF and APRIL are elicited in plasma and mucosal secretions just before the initial rise in the HIV-1 Env-specific IgG and IgA responses during AHI. In acute and recent HIV-1 infection, naive B cells were significantly reduced relative to the total B-cell population in both the blood and terminal ileum.15 Scholz et al.46 demonstrated that inhibition of BAFF can result in decreased numbers of naive B cells and primary immune responses with little effect on memory B cells and long-lived plasma cells. One possibility for the short half-life of the anti-Env IgA response in acute infection is that although innate immune stimulation can release factors that promote B-cell class switching, this occurs through a T-cell-independent pathway.47 A subsequent T-cell-dependent pathway for HIV-1-specific Ab induction may be hampered due to the massive CD4 T-cell depletion that occurs early in infection.10, 48, 49 The transient peak in cytokines that stimulate B-cell responses, and the short half-life of HIV-1 Env IgA coupled with massive CD4 depletion and damage to mucosal generative B-cell environments15 in AHI are consistent with a T-independent pathway for the initial HIV-1 Ab response.
We recently reported on the concentrations and kinetics of the IgG3 subclass of the anti-HIV response during recent HIV-1 infection.31 Anti-gp41 IgG3 appears first among the different antigen-specific IgG3 responses, and the anti-gp41, anti-Gag and anti-RT IgG Abs decline with defined kinetics within the first year of infection, which could be useful as part of an algorithm for measuring HIV-1 incidence.31 The half-life estimates and concentrations of anti-gp41 IgA Abs systemically and in genital fluids during acute infection, as reported here, are additional measurements that have potential as part of an HIV-1 incidence algorithm. There is precedence for the utilization of pathogen-specific IgA in tests for recent infection, such as in the case of dengue,50 which utilizes the short half-life of IgA for determining recent infection. Additional characterization of HIV-1-specific IgA responses at later stages in HIV-1 infection is needed to determine whether the IgA profile can distinguish recent from chronic HIV-1 infection.
The protective properties (e.g., inhibition of transport through mucus, Ab-dependent cellular cytotoxicity, neutralization, phagocytosis) of mucosal IgA Abs with different HIV-1 specificities have not been fully delineated. However, some studies of persistently highly HIV-1 exposed but uninfected individuals demonstrated that HIV-1-specific mucosal Abs could be detected, and in some cases, neutralizing IgA responses were found in both sera and mucosal fluids in the absence of detectable HIV-1-specific IgA responses.51, 52, 53, 54 In addition to having antiviral properties, antigen-specific IgA Abs may block some IgG functions.55, 56 Recent results from the RV144 trial57 raise the hypothesis that plasma anti-gp120 IgA Abs with specific epitope specificities may have potentially blocked anti-gp120 IgG Abs with functional activity. However, it is unknown whether mucosal IgA had a role in observed protection in RV144, as no mucosal samples were collected. In a recent report of a heterologous prime boost, rectal challenge study in non-human primates, rectal anti-Env IgG was among a number of variables that correlated with reduced acquisition risk.58 Moreover, IgA Abs with specificity for gp41 Env have been reported to have potentially protective functional properties;59, 60, 61 thus the focused specificity of IgA Abs to gp41 during acute infection suggests that these easy to elicit Abs at the mucosal surface could potentially have some protective effect if present before HIV-1 transmission and if a durable HIV-1 Env-specific B-cell response can be elicited by HIV-1 vaccination. We previously reported that the initial gp41-specific IgG Abs can capture infectious virions;62 however, additional work is needed to fully determine the spectrum of inhibitory capabilities of both systemic and mucosal IgA and IgG Abs induced in acute infection, including any potential effects of mucosal HIV-1 Abs on transmission.
Finally, our data highlight the need to determine the mechanisms that lead to the induction of long-lived effective mucosal antibody responses. Whereas vaccination for influenza63 and polio64 or infection with rotavirus65 can elicit IgA responses that can be protective, as yet it remains uncertain whether either infection or vaccination with HIV-1 Env can generate protective mucosal or systemic IgA responses.
Methods
Participants and specimen collection. HIV-1-infected individuals from three different acute infection cohorts were examined: 25 HIV-1+ plasma donors (Clade B),20 8 AHI patients from the Trinidad cohort (Clade B),66 and 30 AHI patients from the CHAVI 001 acute infection cohort (Clades B and C from United States, South Africa, and Malawi).4, 62, 67 HIV-1, HBV, and HCV acute infection plasma donor samples and uninfected control plasma donor samples were purchased from Zeptometrix (Franklin, MA).20 Paired blood and seminal plasma or ectocervicovaginal lavage fluids were obtained from the CHAVI 001 acute infection cohort.4, 62, 67 Seminal plasma samples were obtained from ejaculate collected directly into a refrigerated sterile cup containing 2.5 ml of viral transport medium (Roswell Park Memorial Institute medium, 1,000 U penicillin, 1 mg/ml streptomycin, 200 U/ml nystatin). After liquefaction, the sample was transferred to a centrifuge tube containing 2.5 ml of 2 × protease inhibitor buffer and the total volume and dilution factor were calculated. Semen was centrifuged at 800 g for 10 min (18–26 °C), and supernatant was aliquoted and stored (−80 °C). Ectocervicovaginal lavage fluids (cervicovaginal lavage) were obtained through repeated rinsing of the cervix and ectocervix with 10 ml total of saline or buffered saline. Fluid was then transferred to a sterile tube with 100 × protease inhibitor and centrifuged at 600–800 g for 10 min (18–26 °C) to remove cells. The supernatant, including any mucus, was aliquoted and stored at −80 °C. All work performed as part of this study was reviewed and approved by the institutional review boards of each participating center, Duke University Medical Center, and the Division of AIDS, NIH.
Specimen preparation for IgG removal. For detection of IgA and IgM Abs, IgG was removed using protein G columns, as previously described.4, 57
Binding Ab assay. Customized multiplex HIV-1-binding assays (Bio-Plex instrument (Bio-Rad, Hercules, CA)) were performed as previously described4 to determine IgG, IgA, and IgM responses specific for recombinant HIV-1 p55 or p24 Gag (BD Biosciences, San Jose, CA), recombinant HIV-1 gp41 MN (Immunodiagnostics, Woburn, MA), a previously described artificial multi-clade group M consensus gp120 Env protein (Con6 gp120) (ref. 68), HIV-1 p66RT (Protein Sciences, Meriden, CT), HIV-1 recombinant Nef (Genway, San Diego, CA), recombinant HIV-1 Tat (Advanced Bioscience, Kensington, MD), and recombinant HIV-1 p31 Integrase (Genway).4 To determine concentration of Abs binding to the 2F5 epitope, 2F5 monoclonal Abs (IgG, IgA, IgM) (Polymune Scientific, Vienna, Austria) were titrated on gp41-coupled beads to generate a standard curve. In addition, the following peptides (Primm Biotech, Cambridge, MA) were used: (gp41 immunodominant region, RVLAVERYLRDQQLLGIWGCSGKLICTTAVPWNASWSNKSLNKI), SP62 (gp41 MPER, QQEKNEQELLELDKWASLWN). Immunodominant and MPER tetramers were utilized for the detection of epitope-specific responses.69 HIV-1-binding Ab measurements and measurement of IgA Abs to tetanus toxoid and influenza (Fluzone 2007, Sanofi Pasteur) by standard enzyme-linked immunosorbent assay (ELISA) were performed as previously described.42 Total IgG and IgA Ab measurements for calculating specific activity were performed using Bio-Plex Pro human isotyping 7-plex panel (Bio-Rad) according to the manufacturer's instructions. The positivity criteria per HIV-1 antigen per Ab isotype was determined by screening ≥30 seronegative patients. A standardized HIV-1positive (HIV+) control was titrated on each assay (tracked with a Levy-Jennings plot with acceptance of titer only within 3 s.d's of the mean). Standard curves generated using titrated monoclonal Abs and concentrations were used to obtain a four-parameter logistic equation to determine concentrations of Ab in patient samples (SigmaPlot, Systat Software, Chicago, IL). The coefficient of variation per sample was ≤15%. Two negative sera and two HIV-1+control sera were included in each assay to ensure specificity and for maintaining consistency and reproducibility between assays.
BAFF/APRIL ELISA. Concentrations of BAFF and APRIL in plasma and mucosal fluids were measured in duplicate by colorimetric ELISA per manufacturer's instructions (Human BAFF Quantikine ELISA from R&D Systems, Abingdon, UK, and Human APRIL Platinum ELISA from eBioscience, Hatfield, UK).
Statistical analysis. Antibody half-life estimates were obtained with an exponential decay model (as previously described),31 based on aligning samples obtained on or after their observed peak plasma or mucosal IgA Ab response. Briefly, estimates for the Ab half-lives were determined by using the data at or post peak (the highest value) and then computing the half-life by fitting an exponential decay model from peak to the last time point. A single exponential decay model was applied to the data for the mucosal Ab responses:
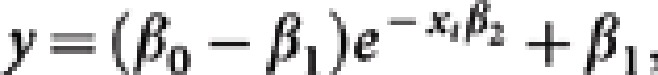 |
although a reduced exponential decay model was fit for the plasma antigens:
 |
Where, β0 is the level at time zero, xt is time, β1 is the lower asymptote, and β2 is the exponential rate of decay such that ln(2)/ β2 is the computed half-life. A random subject effect was assumed. For group comparisons between means, unpaired t tests were performed in Graphpad Prism (La Jolla, CA). Spearman correlations between groups were also performed in Graphpad Prism.
Acknowledgments
We are indebted to the patients and clinical teams of the CHAVI and Trinidad clinical cohorts for their numerous contributions to this study. We thank Drs Salim Abdool Karim, Gift Kamanga, John Crump, Joe Eron, Chuck Hicks of the CHAVI 001 teams and Drs Farley Cleghorn, Noreen Jack, and Michael M. Greenberg of the Trinidad cohort team for design, recruitment, and/or provision of clinical specimens from these acute HIV-1 cohorts. We thank Judith T. Lucas, Robert Parks, Vicki C. Ashley, R. Glenn Overman, and Elizabeth Marshall for expert technical assistance, and Drs Li Qin and Steve Self for T0 measurements for alignment of Ab responses. We also thank Kelly Soderberg and Jenn Kirchherr for CHAVI program management, and Saidi H. Kapiga and Noel E. Sam for oversight of CHAVI clinical studies at the KCMC. We thank Drs Peter Wright, Jri Mestecky, and Pam Kozlowski for helpful advice and guidance in the detection and measurement of HIV-1-specific IgA responses, Dr Hua-xin Liao for provision of consensus envelope antigens, and Dr Marcella Sarzotti-Kelsoe and Dan Ozaki for quality assurance oversight. This work was supported by the National Institutes of Health (NIH/NIAID/ DAIDS): Center for HIV/AIDS Vaccine Immunology Grant (AI067854), HIV-1 Vaccine Trials Network (HVTN)(5U01 AI46725-05), and the Duke University Center for AIDS Research (CFAR) Grant (P30 AI 64518) and the Bill and Melinda Gates Foundation (3830913). PB is a Jenner Institute Investigator.
The authors declared no conflict of interest.
Footnotes
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/mi
Supplementary Material
References
- Cerutti A. The regulation of IgA class switching. Nat Rev Immunol. 2008;8,:421–434. doi: 10.1038/nri2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase A. Protection from HIV: Targeted Intervention Strategies. Whistler Conference Centre: British Columbia; 2011. [Google Scholar]
- Bomsel M., et al. Immunization with HIV-1 gp41 subunit virosomes induces mucosal antibodies protecting nonhuman primates against vaginal SHIV challenges. Immunity. 2011;34,:269–280. doi: 10.1016/j.immuni.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Tomaras G.D., et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J Virol. 2008;82,:12449–12463. doi: 10.1128/JVI.01708-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S., Fauci A.S. B cells in HIV infection and disease. Nat Rev Immunol. 2009;9,:235–245. doi: 10.1038/nri2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMichael A.J., Borrow P., Tomaras G.D., Goonetilleke N., Haynes B.F. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol. 2010;10,:11–23. doi: 10.1038/nri2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauci A.S. Immunopathogenesis of HIV infection. J Acquir Immune Defic Syndr. 1993;6,:655–662. [PubMed] [Google Scholar]
- Lim S.G., et al. Loss of mucosal CD4 lymphocytes is an early feature of HIV infection. Clinical and Exp Immunol. 1993;92,:448–454. doi: 10.1111/j.1365-2249.1993.tb03419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadalupe M., et al. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J Virol. 2003;77,:11708–11717. doi: 10.1128/JVI.77.21.11708-11717.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley J.M., et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004;200,:749–759. doi: 10.1084/jem.20040874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenchley J.M., et al. Preferential infection shortens the life span of human immunodeficiency virus-specific CD4+ T cells in vivo. J Virol. 2006;80,:6801–6809. doi: 10.1128/JVI.00070-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lore K., Smed-Sorensen A., Vasudevan J., Mascola J.R., Koup R.A. Myeloid and plasmacytoid dendritic cells transfer HIV-1 preferentially to antigen-specific CD4+ T cells. J Exp Med. 2005;201,:2023–2033. doi: 10.1084/jem.20042413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane H.C., et al. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N Eng J Med. 1983;309,:453–458. doi: 10.1056/NEJM198308253090803. [DOI] [PubMed] [Google Scholar]
- Moir S., et al. HIV-1 induces phenotypic and functional perturbations of B cells in chronically infected individuals. Proc Natl Acad Sci USA. 2001;98,:10362–10367. doi: 10.1073/pnas.181347898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque M.C., et al. Polyclonal B cell differentiation and loss of gastrointestinal tract germinal centers in the earliest stages of HIV-1 infection. PLoS Med. 2009;6,:e1000107. doi: 10.1371/journal.pmed.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahwa S., Pahwa R., Good R.A., Gallo R.C., Saxinger C. Stimulatory and inhibitory influences of human immunodeficiency virus on normal B lymphocytes. Proc Natl Acad Sci USA. 1986;83,:9124–9128. doi: 10.1073/pnas.83.23.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patke C.L., Shearer W.T. gp120- and TNF-alpha-induced modulation of human B cell function: proliferation, cyclic AMP generation, Ig production, and B-cell receptor expression. J Allergy Clin Immunol. 2000;105,:975–982. doi: 10.1067/mai.2000.105315. [DOI] [PubMed] [Google Scholar]
- He B., et al. HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J Immunol. 2006;176,:3931–3941. doi: 10.4049/jimmunol.176.7.3931. [DOI] [PubMed] [Google Scholar]
- Swingler S., et al. Evidence for a pathogenic determinant in HIV-1 Nef involved in B cell dysfunction in HIV/AIDS. Cell Host Microbe. 2008;4,:63–76. doi: 10.1016/j.chom.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey A.R., et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83,:3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston M.I., Fauci A.S. An HIV vaccine--evolving concepts. N Engl J Med. 2007;356,:2073–2081. doi: 10.1056/NEJMra066267. [DOI] [PubMed] [Google Scholar]
- Wong S.B.J., Siliciano R.F.Biology of early infection and impact on vaccine design AIDS Vaccine Development: Challenges and Opportunities(Koff WC, Kahn P, Gust ID, ( eds)17–22.Caister Academic Press: Hethersett, Norwich, UK; 2007 [Google Scholar]
- Tomaras G.D., Haynes B.F. Strategies for eliciting HIV-1 inhibitory antibodies. Curr Opin HIV AIDS. 2010;5,:421–427. doi: 10.1097/COH.0b013e32833d2d45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElrath M.J., Haynes B.F. Induction of immunity to human immunodeficiency virus type-1 by vaccination. Immunity. 2010;33,:542–554. doi: 10.1016/j.immuni.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebig E.W., et al. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. Aids. 2003;17,:1871–1879. doi: 10.1097/00002030-200309050-00005. [DOI] [PubMed] [Google Scholar]
- Gasper-Smith N., et al. Induction of plasma (TRAIL), TNFR-2, Fas ligand and plasma microparticles after HIV-1 transmission: implications for HIV-1 vaccine design. J Virol. 2008;82,:7700–10. doi: 10.1128/JVI.00605-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M.S., Shaw G.M., McMichael A.J., Haynes B.F. Acute HIV-1 infection. N Engl J Med. 2011;364,:1943–1954. doi: 10.1056/NEJMra1011874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestecky J., et al. (eds)Mucosal Immunology3rd ednElsevier Academic Press: Burlington, MA, USA; 2005 [Google Scholar]
- Mestecky J., Moldoveanu Z., Russell M.W. Immunologic uniqueness of the genital tract: challenge for vaccine development. Am J Reprod Immunol. 2005;53,:208–214. doi: 10.1111/j.1600-0897.2005.00267.x. [DOI] [PubMed] [Google Scholar]
- Mestecky J., et al. Paucity of antigen-specific IgA responses in sera and external secretions of HIV-type 1-infected individuals. AIDS Res Hum Retrovir. 2004;20,:972–988. doi: 10.1089/aid.2004.20.972. [DOI] [PubMed] [Google Scholar]
- Yates N.L., et al. Multiple HIV-1 specific IgG3 responses decline during acute HIV-1: implications for detection of incident HIV infection. AIDS. 2011;25 (17:2089–97. doi: 10.1097/QAD.0b013e32834b348e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleghorn F.R., et al. A distinctive clade B HIV type 1 is heterosexually transmitted in TRINIDAD and Tobago. Proc Natl Acad Sci USA. 2000;97,:10532–10537. doi: 10.1073/pnas.97.19.10532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X., et al. In vivo gp41 antibodies targeting the 2F5 monoclonal antibody epitope mediate human immunodeficiency virus type 1 neutralization breadth. J Virol. 2009;83,:3617–3625. doi: 10.1128/JVI.02631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H.X., et al. Initial antibodies binding to HIV-1 gp41 in acutely infected subjects are polyreactive and highly mutated. J Exp Med. 2011;208,:2237–2249. doi: 10.1084/jem.20110363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haimovici F., Mayer K.H., Anderson D.J. Quantitation of HIV-1-specific IgG, IgA, and IgM antibodies in human genital tract secretions. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;15,:185–191. doi: 10.1097/00042560-199707010-00001. [DOI] [PubMed] [Google Scholar]
- Wolff H., et al. A comparison of HIV-1 antibody classes, titers, and specificities in paired semen and blood samples from HIV-1 seropositive men. J Acquir Immune Defic Syndr. 1992;5,:65–69. [PubMed] [Google Scholar]
- Xu W., et al. HIV-1 evades virus-specific IgG2 and IgA responses by targeting systemic and intestinal B cells via long-range intercellular conduits. Nat Immunol. 2009;10,:1008–1017. doi: 10.1038/ni.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel J.E., Chen W. Balancing acts: the role of TGF-beta in the mucosal immune system. Trends Mol Med. 2011;17,:668–676. doi: 10.1016/j.molmed.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morell A., Skvaril F., Noseda G., Barandun S. Metabolic properties of human IgA subclasses. Clin Exp Immunol. 1973;13,:521–528. [PMC free article] [PubMed] [Google Scholar]
- Morell A., Terry W.D., Waldmann T.A. Metabolic properties of IgG subclasses in man. J Clin Invest. 1970;49,:673–680. doi: 10.1172/JCI106279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghetie V., Ward E.S. Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu Rev Immunol. 2000;18,:739–766. doi: 10.1146/annurev.immunol.18.1.739. [DOI] [PubMed] [Google Scholar]
- Bonsignori M., et al. HIV-1 envelope induces memory B cell responses that correlate with plasma antibody levels after envelope gp120 protein vaccination or HIV-1 infection. J Immunol. 2009;183,:2708–2717. doi: 10.4049/jimmunol.0901068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouda G.G., et al. HIV-specific functional antibody responses in breast milk mirror those in plasma and are primarily mediated by IgG antibodies. J Virol. 2011;85,:9555–9567. doi: 10.1128/JVI.05174-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litinskiy M.B., et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3,:822–829. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Balazs M., Martin F., Zhou T., Kearney J. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. 2002;17,:341–352. doi: 10.1016/s1074-7613(02)00389-8. [DOI] [PubMed] [Google Scholar]
- Scholz J.L., et al. BLyS inhibition eliminates primary B cells but leaves natural and acquired humoral immunity intact. Proc Natl Acad Sci USA. 2008;105,:15517–15522. doi: 10.1073/pnas.0807841105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagarasan S., Kawamoto S., Kanagawa O., Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu Rev Immunol. 2010;28,:243–273. doi: 10.1146/annurev-immunol-030409-101314. [DOI] [PubMed] [Google Scholar]
- Veazey R.S., et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280,:427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- Mattapallil J.J., et al. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434,:1093–1097. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- Nawa M., et al. Immunoglobulin A antibody responses in dengue patients: a useful marker for serodiagnosis of dengue virus infection. Clin Diagn Lab Immunol. 2005;12,:1235–1237. doi: 10.1128/CDLI.12.10.1235-1237.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul R., et al. Mucosal IgA in exposed, uninfected subjects: evidence for a role in protection against HIV infection. AIDS. 2001;15,:431–432. doi: 10.1097/00002030-200102160-00026. [DOI] [PubMed] [Google Scholar]
- Devito C., et al. Mucosal and plasma IgA from HIV-exposed seronegative individuals neutralize a primary HIV-1 isolate. AIDS. 2000;14,:1917–1920. doi: 10.1097/00002030-200009080-00006. [DOI] [PubMed] [Google Scholar]
- Devito C., et al. Mucosal and plasma IgA from HIV-1-exposed uninfected individuals inhibit HIV-1 transcytosis across human epithelial cells. J Immunol. 2000;165,:5170–5176. doi: 10.4049/jimmunol.165.9.5170. [DOI] [PubMed] [Google Scholar]
- Lo Caputo S., et al. Mucosal and systemic HIV-1-specific immunity in HIV-1-exposed but uninfected heterosexual men. AIDS. 2003;17,:531–539. doi: 10.1097/00002030-200303070-00008. [DOI] [PubMed] [Google Scholar]
- Griffiss J.M., Goroff D.K. IgA blocks IgM and IgG-initiated immune lysis by separate molecular mechanisms. J Immunol. 1983;130,:2882–2885. [PubMed] [Google Scholar]
- Komiyama K., Crago S.S., Itoh K., Moro I., Mestecky J. Inhibition of natural killer cell activity by IgA. Cell Immunol. 1986;101,:143–155. doi: 10.1016/0008-8749(86)90193-0. [DOI] [PubMed] [Google Scholar]
- Haynes B.F., et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. 2012;366,:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouch D.H., et al. Vaccine protection against acquisition of neutralization-resistant SIV challenges in rhesus monkeys. Nature. 2012;482,:89–93. doi: 10.1038/nature10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfsen A., Iniguez P., Bouguyon E., Bomsel M. Secretory IgA specific for a conserved epitope on gp41 envelope glycoprotein inhibits epithelial transcytosis of HIV-1. J Immunol. 2001;166,:6257–6265. doi: 10.4049/jimmunol.166.10.6257. [DOI] [PubMed] [Google Scholar]
- Clerici M., et al. Serum IgA of HIV-exposed uninfected individuals inhibit HIV through recognition of a region within the alpha-helix of gp41. AIDS. 2002;16,:1731–1741. doi: 10.1097/00002030-200209060-00004. [DOI] [PubMed] [Google Scholar]
- Tudor D., et al. HIV-1 gp41-specific monoclonal mucosal IgAs derived from highly exposed but IgG-seronegative individuals block HIV-1 epithelial transcytosis and neutralize CD4(+) cell infection: an IgA gene and functional analysis. Mucosal Immunol. 2009;2,:412–426. doi: 10.1038/mi.2009.89. [DOI] [PubMed] [Google Scholar]
- Liu P., et al. Dynamic antibody specificities and virion concentrations in circulating immune complexes in acute to chronic HIV-1 infection. J Virol. 2011;85,:11196–11207. doi: 10.1128/JVI.05601-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshe R.B., et al. Correlates of immune protection induced by live, attenuated, cold-adapted, trivalent, intranasal influenza virus vaccine. J Infect Dis. 2000;181,:1133–1137. doi: 10.1086/315323. [DOI] [PubMed] [Google Scholar]
- Buisman A.M., et al. Preexisting poliovirus-specific IgA in the circulation correlates with protection against virus excretion in the elderly. J Infect Dis. 2008;197,:698–706. doi: 10.1086/527487. [DOI] [PubMed] [Google Scholar]
- Franco M.A., Angel J., Greenberg H.B. Immunity and correlates of protection for rotavirus vaccines. Vaccine. 2006;24,:2718–2731. doi: 10.1016/j.vaccine.2005.12.048. [DOI] [PubMed] [Google Scholar]
- Blattner W.A., et al. Rapid clearance of virus after acute HIV-1 infection: correlates of risk of AIDS. J Inf Dis. 2004;189,:1793–1801. doi: 10.1086/386306. [DOI] [PubMed] [Google Scholar]
- Yates N.L., et al. Multiple HIV-1-specific IgG3 responses decline during acute HIV-1: implications for detection of incident HIV infection. AIDS. 2011;25,:2089–2097. doi: 10.1097/QAD.0b013e32834b348e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F., et al. Antigenicity and immunogenicity of a synthetic human immunodeficiency virus type 1 group m consensus envelope glycoprotein. J Virol. 2005;79,:1154–1163. doi: 10.1128/JVI.79.2.1154-1163.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris L., et al. Isolation of a human anti-HIV gp41 membrane proximal region neutralizing antibody by antigen-specific single B cell sorting. PloS one. 2011;6,:e23532. doi: 10.1371/journal.pone.0023532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.