

Abstract
Alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV) is a rare and lethal developmental disorder of the lung defined by a constellation of characteristic histopathological features. Non-pulmonary anomalies involving organs of gastrointestinal, cardiovascular, and genitourinary systems have been identified in approximately 80% of patients with ACD/MPV. We have collected DNA and pathological samples from more than 90 infants with ACD/MPV and their family members. Since the publication of our initial report of four point mutations and ten deletions, we have identified an additional thirty eight novel nonsynonymous mutations of FOXF1 (nine nonsense, seven frameshift, one inframe deletion, twenty missense, and one no stop). This report represents an up to date list of all known FOXF1 mutations to the best of our knowledge. Majority of the cases are sporadic whereas four familial cases with three showing maternal inheritance, consistent with paternal imprinting of the gene. Twenty five mutations (60%) are located within the putative DNA binding domain, indicating its plausible role in gene regulation. Five mutations map to the second exon. We identified two additional genic and eight genomic deletions upstream to FOXF1. These results corroborate and extend our previous observations and further establish involvement of FOXF1 in ACD/MPV and lung organogenesis.
Keywords: Lung, Development, Angiogenesis, ACD/MPV, FOXF1, Imprinting
Background
Alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV; MIM# 265380) is a rare lethal developmental disorder of the lung and its vasculature [Bishop, et al., 2011]. Affected patients typically develop severe respiratory distress and pulmonary hypertension a few hours after birth, although rare late presentations have also been reported [Ahmed, et al., 2008; Licht, et al., 2004]. Supportive treatment with supplemental oxygen, inhaled nitric oxide, extracorporeal membrane oxygenation (ECMO) and, more recently, sildenafil provide only temporary improvement [Al-Hathlol, et al., 2000; Kitayama, et al., 1997; Plat, et al., 2007]. There is no cure for the disease and affected infants usually die within the first month of their life. The diagnosis of ACD/MPV has been based on the presence of a constellation of histologic abnormalities on lung biopsies or at autopsy and include malposition (misalignment) of pulmonary veins adjacent to small pulmonary arteries, medial thickening of the smooth muscle walls of small pulmonary arteries, deficient pulmonary lobular development, and severe paucity of normally positioned alveolar wall capillaries (Fig. 1) [Wagenvoort, 1986]. About a third of the patients with ACD/MPV also have pulmonary lymphangiectasis [Langston, 1991] and most (approximately 80%) have anomalies of other organs, particularly of the cardiovascular, gastrointestinal, and genitourinary systems [Sen, et al., 2004]. The vast majority of ACD/MPV cases are sporadic although a few familial cases have been reported [Boggs, et al., 1994; Gutierrez, et al., 2000; Sen, et al., 2012].
Figure 1.
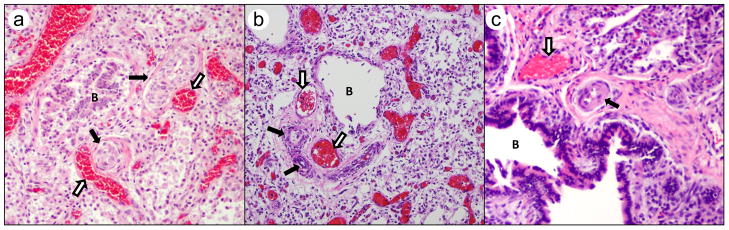
Sections of formalin fixed paraffin embedded lung tissue obtained either at autopsy (Cases 1 and 2) or diagnostic lung biopsy (Case 7) are examined stained with hematoxylin and eosin for the constellation of changes seen in ACD/MPV. These changes are often confirmed with a variety of additional stains including Movat pentachrome, trichrome and elastic stains and immunostains for pulmonary epithelium (CK7), the microvasculature (CD31) and vascular smooth muscle (Smooth Muscle Actin) (not shown). These changes include increased medial smooth muscle in small pulmonary arteries (black arrows), malposition of pulmonary veins (white arrows) adjacent to small pulmonary arteries in their normal parabronchiolar location (B identified bronchioles), and lobular underdevelopment seen in all panels. Other changes include paucity of alveolar capillaries and abnormal capillary size and location, seen in panels “a” and “b” as centrally located thin-walled dilated and congested alveolar wall vessels. Images are from Case 1 (a), Case 2 (b), and Case 7 (c); all H&E stain.
In 2009, we reported four unique heterozygous inactivating point mutations (one nonsense, one no stop, and two frameshift; patients 38–41 in Table 1), and 10 microdeletions involving FOXF1 and its upstream interval in the 16q24 (Fig. 2, D1–D6, D8–D10) region, in about 40% of patients with ACD/MPV analyzed [Stankiewicz, et al., 2009]. Since the publication of that report, we have accumulated material from 53 additional cases of histologically diagnosed ACD/MPV and five cases identified genetically. Recently, we have shown that FOXF1 is incompletely paternally imprinted in humans [Sen, et al., 2012; Szafranski, et al., 2013].
Table 1.
List of mutations inf FOXF1 coding sequence
| Patient ID | Exon/Intron | DNA Variant Mutalyzer | Protein Variant Mutalyzer | Type | Located in DNA binding domain† | Parental origin | Predicted effect on protein | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 | exon 1 | c.145C>T | p.Pro49Ser | missense | √ | de novo | Probably deleterious¶ | This Study |
| 2 | exon 1 | c.89C>A | p.Ser30* | nonsense | de novo | deleterious | This Study | |
| 3 | exon 1 | c.872_879del8 | p.Ser291* | nonsense | de novo | deleterious | This Study | |
| 4 | exon 1 | c.899_903dup5 | p.Gly302Cysfs*79 | frameshift | de novo | deleterious | This Study | |
| 5 | exon 1 | c.191C>A | p.Ser64* | nonsense | √ | de novo | deleterious | This Study |
| 6 | exon 1 | c.691_698del8 | p.Ala231Argfs*61 | frameshift | de novo | deleterious | This Study | |
| 7a | exon 1 | c.235C>T | p.Gln79* | nonsense | √ | de novo | deleterious | This Study |
| 7b | exon 2 | c.988C>T | p.Arg330Trp | missense | de novo | Probably deleterious¶ | This Study | |
| 8 | exon 1 | c.256C>T | p.Arg86Trp | missense | √ | de novo | Probably deleterious¶ | This Study |
| 9 | exon 1 | c.302C>T | p.Ser101Leu | missense | √ | de novo | Probably deleterious¶ | This Study |
| 10 | exon 1 | c.338_358del21 | p.Leu113_Gly119del | inframe deletion | √ | de novo | deleterious | This Study |
| 11 | exon 2 | c.1139G>C | p.*380Serext*73 | no stop | de novo | deleterious | This Study | |
| 12 | exon 1 | c.158A>G | p.Tyr53Cys | missense | √ | de novo | Probably deleterious¶ | This Study |
| 13 | exon 1 | c.936del1 | p.Tyr313Ilefs*66 | frameshift | de novo | deleterious | This Study | |
| 14 | exon 1 | c.899del1 | p. Leu300Argfs*79 | frameshift | de novo | deleterious | This Study | |
| 15 | exon 1 | c.286G>A | p.Val96Met | missense | √ | de novo | Probably deleterious¶ | This study |
| 16 | exon 1 | c.221T>A | pIle74Asn | missense | √ | de novo | Probably deleterious¶ | This Study |
| 17 | exon 1 | c.710dupC | p.His238Alafs*57 | frameshift | de novo | deleterious | This Study | |
| 18 | exon 1 | c.276G>A | p.Trp92* | nonsense | √ | de novo | Probably deleterious¶ | This Study |
| 19 | exon 2 | c.1021_1022insA | p.Arg341Glnfs*70 | frameshift | de novo | deleterious | This Study | |
| 20 | exon 1 | c.155C>T | p.Ser52Phe | missense | √ | de novo | Probably deleterious¶ | This Study |
| 21 | exon 1 | c.272_273delinsAA | p.Gly91Glu | missense | √ | de novo | Probably deleterious¶ | This Study |
| 22 | exon 1 | c.849_850del2 | p.Ile285Glnfs*9 | frameshift | de novo | deleterious | This Study | |
| 23 | exon 1 | c.539C>A | p.Ser180* | nonsense | de novo | deleterious | This Study | |
| 24 | exon 1 | c.290G>A | p.Arg97His | missense | √ | de novo | Probably deleterious¶ | This Study |
| 25 | exon 1 | c.254T>C | p.Phe85Ser | missense | √ | de novo | Probably deleterious¶ | This Study |
| 26 | exon 1 | c.356G>A | p.Gly119Asp | missense | √ | de novo | Probably deleterious¶ | This Study |
| 27 | exon 1 | c.272G>T | p.Gly91Val | missense | √ | de novo | Probably deleterious¶ | This study |
| 28 | exon 1 | c.310G>T | p.Glu104* | nonsense | √ | not known | deleterious | This study |
| 29 | exon 1 | c.810G>A | p.Trp270* | nonsense | de novo | deleterious | This study | |
| 30 | exon 1 | c.862C>T | p.Gln288* | nonsense | de novo | deleterious | This study | |
| 31 | exon 1 | c.146C>A | p.Pro49Gln | missense | √ | not known | Probably deleterious¶ | This study |
| 32 | exon 1 | c.377C>T | p.Pro126Leu | missense | √ | not known | Probably deleterious¶ | This study |
| 33 | exon 1 | c.316T>C | p.Phe106Leu | missense | √ | not known | Probably deleterious¶ | This study |
| 34§ | exon 1 | c.253T>A | p.Phe85Ile | missense | √ | likely germline mosaicism | Probably deleterious¶ | This study |
| 35§ | exon 1 | c.253T>C | p.Phe85Leu | missense | √ | maternal | Probably deleterious¶ | This study |
| 36§ | exon 1 | c.294C>A | p.His98Gln | missense | √ | maternal | Probably deleterious¶ | This Study |
| 37§ | exon 1 | c.416G>T | p.Arg139Leu | missense | √ | maternal | Probably deleterious¶ | Sen et al 2012 |
| 38 | exon 1 | c.225C>A | p.Tyr75* | nonsense | √ | de novo | deleterious | Stankiewicz et al 2009 |
| 39 | exon 1 | c.850dupT | p.Tyr284Leufs*11 | frameshift | de novo | deleterious | Stankiewicz et al 2009 | |
| 40 | exon 2 | c.1031_1032del2 | p.Phe344Cysfs*66 | frameshift | de novo | deleterious | Stankiewicz et al 2009 | |
| 41 | exon 2 | c.1138T>C | p.*380Argext*73 | no stop | de novo | deleterious | Stankiewicz et al 2009 |
DNA binding domain (Mahlapuu et al 1998)
Familial case, present in the mother
Predicted by in silico prediction programs (SIFT; PolyPhen2)
The RefSeq used was NM_001451.2 and the mutations have been named using directions from Mutalyzer (https://humgenprojects.lumc.nl/trac/mutalyzer)
Figure 2.

Genomic and genic deletions identified in the 16q24.1 region in patients with ACD/MPV. Patients D1–D6 and D8–D10 have been reported in Stankiewicz et al 2009 and the remainder except 94.3 and 104.3 have been reported in Szafranski et al (2013). Note that many of the deletions are flanked by Alu repetitive segments. SDR represents the smallest deletion overlap defining the cis-regulatory region mapping 250 kb upstream of FOXF1 (Szafranski et al 2012). The numbers with decimals refer to patients with ACD/MPV in our cohort.
We now report 34 novel de novo and four familial mutations of which three are maternally inherited, in unrelated patients with ACD/MPV that imply a role for FOXF1 DNA-binding domain (DBD) (Table 1). This report represents an up to date list of all known FOXF1 mutations to the best of our knowledge. Our data further substantiate the notion that mutations in FOXF1 lead to manifestation of ACD/MPV and that this transcription factor is involved in the development of the pulmonary, cardiovascular, gastrointestinal, and genitourinary systems. Three maternally inherited cases are consistent with the finding that FOXF1 is paternally imprinted [Sen, et al., 2012; Szafranski, et al., 2013].
Variants
The exact location and other pertinent information for each mutation are detailed in Table 1 and Fig. 3. Patient 7 has two mutations of unknown phase. Twenty nine of the newly identified mutations (patients 1, 2, 5, 7a & b, 8, 9, 11, 12, 15, 16, 18, 20, 21, and 23 – 37) are substitution point mutations (missense, nonsense, and no stop), one generates a premature stop codon at the site of the deletion (patient 3), one is an in frame deletion (patient 10), one is indel (patient 21) and four (patients 6, 13, 14, and 22) are small deletions, and two (patients 4 and 17) are insertions, resulting in a shift in the FOXF1 reading frame. These are all unique mutations and only patient 37 [Sen, et al., 2012] and 38–41 [Stankiewicz, et al., 2009] have been previously reported. Two groups of mutations affect the same codon but involve different nucleotides; thus resulting in different amino acids (Table 1): patients 25,34 and 35 (c.254T>C, p.Phe85Ser, c.253T>A;p.Phe85Ile & c.253T>C, p.Phe85Leu); and a pair of one new (patient 11) and a previously reported (patient 37) mutation change the stop codon while involving separate nucleotides, and extend the protein by 73 amino acids (c.1139G>C, p.*380Ser ext*73& c.11138T>C, p.*380Argext*73).
Figure 3.

A: Schematic representation of FOXF1 at 16q24.1. The upper segment depicts the genomic arrangement of FOXF1 with two exons in the gene shown by open boxes. The non-coding part of the gene is shown in shaded boxes. The numbers underneath show the beginning and end of the coding sequences and the exon intron junctions. The lower segment depicts the protein with the relative location of the DNA binding “Forkhead box” domain (DBD) and, the Cell-type specific activation domain in exon 1 and the general activation domain in exon 2. The numbers underneath indicate the position of amino acids for the domains.
B: Positional representation of all reported and novel mutations in FOXF1. The green solid lines and green shaded boxes represent the first exon while the red solid line and the red shaded box represent the second exon. The first exon contains the DNA binding domain and the cell-type specific activation domain and the second exon contains the general activation domain. The locations of the mutations are shown with arrows. Golden arrows indicate nonsense mutations, purple, blue and green arrows indicate missense mutations, frameshift and indel mutations and no-stop mutations, respectively. The details of the mutations are shown above respective arrows. The black arrow indicates the nonsynonymous SNP.
C: Evolutionary conservation of the amino acid sequence of the DNA binding domain of FOXF1 among vertebrates and conservation of the mutated amino acids within other forkhead family proteins. The identical amino acids are shown in blue while conservative changes are shown in burgundy and non-conservative changes shown in green. Only missense mutations are shown. The resulting amino acids after mutations are shown between the two panels. Although two of the missense mutations in unrelated patients involve the same amino acids the resulting changed amino acids are different in each case. The resulting amino acids are shown one above the other.
The identified mutations are not reported in the dbSNP and are not cited in the Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/), that covers more than 10,000 alleles. There are three known SNPs (rs61740819, rs61753347, and rs8061302) [Common SNPs (135)] in the first exon of FOXF1 outside the DBD. SNPs rs61740819 and rs61753347 map within the cell specific activation domain of FOXF1 (Mahlapuu, et al., 1998; Pierrou, et al., 1994) [Mahlapuu, et al., 1998; Pierrou, et al., 1994], and are both synonymous. The third one, rs8061302, changes a proline to threonine at position 299 (Fig. 3B). Although this proline is conserved among other FOXF1 orthologs, we believe this SNP is neutral because it is present outside the DBD of the protein. The sources of these SNPs are unknown. The reference sequence used in this study is NM_001451.2.
Figure 3B shows the locations and types of all identified mutations. In patient 1 and 31, the nonsynonymous missense mutation is predicted to lead to a proline to serine or glutamine change, respectively, at the 49th position that is within the DBD of FOXF1 and is conserved in members of the forkhead family of proteins (Fig. 3C) [Pierrou, et al., 1994]. A proline residue is known to create a kink in protein structure [Langelaan, et al., 2010; Vanhoof, et al., 1995]. Therefore, these changes in the primary structure of the protein is likely to produce a substantial alteration in its three dimensional structure, especially in the forkhead domain, thus affecting its DNA binding function. Missense mutations in patients 8 (p.Arg86Trp), 9 (p.Ser101Leu), 12 (p.Tyr53Cys), 15 (p.Val96Met), 16 (p.Ile74Asn), 20 (p.Ser52Phe), 21 (p.Gly91Glu), 24 (p.Arg97His), 25 (p.Phe85Ser), 26 (p.Gly119Asp), 27 (p.Gly91Val), 32 (p.Pro126Leu), 33 (p.Phe126Leu), 34 (p.Phe85Ile), 35 (p.Phe85Leu), 36 (p.His98Gln), 37 (p.Arg139Leu) also map within the DBD of the protein. Arginine and serine residues have also been shown to be involved in DNA binding [Kirchler, et al., 2010; Wache, et al., 2005; Wei, et al., 2003]. All of the 15 amino acids within the DBD of FOXF1 that are altered due to missense mutations are conserved in vertebrates and other members of the forkhead family (Fig 3C). These missense mutations are important since they provide information regarding the function of FOXF1 as a transcription factor with DNA binding ability.
Patients 2 (p.Ser30*), 5 (p.Ser64*), 7a (p.Gln79*), 18 (p.W92*), 23 (p.Ser180*), 28 (p.Glu104*), 29 (p.W270*), and 30 (p.Q288*) have nonsense mutations introducing premature stop codons. The mutations in patients 5, 7, and 18 also map to the DBD. In patient 7, an additional variant of unknown significance was identified in the second exon and is predicted to lead to p.Arg330Trp (Table 1) change.
Deletion of eight bases in patient 3 introduces a stop codon at the point of rearrangement. Patients 6 (p.Ala231Argfs*61), 13 (p.Tyr313Ilefs*66), 14 (p. Leu300Argfs*79), 17 (p.His238Alafs*57), and 19 (p.Arg341Glnfs*70) have deletions of various numbers of bases thereby altering the reading frame and introducing premature stop codons. Patient 10 (p.Leu113_G119del) has an inframe deletion of seven amino acids resulting in a shorter polypeptide without altering the reading frame. Patient 21 has a deletion and reciprocal insertion of two bases, thereby maintaining the reading frame with a p.Gly91Glu missense mutation (Table 1). All except one (patient 19) of these deletion mutations are in the first exon of the FOXF1 with the mutation in patients six and 10 being present within the DBD of the protein. Interestingly, in patient 10, the deletion is flanked by a direct repeat sequence AAGGGCC, which might have provided microhomology for the deletion [Boone, et al., 2011; Hastings, et al., 2009].
The insertion of five nucleotides (CCTGT) after 903 bases from the first codon in patient 4 results in no change in the size of the predicted polypeptide although only first 301 amino acids remain unchanged. The mutation in patient 19 also represents an out-of-frame insertion of a single base in the first exon, leading to an extension of 30 amino acids in the protein.
The three familial cases (patients 35, 36 and 37) show maternal inheritance of the mutated allele. Investigation of one of the families (patient 37) showed that the missense mutation in the children arose de novo on the paternal allele of the mother [Sen, et al., 2012]. Patients 34.1 and 34.2 have the same mutation although none of their parents have it. This situation could be explained as an example of germline mosaicism most probably in the mother since the gene is paternally imprinted. However, in patient 36 we could not determine whether the mutation is inherited or de novo on the paternal allele in the mother due to lack of DNA samples from her parents. The familial cases represent almost 10% of the total mutations presented herein. It is likely that other inactivating mutations in FOXF1 might be present in the general population but they do not manifest ACD/MPV and thus remain undetected as they are most likely located on the paternal chromosome 16 of unaffected carriers.
Fifteen nonsense and frameshift mutations of the 42 new and previously reported mutations presented here, map to the first exon of the FOXF1 gene, resulting in premature stop codons. Thus, the aberrant mRNA is likely to be destroyed by the nonsense mediated decay (NMD) surveillance mechanism [Bhuvanagiri, et al., 2010; Nicholson, et al., 2010] and, leading to FOXF1 haploinsufficiency. The frameshift mutations in patients 4, 13, 14, and 19 change the predicted amino acid sequence from their respective point of mutation and while mutation in patient 19 extends the predicted polypeptide sequence, mutations in patients 4, 13 and 14, result in predicted polypeptides that are equal in size with the wild type protein. The missense mutations p.Arg330Trp in patient 7, p.*380Serext*73 in patient 11, the no stop mutation extending the protein with 73 amino acids, and an insertion of a single base c.1021_1022insA in patient 19, are predicted to escape the NMD. We reported another no stop mutation previously [Stankiewicz, et al., 2009].
In addition to the previously reported 12 microdeletions involving FOXF1 and its upstream region [Garabedian et al., 2012; Handrigan et al., 2013; Stankiewicz, et al., 2009; Yu, et al., 2010; Zufferey, et al., 2011], we have recently identified ten novel de novo deletions: two involving FOXF1 and eight mapping upstream to FOXF1(Fig. 2). Interestingly, among deletions for which parental origin could be determined (eight upstream and seven others harboring FOXF1) each arose de novo on the maternal chromosome, consistent with the fact that FOXF1 is paternally imprinted. The deletions enabled identification of a distant cis-regulatory region (Fig. 2; SDR: Smallest Deleted Overlapping Region) mapping at 250 kb upstream to FOXF1 that harbors lncRNA genes, a differentially methylated CpG island that binds GLI2 depending on its methylation status, and physically interacts with and up-regulates the FOXF1 promoter [Szafranski, et al., 2013].
The data presented here corroborate our earlier results of involvement of FOXF1 in ACD/MPV where we had identified point mutations or deletions involving FOXF1 in about 40% of 46 patients studied. We have since identified mutations in FOXF1 in 70% (37/53) of 53 additional patients and also found 10 more deletions. This further proves that FOXF1 haploinsufficiency is the major cause of ACD/MPV and its essential role in the development of the lungs and other organs. With approximately 60% (25/42) of the mutations located in the DBD, our data also indicate that this domain of the protein plays an important role in mediating its function as a transcription factor. All mutations are predicted to be deleterious by bioinformatics analysis (Table 1) [Adzhubei, et al., 2010; Kumar, et al., 2009; Mathe, et al., 2006].
Databases
The mutations have been deposited in the LOVD database (www.lovd.nl/FOXF1).
Consortium
FOXF1 is one of the research interests of a few laboratories (Dr. Vladimir Kalinichenko, Cincinnati Children’s Hospital, Ohio, USA and Dr. Charles Shaw-Smith, Peninsula College of Medicine and Dentistry, Exeter, UK). However, we are the only group that is interested in the genetic abnormalities of FOXF1, related to ACD/MPV, and there are currently no consortia that specifically study this particular entity, although the Childhood Interstitial Lung Disease Research Network has considered inclusion of patients with ACD/MPV in an as yet not activated patient registry. Our group has benefited from the activities of the “ACD-Association (ACDA)” (http://www.acd-association.com), an organization formed in 1996 to support families affected by ACD/MPV. This organization has been a source of information and support for parents of ACD/MPV patients. Recently, “The Breath of Life Project” (http://breathoflifeproject.com/), has provided information about ACD/MPV to physicians and other health professionals as part of a CDC (Centers for Disease Control and Prevention, Atlanta, GA) funded awareness project.
Biological Relevance
The FOXF1 protein is a dosage-sensitive transcription factor that binds to the promoter regions of other genes involved in organogenesis [Madison, et al., 2009; Mahlapuu, et al., 2001; Shaw-Smith, 2010]. It contains a forkhead (DNA binding) domain between amino acids 44–148 [Pierrou, et al., 1994] and two more activation domains, one of which, between amino acids 135 and 260 (encoded by the first exon), has been shown to specifically activate genes in the lung. The last 28 amino acids at the C terminus of the protein constitute the second activation domain that is involved in general gene expression in all cell types (Fig. 2) [Mahlapuu, et al., 1998]. Moreover, it is known that FOXF1 binds as a monomer [Pierrou, et al., 1994]. Further, FOXF1 has recently been shown to be incompletely paternally imprinted in human [Sen, et al., 2012; Szafranski, et al., 2013]. The members of the Fox family of transcription factors (Forkhead box) are a group of ancient proteins that originated in unicellular eukaryotes. Through evolution, they have multiplied to several copies and currently there are approximately 40 such proteins in mammals [Hannenhalli and Kaestner, 2009]. They play important roles in functions as diverse as cell cycle controls, organ development, and language disorders. Some of the Fox proteins have been implicated in various types of cancer [Katoh et al., 2013].
Homozygous Foxf1−/− mice die by embryonic day 8.5 because of defects in the development of extra-embryonic mesoderm [Kalinichenko, et al., 2001a; Mahlapuu, et al., 2001]. Heterozygous knockout mice (Foxf1+/−) showed phenotypes similar to ACD/MPV. Although there is no misalignment of veins, reduction in the number of alveolar capillaries and lymphangiectasis are observed both in mice and humans. Interestingly, half of the newborn mice (Foxf1+/−) died of severe defects in alveolarization, vasculogenesis, and pulmonary hemorrhage [Kalinichenko, et al., 2001b]. Other defects such as fusion of lung lobes, abnormal gall bladder, and esophageal atresia were also observed [Kalinichenko, et al., 2001a; Kalinichenko, et al., 2001b; Mahlapuu, et al., 2001]. The remaining Foxf1+/− mice showed normal lung morphology that was interpreted as “compensation” for the defect [Kalinichenko, et al., 2001b].
Transcription factors bind to specific DNA sequences and thereby affecting gene expression. This is done through a DBD of which there are many different types. Members of the forkhead family have a winged helix-turn-helix domain that is known as the “forkhead domain” that functions as the DBD in them. It is interesting to note that 60% of all the mutations reported here are located within the DBD of FOXF1. Further, 88% of the mutations in the first exon and 95% of all missense mutations are clustered in the DBD of the protein. This clearly suggests that this domain plays a crucial role in the proper functioning of this important transcription factor since changes in this domain of the FOXF1 protein may affect its DNA binding properties resulting in ACD/MPV. Specific in vitro DNA binding experiments with the modified proteins mimicking the missense mutations are required to conclusively determine their role in the DBD of FOXF1.
Foxf1 expression in mice is thought to be regulated by Bmp4 [Astorga and Carlsson, 2007] Gli2 [Astorga and Carlsson, 2007; Tseng, et al., 2004], and by Pten [Tiozzo, et al., 2012]. A reduced expression of Vegfa, Vegfr2, Bmp-4, Tbx, Lklf, Fgf-10, Gli3, and Notch-2 has been observed in Foxf1−/− mice [Hellqvist, et al., 1996; Kalinichenko, et al., 2004; Kalinichenko, et al., 2001b; Lim, et al., 2002; Mahlapuu, et al., 2001].
Clinical Relevance
Patients with ACD/MPV were recruited after having signed consent approved by the IRB of Baylor College of Medicine (BCM). ACD/MPV is a rare and lethal developmental disorder presenting as respiratory distress and persistent pulmonary hypertension of the newborn [Abazov, et al., 2003; Bishop et al., 2011]. Initially, patients with ACD/MPV typically present with a clinical course consistent with persistent pulmonary hypertension without ready explanation. Further, approximately 80% of patients with ACD/MPV also have defects of non-pulmonary organ system [Sen, et al., 2004]. In addition, approximately 65% of the ACD/MPV families reported a history of miscarriages (data unpublished). As FOXF1 is also expressed in the placenta [Hellqvist, et al., 1996; Shaut, et al., 2008], this finding needs to be further investigated. A clinical summary of the presented cases is outlined in Table 2.
Table 2.
Clinical information of the reported cases
| patient # | Gender | Ethnicity | Birth weight (kg) | Mother’s age, pregnancy | Gestation (weeks) | Apgar 1 & 5 min | Delivery | Life span (days) | Diagnosis | Medications | ECMO | Associated anomalies |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | Caucasian | 2.06 | 28y, G2P2 | 37 & 3/7 | 8, 9 | Vaginal | 29 | Autopsy | Sildenafil | No | Incomplete rotation of bowel, high arched palate, hypocalvaria, incomplete lung lobation, incomplete rotation of bowel |
| 2 | F | Hispanic | 2.8 | 26y, G3P2 | 39 & 5/7 | 8, 9 | Vaginal | 18 | Autopsy | Nitric Oxide, Milrinone, Dopamine, Fentanyl | Yes | Malrotation of intestine, imperforate anus, butterfly vertebra |
| 3 | M | Caucasian | 3.3 | 22y, G4P2 | 36 | 9, 9 | Vaginal | 35 | Autopsy | N/A | Yes | Aortic coarctation, Meckel’s diverticulum |
| 4 | F | Hispanic | 3.6 | 28y, G3P2Ab1 | 39 | 9, 9 | Elective c-section | 16 | Autopsy | N/A | Yes | Bilateral incomplete lobation of lungs, lymphangiectasis, limited autopsy |
| 5 | M | Caucasian | 3.0 | 38 & 2/7 | 9, 9 | Elective c-section | 5 | Autopsy | Nitric oxide, Milrinone,, Sildenafil, Epinephrine, Dobutamine, Neuromuscular blockers | Yes | Abnormal lung lobation, malrotation of the intestine, duodenal atresia | |
| 6 | F | Caucasian | 3.2 | 28y, G2P1 | 39 | 8, 9 | Vaginal | 28 | Biopsy | Nitric oxide, Milrinone, Epoprostenol | Yes | No autopsy |
| 7 | M | Hispanic | 2.7 | 25y, G1P0→1 | 36 & 4/7 | 8, 9 | Vaginal | 10 | Biopsy | Nitric oxide, Dopamine, Milrinone, Sildenafil, Epinephrine, hydrocortisone | Yes | None identified |
| 8 | F | Caucasian | 3.3 | 28y, G3P2 | 39 | 6, 9 | Vaginal | 120 | Autopsy | Sildenafil | No | Bilobed right lung |
| 9 | F | Caucasian | 4.02 | 38y, G2P1 | 38 | 8, 9 | C-section for polyhydramnios | 10 | Autopsy | Nitric oxide, morphine, dopamine | Yes | None identified |
| 10 | F | African American | 3.1 | 25y, G2P2 | 40 & 4/7 | 8, 9 | Vaginal | 14 | Autopsy | Nitric oxide, epinephrine, dopamine, epoprostenol, Sildenafil | Yes | None identified, Limited autopsy |
| 11 | F | Caucasian | 3.7naline | 38y, G2P1→2 | 40 | 8, 9 | Vaginal | 95 | Nitric Oxide, Sildenafil, Epoprostenol, Milrinone, Hydrocortisone, Morphine, | Yes | Atrial septal defect | |
| 12 | F | Caucasian | 4.1 | N/A, G1P1 | 42 | N/A | Vaginal | 111 | Biopsy | Nitric Oxide, Sildenafil | Yes | N/A |
| 13 | F | Caucasian | 3.2 | 39y, G1P1 | 39 | 10, 10 | Vaginal | 42 | Biopsy | Sildenafil, Epoprostenol, Milrinone, Dopamine, Epinephnire, Norepinephrine | Yes | No lung lobation |
| 14 | M | Caucasian | 3.3 | 26y, G1p1 | 41 & 2/7 | 9, 10 | Vaginal | 15 | Autopsy | Nitric oxide, epoprostenol, surfactant and catecholeamines | No | None identified |
| 15 | M | Caucasian | 3.0 | 36y, G2P1 | 39 | 8, 9 | C-section (h/o prior c-section) | 7 | Biopsy | Nitric oxide, Milrinone, dopamine, epoprostenol | No | Malrotation of the intestine, atrial septal defect, thickened distended bladder with muscular hypertrophy |
| 16 | M | Caucasian | 2.1 | 31y, G3P2 | 32 | 4, 8 | Vaginal | 0.75 | Autopsy | Dopamine, epinephrine, fentanyl | No | Hyaline membrane disease, patent ductus arteriosus, multiple intestinal atresias, imperforate anus, undescended testis, Hirschprung disease |
| 17 | F | Caucasian | 1.4 | 30y, G1P1 | 33 & 3/7 | 10, 10 | Vaginal | 7 | Autopsy | Epoprostenol | No | Duodenal atresia, polysplenia, annular pancreas and left hydronephrosis |
| 18 | M | Caucasian | 2.8 | 42y, G3P3 | 38 | 9, 10 | Vaginal | 19 | Biopsy | Nitric Oxide, Milrinone, Dopamine, Ambrisentan | Yes | Hydronephrosis, partial stenosis at transition to colon transversum |
| 20 | M | Caucasian | 26y, G1P0→1 | 40 | 8, 8 | Vaginal | 51 | Autopsy | Nitric oxide, dopamine, epinephrine, Fentanyl, | Yes | Malrotation of the intestine, hydronephrosis, bilateral hydroureter, enlarged and thick bladder | |
| 22 | M | Asian | 3.3 | 39y, G2P0→1 | 38 & 2/7 | 8, 9 | C-section | 16 | Clinical characteristic and genetic mutation | Nitric Oxide, epinephrine, dopamine, Fentanyl, Milrinone, Sildenafil | Yes | N/A |
| 23 | F | Caucasian | 2.8 | 15y, G1P1 | 36 | 5, 9 | Vaginal | 15 | Autopsy | Nitric Oxide, dobutamine, norepinephrine, sildenafil, epoprostenol | No | Duodenal stenosis, annular pancreas, malrotation of large intestine |
| 24 | F | Caucasian | 2.9 | 24y, G1P0 | 39 & 4/7 | 4, 7 | Vaginal | 11 | Biopsy | Dobutamine, Dopamine, Epinephrine, Milrinone | Yes | Meckel’s divurticulum, moderate acute cortical involution of thymus |
| 25 | F | Caucasian | 2.7 | 33y, G4P3→4 | 37 & 5/7 | 7, 9 | Vaginal | 19 | Autopsy | Nitric oxide, Noradrenalin, Milrinone, Dobutamine | Yes | Intestinal malrotation |
| 26 | M | Caucasian | 3.5 | 38, G4P1 | 39 | 9, 9 | Vaginal | 21 | Biopsy | Nitric oxide | No | Megaduodenum, pyloric stenosis, annular pancreas |
| 27 | F | Caucasian | 2.7 | 34y, G1P1 | 37 & 2/7 | 8, 8 | Vaginal | 16 | Biopsy | Nitric oxide, Sildenafil, Bosentan | Yes | None identified |
| 28 | M | Caucasian | 2.6 | 34y, G5P1 | 39 | 9, 9 | C-section | 19 | Autopsy | Nitric oxide, Milrinone, Sildenafil, Noeepinephrine | No | Duodenal atresia |
| 29 | F | Caucasian | 2.4 | 20y, G1P1 | 34 &5/7 | 8, 9 | Vaginal | 12 | Biopsy | Nitric oxide, dopamine, dobutamine, hydrocortisone | Yes | Suspected jejuna atresia, no autopsy |
| 30 | F | Caucasian | 2.9 | 34y, G1P0→1 | 39 &6/7 | 7, 9 | Vaginal | 15 | Autopsy | Dopamine, dobutamine, hydrocortisone | Yes | Thickening of the pylorus |
| 31 | M | Asian | 3.5 | 24, G1P1 | 38 | N/A | Vaginal | N/A | Biopsy | Nitric oxide, furosemide, nifedipine | No | Posterior urethral valves |
| 32 | F | African American | 1.7 | 37, G5P3→4 | 34 | 8, 9 | Vaginal | N/A | Biopsy | Nitric oxide, sildenafil, milrinone | No | Atrial septal defects |
| 33 | F | Caucasian | 3.2 | 37, G2P2 | 37 | 7, 9 | C-section | 24 | Biopsy | Nitric oxide | No | Butterfly vertebra T5 |
| 34.1 | M | Caucasian | 0.5 | 29 | Terminated at 23 4/7 | N/A | Vaginal | N/A | Autopsy | N/A | No | Duedenal atresia, malrotation of the large intestine, anal atresia, gallbladder agenesis |
| 34.2 | M | Caucasian | 2.6 | 30 | 39 3/7 | N/A | Vaginal | 17 | Autopsy | Nitric oxide, sildenafil, bosentan, dobutamine, adrenaline, noradrenaline | Yes | None detected |
| 35 | F | Caucasian | 2.8 | 30y, G3 P1 | 38 & 5/7 | 3, 9 | Elective C-section | 50 | Clinical characteristic and genetic mutation | Dobutamine, Milrinone, Bosentan, Sildenafil | No | Atrial septal defect, polyhydramnios |
| 36 | M | Asian/Caucasian | 3.0 | 31y, G3P3 | 37 & 5/7 | 7, 8 | C-section (breech presentation) | 2 | Autopsy | Dobutamine, dopamine, adrenaline, noeepinephrine | No | Atrial septal defect, polyhydramnios |
| 37.1 | F | Caucasian | 3.4 | 25y, G2P1 | 39 | 9, 9 | Vaginal | 17 | Autopsy | Nitric oxide, dopamine, dobutamine, tolazolin | No | N/A |
| 37.2 | M | Caucasian | N/A | 25y | Terminated at 21 weeks | N/A | Vaginal | N/A | Autopsy | N/A | No | Uretral stenosis, hydronephrosis, |
| 37.3 | M | Caucasian | 3.2 | 27y, G4P2 | 37 & 1/7 | 9, 10 | Vaginal | 0.166 | Autopsy | Surfactants & Catecholamines | No | Pulmonary hypoplasia, hydrothorax, polyhydramnios |
| 37.4 | F | Caucasian | 3.2 | 31y, G5P3 | 39 | 9, 9 | Vaginal | 1 | Autopsy | Nitric oxide, Catecholamines | No | Isolated arhinencephaly |
| 37.5 | M | Caucasian | N/A | 36y | Terminated at 22 weeks | N/A | Vaginal | N/A | Autopsy | N/A | No | Obstructive uropathy |
N/A Not available. Clinical information of cases 19 and 21 are not available. The clinical information of cases 38 – 41 have been presented in Stankiewicz et al 2009.
We would like to emphasize that although ACD/MPV is a rare diffuse disorder of the neonatal lung, it is very likely that the diagnosis is missed in a significant number of patients due to lack of biopsies and autopsies. Further, we advocate that FOXF1 testing be done in patients with refractory pulmonary hypertension. This will result in a much larger cohort. Two retrospective studies have confirmed this notion [Eulmesekian, et al., 2005; Tibballs and Chow, 2002].
Diagnostic Relevance
About 200 cases of ACD/MPV have been reported in the literature [Sen, et al., 2010]. As physicians, pathologists, genetic counselors and care givers are becoming more cognizant with the disorder; more diagnoses are being made through histological examination of lung biopsies. A CLIA (Clinical Laboratory Improvement Amendment) certified service for the detection of mutation and deletion in suspected prenatal and postnatal cases is now available through the Medical Genetics Laboratories at the BCM, Houston, TX (https://www.bcm.edu/geneticlabs/). We continue to offer the analyses for genetic alterations of FOXF1 including mutation and deletion on a research basis. Results of such studies will help to provide better genetic counseling and estimates of recurrence risks to facilitate parents of ACD/MPV patients planning for future pregnancies. Very few lung transplants have been performed successfully. However, all of these patients have atypical late presentation of ACD/MPV (personal communication, Jennifer Wambach, Washington University, St. Louis, MO).
Future Prospects
We continue to investigate the molecular bases of lung development with a particular reference to FOXF1 and interacting genes. As a transcription factor, FOXF1 expectedly might regulate the expression of a number of downstream genes that are involved in lung and pulmonary vascular development. The early identification of affected infants by analysis for abnormalities of the FOXF1 gene may aid management decisions for these infants, conserving health care resources.
Acknowledgments
We would like to thank Rosemary Fantozzi for her help in preparation of the manuscript. Part of this work was supported by a NORD grant and Pilot Project Award from the Texas Children’s Hospital, Houston TX to P. Sen and NIH grant 1RO1HL101975-01 to P. Stankiewicz.
Footnotes
The authors declare no conflict of interest.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Ackerman V, Faught P, Langston C. Profound hypoxemia and pulmonary hypertension in a 7-month-old infant: late presentation of alveolar capillary dysplasia. Pediatr Crit Care Med. 2008;9:e43–6. doi: 10.1097/PCC.0b013e31818e383e. [DOI] [PubMed] [Google Scholar]
- Al-Hathlol K, Phillips S, Seshia MK, Casiro O, Alvaro RE, Rigatto H. Alveolar capillary dysplasia. Report of a case of prolonged life without extracorporeal membrane oxygenation (ECMO) and review of the literature. Early Hum Dev. 2000;57:85–94. doi: 10.1016/s0378-3782(99)00065-1. [DOI] [PubMed] [Google Scholar]
- Astorga J, Carlsson P. Hedgehog induction of murine vasculogenesis is mediated by Foxf1 and Bmp4. Development. 2007;134:3753–61. doi: 10.1242/dev.004432. [DOI] [PubMed] [Google Scholar]
- Bhuvanagiri M, Schlitter AM, Hentze MW, Kulozik AE. NMD: RNA biology meets human genetic medicine. Biochem J. 2010;430:365–77. doi: 10.1042/BJ20100699. [DOI] [PubMed] [Google Scholar]
- Bishop NB, Stankiewicz P, Steinhorn RH. Alveolar capillary dysplasia. Am J Respir Crit Care Med. 2011;184:172–9. doi: 10.1164/rccm.201010-1697CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggs S, Harris MC, Hoffman DJ, Goel R, McDonald-McGinn D, Langston C, Zackai E, Ruchelli E. Misalignment of pulmonary veins with alveolar capillary dysplasia: affected siblings and variable phenotypic expression. J Pediatr. 1994;124:125–8. doi: 10.1016/s0022-3476(94)70267-5. [DOI] [PubMed] [Google Scholar]
- Boone PM, Liu P, Zhang F, Carvalho CM, Towne CF, Batish SD, Lupski JR. Alu-specific microhomology-mediated deletion of the final exon of SPAST in three unrelated subjects with hereditary spastic paraplegia. Genet Med. 2011;13:582–92. doi: 10.1097/GIM.0b013e3182106775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulmesekian P, Cutz E, Parvez B, Bohn D, Adatia I. Alveolar capillary dysplasia: a six-year single center experience. J Perinat Med. 2005;33:347–52. doi: 10.1515/JPM.2005.067. [DOI] [PubMed] [Google Scholar]
- Garabedian MJ, Wallerstein D, Medina N, Byrne J, Wallerstein RJ. Prenatal diagnosis of cystic hygroma related to a deletion of 16q24.1 with haploinsuffciency of FOXF1 and FOXC2 genes. Case Rep Genet. 2012 doi: 10.1155/2012/490408. Epub 2012 Aug 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez C, Rodriguez A, Palenzuela S, Forteza C, Rossello JL. Congenital misalignment of pulmonary veins with alveolar capillary dysplasia causing persistent neonatal pulmonary hypertension: report of two affected siblings. Pediatr Dev Pathol. 2000;3:271–6. doi: 10.1007/s100249910035. [DOI] [PubMed] [Google Scholar]
- Handrigan GR, Chitayat D, Lionel AC, Pinsk M, Vaags AK, Marshall CR, Dyack S, Escobar LF, Fernandez BA, Stegman JC, et al. Deletions in 16q24.2 are associated with autism spectrum disorder, intellectual disability and congenital renal malformation. J Med Genet. 2013 doi: 10.1136/jmedgenet-2012-101288. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–64. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellqvist M, Mahlapuu M, Samuelsson L, Enerback S, Carlsson P. Differential activation of lung-specific genes by two forkhead proteins, FREAC-1 and FREAC-2. J Biol Chem. 1996;271:4482–90. doi: 10.1074/jbc.271.8.4482. [DOI] [PubMed] [Google Scholar]
- Kalinichenko VV, Gusarova GA, Kim IM, Shin B, Yoder HM, Clark J, Sapozhnikov AM, Whitsett JA, Costa RH. Foxf1 haploinsufficiency reduces Notch-2 signaling during mouse lung development. Am J Physiol Lung Cell Mol Physiol. 2004;286:L521–30. doi: 10.1152/ajplung.00212.2003. [DOI] [PubMed] [Google Scholar]
- Kalinichenko VV, Lim L, Shin B, Costa RH. Differential expression of forkhead box transcription factors following butylated hydroxytoluene lung injury. Am J Physiol Lung Cell Mol Physiol. 2001a;280:L695–704. doi: 10.1152/ajplung.2001.280.4.L695. [DOI] [PubMed] [Google Scholar]
- Kalinichenko VV, Lim L, Stolz DB, Shin B, Rausa FM, Clark J, Whitsett JA, Watkins SC, Costa RH. Defects in pulmonary vasculature and perinatal lung hemorrhage in mice heterozygous null for the Forkhead Box f1 transcription factor. Dev Biol. 2001b;235:489–506. doi: 10.1006/dbio.2001.0322. [DOI] [PubMed] [Google Scholar]
- Kirchler T, Briesemeister S, Singer M, Schutze K, Keinath M, Kohlbacher O, Vicente-Carbajosa J, Teige M, Harter K, Chaban C. The role of phosphorylatable serine residues in the DNA-binding domain of Arabidopsis bZIP transcription factors. Eur J Cell Biol. 2010;89:175–83. doi: 10.1016/j.ejcb.2009.11.023. [DOI] [PubMed] [Google Scholar]
- Kitayama Y, Kamata S, Okuyama H, Usui N, Sawai T, Kobayashi T, Fukui Y, Okada A. Nitric oxide inhalation therapy for an infant with persistent pulmonary hypertension caused by misalignment of pulmonary veins with alveolar capillary dysplasia. J Pediatr Surg. 1997;32:99–100. doi: 10.1016/s0022-3468(97)90105-6. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Langelaan DN, Wieczorek M, Blouin C, Rainey JK. Improved helix and kink characterization in membrane proteins allows evaluation of kink sequence predictors. J Chem Inf Model. 2010;50:2213–20. doi: 10.1021/ci100324n. [DOI] [PubMed] [Google Scholar]
- Langston C. Misalignment of pulmonary veins and alveolar capillary dysplasia. Pediatr Pathol. 1991;11:163–70. doi: 10.3109/15513819109064753. [DOI] [PubMed] [Google Scholar]
- Licht C, Schickendantz S, Sreeram N, Arnold G, Rossi R, Vierzig A, Mennicken U, Roth B. Prolonged survival in alveolar capillary dysplasia syndrome. Eur J Pediatr. 2004;163:181–2. doi: 10.1007/s00431-003-1385-6. [DOI] [PubMed] [Google Scholar]
- Lim L, Kalinichenko VV, Whitsett JA, Costa RH. Fusion of lung lobes and vessels in mouse embryos heterozygous for the forkhead box f1 targeted allele. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1012–22. doi: 10.1152/ajplung.00371.2001. [DOI] [PubMed] [Google Scholar]
- Madison BB, McKenna LB, Dolson D, Epstein DJ, Kaestner KH. FoxF1 and FoxL1 link hedgehog signaling and the control of epithelial proliferation in the developing stomach and intestine. J Biol Chem. 2009;284:5936–44. doi: 10.1074/jbc.M808103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlapuu M, Enerback S, Carlsson P. Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development. 2001;128:2397–406. doi: 10.1242/dev.128.12.2397. [DOI] [PubMed] [Google Scholar]
- Mahlapuu M, Pelto-Huikko M, Aitola M, Enerback S, Carlsson P. FREAC-1 contains a cell-type-specific transcriptional activation domain and is expressed in epithelial-mesenchymal interfaces. Dev Biol. 1998;202:183–95. doi: 10.1006/dbio.1998.9010. [DOI] [PubMed] [Google Scholar]
- Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–25. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson P, Yepiskoposyan H, Metze S, Zamudio Orozco R, Kleinschmidt N, Muhlemann O. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci. 2010;67:677–700. doi: 10.1007/s00018-009-0177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierrou S, Hellqvist M, Samuelsson L, Enerback S, Carlsson P. Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J. 1994;13:5002–12. doi: 10.1002/j.1460-2075.1994.tb06827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plat G, Rouquette I, Marcoux MO, Bloom MC, Acar P, Dulac Y. [Alveolar capillary dysplasia and persistent pulmonary hypertension of the newborn] Arch Mal Coeur Vaiss. 2007;100:458–61. [PubMed] [Google Scholar]
- Sen P, Choudhury T, Smith EO, Langston C. Expression of angiogenic and vasculogenic proteins in the lung in alveolar capillary dysplasia/misalignment of pulmonary veins: an immunohistochemical study. Pediatr Dev Pathol. 2010;13:354–61. doi: 10.2350/09-04-0640-OA.1. [DOI] [PubMed] [Google Scholar]
- Sen P, Gerychova R, Janku P, Jezova M, Valaskova I, Navarro C, Silva I, Langston C, Welty S, Belmont J, Stankiewicz P. A familial case of alveolar capillary dysplasia with misalignment of pulmonary veins supports paternal imprinting of FOXF1 in human. Eur J Hum Genet. 2012 doi: 10.1038/ejhg.2012.171. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Thakur N, Stockton DW, Langston C, Bejjani BA. Expanding the phenotype of alveolar capillary dysplasia (ACD) J Pediatr. 2004;145:646–51. doi: 10.1016/j.jpeds.2004.06.081. [DOI] [PubMed] [Google Scholar]
- Shaut CA, Keene DR, Sorensen LK, Li DY, Stadler HS. HOXA13 Is essential for placental vascular patterning and labyrinth endothelial specification. PLoS Genet. 2008;4:e1000073. doi: 10.1371/journal.pgen.1000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw-Smith C. Genetic factors in esophageal atresia, tracheo-esophageal fistula and the VACTERL association: roles for FOXF1 and the 16q24.1 FOX transcription factor gene cluster, and review of the literature. Eur J Med Genet. 2010;53:6–13. doi: 10.1016/j.ejmg.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, Wiszniewska J, Driscoll DJ, Maisenbacher MK, Bolivar J, Bauer M, et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet. 2009;84:780–91. doi: 10.1016/j.ajhg.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranski P, Dharmadhikari AV, Brosens E, Gurha P, Kolodziejska KE, Ou Z, Dittwald P, Majewski T, Mohan KN, Chen B, Pearson RE, Tibboel, et al. Small non-coding differentially-methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder. Genome Res. 2013;23:23–33. doi: 10.1101/gr.141887.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibballs J, Chow CW. Incidence of alveolar capillary dysplasia in severe idiopathic persistent pulmonary hypertension of the newborn. J Paediatr Child Health. 2002;38:397–400. doi: 10.1046/j.1440-1754.2002.00014.x. [DOI] [PubMed] [Google Scholar]
- Tiozzo C, Carraro G, Al Alam D, Baptista S, Danopoulos S, Li A, Lavarreda-Pearce M, Li C, De Langhe S, Chan B, Borok Z, Bellusci S, et al. Mesodermal Pten inactivation leads to alveolar capillary dysplasia-like phenotype. J Clin Invest. 2012 doi: 10.1172/JCI61334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng HT, Shah R, Jamrich M. Function and regulation of FoxF1 during Xenopus gut development. Development. 2004;131:3637–47. doi: 10.1242/dev.01234. [DOI] [PubMed] [Google Scholar]
- Vanhoof G, Goossens F, De Meester I, Hendriks D, Scharpe S. Proline motifs in peptides and their biological processing. FASEB J. 1995;9:736–44. [PubMed] [Google Scholar]
- Wache SC, Hoagland EM, Zeigler G, Swanson HI. Role of arginine residues 14 and 15 in dictating DNA binding stability and transactivation of the aryl hydrocarbon receptor/aryl hydrocarbon receptor nuclear translocator heterodimer. Gene Expr. 2005;12:231–43. doi: 10.3727/000000005783991981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenvoort CA. Misalignment of lung vessels: a syndrome causing persistent neonatal pulmonary hypertension. Hum Pathol. 1986;17:727–30. doi: 10.1016/s0046-8177(86)80182-4. [DOI] [PubMed] [Google Scholar]
- Wei G, Liu G, Liu X. Identification of two serine residues important for p53 DNA binding and protein stability. FEBS Lett. 2003;543:16–20. doi: 10.1016/s0014-5793(03)00369-7. [DOI] [PubMed] [Google Scholar]
- Yu S, Shao L, Kilbride H, Zwick DL. Haploinsufficiencies of FOXF1 and FOXC2 genes associated with lethal alveolar capillary dysplasia and congenital heart disease. Am J Med Genet A. 2010;152A:1257–62. doi: 10.1002/ajmg.a.33378. [DOI] [PubMed] [Google Scholar]
- Zufferey F, Martinet D, Osterheld MC, Niel-Butschi F, Giannoni E, Schmutz NB, Xia Z, Beckmann JS, Shaw-Smith C, Stankiewicz P, Langston C, Fellmann F. 16q24.1 microdeletion in a premature newborn: usefulness of array-based comparative genomic hybridization in persistent pulmonary hypertension of the newborn. Pediatr Crit Care Med. 2011;12:e427–32. doi: 10.1097/PCC.0b013e3182192c96. [DOI] [PMC free article] [PubMed] [Google Scholar]