

Abstract
Human African trypanosomiasis (HAT) is a neglected tropical disease caused by the protozoan parasite Trypanosoma brucei. Since drugs in use against HAT are toxic and require intravenous dosing, new drugs are needed. Initiating lead discovery campaigns by using chemical scaffolds from drugs approved for other indications can speed up drug discovery for neglected diseases. We demonstrated recently that the 4-anilinoquinazolines lapatinib (GW572016, 1) and canertinib (CI-1033) kill T. brucei with low micromolar EC50 values. We now report promising activity of analogs of 1, which provided an excellent starting point for optimization of the chemotype. We report our compound optimization that has led to synthesis of several potent 4-anilinoquinazolines, including NEU621, 23a, a highly potent, orally bioavailable inhibitor of trypanosome replication. At the cellular level, 23a blocks duplication of the kinetoplast and arrests cytokinesis, making it a new tool for studying regulation of the trypanosome cell cycle.
Introduction
Neglected tropical diseases (NTDs) represent a significant global health burden, particularly in developing regions of the world. Estimates of as many as one in six in the world population (over 1 billion people) are infected by one or more NTD, with one in three people at risk.1 These diseases are “neglected” since so few research dollars are invested in treating or preventing them, in comparison to those conditions primarily affecting the developed world. As a result, pragmatic and cost-effective approaches for identification of drug leads are needed in order to spawn the discovery of new drugs.
One such approach is to “repurpose” classes of proven molecular targets with essential homologs in the pathogens that cause these NTDs.2 For example, Trypanosoma brucei (which causes human African trypanosomiasis (HAT), T. cruzi (Chagas’ disease), Leishmania spp. (causative agents for leishmaniases) and Plasmodium spp. (malaria) all express kinases and phosphodiesterases (PDEs) that are involved in aspects of cellular signaling.3,4 Indeed, kinases and PDEs represent proven drug target classes in humans for a variety of indications, and, as such, a large amount of data related to medicinal chemistry, toxicology, and structural biology are available that can potentially inform new optimization programs against parasites. Furthermore, the clinical and pre-clinical chemical matter itself can sometimes represent a starting point for new antiparasitic approaches, an approach demonstrated by us5-7 and by others.8,9
Following parasite transmission via an infected tsetse fly, a trypanosome bloodstream infection gives rise to flu-like symptoms that eventually subside. At this point, the parasites invade the central nervous system (CNS), where they establish an infection that leads to sleep disruption, coma and eventually death. Current drugs have less than optimal toxicity profiles, and the dosing regimens can be inconvenient, long, and costly. There is therefore a stated need for new HAT therapeutics that are orally administered, with minimal toxicity, and which are effective against both bloodstream and CNS forms of the disease. To that end, “hit” and “lead” criteria for HAT and other NTDs are clearly described.10
Kinase inhibitors have come to the fore as one of the principal enzyme target classes in drug discovery for a wide variety of indications, including cancer,11 inflammation,12,13 diabetes,14,15 and CNS diseases.16 In particular, a number of tyrosine kinase inhibitors have been approved for clinical use.17 This list includes lapatinib (GW572016, Tykerb, 1), an EGFR inhibitor that gained FDA approval in 2007.18
T. brucei expresses over 180 protein kinases,19,20 some of which (such as glycogen synthase kinase-3,21 phosphoinositol-3-kinases/TOR,7 and Aurora kinase 16) have been targeted in drug discovery efforts already. There is unequivocal chemical data for protein Tyr phosphorylation in the parasite.22,23 However, trypanosomes do not express receptor tyrosine kinases (RTKs),4 and it is widely held that Tyr-phosphorylation must therefore be performed by dual-specificity enzymes (e.g., wee1) that act on Ser/Thr as well as Tyr residues.4 Intriguingly, based on bioinformatics analysis, enzymes with “EGFR-like” kinase domains are present in the parasite.24 Further, inhibitors of human EGFR/HER2 (i.e., canertinib,251,26 and AEE78827) kill T. brucei with EC50 in the low micromolar range.24
Transferrin is a growth factor that T. brucei acquires from its vertebrate host by receptor-mediated endocytosis.28 We discovered that receptor-mediated endocytosis of Tf in the African trypanosome is stimulated by diacylglycerol (DAG).29 In most eukaryotes, effects of DAG on signaling pathways are amplified by the Ser/Thr kinase protein kinase C, which binds to the lipid with its C1-domain. In trypanosomes, DAG signaling pathways have not been studied. To understand the pathway linking DAG and Tf endocytosis in the trypanosome, we tested the effect of inhibitors of Ser/Thr protein kinases (e.g., protein kinase C) or Tyr kinases on DAG-stimulated endocytosis of Tf. Unexpectedly, DAG-stimulated endocytosis of Tf was not blocked by a Ser/Thr protein kinase inhibitor, nor does the genome of T. brucei encode for a classic PKC. Instead, the pathway was inhibited by a Tyr kinase inhibitor Tyrphostin A47, a tyrosine mimic.30 In a related study, we found that 1, similar to Tyrphostin A47, inhibited endocytosis of transferrin.31
These data suggested to us that tyrosine kinase inhibitor drugs approved for treatment of non-parasite human diseases were worth testing as anti-trypanosomal agents. We have evaluated the trypanocidal properties of a panel of EGFR inhibitors provided by GlaxoSmithKline (Table 1), and we describe the subsequent design and synthesis of novel analogs for a structure activity relationship study of a 6-phenyl 4-anilinoquinazoline scaffold, culminating in 23a, a highly selective and potent inhibitor of trypanosome replication in vitro.
Table 1.
Initial screening data of analogs of lapatinib 1.
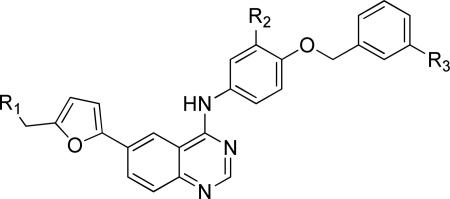
| |||||
|---|---|---|---|---|---|
| Compd | GSK Number | R1 | R2 | R3 | Tbb EC50 (μM)a,b |
| 2 | GW58337A |
|
Cl | H | 0.41 |
| 3 | GW601906A |
|
Cl | F | 0.43 |
| 4 | GW633460A |
|
Cl | F | 0.48 |
| 5 | GW616030X |
|
Cl | F | 0.52 |
| 6 | GW615311X |
|
Cl | F | 0.55 |
| 7 | GW580496A |
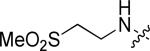
|
Br | H | 0.56 |
| 8 | GW576924A |
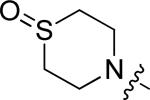
|
F | F | 0.60 |
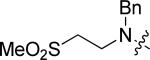
| 9 | GW616907X |
|
Cl | F | 1.51 |
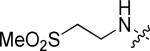
| 1 | lapatinib |
|
Cl | F | 1.54 |
| melarsoprol49 | 0.0063 | ||||
| eflornithine50 | 16.4 | ||||
| pentamidine49 | 0.0035 | ||||
| SCYX-715851 | 0.794 | ||||
All EC50 values are ± 7%.
Concentration giving 50% inhibition of growth of T brucei brucei Lister 427 cells
Results
We obtained nine quinazoline-based EGFR inhibitors (1-9, Table 1)18,32-34 from GlaxoSmithKline and screened them against cultures of T brucei brucei Lister 427. The inhibitors demonstrated a four-fold range in potency (Table 1) within the limited scope in variation of the R1 “tail” region of the scaffold. We noted the EC50 of 1 (1.54 μM) to be four-fold more potent against T. brucei as compared to a HepG2 hepatocarcinoma cell line.35 For comparison, we have also included in Table 1 the published activities of three front-line HAT treatments (eflornithine, melarsoprol, and pentamidine), and of SCYX-7158, currently in clinical trials. The goal of our subsequent optimization efforts have dwelt on improvement of the potency of this chemotype for inhibition of T. brucei replication, and increasing the selectivity ratio over HepG2 cells.
Noting the effect of subtle tail-group changes on parasite growth inhibition, we first focused our attention on broader exploration of replacements for the furan-derived tail. This was achieved by a broad diversity scan utilizing Suzuki chemistry methodology using boronic acids or esters to enumerate a virtual library of analogs of 1 ( Scheme 1). The structures in this 197-membered virtual library were clustered based on a maximum dissimilarity algorithm, and cluster centers were selected for synthesis (PipelinePilot, Scitegic, Inc).
Scheme 1.

Synthesis of analogs 10.a
In anticipation of the parallel synthesis, we prepared the iodoquinazoline 14 by the route shown in Scheme 1. Treatment of the commercially available anthranilic acid 11 with formamide proceeded in 70% yield, followed by chlorination with thionyl chloride to provide the chloroquinazoline 13 in 85% yield. This template was reacted with the requisite aniline (17, Scheme 2), which was prepared by a sequence of alkylation of the nitrophenol 15 with 3-fluorobenzyl bromide followed by nitro group reduction. With the required template 14 in hand, we prepared ten analogs (10a-k) from the selected boronates using standard Suzuki reaction conditions. The structures and biological activities for these compounds are summarized in Table 2. From this series of analogs we identified NEU369 (10a), which was approximately equipotent to 1 against T brucei cells. Further testing of this compound and its analogs showed that, unlike 1, it did not inhibit HepG2 cell growth (EC50 > 15 μM) (Table 3).
Scheme 2.

Synthesis of requisite aniline intermediates 17.a
Table 2.
Trypanosoma brucei growth inhibition data of diverse analogs 10a-10k.
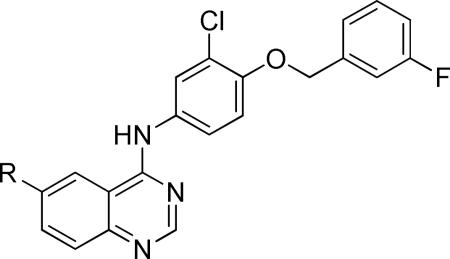
| ||
|---|---|---|
| Compd | R1 | Tbb EC50 (μM)a,b |
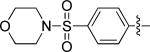
| 10a |
|
1.39 |
| 10b |
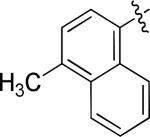
|
2.27 |
| 10c |
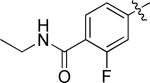
|
3.85 |
| 10d |
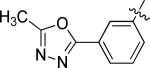
|
4.21 |
| 10e |
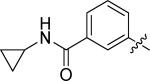
|
4.50 |
| 10f |
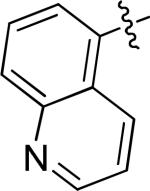
|
4.53 |
| 10g |
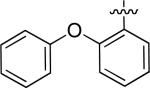
|
5.3 |
| 10h |
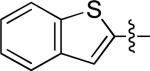
|
5.98 |
| 10i |
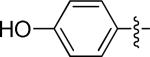
|
6.45 |
| 10j |
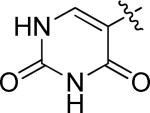
|
22.63 |
All EC50 values are ± 7%.
Concentration giving 50% inhibition of growth of T brucei brucei Lister 427 cells
Table 3.
T. brucei growth inhibition data for headgroup variations of 10a.
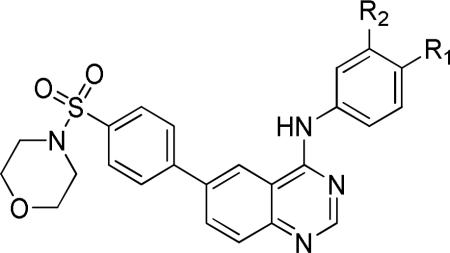
| ||||
|---|---|---|---|---|
| Compd | R1 | R2 | Tbb EC50 (μM)a,b | HepG2 IC50 (uM) |
| 10a |
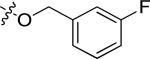
|
Cl | 1.39 | >15 |
| 20a | H | H | 1.44 | > 3 |
| 20b | CH3 | H | 1.15 | > 15 |
| 20c | OH | Cl | 1.06 | > 15 |
| 20d | OCH3 | Cl | 0.82 | > 15 |
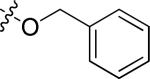
| 20e |
|
Cl | 1.35 | > 15 |
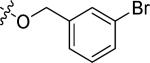
| 20f |
|
Cl | 0.68 | > 15 |
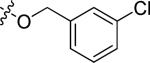
| 20g |
|
Cl | 0.66 | > 15 |
| 20h |
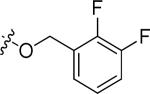
|
Cl | 0.82 | >15 |
| 20i |
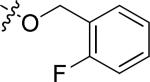
|
Cl | 1.65 | > 15 |
| 20j |
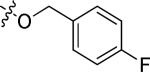
|
Cl | 1.34 | > 15 |
| 20k |
|
Cl | 1.43 | > 15 |
| 20l |
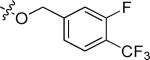
|
Cl | 0.54 | > 15 |
| 20m |
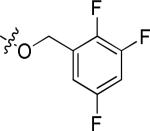
|
Cl | 1.12 | > 3 |
| 20n |
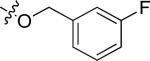
|
H | 0.65 | > 15 |
| 20o |
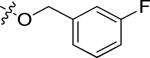
|
OCH3 | 1.88 | > 15 |
All EC50 values are ± 7%.
Concentration giving 50% inhibition of growth of T brucei brucei Lister 427 cells
Keeping the newly identified tail group present in 10a (Table 3), we turned our attention to exploration of the aniline head group region of the molecule. Preparation of the requisite chloroquinazoline 19 (Scheme 3) was achieved by treatment of 12 with the required boronic acid using Suzuki conditions, followed by chlorination with thionyl chloride. This intermediate was reacted with a range of anilines (Scheme 2) to provide analogs 20 (Table 3).
Scheme 3.

Synthesis of head group expansion set 20.a
This library was designed to explore the role of halogen substitutions on the terminal benzyl substituent (R1) headgroup, testing positional isomers of fluoro substitutions and other potential halogen replacements such as methoxy and trifluoromethyl groups. These modifications produced insignificant changes in activity of the compounds against T. brucei. We prepared a few analogs to assess functional group tolerance at the R2 position of the head group, replacing the chlorine atom of 1 with hydrogen and methoxy groups; these changes also resulted in very modest alterations in anti-trypanosomal activity. Interestingly, truncation of the molecule (20a) gave potency approximately equal to 10a, translating to a similar ligand efficiency value (LE of 10a=0.14; 20a=0.18).36 Importantly, HepG2 cell toxicity was minimal with these modifications.
For the next round of analogs, we turned our attention back to further refinement of the tail group region of 1, performing focused modifications of the 6-aryl position of the quinazoline ring that were designed to evaluate steric requirements, as well as the required adornment of polarity in this region of the inhibitor. These compounds (10k-w) were accessed from the corresponding boronic acids using the route shown in Scheme 1. The larger sulfonamide side chains showed some modest preference for meta-substitution (Table 4, entries 1-6), though the methyl sulfones (entries 11-12) showed preference for para-substituents. Comparing the sulfonamide substituents, morpholine was preferred over the other heterocyclics tested.
Table 4.
T. brucei and HepG2 growth inhibition data for focused analogs of 10a.
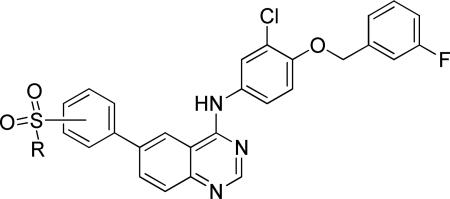
| |||||
|---|---|---|---|---|---|
| Entry | Compd | position | R1 | Tbb EC50 (μM)a,b | HepG2 IC50 (uM) |
| 1 | 10a | p |
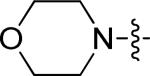
|
1.39 | >15 |
| 2 | 10k | m |
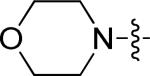
|
0.33 | > 15 |
| 3 | 10l | p |
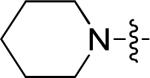
|
0.32 | > 15 |
| 4 | 10m | m |
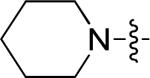
|
0.46 | >15 |
| 5 | 10n | p |
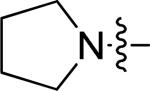
|
0.35 | > 15 |
| 6 | 10o | m |
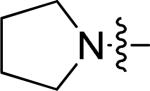
|
0.25 | > 15 |
| 7 | 10p | p |
|
0.53 | > 15 |
| 8 | 10q | p |
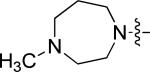
|
0.81 | 1.81 |
| 9 | 10r | p |
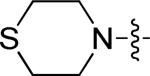
|
0.47 | > 15 |
| 10 | 10s | p |
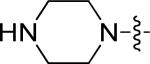
|
0.28 | 3.32 |
| 11 | 10t | p | CH3 | 0.90 | > 15 |
| 12 | 10u | m | CH3 | 3.21 | > 15 |
| 13 | 10v | o | N(CH3)2 | 1.04 | > 15 |
| 14 | 10w | o | NHC(CH3)3 | 4.66 | ndc |
All EC50 values are ± 7%.
Concentration giving 50% inhibition of growth of T brucei brucei Lister 427 cells.
Not determined.
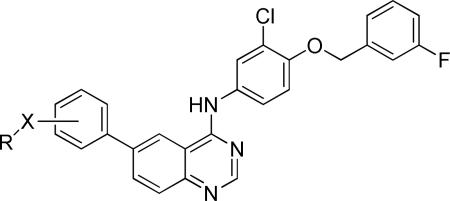
A more focused evaluation of linker and regiochemistry is shown in Table 5. Compounds were synthesized from 14 by reaction with the appropriate boronate ester 22. In the case of the morpholinosulfonamides (entries 1-8), meta- substitution is consistently better than para; the most potent analog, NEU617 (23a), is directly linked to the aromatic ring. For piperidinosulfonamides (entries 9-16), the meta preference is less consistent, and none of these analogs show as potent growth inhibition as 23a. When the tail contains a morpholine (entries 1-8), the linker has little impact on potency (except for 23a, a clear outlier); all meta-substituted analogs are otherwise approximately equipotent. In cases where the morpholine moiety is at the para-position there is little difference resulting from linker variation.
Table 5.
Growth inhibitory potency of regiochemical and linker analogs of 10.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Compd | R | Regio | X | Tbb EC50 (μM)a,b | HepG2 IC50 (μM) |
| 1 | 23a |
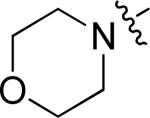
|
m | -- | 0.042 | >20 |
| 2 | 23b | p | -- | 1.91 | 9.6 | |
| 3 | 23c | m | CH2 | 0.36 | ndc | |
| 4 | 23d | p | CH2 | 0.99 | 12.9 | |
| 5 | 23e | m | C=O | 0.55 | nd | |
| 6 | 23f | p | C=O | 1.21 | nd | |
| 7 | 10k | m | SO2 | 0.33 | >20 | |
| 8 | 10a | p | SO2 | 1.39 | >20 | |
| 9 | 23g |
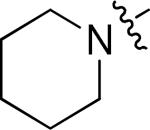
|
m | -- | 0.76 | >15 |
| 10 | 23h | p | -- | 0.38 | nd | |
| 11 | 23i | m | CH2 | 0.65 | nd | |
| 12 | 23j | p | CH2 | 1.5 | nd | |
| 13 | 23k | m | C=O | 0.14 | >6 | |
| 14 | 23l | p | C=O | 0.94 | nd | |
| 15 | 10m | m | SO2 | 0.46 | >20 | |
| 16 | 10l | p | SO2 | 0.32 | >20 | |
All EC50 values are ± 7%.
Concentration giving 50% inhibition of growth of T brucei brucei Lister 427 cells.
Not determined.
With piperidine-substitution (entries 9-16), it appears that a modest preference exists for the meta-substituted amide, with a 6.7-fold loss of activity when moved to the para position (compare entries 13 and 14), though the importance of positional substitution is otherwise less for other examples, within ~2-fold in activity.
Lastly, we sequentially removed the head group substituents of 23a. Removal of one (23m) or both (23n) halogens afforded an approximately 5-fold reduction in potency, and further truncation (23o) significantly reduced antiparasitic activity while restoring HepG2 potency (Table 6).
Table 6.
Headgroup truncation analogs of 23a.
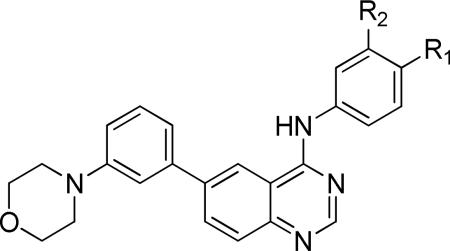
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | Tbb EC50 (μM)a,b | HepG2 IC50 (μM) | LE |
| 23a |
|
Cl | 0.042 | >20 | 0.19 |
| 23m | OBn | Cl | 0.23 | >20 | 0.18 |
| 23n | OBn | H | 0.18 | >20 | 0.19 |
| 23o | H | H | 11 | 7.02 | 0.17 |
All EC50 values are ± 7%.
Concentration giving 50% inhibition of growth of T brucei brucei Lister 427 cells.
Noting its potency against T. brucei and selectivity over HepG2 cells (Table 5), we advanced 23a into a mouse oral pharmacokinetic study. Mice were administered a single oral dose (40 mg/kg) of 23a, and plasma and brain tissue drug levels were measured over 24 hours (Supporting Information Figure S1 and Table S6). Though the CNS fraction was low (5%), the plasma levels were in excess of the EC50 for >12 hours. We determined that 23a was 99.6% plasma protein bound. However, since trypanosomes are noted for their ability to endocytose host plasma proteins,29,37,38 we believed that the oral exposure and in vitro potency warranted in vivo efficacy evaluation.
In a test of efficacy in a mouse model of HAT, mice were infected with T. brucei brucei CA427(104 cells) and after 24 h, were administered a 40 mg/kg dose of 23a once per day. No parasites were detected in the blood of the infected mice for 3 days whereas control mice had trypanosomes in their blood on day 2 post-infection. However drug-related toxicity was observed with 23a in a multi-day regimen at the 40 mg/kg dose. These data point to a need to improve the pharmacokinetic properties of 23a so that it can be more effective in the mouse model of HAT.
Nonetheless, inspired by these initial results, we adjusted the dosing regimen, opting to administer 23a at 10 mg/kg twice per day (total dose of 20 mg/kg/day), either orally or intraperitoneally (i.p.). The results are shown in Figure 1A. We noted three effects: First, i.p. treatment with 23a delayed detection of trypanosomes in the blood of infected mice by 24 h. Whereas all control mice had trypanosomes in their blood on day 3, mice treated with 23a all had parasites in their blood 24 h later, suggestive of either a significant reduction in replication rate (trypanosomes divide every 6 hours) or of parasite killing during this initial phase of infection. Second, on day 8 when all untreated mice had died, the mice dosed i.p. with 23a were all alive (Figure 1B). In the oral administration experiment, mice died in the same time frame as control mice, suggesting insufficient drug exposure at this dosage. Third, i.p administration of 23a led to better control of infection, leading to a doubling of mouse survival life span from 5 days in the control group to 9 days in 23a-treated mice.
Figure 1.

Assessment of 23a in a mouse model of human African trypanosomiasis. Parasitemia was measured each day in untreated (U, red) and treated (T, blue) mice (20 mg/kg).
Although 23a is structurally similar to 1, the biological effects of the two compounds are different. Whereas 1 inhibited endocytosis of transferrin in the trypanosome31 in a manner similar to what we previously observed with tyrphostin,3023a had no effect on receptor-mediated endocytosis of transferrin. Instead, 23a affected the cell cycle in ways not observed with 1 (Figure 2).
Figure 2.

Effects of 23a on the cell cycle of the trypanosome. (A) 23a-treated or control trypanosomes were analyzed by flow cytometry (10,000 events analyzed; see Methods section). Propidium iodide (PI) fluorescence is plotted against cell populations containing 2C or 4C equivalents of DNA. (B) Quantitation of DNA content per cell (calculated with FloJo software). (C) Microscopic analysis of 23a-treated or untreated trypanosomes after staining with DNA-binding dye DAPI. N, nucleus; K, kinetoplast; DIC, differential interference contrast image. (D) Quantitation of organelle (nucleus (N) or kinetoplast (K)) distribution after 23a treatment.
Trypanosomes possess two DNA-containing organelles (nucleus and kinetoplast (mitochondrial nucleoid)). The kinetoplast and nucleus can be tracked by microscopy during the cell cycle, which begins with trypanosomes harboring one nucleus (1N) and one kinetoplast (1K) (i.e., 1K1N cells).39,40 A kinetoplast that is replicating its (DNA) (i.e., kDNA) and increasing in the organelle's mass is observed as an elongated kinetoplast (1Ke). Fission (segregation) of 1Ke kinetoplasts into two daughter kinetoplasts (2K) inside the same cell is coincident with the nuclear S-phase, and produces 2K1N trypanosomes. Mitosis then occurs, yielding trypanosomes containing two kinetoplasts and two nuclei (2K2N). Following cell division each daughter trypanosome has a 1K1N1 configuration of the organelles.41
After a 7h incubation with 23a, the chromosomal DNA profile of T. brucei was altered (Figure 2A); the fraction of cells with 2C-4C equivalents of DNA was reduced from 40 to 25%, whereas the proportion of cells with 4C DNA increased from 25% to 40% (Figure 2B). Single cell microscopy studies were performed to determine whether the changes in DNA per cell caused by 23a (Figure 2B) were reflected in alterations in the number of DNA-containing organelles per cell (Figure 2C). Quantitation of the data obtained in Figure 2C revealed that 23a reduced the number of cells containing one nucleus (i.e., 1K1N, 1Ke1N) but selectively increased the fraction of a group of cells that are not normally detected in the absence of the drug; cells containing two nuclei and one kinetoplast (1K2N) (Figure 2D). This data is consistent with the increase in trypanosomes with 4C equivalent of DNA (Figure 2B). Thus, 23a blocks duplication of the kinetoplast and arrests cytokinesis, without inhibiting division of the trypanosome nucleus.
Discussion and conclusions
While some efforts to find new anti-trypanosomal lead compounds involve screening of large compound libraries against cell cultures,42 these can be complemented with lead discovery programs that target important physiological pathways in the parasite. Further, since many of these pathways employ enzymes of functional similarity to drug targets in humans, a privileged set of starting compounds already exist that can represent useful starting points for lead discovery without massive drug screening initiatives.
In this report we have described the discovery of potent trypanocidal compounds that are based on established human EGFR inhibitor chemotypes. By starting with such “privileged” lead compounds, we have accelerated our anti-trypanosomal lead discovery program by circumventing a need to run high-throughput screens. Indeed, in only a few optimization cycles we have identified a potent chemotype as a lead series for trypanosomiasis. Though compound 23a has high calculated lipophilicity and molecular weight, its oral bioavailability lent the compound to further assessment in a mouse model of HAT, where we observed modest effects in controlling parasitemia, with concomitant life extension of infected mice. Since the pharmacokinetic experiments suggest acceptable plasma levels in mice following oral dosing, we hypothesize that the high plasma protein binding (99.6%) observed for 23a is the cause of the lower-than-expected effect on in vivo parasite loads.
Trypanosome physiology analysis indicates that 23a it acts via a mechanism different from 1 and tyrphostin. Work is in progress to identify the molecular targets of our lead compound series. Nonetheless, we note that utilization of cell replication assays has led to promising outcomes that are worthy of future lead optimization studies.
Besides working to reduce plasma protein binding, further optimization of 23a is needed to improve predicted central nervous system exposure. A recent report by Wager et al. described an analysis that correlated compound properties to CNS exposure using data for both experimental and marketed drugs,43 and devised a scoring paradigm for prediction of CNS exposure.44 This protocol, termed multi-parameter optimization, or MPO, allows prediction of likely CNS exposure for a given compound based on how closely it meets desired property ranges for MW, cLogP, LogD, pKa, and TPSA with a maximal MPO score of 6.0, and compounds >4.0 predicted to be CNS-penetrant. As shown in Table 7, compound 23a has significant shortcomings in terms of properties that have been shown relevant to CNS activity, particularly due to high calculated lipophilicity (cLogP, cLogD=7.1) and molecular weight (541 Da). Future efforts will be focused on reduction of molecular size and lipophilicity to improve the likelihood of CNS penetration, a pivotal requirement for any new therapeutic for HAT.
Table 7.
Desirable ranges for CNS penetration and the MPO scoring for 23a.
| Prop | Targeted Value | Properties of 23a | MPO Score |
|---|---|---|---|
| cLogP | ≤ 3 | 7.1 | 0 |
| cLogD | ≤ 2 | 7.1 | 0 |
| TPSA | 40 < X ≤ 90 | 59.5 | 1.00 |
| MW | ≤ 360 | 541 | 0 |
| HBD | ≤ 0.5 | 1 | 0.8 |
| pKa | ≤ 8 | 3.05 | 1.0 |
| MPO score | 2.8 | ||
In summary, we have identified a potent trypanosomal growth inhibitor (23a) based on the 6-furanyl 4-anilinoquinazoline scaffold of 1. The lead displays good selectivity over HepG2 cells, is orally bioavailable, and is modestly efficacious in a mouse model of HAT, such that it meets “Lead Criteria” as defined by The Special Programme for Research and Training in Tropical Diseases (TDR).10 Further optimization of this scaffold for increased effectiveness in the mouse model will be reported in due course. Our ability to (i) identify trypanosome replication inhibitors based on a basic biology principles using established kinase drug chemotypes, and (ii) perform rapid hit-to-lead medicinal chemistry supports our approach of re-optimizing existing drug scaffolds for anti-trypanosome lead discovery.
Experimental Section
Chemical synthesis
Unless otherwise noted, reagents were obtained from Sigma-Aldrich, Inc. (St. Louis, MO), or Frontier Scientific Services, Inc. (Newark, DE) and used as received. Boronic acids and aniline reagents were purchased, unless the synthesis is specifically described below. Reaction solvents were purified by passage through alumina columns on a purification system manufactured by Innovative Technology (Newburyport, MA). NMR spectra were obtained with Varian NMR systems, operating at 400 or 500 MHz for 1H acquisitions as noted. LCMS analysis was performed using a Waters Alliance reverse-phase HPLC, with single-wavelength UV-visible detector and LCT Premier time-of-flight mass spectrometer (electrospray ionization). All newly synthesized compounds were that were submitted for biological testing were deemed >95% pure by LCMS analysis (UV and ESI-MS detection) prior to submission for biological testing. Preparative LCMS was performed on a Waters FractionLynx system with a Waters MicroMass ZQ mass spectrometer (electrospray ionization) and a single-wavelength UV-visible detector, using acetonitrile/water gradients with 0.1% formic acid. Fractions were collected on the basis of triggering using UV and mass detection. Yields reported for products obtained by preparative HPLC represent the amount of pure material isolated; impure fractions were not repurified
4-chloro-6-iodoquinazoline hydrochloride (13).45
Yield: 85%. 1H NMR (500 MHz, DMSO-d6) δ: 8.39 (d, J = 1.95 Hz, 1H), 8.29 (s, 1H), 8.13 (dd, J = 1.95, 8.30 Hz, 1H), 7.49 (d, J = 8.30 Hz, 1H). MS: m/z = 290.83 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-iodoquinazolin-4-amine hydrochloride (14).46
Yield: 84%. 1H NMR (500 MHz, DMSO-d6) δ: 11.21 (br. s., 1H), 9.16 (s, 1H), 8.92 (s, 1H), 8.34 (d, J = 8.79 Hz, 1H), 7.93 (d, J = 2.44 Hz, 1H), 7.64 - 7.68 (m, 2H), 7.46 - 7.51 (m, 1H), 7.30 - 7.37 (m, 2H), 7.20 (dt, J = 2.44, 8.79 Hz, 1H), 5.30 (s, 2H). MS: m/z = 505.85 (M+H)+.
Libraries of 10 were synthesized by Suzuki coupling of 14 with respective boronic acid/esters following General procedure A. Into glass vials was combined N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-iodoquinazolin-4-amine (14, 100 μM), boronic acids/esters (120 μmol) and tetrakis(triphenylphosphine)palladium(0) (7 μmol). To the reaction mixture was added 1,2-dimethoxyethane (2 mL), ethanol (1.33 mL) and a 2M aqueous solution of sodium carbonate (0.301 mL, 600 μM). The vials were capped and shaken at 80 °C for 18 h. The progress of the reaction was followed by LC-MS. Reaction mixture was evaporated to dryness. Crude products were purified using flash column chromatography, or by dissolving in DMSO and purifying by reverse phase HPLC using a gradient of 30-100% acetonitrile in water containing 0.1% formic acid.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine (10a)
Yield: 48.1%. 1H NMR (500 MHz, DMSO-d6) δ: 10.00 (s, 1H), 8.92 (d, J = 1.46 Hz, 1H), 8.64 (s, 1H), 8.28 (dd, J = 1.95, 8.79 Hz, 1H), 8.17 (d, J = 8.79 Hz, 2H), 8.04 (d, J = 2.44 Hz, 1H), 7.92 (d, J = 8.79 Hz, 3H), 7.76 (dd, J = 2.45, 8.80 Hz, 1H), 7.46 - 7.50 (m, 1H), 7.30 - 7.36 (m, 3H), 7.19 - 7.23 (m, 1H), 5.28 (s, 2H), 3.66 - 3.68 (m, 4H), 2.93 - 2.95 (m, 4H). MS: m/z = 605.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-methylnaphthalen-1-yl)quinazolin-4-amine, (10b)
Yielded 1 mg (2.6%) as a yellow film. 1H NMR (400 MHz, DMSO-d6) δ: 9.82 (s, 1H), 8.65 (s, 2H), 8.15 (d, J = 8.8 Hz, 1H), 8.03 (d, J = 2.2 Hz, 1H), 7.87-7.96 (m, 2H), 7.81 (d, J = 8.8 Hz, 1H), 7.71-7.76 (m, 1H), 7.63 (t, J = 8.0 Hz, 1H), 7.49-7.57 (m, 2H), 7.42-7.49 (m, 2H), 7.31 (t, J = 6.0 Hz, 2H), 7.25 (d, J = 8.8 Hz, 1H), 7.17 (t, J = 7.3 Hz, 1H), 5.24 (s, 2H), 2.74 (s, 3H). MS: m/z = 520.1 (M+H)+.
4-(4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)quinazolin-6-yl)-N-ethyl-2-fluorobenzamide (10c)
Yielded 1 mg (2.4%) as a yellow film. 1H NMR (400 MHz, DMSO-d6) δ: 9.98 (s, 1H), 8.87 (s, 1H), 8.58-8.61 (s, 1H), 8.35-8.42 (m, 1H), 8.26-8.34 (m, 1H), 8.01 (s, 1H), 7.87 (d, J = 4.4 Hz, 1H), 7.84 (m, 2H), 7.79 (d, J = 8.1 Hz, 1H), 7.70-7.76 (m, 1H), 7.47 (q, J = 7.3 Hz, 1H), 7.32 (dd, JA = 13.2 Hz, JB = 7.3 Hz, 3H), 7.18 (t, J = 8.8 Hz, 1H), 5.27 (s, 2H), 3.29 (q, J = 8.0 Hz, 2H), 1.14 (t, J = 7.0 Hz, 3H).: MS: m/z = 545.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(3-(5-methyl-1,3,4-oxadiazol-2-yl)phenyl)quinazolin-4-amine (10d)
Obtained 1 mg (2.5% yield) as a yellow oil. 1H NMR (400 MHz, DMSO-d6) δ: 9.09-10.04 (brs, 1H), 8.88 (s, 1H), 8.61 (s, 1H), 8.42 (s, 1H), 8.37 (s, 1H), 8.26 (d, J = 8.8 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 8.0 (m, 2H), 7.89 (d, J = 8.8 Hz, 1H), 7.72-7.81 (m, 2H), 7.43-7.51 (m, 1H), 7.28-7.36 (m, 2H), 7.14-7.22 (m, 1H) 5.26 (s, 2H), 2.65 (s, 3H). MS: m/z = 538.1 (M+H)+.
3-(4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)quinazolin-6-yl)-N-cyclopropylbenzamide (10e)
Yielded 0.8 mg (2.0%) as a yellow film. 1H NMR (400 MHz, DMSO-d6) δ: 10.00 (s, 1H), 8.83 (s, 1H), 8.63 (d, J = 3.7 Hz, 1H), 8.59 (s, 1H), 8.35 (s, 1H), 8.26 (d, J = 5.1 Hz, 1H), 8.22 (s, 1H), 7.97-8.05 (m, 1H), 7.87 (t, J = 8.1Hz, 2H), 7.74 (dd, JA = 8.8 Hz, JB = 2.0 Hz, 1H), 7.63 (t, J = 7.7 Hz, 1H), 7.43-7.52 (m, 1H), 7.25-7.35 (m, 2H), 7.17 (t, J = 8.0 Hz, 1H), 5.27 (s, 2H), 2.85-2.92 (m, 1H), 0.69-0.76 (m, 2H), 0.58-0.65 (m, 2H). MS: m/z = 539.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(quinolin-5-yl)quinazolin-4-amine (10f)
Yielded 3.2 mg (8.4%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ: 9.81 (s, 1H), 8.98 (d, J = 2.9 Hz, 1H), 8.70 (s, 1H), 8.68 (s, 1H), 8.21-8.27 (m, 2H), 8.14 (d, J = 8.1 Hz, 1H), 8.03 (d, J = 2.2 Hz, 1H), 7.95-8.00 (m, 1H), 7.88-7.95 (m, 3H),7.68-7.96 (m, 2H), 7.53-7.59 (dd, JA = 8.4 Hz, JB = 4.0 Hz, 1H), 7.46 (q, J = 8.0 Hz, 1H), 7.22-7.33 (m, 3H), 7.17 (t, J = 8.8 Hz, 1H), 5.23 (s, 2H). MS: m/z = 508.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(2-phenoxyphenyl)quinazolin-4-amine (10g)
Yielded 1.4 mg (2.4%) as an orange oil. 1H NMR (400 MHz, DMSO-d6) δ: 9.85 (s, 1H), 8.65 (s, 1H), 8.58 (s, 1H), 8.01-8.06 (m, 2H), 7.72-7.78 (m, 2H), 7.67 (d, J = 6.6 Hz, 1H), 7.42-7.51 (m, 2H), 7.24-7.39 (m, 6H), 7.19 (t, J = 7.3 Hz, 1H), 7.00-7.09 (m, 2H), 6.94 (d, J = 8.1 Hz, 2H), 5.27 (s, 2H). MS: m/z = 548.1 (M+H)+.
6-(benzo[b]thiopen-2-yl)-N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl) quinazolin-4-amine (10h)
Obtained 5.4 mg (14% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ: 8.75 (s, 1H), 8.15 (t, J = 8.8 Hz, 1H), 8.03 (m, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.87 (d, J = 2.1 Hz, 2H), 7.82 (d, J = 7.3 Hz, 1H), 7.68 (s, 1H), 7.51-7.59 (m, 1H), 7.49 (s, 1H), 7.32-7.44 (m, 3H), 7.19-7.25 (m, 1H), 6.91-7.09 (m, 2H), 5.27 (s, 2H). MS: m/z = 512.0 (M+H)+.
4-(4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)quinazolin-6-yl)phenol (10i)
Yielded 1 mg (2.4%) as a yellow film. 1H NMR (400 MHz, DMSO-d6) δ: 10.09 (s, 1H), 8.98 (s, 1H), 8.63 (s, 2H), 8.45 (s, 1H), 8.42 (s, 1H), 8.05 (s, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.82 (d, J = 9.5 Hz, 1H), 7.43-7.51 (m, 2H), 7.27-7.36 (m, 2H), 7.14-7.22 (m, 1H), 6.66-6.72 (m, 2H), 5.27 (s, 2H). MS: m/z = 472.1 (M+H)+.
5-(4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)quinazolin-6-yl)pyrimidine-2,4(1H,3H)-dione (10j)
Yielded 2.6 mg (7.1%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ: 9.76-9.82 (brs, 1H), 8.47-8.59 (m, 2H), 8.30 (s, 1H), 8.04 (m, 2H), 7.81-7.88 (m, 1H), 7,71-7.80 (m. 2H), 7.59-7.67 (m, 1H), 7.43-7.50 (m, 1H), 7.31-7.36 (m, 1H), (m, 7.23-7.31 (m, 2H), 7.18 (t, J = 8.4 Hz, 1H), 5.26 (s, 2H). MS: m/z = 490.0 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(3-(morpholinosulfonyl)phenyl)quinazolin-4-amine (10k)
Yield: 30.4%. 1H NMR (500 MHz, DMSO-d6) δ: 10.01 (s, 1H), 8.85 (d, J = 1.95 Hz, 1H), 8.63 (s, 1H), 8.24 - 8.27 (m, 2H), 8.12 (t, J = 1.71 Hz, 1H), 8.03 (d, J = 2.93 Hz, 1H), 7.86 - 7.92 (m, 2H), 7.80 - 7.85 (m, 1H), 7.73 (dd, J = 2.44, 8.79 Hz, 1H), 7.46 - 7.52 (m, 1H), 7.30 - 7.35 (m, 3H), 7.17 - 7.22 (m, 1H), 5.28 (s, 2H), 3.66 (t, J = 4.90 Hz, 4H), 2.95 (t, J = 4.40 Hz, 4H). MS: m/z = 605.1 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-(piperidin-1-ylsulfonyl)phenyl)quinazolin-4-amine (10l)
Yield: 14.6%. 1H NMR (500 MHz, DMSO-d6) δ: 10.00 (s, 1H), 8.91 (d, J = 1.46 Hz, 1H), 8.64 (s, 1H), 8.27 (dd, J = 1.95, 8.30 Hz, 1H), 8.14 (d, J = 8.30 Hz, 2H), 8.04 (d, J = 2.93 Hz, 1H), 7.89 - 7.92 (m, 3H), 7.76 (dd, J = 2.69, 9.03 Hz, 1H), 7.46 - 7.51 (m, 1H), 7.31 - 7.36 (m, 3H), 7.20 (dt, J = 2.44, 8.55 Hz, 1H), 5.28 (s, 2H), 2.95 - 2.98 (m, 4H), 1.55 - 1.60 (m, 4H), 1.39 - 1.40 (m, 2H). MS: m/z = 603.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(3-(piperidin-1-ylsulfonyl)phenyl)quinazolin-4-amine (10m)
Yield: 24.8%. 1H NMR (500 MHz, DMSO-d6) δ: 10.01 (s, 1H), 8.84 (d, J = 1.95 Hz, 1H), 8.62 (s, 1H), 8.24 (dd, J = 1.95, 8.79 Hz, 1H), 8.21 (d, J = 7.35 Hz, 1H), 8.11 (t, J = 1.71 Hz, 1H), 8.02 (d, J = 2.44 Hz, 1H), 7.90 (d, J = 8.30 Hz, 1H), 7.82 - 7.86 (m, 1H), 7.79 - 7.81 (m, 1H), 7.72 (dd, J = 2.69, 9.03 Hz, 1H), 7.45 - 7.51 (m, 1H), 7.29 - 7.36 (m, 3H), 7.19 (dt, J = 2.44, 8.55 Hz, 1H), 5.27 (s, 2H), 2.96 (t, J = 5.4 Hz, 4Hm, 4H), 1.52 - 1.60 (m, 4H), 1.33 - 1.40 (m, 2H). MS: m/z = 603.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-(pyrrolidin-1-ylsulfonyl)phenyl)quinazolin-4-amine (10n)
Yield: 19.6%. 1H NMR (500 MHz, DMSO-d6) δ: 10.00 (s, 1H), 8.91 (d, J = 1.95 Hz, 1H), 8.63 (s, 1H), 8.27 (dd, J = 1.95, 8.79 Hz, 1H), 8.13 (d, J = 8.30 Hz, 2H), 8.03 (d, J = 2.44 Hz, 1H), 7.98 (d, J = 8.79 Hz, 2H), 7.90 (d, J = 8.79 Hz, 1H), 7.76 (dd, J = 2.44, 8.79 Hz, 1H), 7.45 - 7.52 (m, 1H), 7.29 - 7.36 (m, 3H), 7.20 (dt, J = 2.44, 8.55 Hz, 1H), 5.28 (s, 2H), 3.22 (t, J = 6.84 Hz, 4H), 1.66 - 1.72 (m, 4H). MS: m/z = 589.1 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(3-(pyrrolidin-1-ylsulfonyl)phenyl)quinazolin-4-amine (10o)
Yield: 28.2%. 1H NMR (500 MHz, DMSO-d6) δ: 10.03 (s, 1H), 8.85 (d, J = 1.46 Hz, 1H), 8.62 (s, 1H), 8.26 (dd, J = 1.95, 8.79 Hz, 1H), 8.18 - 8.23 (m, 2H), 8.03 (d, J = 2.44 Hz, 1H), 7.88 - 7.93 (m, 2H), 7.81 - 7.87 (m, 1H), 7.73 (dd, J = 2.44, 8.79 Hz, 1H), 7.45 - 7.52 (m, 1H), 7.30 - 7.37 (m, 3H), 7.20 (dt, J = 2.44, 8.55 Hz, 1H), 5.28 (s, 2H), 3.23 (t, J = 6.84 Hz, 4H), 1.68 (td, J = 3.54, 6.59 Hz, 4H). MS: m/z = 589.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-((4-methylpiperazin-1-yl)sulfonyl)phenyl)quinazolin-4-amine (10p)
Yield: 30.6%. 1H NMR (500 MHz, DMSO-d6) δ: 9.95 (s, 1H), 8.87 (d, J = 1.46 Hz, 1H), 8.59 (s, 1H), 8.23 (dd, J = 1.95, 8.79 Hz, 1H), 8.11 (d, J = 8.30 Hz, 2H), 7.99 (d, J = 2.44 Hz, 1H), 7.83 - 7.90 (m, 3H), 7.71 (dd, J = 2.69, 9.03 Hz, 1H), 7.40 - 7.47 (m, 1H), 7.25 - 7.32 (m, 3H), 7.15 (dt, J = 2.44, 8.55 Hz, 1H), 5.23 (s, 2H), 2.91 (br. s., 4H), 2.34 (br. s., 4H), 2.11 (s, 3H). MS: m/z = 618.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-((4-methyl-1,4-diazepan-1-yl)sulfonyl)phenyl)quinazolin-4-amine (10q)
Yield: 42.8%. 1H NMR (500 MHz, DMSO-d6) δ: 10.00 (s, 1H), 8.91 (d, J = 1.95 Hz, 1H), 8.63 (s, 1H), 8.27 (dd, J = 1.95, 8.79 Hz, 1H), 8.12 (d, J = 8.80 Hz, 2H), 8.03 (d, J = 2.44 Hz, 1H), 7.96 (d, J = 8.30 Hz, 2H), 7.90 (d, J = 8.79 Hz, 1H), 7.76 (dd, J = 2.69, 9.03 Hz, 1H), 7.45 - 7.50 (m, 1H), 7.30 - 7.36 (m, 3H), 7.20 (dt, J = 2.44, 8.55 Hz, 1H), 5.28 (s, 2H), 3.37 - 3.39 (m, 2H), 3.34 (t, J = 6.10 Hz, 3H), 2.61 - 2.64 (m, 2H), 2.54 - 2.58 (m, 2H), 2.28 (s, 3H), 1.74 - 1.80 (m, 2H). MS: m/z = 632.2 (M+H)+.
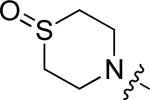
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-(thiomorpholinosulfonyl)phenyl)quinazolin-4-amine (10r)
Yield: 5.6%. 1H NMR (500 MHz, DMSO-d6) δ: 10.00 (s, 1H), 8.92 (d, J = 1.47 Hz, 1H), 8.64 (s, 1H), 8.28 (dd, J = 1.95, 8.79 Hz, 1H), 8.16 (d, J = 8.79 Hz, 2H), 8.04 (d, J = 2.44 Hz, 1H), 7.89 - 7.95 (m, 3H), 7.76 (dd, J = 2.45, 8.80 Hz, 1H), 7.46 - 7.52 (m, 1H), 7.30 - 7.37 (m, 3H), 7.20 (dt, J = 2.20, 8.67 Hz, 1H), 5.28 (s, 2H), 3.28 (t, J = 4.35 Hz, 4H), 2.71 (t, J = 5.35 Hz, 4H). MS: m/z = 621.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-(piperazin-1-ylsulfonyl)phenyl)quinazolin-4-amine (10s)
To glass vials was weighed 46 mg of 14 (0.085 mmoL) and tert-butyl 4-((4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)sulfonyl)piperazine-1-carboxylate (38.4 mg 0.085 mmol) and tetrakis(triphenylphosphine)palladium(0) (0.006 mmol). To the reaction mixture was added 1,2-dimethoxyethane (0.4 mL), ethanol (0.3 mL) and a 2M aqueous solution of sodium carbonate (0.255 mL, 0.51 mmol). The vials were capped and shaken at 85 °C for 12, and progress of the reaction was monitored by LC-MS. The reaction mixture was evaporated to dryness, and the residue was dissolved in DMSO and purified by reverse phase HPLC using a gradient of 5-100% acetonitrile in water containing 0.1% formic acid providing the Boc-protected compound in 23.7% yield. 1H NMR (500 MHz, DMSO-d6) δ: 9.99 (s, 1H), 8.91 (d, J = 1.95 Hz, 1H), 8.64 (s, 1H), 8.28 (dd, J = 1.95, 8.79 Hz, 1H), 8.16 (d, J = 8.79 Hz, 2H), 8.04 (d, J = 2.44 Hz, 1H), 7.89 - 7.93 (m, 3H), 7.76 (dd, J = 2.44, 8.79 Hz, 1H), 7.46 - 7.52 (m, 1H), 7.30 - 7.36 (m, 3H), 7.20 (dt, J = 2.44, 8.79 Hz, 1H), 5.28 (s, 2H), 3.41 - 3.46 (m, 4H), 2.94 (t, J = 4.64 Hz, 4H), 1.34 (s, 9H). MS: m/z = 704.3 (M+H)+. To a solution of this compound (0.015 mmol) in 0.4 mL of dichloromethane was added trifluoroacetic acid (200 μmol, 0.154 mL). The reaction mixture was stirred for 12 h at 25 °C. Volatiles were removed in vacuo, and the crude product was triturated with hexanes to afford a crude solid that was purified via flash column chromatography (0-10% MeOH-DCM) to afford the desired compound 10t. Yield: 78%. 1H NMR (500 MHz, DMSO-d6) δ: 9.01 (s, 1H), 8.80 (br. s., 1H), 8.59 (br. s., 2H), 8.39 (d, J = 7.81 Hz, 1H), 8.22 (d, J = 8.30 Hz, 2H), 7.92 - 8.03 (m, 4H), 7.72 (dd, J = 2.44, 8.79 Hz, 1H), 7.47 - 7.52 (m, 1H), 7.32 - 7.37 (m, 3H), 7.21 (dt, J = 2.20, 8.67 Hz, 1H), 5.31 (s, 2H), 3.25 (br. s., 4H), 3.18 (br. s., 4H). MS: m/z = 604.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-(methylsulfonyl)phenyl)quinazolin-4-amine (10t)
Yield: 31.8%. 1H NMR (500 MHz, DMSO-d6) δ: 10.00 (s, 1H), 8.92 (d, J = 1.95 Hz, 1H), 8.64 (s, 1H), 8.29 (dd, J = 1.95, 8.79 Hz, 1H), 8.14 (d, J = 8.80 Hz, 2H), 8.11 (d, J = 8.30 Hz, 2H), 8.03 (d, J = 2.44 Hz, 1H), 7.92 (d, J = 8.79 Hz, 1H), 7.76 (dd, J = 2.44, 8.79 Hz, 1H), 7.45 - 7.53 (m, 1H), 7.31 - 7.37 (m, 3H), 7.20 (dt, J = 2.20, 8.42 Hz, 1H), 5.28 (s, 2H), 3.31 (s, 3H). MS: m/z = 534.1 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(3-(methylsulfonyl)phenyl)quinazolin-4-amine (10u)
Yield: 34.9%. 1H NMR (500 MHz, DMSO-d6) δ: 10.02 (s, 1H), 8.88 (s, 1H), 8.63 (s, 1H), 8.38 (s, 1H), 8.30 (dd, J = 1.46, 8.79 Hz, 1H), 8.25 (d, J = 8.30 Hz, 1H), 7.99 - 8.06 (m, 2H), 7.92 (d, J = 8.79 Hz, 1H), 7.84 - 7.87 (m, 1H), 7.74 (dd, J = 2.44, 8.79 Hz, 1H), 7.45 - 7.53 (m, 1H), 7.30 - 7.36 (m, 3H), 7.20 (dt, J = 1.71, 8.67 Hz, 1H), 5.28 (s, 2H), 3.35 (s, 3H). MS: m/z = 534.2 (M+H)+.
2-(4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)quinazolin-6-yl)-N,N-dimethylbenzenesulfonamide (10v)
Yield: 35%. 1H NMR (500 MHz, DMSO-d6) δ: 9.78 (s, 1H), 8.65 (s, 1H), 8.52 (d, J = 0.98 Hz, 1H), 8.06 (d, J = 2.44 Hz, 1H), 8.01 (dd, J = 0.98, 7.81 Hz, 1H), 7.83 (dt, J = 1.46, 9.03 Hz, 1H), 7.76 - 7.81 (m, 3H), 7.72 (dt, J = 1.22, 7.69 Hz, 1H), 7.54 (dd, J = 0.98, 7.81 Hz, 1H), 7.45 - 7.50 (m, 1H), 7.30 - 7.34 (m, 2H), 7.27 (d, J = 8.79 Hz, 1H), 7.19 (dt, J = 2.44, 8.55 Hz, 1H), 5.26 (s, 2H), 2.45 (s, 6H). MS: m/z = 563.2 (M+H)+.
N-(tert-butyl)-2-(4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)quinazolin-6-yl)benzenesulfonamide (10w)
Yield: 12.5%. 1H NMR (500 MHz, DMSO-d6) δ: 9.79 (s, 1H), 8.64 (s, 1H), 8.53 (d, J = 1.95 Hz, 1H), 8.11 (dd, J = 1.22, 8.06 Hz, 1H), 8.05 (d, J = 2.44 Hz, 1H), 7.86 (dd, J = 1.71, 8.55 Hz, 1H), 7.70 - 7.79 (m, 3H), 7.63 - 7.67 (m, 1H), 7.45 - 7.50 (m, 2H), 7.30 - 7.35 (m, 2H), 7.27 (d, J = 9.28 Hz, 1H), 7.19 (dt, J = 2.44, 8.55 Hz, 1H), 6.90 (s, 1H), 5.26 (s, 2H), 1.04 (s, 9H). MS: m/z = 591.2(M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(3-morpholinophenyl)quinazolin-4-amine (23a)
Yield: 38%. 1H NMR (500 MHz, DMSO-d6) δ: 9.90 (s, 1H), 8.76 (d, J = 1.95 Hz, 1H), 8.60 (s, 1H), 8.18 (dd, J = 1.71, 8.55 Hz, 1H), 8.04 (d, J = 2.93 Hz, 1H), 7.84 (d, J = 8.79 Hz, 1H), 7.76 (dd, J = 2.44, 8.79 Hz, 1H), 7.45 - 7.51 (m, 1H), 7.39 - 7.42 (m, 1H), 7.28 - 7.35 (m, 5H), 7.19 (dt, J = 2.44, 8.55 Hz, 1H), 7.03 (dd, J = 1.95, 8.30 Hz, 1H), 5.27 (s, 2H), 3.79 (t, J = 4.9 Hz, 4H), 3.24 (t, J = 4.86 Hz, 4H). MS: m/z = 541.2 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-morpholinophenyl)quinazolin-4-amine (23b)
Yield: 35.2%. 1H NMR (500 MHz, DMSO-d6) δ: 9.87 (s, 1H), 8.72 (d, J = 1.95 Hz, 1H), 8.55 (s, 1H), 8.16 (dd, J = 1.95, 8.79 Hz, 1H), 8.03 (d, J = 2.93 Hz, 1H), 7.78 - 7.82 (m, 3H), 7.76 (dd, J = 2.44, 8.79 Hz, 1H), 7.45 - 7.51 (m, 1H), 7.28 - 7.35 (m, 3H), 7.19 (dt, J = 2.69, 8.67 Hz, 1H), 7.11 (d, J = 8.79 Hz, 2H), 5.27 (s, 2H), 3.76 - 3.80 (m, 4H), 3.19 - 3.23 (m, 4H). MS: m/z = 541.04 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(3-(morpholinomethyl)phenyl)quinazolin-4-amine (23c)
Yield: 54.5%. 1H NMR (500 MHz, DMSO-d6) δ: 9.92 (s, 1H), 8.77 (s, 1H), 8.59 (s, 1H), 8.15 (d, J = 8.79 Hz, 1H), 8.03 (d, J = 1.95 Hz, 1H), 7.85 (d, J = 8.79 Hz, 1H), 7.72 - 7.80 (m, 3H), 7.43 - 7.54 (m, 2H), 7.39 (d, J = 7.32 Hz, 1H), 7.25 - 7.35 (m, 3H), 7.14 - 7.22 (m, 1H), 5.26 (s, 2H), 3.52-3.68 (m, 6H), 2.40 (br. s., 4H). MS: m/z = 554.3060 (M)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-(morpholinomethyl)phenyl)quinazolin-4-amine (23d)
Yield: 36.8%. 1H NMR (500 MHz, DMSO-d6) δ: 9.92 (s, 1H), 8.80 (d, J = 1.95 Hz, 1H), 8.60 (s, 1H), 8.20 (dd, J = 1.95, 8.79 Hz, 1H), 8.04 (d, J = 2.44 Hz, 1H), 7.84 - 7.87 (m, 3H), 7.77 (dd, J = 2.69, 9.03 Hz, 1H), 7.46 - 7.54 (m, 3H), 7.28 - 7.36 (m, 3H), 7.20 (dt, J = 1.95, 8.55 Hz, 1H), 5.28 (s, 2H), 3.61 (t, J = 4.64 Hz, 4H), 3.56 (s, 2H), 2.40 (br. s., 4H). MS: m/z = 555.2 (M+H)+.
(3-(4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)quinazolin-6-yl)phenyl)(morpholino)methanone (23e)
Yield: 53.2%. 1H NMR (500 MHz, DMSO-d6) δ: 9.99 (br. s., 1H), 8.83 (d, J = 1.46 Hz, 1H), 8.61 (s, 1H), 8.23 (dd, J = 1.46, 8.79 Hz, 1H), 8.01 (d, J = 2.44 Hz, 1H), 7.97 (d, J = 7.81 Hz, 1H), 7.92 (s, 1H), 7.86 (d, J = 8.79 Hz, 1H), 7.73 (dd, J = 2.44, 8.79 Hz, 1H), 7.63 (t, J = 7.81 Hz, 1H), 7.47 (q, J = 7.65 Hz, 2H), 7.26 - 7.36 (m, 3H), 7.18 (dt, J = 2.20, 8.67 Hz, 1H), 5.26 (s, 2H), 3.20 - 3.78 (m, 8H). MS: m/z = 568.2021 (M)+.
(4-(4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)quinazolin-6-yl)phenyl)(morpholino)methanone (23f)
Yield: 58.9%. 1H NMR (500 MHz, DMSO-d6) δ: 9.93 (br. s., 1H), 8.84 (br. s., 1H), 8.60 (s, 1H), 8.21 (d, J = 8.30 Hz, 1H), 7.93 - 8.05 (m, 3H), 7.86 (d, J = 8.30 Hz, 1H), 7.76 (d, J = 7.81 Hz, 1H), 7.60 (d, J = 7.81 Hz, 2H), 7.43 - 7.51 (m, 1H), 7.26 - 7.36 (m, 3H), 7.18 (t, J = 8.06 Hz, 1H), 5.26 (s, 2H), 3.37 - 3.80 (m, 8H). MS: m/z = 568.2183 (M).
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(3-(piperidin-1-yl)phenyl)quinazolin-4-amine (23g)
Yield: 21%. 1H NMR (500 MHz, DMSO-d6) δ: 9.90 (s, 1H), 8.74 (d, J = 1.95 Hz, 1H), 8.58 (s, 1H), 8.16 (dd, J = 1.71, 8.55 Hz, 1H), 8.03 (d, J = 2.44 Hz, 1H), 7.83 (d, J = 8.79 Hz, 1H), 7.75 (dd, J = 2.44, 8.79 Hz, 1H), 7.44 - 7.51 (m, 1H), 7.27 - 7.39 (m, 5H), 7.23 (d, J = 7.81 Hz, 1H), 7.19 (dt, J = 2.44, 8.55 Hz, 1H), 7.00 (dd, J = 1.95, 8.30 Hz, 1H), 5.27 (s, 2H), 3.23 - 3.27 (m, 4H), 1.64 - 1.69 (m, 4H), 1.53 - 1.59 (m, 2H). MS: m/z = 539.16 (M+H)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-(piperidin-1-yl)phenyl)quinazolin-4-amine (23h)
Yield: 41.4%. 1H NMR (500 MHz, DMSO-d6) δ: 9.87 (s, 1H), 8.69 (d, J = 0.98 Hz, 1H), 8.55 (s, 1H), 8.12 (dd, J = 1.71, 8.55 Hz, 1H), 8.03 (d, J = 2.44 Hz, 1H), 7.72 - 7.81 (m, 4H), 7.43 - 7.51 (m, 1H), 7.25 - 7.37 (m, 3H), 7.18 (dt, J = 2.44, 8.55 Hz, 1H), 7.06 (d, J = 8.79 Hz, 2H), 5.26 (s, 2H), 3.18 - 3.26 (m, 4H), 1.49 - 1.70 (m, 6H). MS: m/z = 532.3831 (M)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(3-(piperidin-1-ylmethyl)phenyl)quinazolin-4-amine (23i)
Yield: 66.7%. 1H NMR (500 MHz, DMSO-d6) δ: 9.91 (br. s., 1H), 8.81 (d, J = 1.95 Hz, 1H), 8.60 (s, 1H), 8.19 (dd, J = 1.95, 8.79 Hz, 1H), 8.01 (d, J = 2.93 Hz, 1H), 7.87 - 7.97 (m, 3H), 7.74 (dd, J = 2.90, 9.25 Hz, 1H), 7.63 (t, J = 7.57 Hz, 1H), 7.52 (d, J = 7.32 Hz, 1H), 7.46 (dt, J = 6.35, 8.06 Hz, 1H), 7.26 - 7.35 (m, 3H), 7.17 (dt, J = 2.44, 8.55 Hz, 1H), 5.25 (s, 2H), 4.22 (br. s., 2H), 3.00 (d, J = 5.86 Hz, 4H), 1.42 - 1.77 (m, 6H). MS: m/z = 552.3265 (M)+.
N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(4-(piperidin-1-ylmethyl)phenyl)quinazolin-4-amine (23j)
Yield: 52.3%. 1H NMR (500 MHz, DMSO-d6) δ: 9.89 (br. s., 1H), 8.80 (d, J = 1.46 Hz, 1H), 8.58 (s, 1H), 8.19 (dd, J = 1.95, 8.79 Hz, 1H), 8.01 (d, J = 2.44 Hz, 1H), 7.90 (d, J = 8.30 Hz, 2H), 7.85 (d, J = 8.79 Hz, 1H), 7.74 (dd, J = 2.44, 8.79 Hz, 1H), 7.56 (d, J = 8.30 Hz, 2H), 7.41 - 7.50 (m, 1H), 7.25 - 7.35 (m, 3H), 7.17 (dt, J = 2.20, 8.67 Hz, 1H), 5.25 (s, 2H), 3.93 (br. s., 2H), 2.74 (br. s., 4H), 1.56 - 1.69 (m, 4H), 1.45 (br. s., 2H). MS: m/z = 552.3265 (M)+.
(3-(4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)quinazolin-6-yl)phenyl)(piperidin-1-yl)methanone (23k)
Yield: 66.3%. 1H NMR (500 MHz, DMSO-d6) δ: 8.91 (s, 1H), 8.76 (br. s., 1H), 8.35 (d, J = 9.28 Hz, 1H), 7.96 - 8.00 (m, 2H), 7.87 - 7.93 (m, 2H), 7.70 (dd, J = 2.45, 8.80 Hz, 1H), 7.59 - 7.65 (m, 2H), 7.44 - 7.52 (m, 2H), 7.32 - 7.36 (m, 3H), 7.20 (dt, J = 2.44, 8.79 Hz, 1H), 5.30 (s, 2H), 3.65 (br. s., 3H), 1.41 - 1.70 (m, 7H). MS: m/z = 567.3 (M+H)+.
(4-(4-((3-chloro-4-((3-fluorobenzyl)oxy)phenyl)amino)quinazolin-6-yl)phenyl)(piperidin-1-yl)methanone (23l)
Yield: 73.3%. 1H NMR (500 MHz, DMSO-d6) δ: 9.93 (s, 1H), 8.83 (s, 1H), 8.59 (s, 1H), 8.21 (dd, J = 1.95, 8.30 Hz, 1H), 8.01 (d, J = 2.44 Hz, 1H), 7.93 (d, J = 8.30 Hz, 2H), 7.86 (d, J = 8.79 Hz, 1H), 7.74 (dd, J = 2.44, 8.79 Hz, 1H), 7.53 (d, J = 8.30 Hz, 3H), 7.43 - 7.49 (m, 1H), 7.27 - 7.34 (m, 3H), 7.17 (dt, J = 1.95, 8.55 Hz, 1H), 5.25 (s, 2H), 3.60 (br. s., 2H), 1.38 - 1.67 (m, 7H). MS: m/z = 567.3 (M+H)+.
N-(4-(benzyloxy)-3-chlorophenyl)-6-(3-morpholinophenyl)quinazolin-4-amine (23m)
Yield: 38%. 1H NMR (500 MHz, DMSO-d6) δ: 9.88 (s, 1H), 8.75 (d, J = 1.95 Hz, 1H), 8.58 (s, 1H), 8.18 (dd, J = 1.95, 8.79 Hz, 1H), 8.01 (d, J = 2.93 Hz, 1H), 7.84 (d, J = 8.30 Hz, 1H), 7.74 (dd, J = 2.69, 9.03 Hz, 1H), 7.50 (d, J = 7.32 Hz, 2H), 7.39 - 7.45 (m, 3H), 7.33 - 7.37 (m, 2H), 7.30 (d, J = 8.79 Hz, 2H), 7.03 (dd, J = 1.95, 8.30 Hz, 1H), 5.24 (s, 2H), 3.77 - 3.81 (m, 4H), 3.22 - 3.25 (m, 4H). MS: m/z = 523.07 (M+H)+.
N-(4-(benzyloxy)phenyl)-6-(3-morpholinophenyl)quinazolin-4-amine (23n)
Yield: 18%. 1H NMR (500 MHz, DMSO-d6) δ: 9.84 (s, 1H), 8.76 (d, J = 1.46 Hz, 1H), 8.51 (s, 1H), 8.16 (dd, J = 1.95, 8.79 Hz, 1H), 7.81 (d, J = 8.30 Hz, 1H), 7.68 (d, J = 9.30 Hz, 2H), 7.48 (d, J = 7.32 Hz, 2H), 7.38 - 7.43 (m, 3H), 7.28 - 7.37 (m, 3H), 7.07 (d, J = 8.80 Hz, 2H), 7.02 (dd, J = 1.95, 7.80 Hz, 1H), 5.14 (s, 2H), 3.78 (t, J = 4.87 Hz, 4H), 3.24 (t, J = 4.87 Hz, 4H). MS: m/z = 489.11 (M+H)+.
6-(3-morpholinophenyl)-N-phenylquinazolin-4-amine (23o)
Yield: 57.5%. 1H NMR (500 MHz, DMSO-d6) δ: 9.92 (s, 1H), 8.80 (d, J = 1.46 Hz, 1H), 8.58 (s, 1H), 8.19 (dd, J = 1.95, 8.79 Hz, 1H), 7.82 - 7.87 (m, 3H), 7.39 - 7.45 (m, 3H), 7.36 (d, J = 2.44 Hz, 1H), 7.30 - 7.33 (m, 1H), 7.15 - 7.18 (m, 1H), 7.03 (dd, J = 2.20, 8.06 Hz, 1H), 3.79 (t, J = 4.85 Hz, 4H), 3.24 (t, J = 4.85 Hz, 4H). MS: m/z = 383.06 (M+H)+.
Aniline synthesis
Anilines 17 were synthesized using General procedure B. In a 20 mL glass vial a solution of substituted 4-nitrophenol (1.5 mmol) in 5 mL acetonitrile was combined with potassium carbonate (3 mmol) and the appropriate benzyl bromide (1.5 mmol) at 25 °C. The reaction mixture was stirred at 50 °C for 12 h. After completion of the alkylation reaction, the reaction mixture was triturated with 10 mL of 10% MeOH/DCM and was filtered through a silica gel plug. The organic filtrate was evaporated and re-dissolved in 5 mL of MeOH:H2O (5:1). To this solution was added Zinc (0.29 g, 4.5 mmol) followed by ammonium chloride (0.48 g, 9 mmol) at 25 °C. The temperature was raised to 50 °C and the stirring was continued 6 h. Progress of the reaction was followed by LC-MS. Upon completion of the reaction, 15 mL of DCM:MeOH (1:1) was added to the reaction mixture, inorganic residues were removed by filtration. The filtrate was evaporated and the residue was purified via flash chromatography (0-50% EtOAc/hexanes) to afford the desired substituted anilines.
4-(benzyloxy)-3-chloroaniline (17e)
Yield: 24.3%. 1H NMR (500 MHz, CDCl3) δ: 7.47 (d, J = 7.32 Hz, 2H), 7.37 - 7.41 (m, 2H), 7.30 - 7.35 (m, 1H), 6.81 (d, J = 8.79 Hz, 1H), 6.76 (d, J = 2.93 Hz, 1H), 6.51 (dd, J = 2.69, 8.55 Hz, 1H), 5.06 (s, 2H), 3.49 (br. s., 2H). MS: m/z = 234.02 (M+H)+.
4-((3-bromobenzyl)oxy)-3-chloroaniline (17f)
Yield: 25%. 1H NMR (500 MHz, CDCl3) δ: 7.62 (s, 1H), 7.45 (d, J = 8.30 Hz, 1H), 7.39 (d, J = 7.81 Hz, 1H), 7.22 - 7.26 (m, 1H), 6.75 - 6.80 (m, 2H), 6.52 (dd, J = 2.93, 8.79 Hz, 1H), 5.01 (s, 2H), 3.52 (br. s., 2H). MS: m/z = 311.89 (M+H)+.
3-chloro-4-((3-chlorobenzyl)oxy)aniline (17g)
Yield: 38%. 1H NMR (500 MHz, CDCl3) δ: 7.47 (s, 1H), 7.29 - 7.36 (m, 3H), 6.78 (d, J = 8.30 Hz, 1H), 6.75 (d, J = 2.44 Hz, 1H), 6.50 (dd, J = 2.93, 8.79 Hz, 1H), 5.01 (s, 2H), 3.52 (br. s., 2H). MS: m/z = 267.96 (M+H)+.
3-chloro-4-((2,3-difluorobenzyl)oxy)aniline (17h)
Yield: 31%. 1H NMR (500 MHz, CDCl3) δ: 7.35 (t, J = 6.59 Hz, 1H), 7.07 - 7.16 (m, 2H), 6.83 (d, J = 8.79 Hz, 1H), 6.75 (d, J = 2.93 Hz, 1H), 6.52 (dd, J = 2.69, 8.55 Hz, 1H), 5.12 (s, 2H), 3.51 (br. s., 2H). MS: m/z = 270.0 (M+H)+.
3-chloro-4-((2-fluorobenzyl)oxy)aniline (17i)
Yield: 37%. 1H NMR (500 MHz, CDCl3) δ: 7.58 (dt, J = 1.71, 7.45 Hz, 1H), 7.27 – 7.32 (m, 1H), 7.16 (dt, J = 0.98, 7.57 Hz, 1H), 7.04 - 7.08 (m, 1H), 6.83 (d, J = 8.79 Hz, 1H), 6.75 (d, J = 2.93 Hz, 1H), 6.51 (dd, J = 2.69, 8.55 Hz, 1H), 5.12 (s, 2H), 3.50 (br. s., 2H). MS: m/z = 251.99 (M+H)+.
3-chloro-4-((4-fluorobenzyl)oxy)aniline (17j)
Yield: 22%. 1H NMR (500 MHz, CDCl3) δ: 7.40 - 7.44 (m, 2H), 7.03 - 7.10 (m, 2H), 6.78 (d, J = 8.79 Hz, 1H), 6.75 (d, J = 2.44 Hz, 1H), 6.50 (dd, J = 2.93, 8.79 Hz, 1H), 5.00 (s, 2H), 3.50 (br. s., 2H). MS: m/z = 252.0 (M+H)+.
3-chloro-4-((3-methoxybenzyl)oxy)aniline (17k)
Yield: %. 1H NMR (500 MHz, CDCl3) δ: 7.27 - 7.32 (m, 1H), 7.01 - 7.05 (m, 2H), 6.86 (dd, J = 2.69, 8.06 Hz, 1H), 6.79 (d, J = 8.79 Hz, 1H), 6.74 (d, J = 2.93 Hz, 1H), 6.49 (dd, J = 2.45, 8.80 Hz, 1H), 5.04 (s, 2H), 3.83 (s, 3H), 3.44 (br. s., 2H). MS: m/z = 264.01 (M+H)+.
3-chloro-4-((3-fluoro-4-(trifluoromethyl)benzyl)oxy)aniline (17l)
Yield: 35.2%. 1H NMR (500 MHz, CDCl3) δ: 7.60 (t, J = 7.81 Hz, 1H), 7.29 - 7.36 (m, 2H), 6.77 (d, J = 4.39 Hz, 1H), 6.76 (d, J = 1.47 Hz, 1H), 6.51 (dd, J = 2.93, 8.79 Hz, 1H), 5.06 (s, 2H), 3.54 (br. s., 2H). MS: m/z = 319.93 (M+H)+.
3-chloro-4-((2,3,5-trifluorobenzyl)oxy)aniline (17m)
Yield: 22%. 1H NMR (500 MHz, CDCl3) δ: 7.13 - 7.18 (m, 1H), 6.85 - 6.93 (m, 1H), 6.81 (d, J = 8.30 Hz, 1H), 6.76 (d, J = 2.93 Hz, 1H), 6.53 (dd, J = 2.93, 8.79 Hz, 1H), 5.09 (s, 2H), 3.54 (br. s., 2H). MS: m/z = 287.96 (M+H)+.
4-((3-fluorobenzyl)oxy)aniline (17n)
Yield: 28%. 1H NMR (500 MHz, CDCl3) δ: 7.34 (m, 1H), 7.14 - 7.21 (m, 2H), 7.01 (dt, J = 2.44, 8.55 Hz, 1H), 6.81 (d, J = 8.30 Hz, 2H), 6.64 (d, J = 8.30 Hz, 2H), 4.99 (s, 2H), 3.32 (br. s., 2H). MS: m/z = 218.07 (M+H)+.
4-((3-fluorobenzyl)oxy)-3-methoxyaniline (17o)
Yield: 7.52%. 1H NMR (500 MHz, CDCl3) δ: 7.28 - 7.33 (m, 1H), 7.14 - 7.21 (m, 2H), 6.97 (dt, J = 2.20, 8.67 Hz, 1H), 6.70 (d, J = 8.30 Hz, 1H), 6.32 (d, J = 2.93 Hz, 1H), 6.16 (dd, J = 2.69, 8.55 Hz, 1H), 5.02 (s, 2H), 3.84 (s, 3H), 3.51 (br. s., 2H). MS: m/z = 248.06 (M+H)+.
6-(4-(morpholinosulfonyl)phenyl)quinazolin-4(3H)-one (18)
A mixture of 6-iodoquinazolin-4(3H)-one (4.5 g, 16.54 mmol), (4-(morpholinosulfonyl)phenyl)boronic acid (4.93 g, 18.20 mmol), sodium carbonate (10.52 g, 99 mmol) and tetrakis(triphenylphosphine)palladium(0) (1.338 g, 1.158 mmol) were combined in a flask, and 400 mL of 1,2-dimethoxyethane was added, with ethanol (26.7 mL) and water (33.3 ml). The reaction mixture was heated with stirring at 80 °C for 30 h. The reaction progress was monitored by LC-MS. Upon completion of the reaction, the mixture was cooled to room temperature, and the product precipitated. Solids were collected by filtration, washed with cold water and air-dried, affording 18 (5.35 g, 14.40 mmol, 87 % yield). 1H NMR (500 MHz, DMSO-d6) δ: 12.40 (br. s., 1H), 8.44 (d, J = 2.44 Hz, 1H), 8.22 (dd, J = 2.20, 8.55 Hz, 1H), 8.16 (d, J = 3.42 Hz, 1H), 8.07 (d, J = 8.30 Hz, 2H), 7.84 (d, J = 8.30 Hz, 2H), 7.80 (d, J = 8.79 Hz, 1H), 3.64 (t, J = 4.65 Hz, 4H), 2.91 (t, J = 4.65 Hz, 4H). MS: m/z = 372.2 (M+H)+.
4-((4-(4-chloroquinazolin-6-yl)phenyl)sulfonyl)morpholine hydrochloride (19)
Thionyl chloride (9.83 ml, 135 mmol) was added slowly to 1.0 g of 18 (1 g, 2.69 mmol), followed by N,N-dimethylformamide (2.085 μl, 0.027 mmol). The reaction mixture was refluxed for 36 h, monitoring reaction progress with LC-MS. The volatile components were removed via distillation, providing 19 (1.12 g, 1.683 mmol, 80% pure, 78% yield), which was used for subsequent reactions without further purification. 1H NMR (500 MHz, DMSO-d6) δ: 8.45 (d, J = 1.95 Hz, 1H), 8.32 (s, 1H), 8.26 (dd, J = 2.20, 8.55 Hz, 1H), 8.07 (d, J = 8.79 Hz, 2H), 7.79 - 7.86 (m, 3H), 3.65 (t, J = 4.60 Hz, 4H), 2.92 (t, J = 4.65 Hz, 4H). MS: m/z = 390.04 (M+H)+.
6-(4-(morpholinosulfonyl)phenyl)-N-arylquinazolin-4-amines hydrochloride 20 were synthesized following General procedure C. To a solution of 19 (100 μmol) in N,N-dimethylformamide (0.5 mL) was added aryl amine (110 μmol) and the mixture was heated on a shaker plate at 80 °C for 12 h. After cooling the reaction mixture to room temperature, 0.5 mL of isopropanol was added. The resulting yellowish precipitate was filtered and washed with 2mL of isopropanol, affording the amines 20.
6-(4-(morpholinosulfonyl)phenyl)-N-phenylquinazolin-4-amine hydrochloride. (20a)
Yield: 50.8%. 1H NMR (500 MHz, DMSO-d6) δ: 11.51 (br. s., 1H), 9.20 (s, 1H), 8.93 (s, 1H), 8.50 (d, J = 8.79 Hz, 1H), 8.21 (d, J = 8.30 Hz, 2H), 8.01 (d, J = 8.30 Hz, 1H), 7.95 (d, J = 8.30 Hz, 2H), 7.75 (d, J = 8.30 Hz, 2H), 7.51 - 7.55 (m, 2H), 7.34 - 7.37 (m, 1H), 3.65 - 3.67 (br. m, 4H), 2.92 - 2.95 (br. m., 4H). MS: m/z = 447.2 (M+H)+.
6-(4-(morpholinosulfonyl)phenyl)-N-(p-tolyl)quinazolin-4-amine hydrochloride (20b)
Yield: 54.9%. 1H NMR (500 MHz, DMSO-d6) δ: 11.62 (br. s., 1H), 9.24 (s, 1H), 8.93 (s, 1H), 8.51 (d, J = 8.30 Hz, 1H), 8.22 (d, J = 8.30 Hz, 3H), 8.03 (d, J = 8.79 Hz, 1H), 7.95 (d, J = 8.79 Hz, 2H), 7.63 (d, J = 7.81 Hz, 2H), 7.34 (d, J = 7.81 Hz, 2H), 3.67 (t, J = 4.4 Hz, 4H ), 2.95 (t, J = 4.4 Hz, 4H ), 2.38 (s, 3H). MS: m/z = 496.2 (M+H)+.
2-chloro-4-((6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-yl)amino)phenol hydrochloride (20c)
Yield: 52.7%. 1H NMR (500 MHz, DMSO-d6) δ: 11.60 (br. s., 1H), 10.55 (br. s., 1H), 9.22 (s, 1H), 8.96 (s, 1H), 8.51 (dd, J = 1.47, 8.79 Hz, 1H), 8.22 (d, J = 8.30 Hz, 2H), 8.02 (d, J = 8.79 Hz, 1H), 7.95 (d, J = 8.30 Hz, 2H), 7.81 (d, J = 2.93 Hz, 1H), 7.52 (dd, J = 2.44, 8.79 Hz, 1H), 7.12 (d, J = 8.79 Hz, 1H), 3.7 (t, J = 4.40 Hz, 4H), 2.95 (t, J = 4.4 Hz, 4H). MS: m/z = 497.2 (M+H)+.
N-(3-chloro-4-methoxyphenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine hydrochloride (20d)
Yield: 43.4%. 1H NMR (500 MHz, DMSO-d6) δ: 11.34 (br. s., 1H), 9.14 (s, 1H), 8.94 (s, 1H), 8.49 (d, J = 8.79 Hz, 1H), 8.20 (d, J = 8.79 Hz, 2H), 8.00 (d, J = 8.79 Hz, 1H), 7.96 (d, J = 8.30 Hz, 2H), 7.93 (d, J = 2.44 Hz, 1H), 7.70 (dd, J = 2.44, 8.79 Hz, 1H), 7.31 (d, J = 9.28 Hz, 1H), 3.93 (s, 3H), 3.67 (t, J = 4.6 Hz, 4H), 2.95 (t, J = 4.6 Hz, 4H). MS: m/z = 511.1 (M+H)+.
N-(4-(benzyloxy)-3-chlorophenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine hydrochloride (20e)
Yield: 70.1%. 1H NMR (500 MHz, DMSO-d6) δ: 11.49 (br. s., 1H), 9.18 (s, 1H), 8.95 (s, 1H), 8.50 (dd, J = 1.71, 8.55 Hz, 1H), 8.20 - 8.22 (m, 2H), 8.01 (d, J = 8.79 Hz, 1H), 7.93 - 7.96 (m, 3H), 7.68 (dd, J = 2.69, 9.03 Hz, 1H), 7.51 - 7.53 (m, 2H), 7.35 - 7.46 (m, 4H), 5.30 (s, 2H), 3.66 - 3.68 (t, J = 4.9 Hz, 4H), 2.5 (t, J = 4.4 Hz, 4H). MS: m/z = 587.2 (M+H)+.
N-(4-((3-bromobenzyl)oxy)-3-chlorophenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine hydrochloride (20f)
Yield: 51.6%. 1H NMR (500 MHz, DMSO-d6) δ: 11.45 (br. s., 1H), 9.17 (s, 1H), 8.95 (s, 1H), 8.50 (d, J = 8.79 Hz, 1H), 8.21 (d, J = 8.79 Hz, 2H), 8.01 (d, J = 8.30 Hz, 1H), 7.94 - 7.97 (m, 3H), 7.72 (s, 1H), 7.69 (dd, J = 2.69, 9.03 Hz, 1H), 7.58 (d, J = 7.81 Hz, 1H), 7.52 (d, J = 7.81 Hz, 1H), 7.37 - 7.43 (m, 2H), 5.31 (s, 2H), 3.67 (t, J = 4.4 Hz, 4H), 2.95 (t, J = 4.35 Hz, 4H). MS: m/z = 665.1 (M+H)+.
N-(3-chloro-4-((3-chlorobenzyl)oxy)phenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine hydrochloride (20g)
Yield: 49.1%. 1H NMR (500 MHz, DMSO-d6) δ: 11.54 (br. s., 1H), 9.19 (br. s., 1H), 8.96 (br. s., 1H), 8.50 (d, J = 8.79 Hz, 1H), 8.21 (d, J = 8.30 Hz, 2H), 7.98 - 8.08 (m, 1H), 7.94 - 7.96 (m, 3H), 7.69 (dd, J = 2.44, 8.79 Hz, 1H), 7.58 (br. s., 1H), 7.37 - 7.50 (m, 4H), 5.31 (br. s., 2H), 3.67 (br. s., 4H), 2.95 (br. s., 4H). MS: m/z = 621.1 (M+H)+.
N-(3-chloro-4-((2,3-difluorobenzyl)oxy)phenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine hydrochloride (20h)
Yield: 53.3%. 1H NMR (500 MHz, DMSO-d6) δ: 11.58 (br. s., 1H), 9.22 (s, 1H), 8.97 (s, 1H), 8.51 (dd, J = 1.46, 8.79 Hz, 1H), 8.22 (d, J = 8.30 Hz, 2H), 8.03 (d, J = 8.79 Hz, 1H), 7.93 - 7.97 (m, 3H), 7.73 (dd, J = 2.44, 8.79 Hz, 1H), 7.43 - 7.53 (m, 3H), 7.28 - 7.34 (m, 1H), 5.38 (s, 2H), 3.67 (t, J = 4.4 Hz, 4H), 2.95 (t, J = 4.6 Hz, 4H). MS: m/z = 623.2 (M+H)+.
N-(3-chloro-4-((2-fluorobenzyl)oxy)phenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine hydrochloride (20i)
Yield: 41.2%. 1H NMR (500 MHz, DMSO-d6) δ: 11.32 (br. s., 1H), 9.12 (br. s., 1H), 8.93 (br. s., 1H), 8.47 (d, J = 8.79 Hz, 1H), 8.18 (d, J = 8.30 Hz, 2H), 7.98 (d, J = 8.79 Hz, 1H), 7.92 - 7.96 (m, 3H), 7.70 (dd, J = 2.44, 8.79 Hz, 1H), 7.60 - 7.63 (m, 1H), 7.43 - 7.46 (m, 2H), 7.25 - 7.32 (m, 2H), 5.31 (s, 2H), 3.65 (t, J = 4.6 Hz, 4H), 2.93 (t, J = 4.6 Hz, 4H). MS: m/z = 605.2 (M+H)+.
N-(3-chloro-4-((4-fluorobenzyl)oxy)phenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine hydrochloride (20j)
Yield: 67.1%. 1H NMR (500 MHz, DMSO-d6) δ: 11.66 (br. s., 1H), 9.24 (br. s., 1H), 8.97 (s, 1H), 8.52 (d, J = 8.79 Hz, 1H), 8.23 (d, J = 8.30 Hz, 2H), 8.03 (d, J = 8.79 Hz, 1H), 7.94 - 7.96 (m, 3H), 7.70 (dd, J = 2.44, 8.79 Hz, 1H), 7.55 - 7.58 (m, 2H), 7.41 (d, J = 8.79 Hz, 1H), 7.26 - 7.30 (m, 2H), 5.28 (s, 2H), 3.7 (t, J = 4.9 Hz, 4H), 2.95 (t, J = 4.6 Hz, 4H). MS: m/z = 605.2 (M+H)+.
N-(3-chloro-4-((3-methoxybenzyl)oxy)phenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine hydrochloride (20k)
Yield: 43.1%. 1H NMR (500 MHz, DMSO-d6) δ: 11.40 (br. s., 1H), 9.15 (s, 1H), 8.94 (s, 1H), 8.49 (d, J = 8.30 Hz, 1H), 8.20 (d, J = 8.30 Hz, 2H), 8.00 (d, J = 8.79 Hz, 1H), 7.93 - 7.97 (m, 3H), 7.68 (dd, J = 2.44, 8.79 Hz, 1H), 7.33 - 7.39 (m, 2H), 7.06 - 7.08 (m, 2H), 6.91 - 6.96 (m, 1H), 5.27 (s, 2H), 3.78 (s, 3H), 3.67 (t, J = 4.9 Hz, 4H), 2.95 (t, J = 4.4 Hz, 4H). MS: m/z = 617.2 (M+H)+.
N-(3-chloro-4-((3-fluoro-4-(trifluoromethyl)benzyl)oxy)phenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine hydrochloride (20l)
Yield: 40.9%. 1H NMR (500 MHz, DMSO-d6) δ: 11.74 (br. s., 1H), 9.29 (s, 1H), 8.98 (s, 1H), 8.52 (dd, J = 1.46, 8.79 Hz, 1H), 8.24 (d, J = 8.79 Hz, 2H), 8.05 (d, J = 8.79 Hz, 1H), 7.99 (d, J = 2.44 Hz, 1H), 7.95 (d, J = 8.30 Hz, 2H), 7.89 (t, J = 7.81 Hz, 1H), 7.72 (dd, J = 2.44, 8.79 Hz, 1H), 7.63 (d, J = 11.23 Hz, 1H), 7.55 (d, J = 7.81 Hz, 1H), 7.38 (d, J = 8.79 Hz, 1H), 5.43 (s, 2H), 3.67 (t, J = 4.4 Hz, 4H), 2.95 (t, J = 4.4 Hz, 4H). MS: m/z = 673.2 (M+H)+.
N-(3-chloro-4-((2,3,5-trifluorobenzyl)oxy)phenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine hydrochloride (20m)
Yield: 42%. 1H NMR (500 MHz, DMSO-d6) δ: 11.49 (br. s., 1H), 9.19 (s, 1H), 8.96 (s, 1H), 8.51 (d, J = 9.77 Hz, 1H), 8.21 (d, J = 8.30 Hz, 2H), 8.02 (d, J = 8.79 Hz, 1H), 7.95 - 7.98 (m, 3H), 7.73 (dd, J = 2.69, 9.03 Hz, 1H), 7.56- 7.65 (m, 1H), 7.48 (d, J = 8.79 Hz, 1H), 7.34 - 7.36 (m, 1H), 5.38 (s, 2H), 3.67 (t, J = 4.9 Hz, 4H), 2.95 (t, J = 4.4 Hz, 4H). MS: m/z = 641.1 (M+H)+.
N-(4-((3-fluorobenzyl)oxy)phenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine (20n)
Yield: 45.8%. 1H NMR (500 MHz, DMSO-d6) δ: 11.40 (br. s., 1H), 9.15 (s, 1H), 8.88 (s, 1H), 8.49 (d, J = 8.79 Hz, 1H), 8.20 (d, J = 8.79 Hz, 2H), 7.92 - 8.02 (m, 3H), 7.65 (d, J = 9.30 Hz, 2H), 7.45 - 7.51 (m, 1H), 7.31 - 7.35 (m, 2H), 7.16 - 7.22 (m, 3H), 5.21 (s, 2H), 3.67 (t, J = 4.4 Hz, 4H), 2.95 (t, J = 4.4 Hz, 4H). MS: m/z = 571.2 (M+H)+.
N-(4-((3-fluorobenzyl)oxy)-3-methoxyphenyl)-6-(4-(morpholinosulfonyl)phenyl)quinazolin-4-amine (20o)
Yield: 18%. 1H NMR (500 MHz, DMSO-d6) δ: 11.32 (br. s., 1H), 9.14 (s, 1H), 8.90 (s, 1H), 8.49 (d, J = 8.30 Hz, 1H), 8.20 (d, J = 8.79 Hz, 2H), 7.99 (d, J = 8.79 Hz, 1H), 7.96 (d, J = 8.30 Hz, 2H), 7.45 - 7.50 (m, 1H), 7.41 (d, J = 2.44 Hz, 1H), 7.28 - 7.34 (m, 3H), 7.15 - 7.22 (m, 2H), 5.19 (s, 2H), 3.84 (s, 3H), 3.67 (t, J = 4.9 Hz, 4H), 2.95 (t, J = 4.4 Hz, 4H). MS: m/z = 601.1 (M+H)+.
Trypanosome replication assays
Bloodstream T. brucei brucei Lister 427 cells were seeded at a density of 2 × 103 cells /ml and cultured in HMI-9 medium 47 in a 24 well plate. Two microliters of DMSO (control) or different concentrations of drugs prepared from 200X DMSO stocks were added to the cultures. Cells were incubated at 37°C for 48 h, and counted with a hemocytometer. Drugs were initially tested at concentrations of 10 μM, 1 μM, 100 nM and 10 nM to determine the range of potency of each compound. Thereafter, a set of 5 concentrations centered around a dose that prevented replication of 50% of cells (compared to DMSO) was used to establish the EC50. All experiments were repeated thrice in independent studies where each dose was administered in duplicate (total n = 6).
HepG2 Cell Toxicity assay
The 384 well MTT cytotoxicity assay is a modification of the MTT method described by Ferrari et al.48 optimized for 384 well throughput, with modifications described in detail in the Supporting Information. The 50% inhibitory concentrations (IC50) were generated for each toxicity dose response test using GraphPad Prism (GraphPad Software Inc., San Diego, CA) using the nonlinear regression (sigmoidal dose-response/variable slope) equation.
Supplementary Material
Scheme 4.

Synthesis of analogs 23.a
Acknowledgements
Funding from the National Institutes of Health (R01AI082577 to MP, and R21AI076647 to KM-W), the National Science Foundation (MCB-0843603), Northeastern University, and a gift of compounds (Table 1) from GlaxoSmithKline, Inc. is gratefully acknowledged. We appreciate helpful discussions with Dr. Richard Sciotti at Walter Reed Army Institute of Research and Dr. Franco Lombardo at Novartis.
Abbreviations
- EGFR
epidermal growth factor receptor
- EC50
concentration at which cell growth is inhibited by 50%
- FDA
food and drug administration
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PDE
phosphodiesterase
Footnotes
Author contributions
MPP developed the medicinal chemistry strategy and contributed to compound designs. KM-W initiated the project. GP, CEK, PE, contributed to compound designs and performed chemical synthesis and characterization. RB, PG, and CS performed trypanosome assays, and NER performed the HepG2 assays.
Supporting Information.
Synthesis of selected boronates, and a tabulation of all the inhibitors, with their Northeastern registry numbers and screening data is in the Supporting Information, and the screening data has been made freely available as a shared data set at www.collaborativedrugdiscovery.com. Also included are biological assay details, data from pharmacokinetic and in vivo efficacy studies, plasma protein binding, and results from cell cycle analysis following inhibitor treatment. This supporting information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1. [12 April 2013];Uniting to Combat NTDs Homepage. www.unitingtocombatntds.org.
- 2.Pollastri MP, Campbell RK. Target repurposing for neglected diseases. Future Med Chem. 2011;3:1307–1315. doi: 10.4155/fmc.11.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gould MK, de Koning HP. Cyclic-nucleotide signalling in protozoa. FEMS Microbiol Rev. 2011;35:515–541. doi: 10.1111/j.1574-6976.2010.00262.x. [DOI] [PubMed] [Google Scholar]
- 4.Parsons M, Worthey EA, Ward PN, Mottram JC. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics. 2005;6:127. doi: 10.1186/1471-2164-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bland ND, Wang C, Tallman C, Gustafson AE, Wang Z, Ashton TD, Ochiana SO, McAllister G, Cotter K, Fang AP, Gechijian L, Garceau N, Gangurde R, Ortenberg R, Ondrechen MJ, Campbell RK, Pollastri MP. Pharmacological validation of Trypanosoma brucei phosphodiesterases B1 and B2 as druggable targets for African sleeping sickness. J Med Chem. 2011;54:8188–8194. doi: 10.1021/jm201148s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ochiana SO, Pandarinath V, Wang Z, Kapoor R, Ondrechen MJ, Ruben L, Pollastri MP. The human Aurora kinase inhibitor danusertib is a lead compound for anti-trypanosomal drug discovery via target repurposing. Eur J Med Chem. 2012 doi: 10.1016/j.ejmech.2012.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diaz-Gonzalez R, Kuhlmann FM, Galan-Rodriguez C, Madeira da Silva L, Saldivia M, Karver CE, Rodriguez A, Beverley SM, Navarro M, Pollastri MP. The susceptibility of trypanosomatid pathogens to PI3/mTOR kinase inhibitors affords a new opportunity for drug repurposing. PLoS Negl Trop Dis. 2011;5:e1297. doi: 10.1371/journal.pntd.0001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Koning HP, Gould MK, Sterk GJ, Tenor H, Kunz S, Luginbuehl E, Seebeck T. Pharmacological validation of Trypanosoma brucei phosphodiesterases as novel drug targets. J Infect Dis. 2012;206:229–237. doi: 10.1093/infdis/jir857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beghyn TB, Charton J, Leroux F, Laconde G, Bourin A, Cos P, Maes L, Deprez B. Drug to genome to drug: discovery of new antiplasmodial compounds. J Med Chem. 2011;54:3222–3240. doi: 10.1021/jm1014617. [DOI] [PubMed] [Google Scholar]
- 10.Nwaka S, Hudson A. Innovative lead discovery strategies for tropical diseases. Nat Rev Drug Discov. 2006;5:941–955. doi: 10.1038/nrd2144. [DOI] [PubMed] [Google Scholar]
- 11.Chahrour O, Cairns D, Omran Z. Small molecule kinase inhibitors as anti-cancer therapeutics. Mini Rev Med Chem. 2012;12:399–411. doi: 10.2174/138955712800493915. [DOI] [PubMed] [Google Scholar]
- 12.Page TH, Smolinska M, Gillespie J, Urbaniak AM, Foxwell BM. Tyrosine kinases and inflammatory signalling. Curr Mol Med. 2009;9:69–85. doi: 10.2174/156652409787314507. [DOI] [PubMed] [Google Scholar]
- 13.Ito K, Caramori G, Adcock IM. Therapeutic Potential of Phosphatidylinositol 3-Kinase Inhibitors in Inflammatory Respiratory Disease. J Pharmacol Exp Therap. 2007;321:1–8. doi: 10.1124/jpet.106.111674. [DOI] [PubMed] [Google Scholar]
- 14.Louvet C, Szot GL, Lang J, Lee MR, Martinier N, Bollag G, Zhu S, Weiss A, Bluestone JA. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0810246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagman AS, Johnson KW, Bussiere DE. Discovery and development of GSK3 inhibitors for the treatment of type 2 diabetes. Curr Pharm Des. 2004;10:1105–1137. doi: 10.2174/1381612043452668. [DOI] [PubMed] [Google Scholar]
- 16.Chico LK, Van Eldik LJ, Watterson DM. Targeting protein kinases in central nervous system disorders. Nat Rev Drug Discov. 2009;8:892–909. doi: 10.1038/nrd2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Natoli C. Tyrosine Kinase Inhibitors. Current Cancer Drug Targets. 2010;10:462. doi: 10.2174/156800910791517208. [DOI] [PubMed] [Google Scholar]
- 18.Lackey KE. Lessons from the drug discovery of lapatinib, a dual ErbB1/2 tyrosine kinase inhibitor. Curr Top Med Chem. 2006;6:435–460. doi: 10.2174/156802606776743156. [DOI] [PubMed] [Google Scholar]
- 19.Johannessen LE, Ringerike T, Molnes J, Madshus IH. Epidermal growth factor receptor efficiently activates mitogen-activated protein kinase in HeLa cells and Hep2 cells conditionally defective in clathrin-dependent endocytosis. Exp Cell Res. 2000;260:136–145. doi: 10.1006/excr.2000.5004. [DOI] [PubMed] [Google Scholar]
- 20.Naula C, Parsons M, Mottram JC. Protein kinases as drug targets in trypanosomes and Leishmania. Biochim Biophys Acta. 2005;1754:151–159. doi: 10.1016/j.bbapap.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oduor RO, Ojo KK, Williams GP, Bertelli F, Mills J, Maes L, Pryde DC, Parkinson T, Van Voorhis WC, Holler TP. Trypanosoma brucei Glycogen Synthase Kinase-3, A Target for Anti-Trypanosomal Drug Development: A Public-Private Partnership to Identify Novel Leads. PLoS Negl Trop Dis. 2011;5:e1017. doi: 10.1371/journal.pntd.0001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nett IR, Martin DM, Miranda-Saavedra D, Lamont D, Barber JD, Mehlert A, Ferguson MA. The phosphoproteome of bloodstream form Trypanonosoma brucei, causative agent of African sleeping sickness. Mol Cell Proteomics. 2009;8:1527–1538. doi: 10.1074/mcp.M800556-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nett IR, Davidson L, Lamont D, Ferguson MA. Identification and specific localization of tyrosine-phosphorylated proteins in Trypanosoma brucei. Eukaryot Cell. 2009;8:617–626. doi: 10.1128/EC.00366-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katiyar S, Kufareva I, Behera R, Ogata Y, Pollastri MP, Abagyan R, Mensa-Wilmot K. Lapatinib-binding Protein Kinases in the African Trypanosome: A General Method for Identification of Targets for Kinase-Directed Drugs in a Cell. PLoS ONE. 2013;8:e56150. doi: 10.1371/journal.pone.0056150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slichenmyer WJ, Elliott WL, Fry DW. CI-1033, a pan-erbB tyrosine kinase inhibitor. Semin Oncol. 2001;28:80–85. doi: 10.1016/s0093-7754(01)90285-4. [DOI] [PubMed] [Google Scholar]
- 26.Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, Owens G, Alligood KJ, Spector NL. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. 2002;21:6255–6263. doi: 10.1038/sj.onc.1205794. [DOI] [PubMed] [Google Scholar]
- 27.Traxler P, Allegrini PR, Brandt R, Brueggen J, Cozens R, Fabbro D, Grosios K, Lane HA, McSheehy P, Mestan J, Meyer T, Tang C, Wartmann M, Wood J, Caravatti G. AEE788: a dual family epidermal growth factor receptor/ErbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2004;64:4931–4941. doi: 10.1158/0008-5472.CAN-03-3681. [DOI] [PubMed] [Google Scholar]
- 28.Steverding D. The transferrin receptor of Trypanosoma brucei. Parasitol Int. 2000;48:191–198. doi: 10.1016/s1383-5769(99)00018-5. [DOI] [PubMed] [Google Scholar]
- 29.Subramanya S, Hardin CF, Steverding D, Mensa-Wilmot K. Glycosylphosphatidylinositol-specific phospholipase C regulates transferrin endocytosis in the African trypanosome. Biochem J. 2009;417:685–694. doi: 10.1042/BJ20080167. [DOI] [PubMed] [Google Scholar]
- 30.Subramanya S, Mensa-Wilmot K. Diacylglycerol-stimulated endocytosis of transferrin in trypanosomatids is dependent on tyrosine kinase activity. PLoS ONE. 2010;5:e8538. doi: 10.1371/journal.pone.0008538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Behera R, Guyett P, Thomas S, Katiyar S, Ogata Y, Mensa-Wilmot K. unpublished results.
- 32.Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, Keith BR, Murray DM, Knight WB, Mullin RJ, Gilmer TM. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1:85–94. [PubMed] [Google Scholar]
- 33.Petrov KG, Zhang YM, Carter M, Cockerill GS, Dickerson S, Gauthier CA, Guo Y, Mook RA, Jr., Rusnak DW, Walker AL, Wood ER, Lackey KE. Optimization and SAR for dual ErbB-1/ErbB-2 tyrosine kinase inhibition in the 6-furanylquinazoline series. Bioorg Med Chem Lett. 2006;16:4686–4691. doi: 10.1016/j.bmcl.2006.05.090. [DOI] [PubMed] [Google Scholar]
- 34.Rusnak DW, Affleck K, Cockerill SG, Stubberfield C, Harris R, Page M, Smith KJ, Guntrip SB, Carter MC, Shaw RJ, Jowett A, Stables J, Topley P, Wood ER, Brignola PS, Kadwell SH, Reep BR, Mullin RJ, Alligood KJ, Keith BR, Crosby RM, Murray DM, Knight WB, Gilmer TM, Lackey K. The characterization of novel, dual ErbB-2/EGFR, tyrosine kinase inhibitors: potential therapy for cancer. Cancer Res. 2001;61:7196–7203. [PubMed] [Google Scholar]
- 35.Cai X, Zhai H-X, Wang J, Forrester J, Qu H, Yin L, Lai C-J, Bao R, Qian C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. J Med Chem. 2010;53:2000–2009. doi: 10.1021/jm901453q. [DOI] [PubMed] [Google Scholar]
- 36.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 37.Webster P, Fish WR. Endocytosis by African trypanosomes. II. Occurrence in different life-cycle stages and intracellular sorting. Eur J Cell Biol. 1989;49:303–310. [PubMed] [Google Scholar]
- 38.Hall B, Allen CL, Goulding D, Field MC. Both of the Rab5 subfamily small GTPases of Trypanosoma brucei are essential and required for endocytosis. Mol Biochem Parasitol. 2004;138:67–77. doi: 10.1016/j.molbiopara.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 39.Hammarton TC. Cell cycle regulation in Trypanosoma brucei. Mol Biochem Parasitol. 2007;153:1–8. doi: 10.1016/j.molbiopara.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siegel TN, Hekstra DR, Cross GA. Analysis of the Trypanosoma brucei cell cycle by quantitative DAPI imaging. Mol Biochem Parasitol. 2008;160:171–174. doi: 10.1016/j.molbiopara.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammarton TC, Monnerat S, Mottram JC. Cytokinesis in trypanosomatids. Curr Opin Microbiol. 2007;10:520–527. doi: 10.1016/j.mib.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 42.Mackey ZB, Baca AM, Mallari JP, Apsel B, Shelat A, Hansell EJ, Chiang PK, Wolff B, Guy KR, Williams J, McKerrow JH. Discovery of trypanocidal compounds by whole cell HTS of Trypanosoma brucei. Chem Biol Drug Des. 2006;67:355–363. doi: 10.1111/j.1747-0285.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- 43.Wager TT, Chandrasekaran RY, Hou X, Troutman MD, Verhoest PR, Villalobos A, Will Y. Defining Desirable Central Nervous System Drug Space through the Alignment of Molecular Properties, in Vitro ADME, and Safety Attributes. ACS Chem Neurosci. 2010;1:420–434. doi: 10.1021/cn100007x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties. ACS Chem Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iino T, Sasaki Y, Bamba M, Mitsuya M, Ohno A, Kamata K, Hosaka H, Maruki H, Futamura M, Yoshimoto R, Ohyama S, Sasaki K, Chiba M, Ohtake N, Nagata Y, Eiki J.-i., Nishimura T. Discovery and structure–activity relationships of a novel class of quinazoline glucokinase activators. Bioorg Med Chem Lett. 2009;19:5531–5538. doi: 10.1016/j.bmcl.2009.08.064. [DOI] [PubMed] [Google Scholar]
- 46.Mahboobi S, Sellmer A, Winkler M, Eichhorn E, Pongratz H, Ciossek T, Baer T, Maier T, Beckers T. Novel Chimeric Histone Deacetylase Inhibitors: A Series of Lapatinib Hybrides as Potent Inhibitors of Epidermal Growth Factor Receptor (EGFR), Human Epidermal Growth Factor Receptor 2 (HER2), and Histone Deacetylase Activity. J Med Chem. 2010;53:8546–8555. doi: 10.1021/jm100665z. [DOI] [PubMed] [Google Scholar]
- 47.Hirumi H, Hirumi K. Axenic culture of African trypanosome bloodstream forms. Parasitol.Today. 1994;10:80–84. doi: 10.1016/0169-4758(94)90402-2. [DOI] [PubMed] [Google Scholar]
- 48.Ferrari M, Fornasiero MC, Isetta AM. MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. J Immunol Methods. 1990;131:165–172. doi: 10.1016/0022-1759(90)90187-z. [DOI] [PubMed] [Google Scholar]
- 49.Sykes ML, Baell JB, Kaiser M, Chatelain E, Moawad SR, Ganame D, Ioset J-R, Avery VM. Identification of Compounds with Anti-Proliferative Activity against Trypanosoma brucei brucei Strain 427 by a Whole Cell Viability Based HTS Campaign. PLoS Negl Trop Dis. 2012;6:e1896. doi: 10.1371/journal.pntd.0001896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sokolova AY, Wyllie S, Patterson S, Oza SL, Read KD, Fairlamb AH. Cross-resistance to nitro drugs and implications for treatment of human African trypanosomiasis. Antimicrob Agents Chemother. 2010;54:2893–2900. doi: 10.1128/AAC.00332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jacobs RT, Nare B, Wring SA, Orr MD, Chen D, Sligar JM, Jenks MX, Noe RA, Bowling TS, Mercer LT, Rewerts C, Gaukel E, Owens J, Parham R, Randolph R, Beaudet B, Bacchi CJ, Yarlett N, Plattner JJ, Freund Y, Ding C, Akama T, Zhang YK, Brun R, Kaiser M, Scandale I, Don R. SCYX-7158, an Orally-Active Benzoxaborole for the Treatment of Stage 2 Human African Trypanosomiasis. PLoS Negl Trop Dis. 2011;5:e1151. doi: 10.1371/journal.pntd.0001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.