

Abstract
Although the recreational drug 3,4-methylenedioxymethamphetamine (MDMA) is often described as a selective serotonergic neurotoxin, some research has challenged this view. The objective of this study was to determine the influence of MDMA on subsequent levels of two different markers of dopaminergic function, the dopamine transporter (DAT) as well as dopamine and its major metabolites. In experiment I, adult male Sprague–Dawley rats were administered either a low or moderate dose MDMA binge (2.5 or 5.0 mg/kg × 4 with an inter-dose interval of 1 h) or saline, and were killed 1 week later. The moderate dose dramatically reduced [3H]WIN 35,428 binding to striatal DAT by 73.7% (P ≤ 0.001). In experiment II, animals were binged with a higher dose of MDMA (10 mg/kg × 4) to determine the drug’s effects on concentrations of serotonin (5-HT), dopamine, and their respective major metabolites 5-hydroxyindoleacetic acid (5-HIAA), dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) in the striatum and frontal cortex 1 week later. As expected, MDMA significantly reduced 5-HT and 5-HIAA (≥ 50%) in these structures, while only a marginal decrease in dopamine was noted in the striatum. In contrast, levels of DOPAC (34.3%, P < 0.01) and HVA (33.5%, P < 0.001) were reduced by MDMA treatment, suggesting a decrease in dopamine turnover. Overall, these findings indicate that while serotonergic markers are particularly vulnerable to MDMA-induced depletion, significant dopaminergic deficits may also occur under some conditions. Importantly, DAT expression may be more vulnerable to perturbation by MDMA than dopamine itself.
Keywords: 3,4-methylenedioxymethamphetamine; Neurotoxicity; Dopamine transporter; Dihydroxyphenylacetic acid; Homovanillic acid; Serotonin
1. Introduction
The illicit substance 3,4-methylenedioxymethamphetamine (colloquially known as Ecstasy) is a commonly used amphetamine derivative known to cause long term deficits to serotonergic function (Lyles and Cadet, 2003). These effects have been documented in several species, including rats (Xie et al., 2006), guinea pigs (Commins et al., 1987), non-human primates (Ricaurte and McCann, 1992), and humans (Kish et al., 2000; McCann et al., 1994, 2000, 2008), and include prolonged decreases in serotonin (5-hydroxytryptamine; 5-HT) levels, activity of tryptophan hydroxylase, and serotonin transporter expression. In contrast, rats typically show few long-term effects of MDMA on catecholamine neurons, as exemplified by an absence of changes in levels of dopamine or norepinephrine (Colado et al., 2004), the dopamine transporter (DAT) (Battaglia et al., 1987; Lew et al., 1996), or catecholaminergic fiber density when these projections were stained for tyrosine hydroxylase (O’Hearn et al., 1988). For this reason, MDMA is often described as a selective 5-HT neurotoxin (Cole and Sumnall, 2003; Ricaurte et al., 1988).
Despite this prevailing view that the adverse effects of MDMA are mainly limited to serotonergic neurons, several studies have found evidence for significant, drug-induced deficits in dopamine neurochemistry (reviewed in Colado et al., 2004; Piper, 2007). For example, Commins et al. (1987) reported decreased dopamine content in the striatum two weeks following high, repeated doses of MDMA in rats, while a similar decrease in levels of striatal dopamine was also noted at either 1 (Able et al., 2006) or 3 months (McGregor et al., 2003) following repeated drug administration. Another study showed that the extent of mazindol binding to DAT was decreased in the midbrain, but not the striatum, of rats given a similar dosing regimen (Battaglia et al., 1987). A postmortem case study determined that the dopamine content of an MDMA abuser was approximately half that in comparison subjects in the nucleus accumbens, while abnormal quantities of DOPAC were noted in the putamen (Kish et al., 2000). Additionally, McCann et al. (1994) identified a reduction in the dopamine metabolite homovanillic acid (HVA) in the cerebral spinal fluid of female, but not male, Ecstasy users. Importantly, two investigations have documented MDMA-induced decreases in the vesicular monoamine transporter 2 (VMAT-2), a protein that primarily marks dopaminergic terminals in the striatum (Nirenberg et al., 1997). Levels of VMAT-2 were reduced in the caudate/putamen and frontal cortex of baboons previously treated with multiple doses of MDMA (Ricaurte et al., 2000), with similar deficits also observed in the striatum 1 day following an MDMA binge in rats (Hansen et al., 2002). Taken together, these findings highlight the possibility that MDMA may have the potential to cause long-term neurochemical changes to dopaminergic neurons.
Given that all documented cases of dopaminergic deficits following MDMA exposure involved administration of high, repeated doses of the compound, this particular dosing pattern may prove to be important in unmasking the effects of MDMA on this population of neurons. To test this hypothesis, we administered four doses of MDMA (1 h apart) to rats and measured two different indices of dopaminergic neuron function, either DAT binding (experiment I) or dopamine and its major metabolites (experiment II) 1 week later. In experiment II, to show that this binge paradigm yielded the anticipated deficits in serotonergic markers, we also measured levels of 5-HT and its major metabolite 5-hydroxyindoleacetic acid (5-HIAA).
2. Methods
2.1. Animals
Adult male Sprague–Dawley rats were obtained from Charles River Laboratories (Kingston, NY). Animals were pair-housed in standard plastic tubs under a reverse 12 h light/dark cycle with food and water freely available. All animals were habituated to experimenters by gentle handling for at least 3 days prior to drug administration, and all care was in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996). Experimental protocols were approved by the University of Massachusetts—Amherst Institutional Animal Care and Use Committee.
2.2. MDMA administration
The cohort of animals in experiment I was also part of a separate investigation examining the potential for various pretreatments to influence the extent of serotonergic marker depletion following a subsequent MDMA binge (Piper et al., 2010). For simplicity, only the animals that were pretreated with saline by twice-daily injections of the vehicle (4 h apart) are presented here. This regimen was repeated every fifth day from postnatal day (PD) 60–85, after which animals were assigned to one of three binge groups (N = 7–10/group) on PD92. The binge consisted of four subcutaneous doses with an inter-dose interval of 1 h of either 0.0, 2.5, or 5.0 mg/kg ± MDMA HCl (Research Triangle Institute, Research Triangle Park, NC) in 0.9% NaCl vehicle. All animals were killed for DAT binding 1 week later.
Given the controversy surrounding the potential of MDMA to adversely affect the dopamine system (see Ricaurte et al., 2002 and subsequent retraction), it was important to investigate whether our MDMA binge regimen could cause similar changes in other markers of dopaminergic neuron function. As such, additional information regarding the effects of a similar binge regimen on levels of dopamine and its major metabolites was determined in a separate cohort of animals. In experiment II, adult rats were binged with MDMA or saline as before (PD92) without undergoing saline pretreatment. In this study, however, animals (N = 10/group) were given a higher dose of the compound (10 mg/kg × 4, also with an inter-dose interval of 1 h) to accentuate any potential dopaminergic deficits arising from MDMA exposure. As in experiment I, all animals were killed a week later for endpoint examination.
2.3. Core temperature analysis
Temperature measurements were taken using a rectal probe (RET-2, Physitemp Instruments, Clifton, NJ) connected to a digital thermometer (Thermalert TH-5, Physitemp Instruments, Clifton, NJ) 30 min prior to, and then every 30 min during and following drug administration, terminating at 3 h after the final injection of the MDMA binge. All rats exceeding 40.5 °C during dosing were briefly cooled, either by temporary placement in a 4 °C cold-room (experiment I) or by application of ice packs to both sides of the animal (experiment I and II).
2.4. Dopamine transporter binding
One week following the MDMA binge (PD99), animals in experiment I were lightly anesthetized with CO2 and decapitated. Striatal samples were rapidly dissected over ice according to Piper et al. (2005) and stored at −70 °C for later analysis of DAT binding. On the day of the assay, frozen tissues were homogenized in 100 vols of homogenization buffer (20 mM phosphate buffer, 0.32 M sucrose, pH 7.4) using a Polytron. Homogenates were centrifuged at 20,000 × g for 20 min at 4 °C, the supernatant was decanted, and the pellet was resuspended in 50 vols of homogenization buffer. Centrifugation and resuspension were repeated twice more to yield a crude washed membrane fraction. Transporter binding was measured using tritiated (−)-2-beta-carbomethoxy-3-beta-(4-fluorophenyl)tropane 1,5-napthalenedisulfonate([3H]WIN 35,428; 87.0 Ci/mmol, Perkin Elmer, Boston, MA) at a final concentration of 5.0 nM. For nonspecific binding measurements, cocaine HCl was additionally present at a final concentration of 30 μM. Incubations were performed at a final volume of 500 μL, including 100 μL of membrane suspension, for 90 min at 4 °C on a shaking platform. The reaction was terminated by rapid filtration using Whatman GF/B filters presoaked in 0.05% polyethyleneimine, followed by two 5.0-mL rinses with phosphate buffer. Filters were then placed in scintillation vials with 4.7 mL of ScintiSafe cocktail (Perkin Elmer, Waltham, MA) and counted the following day using a Packard 1900CA liquid scintillation analyzer. The protein concentration in each sample was determined by means of the Bio-Rad© DC protein assay (Hercules, CA) using bovine gamma globulin as the standard. Mean data were expressed as a percentage of the saline group.
2.5. High-performance liquid chromatography
One week following the MDMA binge, animals in experiment II were decapitated and a 2-mm slice beginning 1 mm from the anterior pole was removed, encompassing the entire extent of the frontal cortex up to the most rostral boundary of the caudate-putamen (Paxinos and Watson, 1998). The striatum was dissected as above. All samples were immediately frozen at −70 °C until analysis. Concentration of 5-HT and dopamine and their metabolites 5-hydroxyindole acetic acid (5-HIAA), dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) were quantified by a modified method of high-performance liquid chromatography (HPLC) combined with electrochemical detection (EC) as described by Ali et al. (1994). Briefly, each tissue sample was weighed in a volume (20% weight/volume) of 0.2 N perchloric acid containing 100ng/mg of the internal standard 3,4-dyhydroxybenzylamine (DHBA). Tissue was disrupted by ultrasonication, centrifuged at 15,000 × g for 7 min, and 150 μL of the supernatant was removed and filtered through 0.2 μm Nylon-66 microfilter (MF-1 centrifugal filter, Bioanalytical Systems, West Lafayette, IN). Aliquots of 25 uL representing 2.5 mg of brain tissue were injected directly onto the HPLC/EC system for the separation of 5-HT, dopamine, and their metabolites 5-HIAA DOPAC, and HVA Group data were expressed as a percentage of the saline condition. The analytical system included a Waters Associates model 510 liquid chromatographic pump (Milford, MA), a Rheodyne model 7125 injector (Rheodyne Inc., Cotati, CA), a SupelcoSupelcosil LC-18, 3 μm (7.5 cm × 4.6 mm) analytical column, an LC-4B amperometric detector and LC-17 oxidative flow cells (BAS) consisting of a glassy carbon electrode (TL-5) versus a Ag–AgCl reference electrode maintained at a potential of 0.75 V. The mobile phase consisted of 0.07 M potassium phosphate, pH 3.0, 8% methanol and an ion pairing reagent of 1.02 mM 1-heptane sulfonic acid. Chromatograms were recorded and integrated on a Perkin–Elmer LCI-100 integrator (Perkin–Elmer Corp., Norwalk, CT). The concentrations of 5-HT, dopamine, 5-HIAA, DOPAC and HVA were calculated using standard curves generated by determining in triplicate the ratio between three different known amounts of each amine or its metabolites and a constant amount of internal standard. Mean data were expressed as a percentage of the saline condition.
3. Results
3.1. Experiment I: Effects of MDMA on dopamine transporter binding
Animals in this experiment were administered four doses every hour of either 0, 2.5, or 5 mg/kg MDMA dissolved in a saline vehicle. During the MDMA or saline binge, animals in the MDMA-treated group experienced significant hyperthermia when compared with saline-treated controls (data not shown). One week following dosing, animals given MDMA showed a dose-dependent reduction in striatal DAT binding compared with the saline controls, with the highest drug dose (5 mg/kg × 4) leading to a statistically significant (73.7%) decrease in binding (Fig. 1).
Fig. 1.
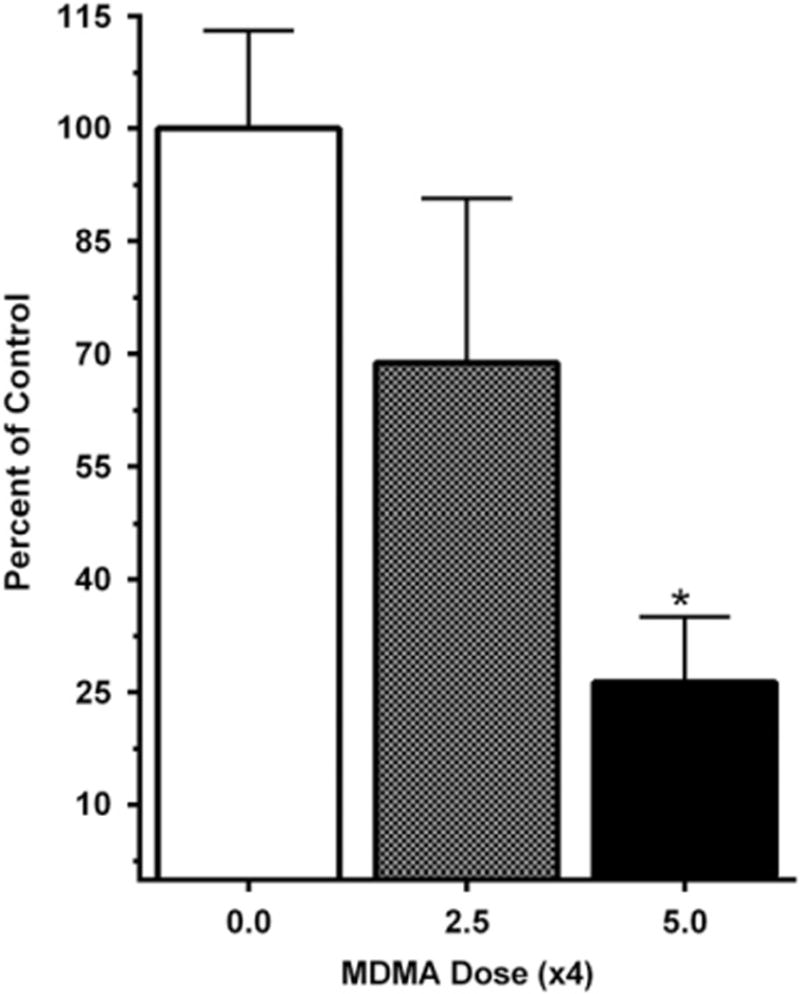
Percentage change in [3H]WIN 35,428 binding to the dopamine transporter in the striatum of adult rats given MDMA (0.0, 2.5, or 5.0 mg/kg × 4 with an inter-dose interval of 1 h) 1 week earlier. Mean binding for the saline control group was 313.6 (±41.0 S.E.M.) fmol/mg protein (***P ≤ 0.001).
3.2. Experiment II: Effects of MDMA on Levels of dopamine and dopamine metabolites
Rats in this experiment were administered either 10 mg/kg MDMA or a saline vehicle every hour for a total of four doses. As in the previous experiment, all animals given MDMA showed a significant rise in body temperature. Core temperature increased an average of +2.3 (±0.3) °C in the MDMA group relative to a −0.2 (±0.3) °C change following saline at 3 h after the first injection (t(14.8) = 7.79, P < 0.0005, see Supplemental Fig. 1 for further details). One week following the binge, the MDMA-treated group showed marked reductions in levels of 5-HT in both the striatum (Fig. 2A) and the frontal cortex (Fig. 2B), while a significant reduction in 5-HIAA content was observed in only the striatum (Fig. 2A). Animals in the MDMA group showed no reduction in dopamine levels in the frontal cortex (Fig. 2B), though a trend towards a significant reduction in dopamine content was noted the striatum (t(9.7) = 2.04, P = 0.070, Fig. 2A). Compared with the saline group, levels of both DOPAC and HVA were significantly reduced in the striatum of MDMA-treated animals (Fig. 2A), while only the HVA content showed a trend towards reduction in the frontal cortex (t(18) = 1.91, P = 0.072, Fig. 2B).
Fig. 2.
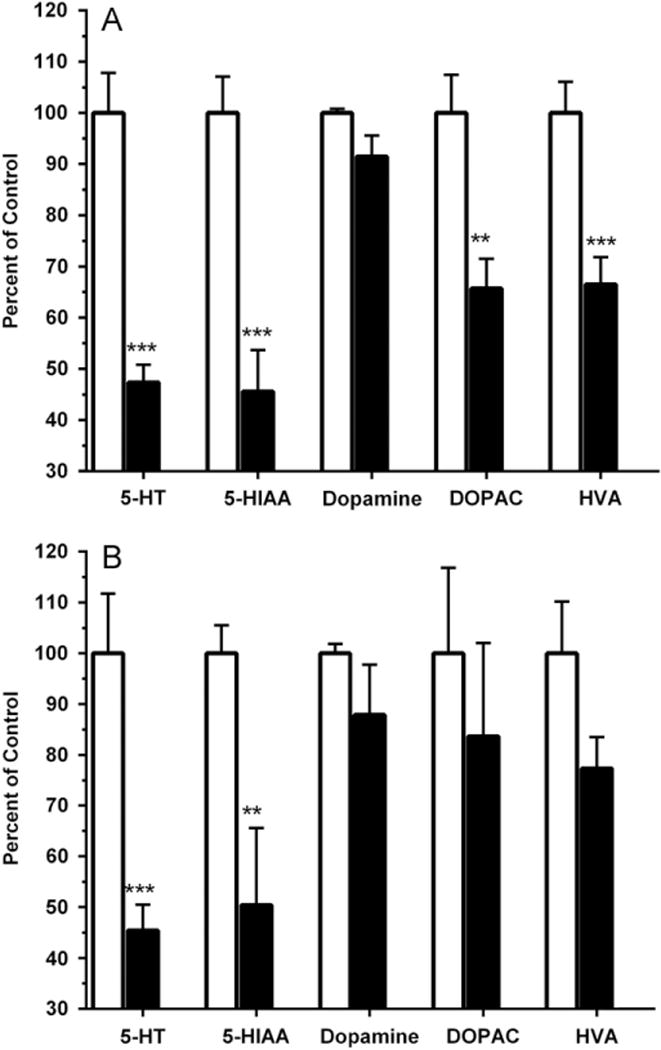
Percentage change in monoamine and respective metabolite levels in the striatum (A) or frontal cortex (B) of rats that received saline (open bars) or MDMA (10mg/kg × 4, filled bars). For the saline group, mean (± S.E.M.) quantities (ng/100 mg tissue) of 5-HT, 5-HIAA, dopamine, DOPAC, and HVA were 22.0 (±1.7), 14.0 (±1.0), 504.9 (±4.0), 82.4 (±6.1), and 53.8 (±3.3) in the striatum, and 18.7 (±2.2), 9.8 (±0.5), 23.9 (±0.4), 9.3 (±0.6), and 5.4 (±0.6) in the frontal cortex (***P ≤ 0.001 or **P ≤ 0.01).
4. Discussion
In this study, we show evidence that repeated administration of MDMA during a short time interval (every hour) has the potential to alter certain aspects of dopaminergic neurochemistry in the striatum of rats. This was demonstrated by a stark decrease in striatal DAT binding as well as significant reductions in dopamine metabolite levels in this brain area, changes accompanied by only a modest, non-significant reduction in dopamine content. Importantly, this MDMA dosing regimen caused a marked depletion in regional 5-HT and 5-HIAA, indicating the presence of the typical pattern of serotonergic neurotoxicity known to occur following MDMA exposure in rats (Green et al., 2003). As such, our findings suggest that MDMA can alter not only the 5-HT system but also another neuromodulatory system typically considered to be unaffected by the compound.
Several previous reports have failed to find alterations in striatal DAT binding following MDMA administration (Lew et al., 1996; Sabol et al., 1996). Interestingly, each of these studies used a dosing regimen consisting of 20 mg/kg MDMA b.i.d. for four consecutive days with a long inter-dose interval of 12 h. In contrast, we documented profound reductions in striatal DAT levels following hourly administration of moderate doses (5 mg/kg × 4) of the compound, a variable which may have contributed to an unmasking of MDMA effects on expression of this transporter. Given the complex metabolism of MDMA (de la Torre and Farré, 2004), it is certainly possible that a more frequent dosing schedule results in higher levels of metabolites that are neurotoxic to the dopaminergic system. Although this hypothesis requires further testing, it is interesting to note that intra-striatal administration of the N-acetylcysteine conjugate of α-methyl dopamine, a metabolite of MDMA, caused significant reductions in both striatal and cortical dopamine in addition to the expected decreases in 5-HT and 5-HIAA (Jones et al., 2005). Additionally, Elayan et al. (1992) reported decreased striatal tyrosine hydroxylase activity 1 week following intracerebroventricular infusion of the MDMA metabolite 2,4,5-trihydroxyamphetamine. Although a caveat to these studies is that they documented dopaminergic deficits following high molar concentrations of these metabolites, they nevertheless highlight the possibility that binge administration of MDMA may result in significant formation of these and other bioactive breakdown products with the potential to alter dopaminergic function.
A second important methodological factor may have been the age of the animals. Most rat studies of MDMA neurotoxicity have used young adult or adolescent animals, whereas animals in the present study were dosed in full adulthood (i.e., PD92). We previously demonstrated that rats at this age are much more sensitive to the toxic effects of MDMA than young adult rats, thus requiring a reduction in binge dose from 10 mg/kg/injection to 5 mg/kg/injection to avoid excessive mortality (Piper et al., 2010). It is interesting to speculate that striatal DAT may be more vulnerable to dysregulation by MDMA in these older animals.
Many studies have also reported no long term reductions in dopamine concentrations in MDMA-treated rats (Colado et al., 2004; Sanchez et al., 2004; Schmidt and Kehne, 1990). On the other hand, there are also several reports of reduced striatal dopamine levels, usually following repeated high doses of MDMA (Able et al., 2006; Commins et al., 1987; Mayerhofer et al., 2001; McGregor et al., 2003). Although the present study found only a non-significant trend towards a decrease in striatal dopamine 1 week after binge MDMA treatment, there were substantial reductions in both DOPAC and HVA that are suggestive of a drug-induced decrease in dopamine turnover.
The present pattern of reduced striatal DAT binding without a concomitant decrease in dopamine levels is most consistent with a change in DAT expression and/or trafficking rather than a degeneration of striatal dopamine nerve terminals. In support of this interpretation, O’Hearn et al. (1988) showed no effect of MDMA on the density of striatal tyrosine hydroxylase-immunoreactive fibers, which primarily mark dopamine axons and terminals in this brain area. Given that our binding assay mainly measured levels of DAT embedded in the plasma membrane, it is possible that the results are due to a persistent MDMA-induced increase in endosomal DAT trafficking (Hansen et al., 2002). Alternatively, our MDMA treatment regimen may have reduced DAT protein levels by compromising DAT gene expression (see Biezonski and Meyer, 2010, for data showing MDMA down-regulation of 5-HT transporter and VMAT-2 gene expression) or by increasing the degradation rate of the transporter. Further research is needed to test these hypotheses regarding the mechanisms by which MDMA may lead to reductions in plasmalemmal DAT expression.
Although the aim of our experiments was to determine the effect of binge MDMA administration on two different markers of dopaminergic neuron function, given that dopamine relies on DAT for recycling back into the terminal, the possibility should be acknowledged that drug-induced changes in one of these markers may have affected levels of the other. For example, if reduced plasmalemmal DAT content following MDMA exposure affected the rate of dopamine reuptake, it is possible that the minor decrease in striatal dopamine levels, measured in axons and terminals in our study, may have simply resulted from hindered recycling of synaptic dopamine back into the terminal. Additionally, this scenario may in part explain our finding of reduced levels of dopamine metabolites in the striatum following MDMA exposure, as reduced dopamine reuptake would limit metabolism of this catecholamine. Alternatively, however, it remains equally possible that MDMA may have altered levels of dopamine metabolism independent of its effects on DAT by reducing the expression or activity of either type A or B monoamine oxidase enzymes, catechol-O-methyl transferase, or both. Additional studies are therefore needed to clarify whether or not the effects of an MDMA binge on DAT levels and dopamine metabolism are mutually exclusive.
In summary, a short-interval binge pattern of MDMA administered to fully adult male rats led to a marked reduction in striatal DAT binding along with decreased dopamine metabolite concentrations suggestive of reduced dopamine turnover. Given that dopamine plays an important role in the stimulatory, rewarding, and thermoregulatory effects of MDMA exposure (Daniela et al., 2004; Green et al., 2004), our findings suggest that the drug could potentially disrupt these systems in humans consuming Ecstasy using the “stacking” and “boosting” pattern modeled in the current study (Parrott, 2005). Additionally, since the dopamine system is also known to underlie working memory (Robbins and Roberts, 2007), it is possible that MDMA-induced deficits in dopaminergic function may also in part explain several documented reports of disruptions in neurocognitive processes in rats given MDMA and in Ecstasy-using humans (Able et al., 2006; Kalechstein et al., 2007; Morley et al., 2001; Piper et al., 2007). Nevertheless, caution should be taken in extracting clinical significance from our findings due to the uncertainties inherent in extrapolating MDMA dosing regimens from experimental animals to humans (de la Torre and Farré, 2004). Alternatively, abnormalities in the dopamine system have been identified in former Ecstasy users (Tai et al., 2011). Further research is needed to determine how the dopamine system is affected by age and by MDMA dosing regimen including the duration of drug abstinence in rats as well as in other species. Such information should be helpful in developing animal models with greater relevance for understanding the neurotoxic and behavioral consequences of human Ecstasy use.
Supplementary Material
Acknowledgments
MDMA was provided by Research Triangle Institute and Lauren Daniels and Charles Betts provided technical assistance. BJP was supported by the National Institute of Drug Abuse (Grant no. L30DA027582) during the latter stages of manuscript preparation.
Appendix A. Supplementary information
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.ejphar.2012.12.024.
References
- Able JA, Gudelsky GA, Vorhees CV, Williams MT. 3,4-Methylenedioxymethamphetamine in adult rats produces deficits in path integration and spatial reference memory. Biol Psychiatry. 2006;59:1219–1226. doi: 10.1016/j.biopsych.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SF, Newport GD, Holson RR, Slikker W, Bowyer JF. Low environmental temperature or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxicity in mice. Brain Res. 1994;658:33–38. doi: 10.1016/s0006-8993(09)90007-5. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Yeh SY, O’Hearn E, Molliver ME, Kuhar MJ, De Souza EB. 3,4-Methylenedioxymethamphetamine and 3,4-methylenedioxyamphetamine destroy serotonin terminals in rat brain: quantification of neurodegeneration by measurement of [3H]paroxetine-labeled serotonin uptake sites. J Pharmacol Exp Ther. 1987;242:911–916. [PubMed] [Google Scholar]
- Biezonski DK, Meyer JS. Effects of 3,4-methylenedioxymethamphetamine (MDMA) on serotonin transporter and vesicular monoamine transporter 2 protein and gene expression in rats: implications for MDMA neurotoxicity. J Neurochem. 2010;112:951–962. doi: 10.1111/j.1471-4159.2009.06515.x. [DOI] [PubMed] [Google Scholar]
- Colado MI, O’Shea E, Green AR. Acute and long-term effects of MDMA on cerebral dopamine biochemistry and function. Psychopharmacology. 2004;173:249–263. doi: 10.1007/s00213-004-1788-8. [DOI] [PubMed] [Google Scholar]
- Cole JC, Sumnall HR. The pre-clinical behavioural pharmacology of 3,4-methylenedioxymethamphetamine (MDMA) Neurosci Biobehav Rev. 2003;27:199–217. doi: 10.1016/s0149-7634(03)00031-9. [DOI] [PubMed] [Google Scholar]
- Commins DL, Vosmer G, Virus RM, Woolverton WL, Schuster CR, Seiden LS. Biochemical and histological evidence that methylenedioxymethamphetamine is toxic to neurons in the rat brain. J Pharmacol Exp Ther. 1987;241:338–345. [PubMed] [Google Scholar]
- Daniela E, Brennan K, Gittings D, Hely L, Schenk S. Effect of SCH 23390 on (±)-3,4-methylenedioxymethamphetamine hyperactivity and self-administration in rats. Pharmacol Biochem Behav. 2004;77:745–750. doi: 10.1016/j.pbb.2004.01.008. [DOI] [PubMed] [Google Scholar]
- de la Torre R, Farré M. Neurotoxicity of MDMA (ecstasy): the limitations of scaling from animals to humans. Trends Pharmacol Sci. 2004;25:505–508. doi: 10.1016/j.tips.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Elayan I, Gibb JW, Hanson GR, Foltz RL, Lim HK, Johnson M. Long-term alteration in the central monoaminergic systems of the rat by 2,4,5-trihydroxyamphetamine but not by 2-hydroxy-4,5-methylenedioxymethamphetamine or 2-hydroxy-4,5-methylenedioxyamphetamine. Eur J Pharmacol. 1992;221:281–288. doi: 10.1016/0014-2999(92)90714-f. [DOI] [PubMed] [Google Scholar]
- Green AR, Mechan AO, Elliott JM, O’Shea E, Colado MI. The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) Pharmacol Rev. 2003;55:463–508. doi: 10.1124/pr.55.3.3. [DOI] [PubMed] [Google Scholar]
- Green AR, O’Shea E, Colado MI. A review of the mechanisms involved in the acute MDMA (ecstasy)-induced hyperthermic response. Eur J Pharmacol. 2004;500:3–13. doi: 10.1016/j.ejphar.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Hansen JP, Riddle EL, Sandoval V, Brown JM, Gibb JW, Hanson GR, Fleckenstein AE. Methylenedioxymethamphetamine decreases plasmalemmal and vesicular dopamine transport: mechanisms and implications for neurotoxicity. J Pharmacol Exp Ther. 2002;300:1093–1100. doi: 10.1124/jpet.300.3.1093. [DOI] [PubMed] [Google Scholar]
- Jones DC, Duvauchelle C, Ikegami A, Olsen CM, Lau SS, de la Torre R, Monks TJ. Serotonergic neurotoxic metabolites of ecstasy identified in rat brain. J Pharmacol Exp Ther. 2005;313:422–431. doi: 10.1124/jpet.104.077628. [DOI] [PubMed] [Google Scholar]
- Kalechstein AD, De La Garza R, Mahoney JJ, Fantegrossi WE, Newton TF. MDMA use and neurocognition: a meta-analytic review. Psychopharmacology. 2007;189:531–537. doi: 10.1007/s00213-006-0601-2. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Furukawa Y, Ang L, Vorce SP, Kalasinsky KS. Striatal serotonin is depleted in brain of a human MDMA (Ecstasy) user. Neurology. 2000;55:294–296. doi: 10.1212/wnl.55.2.294. [DOI] [PubMed] [Google Scholar]
- Lew R, Sabol KE, Chou C, Vosmer GL, Richards J, Seiden LS. Methylenedioxymethamphetamine-induced serotonin deficits are followed by partial recovery over a 52-week period. Part II: radioligand binding and autoradiography studies. J Pharmacol Exp Ther. 1996;276:855–865. [PubMed] [Google Scholar]
- Lyles L, Cadet JL. Methylenedioxymethamphetamine (MDMA, Ecstasy) neurotoxicity: cellular and molecular mechanisms. Brain Res Rev. 2003;42:155–168. doi: 10.1016/s0165-0173(03)00173-5. [DOI] [PubMed] [Google Scholar]
- Mayerhofer A, Kovar KA, Schmidt WJ. Changes in serotonin, dopamine and noradrenalin levels in striatum and nucleus accumbens after repeated administration of the abused drug MDMA in rats. Neurosci Lett. 2001;308:99–102. doi: 10.1016/s0304-3940(01)01992-9. [DOI] [PubMed] [Google Scholar]
- McCann UD, Ridenour A, Shaham Y, Ricaurte GA. Serotonin neurotoxicity after (±)3,4-methylenedioxymethamphetamine (MDMA; “Ecstasy”): a controlled study in humans. Neuropsychopharmacology. 1994;10:129–138. doi: 10.1038/npp.1994.15. [DOI] [PubMed] [Google Scholar]
- McCann UD, Eligulashvili V, Ricaurte GA. (±)3,4-Methylenedioxymethamphetamine (‘Ecstasy’)-induced serotonin neurotoxicity: clinical studies. Neuropsychobiology. 2000;42:11–16. doi: 10.1159/000026665. [DOI] [PubMed] [Google Scholar]
- McCann UD, Szabo Z, Vranesic M, Palermo M, Mathews WB, Ravert HT, Dannals RF, Ricaurte GA. Positron emission tomographic studies of brain dopamine and serotonin transporters in abstinent (±)3,4-methylenedioxymethamphetamine (“ecstasy”) users: relationship to cognitive performance. Psychopharmacology. 2008;200:439–450. doi: 10.1007/s00213-008-1218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor IS, Gurtman CG, Morley KC, Clemens KJ, Blokland A, Li KM, Cornish JL, Hunt GE. Increased anxiety and “depressive” symptoms months after MDMA (“Ecstasy”) in rats: drug-induced hyperthermia does not predict long-term outcomes. Psychopharmacology. 2003;168:465–474. doi: 10.1007/s00213-003-1452-8. [DOI] [PubMed] [Google Scholar]
- Morley KC, Gallate JE, Hunt GE, Mallet PE, McGregor IS. Increased anxiety and impaired memory in rats 3 months after administration of 3,4-methylenedioxymethamphetamine (“ecstasy”) Eur J Pharmacol. 2001;433:91–99. doi: 10.1016/s0014-2999(01)01512-6. [DOI] [PubMed] [Google Scholar]
- Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. Vesicular monoamine transporter-2: immunogold localization in striatal axons and terminals. Synapse. 1997;26:194–198. doi: 10.1002/(SICI)1098-2396(199706)26:2<194::AID-SYN10>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- O’Hearn E, Battaglia G, De Souza EB, Kuhar MJ, Molliver ME. Methylenedioxyamphetamine (MDA) and methylenedioxymethamphetamine (MDMA) cause selective ablation of serotonergic axon terminals in forebrain: immunocytochemical evidence for neurotoxicity. J Neurosci. 1988;8:2788–2803. doi: 10.1523/JNEUROSCI.08-08-02788.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott AC. Chronic tolerance to recreational MDMA (3.4,methylenedioxymethamphetamine) or Ecstasy. J Psychopharmacol. 2005;19:71–83. doi: 10.1177/0269881105048900. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego, CA: 1998. [Google Scholar]
- Piper BJ. A developmental comparison of the neurobehavioral effects of ecstasy (MDMA) Neurotoxicol Teratol. 2007;29:288–300. doi: 10.1016/j.ntt.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper BJ, Ali SF, Daniels LD, Meyer J. Repeated intermittent methylenedioxymethamphetamine exposure protects against the behavioral and neurotoxic, but not hyperthermic, effects of an MDMA binge in adult rats. Synapse. 2010;64:421–431. doi: 10.1002/syn.20744. [DOI] [PubMed] [Google Scholar]
- Piper BJ, Fraiman JB, Meyer JS. Repeated MDMA (“Ecstasy”) exposure in adolescent male rats alters temperature regulation, spontaneous motor activity, attention, and serotonin transporter binding. Dev Psychobiol. 2005;47:145–157. doi: 10.1002/dev.20085. [DOI] [PubMed] [Google Scholar]
- Piper BJ, Fraiman JB, Owens CB, Ali SF, Meyer JS. Dissociation of the neurochemical and behavioral consequences of MDMA (“Ecstasy”) by citalopram. Neuropsychopharmacology. 2007;33:1192–1205. doi: 10.1038/sj.npp.1301491. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Forno LS, Wilson MA, DeLanney LE, Irwin I, Molliver ME, Langston JW. (±)3,4-Methylenedioxymethamphetamine selectively damages central serotonergic neurons in nonhuman primates. J Am Med Assoc. 1988;260:51–55. [PubMed] [Google Scholar]
- Ricaurte GA, McCann UD. Neurotoxic amphetamine analogues: effects in monkeys and implications for humans. Ann NY Acad Sci. 1992;648:371–382. doi: 10.1111/j.1749-6632.1992.tb24586.x. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Yuan J, Hatzidimitriou G, Cord BJ, McCann UD. Severe dopaminergic neurotoxicity in primates after a common recreational dose regimen of MDMA (“ecstasy”) Science. 2002;297:2260–2263. doi: 10.1126/science.1074501. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Yuan J, McCann UD. (±)3,4-Methylenedioxymethamphetamine (‘Ecstasy’)-induced serotonin neurotoxicity: studies in animals. Neuropsychobiology. 2000;42:5–10. doi: 10.1159/000026664. [DOI] [PubMed] [Google Scholar]
- Robbins TW, Roberts AC. Differential regulation of fronto-executive function by the monoamines and acetylcholine. Cereb Cortex. 2007;17:151–160. doi: 10.1093/cercor/bhm066. [DOI] [PubMed] [Google Scholar]
- Sabol KE, Lew R, Richards JB, Vosmer GL, Seiden LS. Methylenedioxymethamphetamine-induced serotonin deficits are followed by partial recovery over a 52-week period. Part I: synaptosomal uptake and tissue concentrations. J Pharmacol Exp Ther. 1996;276:846–854. [PubMed] [Google Scholar]
- Sanchez V, O’shea E, Saadat KS, Elliott JM, Colado MI, Green AR. Effect of repeated (‘binge’) dosing of MDMA to rats housed at normal and high temperature on neurotoxic damage to cerebral 5-HT and dopamine neurons. J Psychopharmacol. 2004;18:412–416. doi: 10.1177/026988110401800312. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Kehne JH. Neurotoxicity of MDMA: neurochemical effects. Ann NY Acad Sci. 1990;600:665–680. doi: 10.1111/j.1749-6632.1990.tb16917.x. [DOI] [PubMed] [Google Scholar]
- Tai YF, Hoshi R, Brignell CM, Cohen L, Brooks DJ, Curran HV, Piccini P. Persistent nigrostriatal dopaminergic abnormalities in users of MDMA (‘Ecstasy’): an 18F-dopa PET study. Neuropsychopharmacology. 2011;36:735–743. doi: 10.1038/npp.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Tong L, McLane MW, Hatzidimitriou G, Yuan J, McCann U, Ricaurte G. Loss of serotonin transporter protein after MDMA and other ring-substituted amphetamines. Neuropsychopharmacology. 2006;31:2639–2651. doi: 10.1038/sj.npp.1301031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.