

Abstract
Hexavalent Chromium [Cr(VI)] is an important human carcinogen associated with pulmonary diseases and lung cancer. Exposure to Cr(VI) induces DNA damage, cell morphological change and malignant transformation in human lung epithelial cells. Despite extensive studies, the molecular mechanisms remain elusive, it is also not known if Cr(VI)-induced transformation might accompany with invasive properties to facilitate metastasis. We aimed to study Cr(VI)-induced epithelial–mesenchymal transition (EMT) and invasion during oncogenic transformation in lung epithelial cells. The results showed that Cr(VI) at low doses represses E-cadherin mRNA and protein expression, enhances mesenchymal marker vimentin expression and transforms the epithelial cell into fibroblastoid morphology. Cr(VI) also increases cell invasion and promotes colony formation. Further studies indicated that Cr(VI) uses multiple mechanisms to repress E-cadherin expression, including activation of E-cadherin repressors such as Slug, ZEB1, KLF8 and enhanced binding of HDAC1 in E-cadherin gene promoter, but DNA methylation is not responsible for the loss of E-cadherin. Catalase reduces Cr(VI)-induced E-cadherin and vimentin protein expression, attenuates cell invasion in matrigel and colony formation on soft agar. These results demonstrate that exposure to a common human carcinogen, Cr(VI), induces EMT and invasion during oncogenic transformation in lung epithelial cells and implicate in cancer metastasis and prevention.
Keywords: Hexavalent Chromium, Oncogenic transformation, Epithelial-Mesenchymal Transition, Histone, cancer signaling, epithelial cells
Introduction
Hexavalent chromium [Cr(VI)] is a potent human mutagen and carcinogen, widely used in industry and present in fossil fuel and cigarette smoking (Gibb et al., 2000; O'Brien et al., 2003; Nickens et al., 2010). Exposure of Cr(VI) compounds are associated with increased inflammation and cancer risk, particularly the carcinoma of the lung (Gibb et al., 2000; Nickens et al., 2010). The diseases that are associated with chromium exposure include nasal ulcer, lung inflammation, fibrosis, fibrosarcomas, adenocarcinomas, and squamous cell carcinomas of lung (Gibb et al., 2000; Takahashi et al., 2005b; Beaver et al., 2009).
One important characteristic of chromium is its oxidative property. In cellular system, Cr(VI) is reduced via Cr(V) and Cr(IV) intermediate oxidation states to stable Cr(III). During the cellular reduction process, reactive oxygen species (ROS) are generated and cause cytotoxicity (Liu et al., 1994; Shi and Dalal, 1994). Several mechanisms have been suggested for Cr(VI)-induced host cell pathogenesis. These include DNA-strand breaks (Stearns et al., 1995), DNA–protein crosslinks, DNA inter- and intrastrand crosslinks (O'Brien et al., 2001), interrupted DNA replication and transcription (Snow, 1994), cell cycle checkpoints dysfunction (Ceryak et al., 2004; Wise et al., 2006), impaired DNA damage repair, microsatelite instability (Takahashi et al., 2005a), activation of oncogenic pathways (O'Hara et al., 2007), all of which may contribute to the imbalance of cell death, survival, and carcinogenesis.
Chromium is known to induce oncogenic transformation in lung epithelial cells as well as in rodent models, with the water-insoluble or “particulate” compounds pose the greatest carcinogenic risk and water soluble form the least one (O'Brien et al., 2003; Nickens et al., 2010). Tumor initiation involves extensive genomic re-arrangements, numerous intracellular signaling alternations, activation of oncogene pathways, and acquiring migratory, invasive properties. Cancer cell invasion is a critical step to establish fatal distance metastasis, which accounts for a large portion of cancer related death especially for lung cancer. An essential and initial process leading to the tumor invasion is epithelial–mesenchymal transition (EMT) (Thiery and Sleeman, 2006). During the EMT process, cells lose their epithelial properties such as cell polarity, normal cell–cell contact, acquire mesenchymal properties presented as fibroblastic morphology, invasion and express mesenchymal markers including vimentin, N-cadherin (Perl et al., 1998; van Roy and Berx, 2008).
Although it is known that Cr(VI) chronic exposure induces oncogenic transformation, the molecular mechanism is not clear. Furthermore, little information is available regarding if a metal compound such as chromium might induce EMT and invasion. We therefore use chronic treatment of chromium with lung epithelial cell model to induce transformation and investigate the molecular mechanisms that involve EMT, invasion during oncogenic transformation processes.
We demonstrate in this work, a common human metal carcinogen, chromium, induces EMT and invasion during oncogenic transformation in human lung epithelial cells. These effects involve catalase/ROS-mediated mechanism and suggest complex interaction of chromium with human lung epithelial cells that lead to EMT, invasion and oncogenesis.
Materials and Methods
Cell lines, cell culture, and reagents
Immortalized normal human bronchial epithelial cell line, BEAS-2B, human lung cancer cell line A549 were purchased from American Type Culture Collection (ATCC, Manassas, VA). BEAS-2B cells that stably express catalase were generated by integration of a catalase expression vector (OriGene, Rockville, MD) and selected with G418. Catalase protein expression and ROS scavenging effects were also confirmed from the selected cells (Wang et al., 2011). Two Cr(VI) transformed BEAS-2B cell lines, CrTF1 and CrTF2 that were isolated from soft agar by chronic exposure of Cr(VI) to BEAS-2B cells in our laboratory were also included in the present study (Wang et al., 2011). Tissue culture reagents were purchased from GIBCO (Invitrogen, Carlsbad, CA). Cells were maintained in DMEM medium supplemented with 10% fetal bovine serum (FBS) and antibiotics at 37°C in a humidified 10% CO2 incubator. Medium was replaced twice a week in the presence or absence of Cr(VI).
Potassium dichromate (Cat.# 483044), specific histone deacetylase (HDAC) inhibitor trichostatin A (TSA, Cat.# T8552) and DNA methytranferase (DNMT) inhibitor 5-aza-2′-deoxycytidine (AZA, Cat.# A3656) were purchased from Sigma Chemical Company (St. Louis, MO). TSA and AZA were prepared in dimethyl sulfoxide solution.
Monoclonal antibody for E-cadherin (Cat.# 610182) was purchased from BD Biosciences (San Jose, CA). Antibodies for vimentin (sc-73259), β-actin (sc-47778), fibronectin (sc-8422), MMP-9 (sc-21733), NF-κB p65 (sc-109), lamin A/C (sc-6215), Twist (sc-81417) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for β-catenin (Cat.# 9562), N-cadherin (Cat.# 4061), HDAC1 (Cat.# 5356), Snail (Cat.# 3879), Slug (Cat.# 9585) were purchased from Cell Signaling Technology (Danvers, MA) Antibodies for Chromatin immunoprecipitation including anti-HDAC1 (Cat.# 17-608), anti-H3K9Ac (Cat.# 07-352) were from Millipore (Temecula, CA).
To understand the effects of co-incubation of Cr(VI) with BEAS-2B cells, Cr(VI) was added to culture medium and incubated with BEAS-2B cells for various periods of time dependent upon different biochemical assays. They range from several hours to 1, 2, 3, 4, 6, 8, and 10 weeks. The cell lysates was collected for various protein analyses by Western blot. Other assays such as cell morphological change, migration, invasion, and transformation were each examined using different methods at specific time course as described below and in the Results.
Western blot analysis of protein expression
BEAS-2B, CrTF1, CrTF2, A549 or BEAS-2B-catalase-stable expressing cells (1 × 106) were treated with or without Cr(VI) for various periods of time. Cells were washed three times with PBS, and lysed with cell lysis buffer for nuclear and cytosolic protein extraction as described previously (Ding et al., 2004). In some experiment, TSA, AZA were added to study their effects. 20 μg of proteins were separated on 10% SDS polyacrylamide gels. Proteins were subsequently transferred from gels onto nitrocellulose membranes (Bio-Rad, Hercules, CA) and blocked for 1 hour at room temperature in Tris-buffered saline plus 0.025% Tween-20 (TBS-T) with 5% nonfat dry milk (pH 7.4). Various antibodies diluted at 1:1000 in TBS-T with 5% nonfat dry milk solution were incubated with membranes at 4°C overnight and washed three times with TBS-T. The secondary antibody, horseradish peroxidase (HRP)-conjugated antibody, diluted at 1:2500 in TBS-T with 5% nonfat dry milk was incubated with membranes at room temperature for 2-3 hours. Images were acquired with an enhanced chemiluminescence detection kit (Perkin Elmer Life Sciences, Boston, MA). In each experiment, either anti-β-actin or anti-lamin A/C antibody was reprobed to monitor protein loading.
Quantitative RT-PCR for gene expression
BEAS-2B cells were treated with or without Cr(VI) at 0.5 μM for three weeks, total RNA from cells were extracted and purified using RNeasy Mini Kit (Qiagen, Valencia, CA) as described previously (Ding et al., 2010). Reverse transcription of 0.5 μg of total cellular RNA was performed in a final volume of 20 μl containing 5× first strand buffer (Invitrogen), 1 mM of each dNTP, 20 units of placental RNase inhibitor, 5 μM random hexamer, and 9 units of Moloney murine leukemia virus reverse transcriptase (Invitrogen). After incubation at 37°C for 45 minutes, the samples were heated for 5 minutes at 92°C to end the reaction, diluted at 1:4 and stored at – 20°C until PCR use. Two μl of cDNA was subjected to real-time quantitative PCR using the Opticon system (MJ Research, Waltham, MA) with SYBR Green I (Molecular Probes, Eugene, OR) as a fluorescent reporter. Threshold cycle number of duplicate reactions was determined using the Opticon software. Levels of selected gene mRNA expression were normalized to hypoxanthine phsophoribosyltransferase (HPRT) levels using the formula 2(Rt–Et); where Rt is the mean threshold cycle for the reference gene HPRT and Et is the mean threshold cycle for the experimental gene. Data are presented as arbitrary units and fold changes are adjusted to the non-stimulated control cells. Primer sequences are provided in supplementary Table 1.
Immunofluorescence staining
To analyze cellular distribution of E-cadherin and vimentin, BEAS-2B cells (1 × 104) treated with or without Cr(VI) at 0.5 μM for 6 weeks were seeded on 8-well chamber slides (Nunc, Rochester, NY), fixed with 1% formaline, permeabilized with Triton X-100 and probed with E-cadherin and vimentin antibodies. The cells were subsequently incubated with secondary-Alexa Fluor 488-conjugated antibody (Molecular Probes, OR). Cells were then washed, and visualized using Zeiss Axio Observer inverted immunofluorescence microscope (Carl Zeiss MicroImaging GmbH, Gottingen, Germany).
Cell migration and invasion assay
BEAS-2B cells cultured in 100 mm dishes were treated with or without Cr(VI) at 0.5 μM for 6 weeks. For trans-well migration assay, 5 × 104 cells were seeded to the top chambers of 24-well trans-well plates insert (8.0 μM pore size membrane, BD, Frankline Lakes, NJ), cells were fixed, stained, and counted by 24 hours in the bottom (migrated) chamber. For scratch wound closure assays, cells seeded in 6-well culture plate (1 × 106/per well) 24 h prior to the wound was incised in the central area using a pipet tip, detached cells were washed away and cell migration was evaluated 24 hours post wounding. For matrigel invasion assay, cells (1 × 105) were seeded to the top chambers of 24-well trans-well plates insert (BD), the insert were coated with a thin layer of matrigel (20 μl) and incubated for 24, 48 and 72 hours. Cells in top chamber (non-migrated) were removed, and cells on bottom of filter insert (migrated) were fixed, stained with paraformaldehyde-ethanol-crystal violate solution and counted under microscope. Individual experiment was performed in duplicate and repeated 3 times.
siRNA transfection
BEAS-2B cells treated with or without Cr(VI) at 0.5 μM for three weeks, siRNAs for HDAC1, 2, 3 (Ambion/Life Technologies Corp., Carlsbad, CA) were transfected to cell with Lipofectamine 2000 in Opti-MEM1 media following the manufacturer recommended protocol. siRNAs final concentration for HDAC1, 2, 3 (Catalog No. s73, s6493, s16878) and the negative control was 10 nM. Fourty-eight hours post-transfection, cytosolic and nuclear protein was extracted for Western blot analysis as mentioned above.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (Ding et al., 2010). Briefly, BEAS-2B cells (5 × 106) treated with or without Cr(VI) at 0.5 μM for 6 weeks were plated in 150 mm dish, washed three times with PBS, and cross-linked with 1% formaldehyde (Sigma) for 10 minutes. Cells were harvested in cell lysis buffer, and sonicated with 6 × 10 second pulses to generate 0.4-1.2 Kb size. The sonicated chromatin was then split equally into four parts; one part served as control, the other 3 parts were immunoprecipitated with rabbit polyclonal HDAC1, H3K9Ac antibodies, and non-immune isogenic IgG respecitvely, at 4°C for 12 hours. DNA/protein complexes were captured by Protein A agarose beads mixed with salmon sperm DNA (Millipore). The samples were then washed, eluted in SDS elution buffer, and the cross-links reversed by overnight incubation at 65°C. DNA was purified using Qiaquick PCR purification kit (Qiagen). Quantitative real-time PCR was performed from input and ChIP material. DNA content in immunoprecipitation (IP) samples was measured relative to the total input and/or housekeeping gene GAPDH level. Data are presented as ratio of IP/GAPDH in each condition and adjusted as fold changes of no antibody control. Non-immune isogenic IgG was used as monitoring control. Primers for both ChIP and RT-PCR are listed in supplementary Table 1.
Colony formation assay
To measure anchorage-independent growth in soft agar, BEAS-2B and BEAS-2B-catalase-stable expressing cells (2.5 × 103) were treated with or without Cr(VI) at 0.5 μM for 8 weeks. Cells were suspended in 0.3% agar in DMEM supplemented with 1% penicillin/streptomycin, 10% FBS and overlaid on 0.5% agar in the same medium in six-well plates for additional 4-12 weeks to allow colony formation. Colonies were stained with 0.005% crystal violet, counted and photographed with a dissection microscope.
Statistical analysis
All quantitative data are presented as mean ± SEM. Paired or unpaired Student “t” test was used for intergroup comparisons, and differences were considered significant if P values <0.05.
Results
Chromium represses E-cadherin, enhances vimentin and differentially regulates E-cadherin suppressor expression in BEAS-2B cells
To characterize the role of chromium in inducing lung epithelial cell pathophysiology, we first incubated BEAS-2B cell at different doses for various periods of time and observed the cell viability and growth. The initial results indicated that Cr(VI) at the dose above 1 μM resulted in significant cell death and cell cycle arrest during prolonged exposure as reported previously (O'Hara et al., 2007; Costa et al., 2010) and data not shown. We therefore incubated low dose of Cr(VI) at 0.25, 0.5 μM with BEAS-2B cells as indicated in Figure 1. The results showed that in short time exposure from 10 h (Fig. 1A) to 1 week (Fig. 1B), Cr(VI) did not significantly repress E-cadherin expression even at higher doses. Cr(VI) enhances E-cadherin repressor Snail expression at 10 hours, but this effect disappeared after longer time incubation by four days and one week (Fig. 1A, B). Another E-cadherin repressor, Twist protein expression was not changed (data not shown).
Figure 1. Expression of E-cadherin and its suppressor after acute chromium exposure.
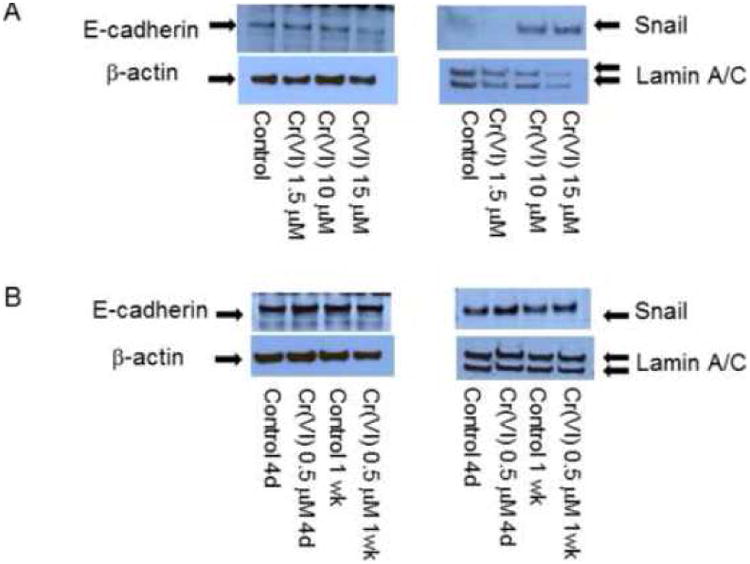
BEAS-2B cells (1 × 106) were treated with medium alone, Cr(VI) at 1.5, 10, 15 μM for 10 hours (A) and 0.5 μM for up to 1 week (B). Cells were washed, cytosolic and nuclear proteins were extracted and separated on 10% SDS-polyacrylamide gel to detect E-cadherin (E-cad) or Snail protein expression. Blots are representative of three separate experiments with similar results, arrows indicate specific band.
As Cr(VI)-BEAS-2B cell co-incubation proceed, we noticed reduced E-cadherin expression and enhanced vimentin expression by third week and throughout the ten weeks study period, but not earlier. Therefore, this time frame was used for all later experiment to study E-cadherin/vimentin expression as well as related cellular events. Fig. 2 A-C indicated representative blots of Cr(VI)-induced E-cadherin repression and vimentin up-regulation from three to ten weeks in BEAS-2B cells, and this effect was Cr(VI) dose-dependent (Fig. 2C). Meanwhile, both Snail and Twist protein expression were surprisingly decreased (Fig. 2D). The presence of serum had no effect on Cr(VI)-induced E-cadherin repression, but affected vimentin protein expression (Fig. 2B). Therefore all subsequent experiments to test vimentin protein expression were performed by replacing the media with serum-free media 24 hours prior to cell lysate collection.
Figure 2. Chronic chromium exposure represses E-cadherin and enhances vimentin protein expression.
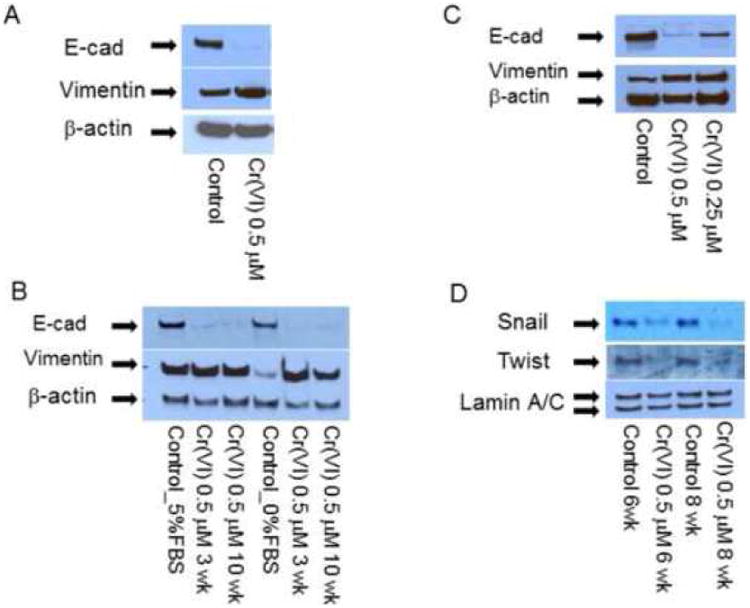
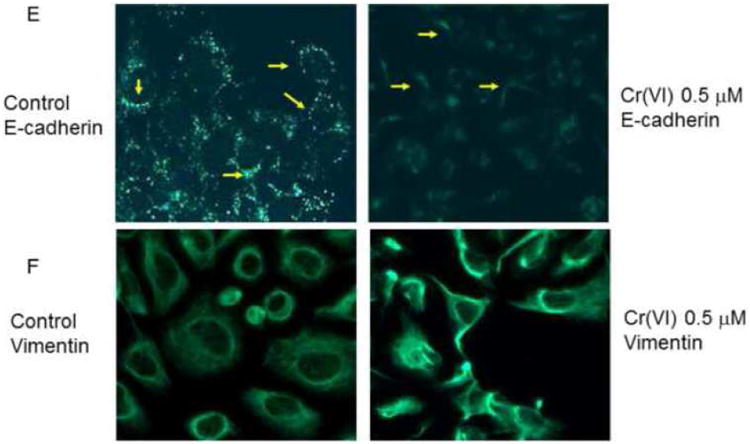
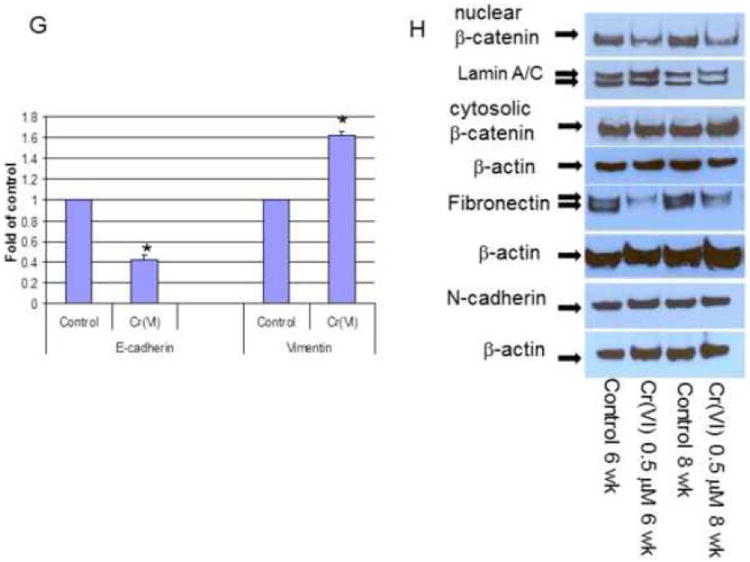
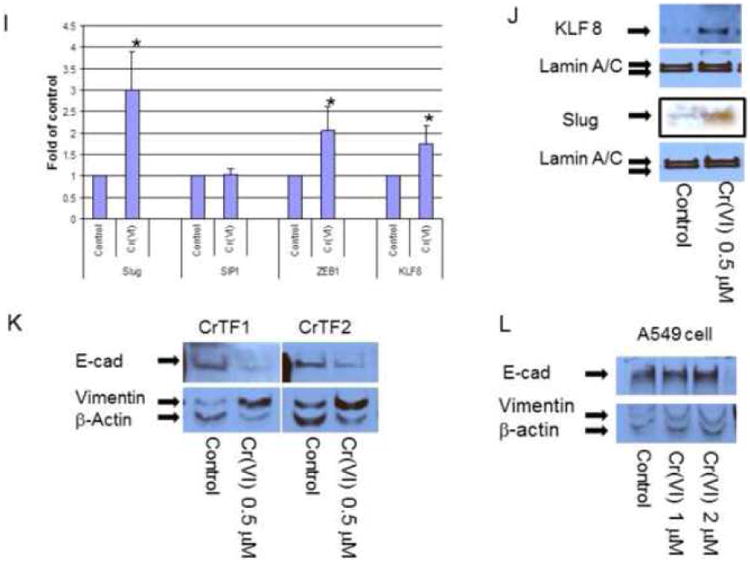
To evaluate E-cadherin related protein expression, BEAS-2B cells were treated with or without Cr(VI) at 0.5 μM for 3-10 weeks (A-J). Cytosolic and nuclear proteins were extracted and separated on 10% SDS-polyacrylamide gel to detect E-cadherin (E-cad), vimentin, Snail, Twist, β-catenin, N-cadherin, fibronectin, krupple-like factor 8 (KLF 8) and Slug protein expression (A-D, H, J). Blots are representative of three separate experiments with similar results, arrows indicate specific band. For immunofluorescence staining of E-cadherin and vimentin, cells were seeded at 1 × 104 in 8 well chamber, fixed with 1% formalin, washed and stained with E-cadherin and vimentin antibody overnight at 4°C. After washing and secondary antibody conjugation, cells were visualized with immunofluorescence microscope (E, F). For gene expression assay, total RNA was collected from above cells and real-time PCR was performed to detected E-cadherin, vimentin, Slug, SIP1, ZEB1 and KLF8 mRNA expression (G, I). Data are mean±SEM from three separate experiments, *P<0.01 when compared with controls. To determine E-cadherin and vimentin protein expression in tumor cells, CrTF1, CrTF2 and A549 cells were treated with or without Cr(VI) at 0.5 μM for 3 weeks, cell lysates were extracted to detect E-cadherin and vimentin protein expression (K, L) as described above.
Immunofluorescence staining using E-cadherin and vimentin antibody also confirmed the disappearance of E-cadherin in the cell adherent junctions and enhanced overall vimentin expression in BEAS-2B cells (Fig. 2E-F).
To further examine if Cr(VI)-induced E-cadherin and vimentin protein expression also reflect their mRNA expression level, we performed real-time PCR, and confirmed the similar pattern of alterations at mRNA level (Fig. 2G), suggesting transcriptional repression/up-regulation mechanism.
In order to check if these changes could also happen to other EMT markers in BEAS-2B cells such as β-catenin, N-cadherin, fibronectin (Thiery and Sleeman, 2006), and EMT suppressors such as Slug (Bolos et al., 2003), SIP1 (Comijn et al., 2001), ZEB1 (Onder et al., 2008) and KLF8 (Wang et al., 2007), Western blot or real-time PCR were performed to detect their protein or mRNA expression. The results revealed that there were no obvious changes in N-cadherin, but reduced fibronectin and nuclear β-catenin expression (Fig. 2H), and increased Slug, ZEB1 and KLF8 mRNA expression (Fig. 2I). Among them, Slug and KLF8 mRNA expression were confirmed by Western blot analysis (Fig. 2J).
Furthermore, to evaluate if these changes might also occur after transformation or in tumor cells, CrTF1, CrTF2 and A549 cells were treated with Cr(VI) for 3 weeks, and the cell lysates were analyzed by Western blot, we detected similar E-cadherin repression and vimentin up-expression in CrTF1, CrTF2 cells, but not in A549 cell (Fig. 2K, L).
BEAS-2B cell morphological changes, migration and invasion induced by chromium
Prolonged co-incubation of Cr(VI) with BEAS-2B cells resulted in a number of cell morphological changes; cells lost their original cobblestone-like appearance, had loose intercellular contact and increased intercellular space when compared with control cells (Fig. 3A). By six weeks but not earlier, these cells started to show fibroblastoid morphology, presented as elongated, spindle-shape appearances (Fig. 3B). By eight weeks, they started scattering and drafted toward all directions randomly (Fig 3C, suppl. Fig. 1A, B).
Figure 3. Chromium induces BEAS-2B cell sequential morphological changes.
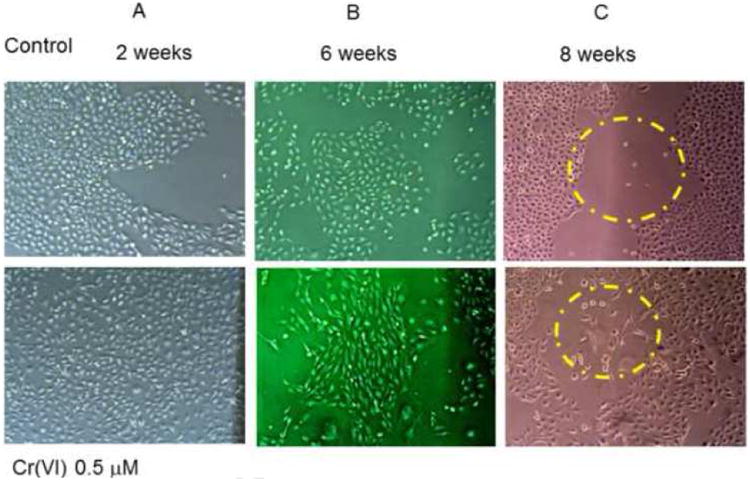
BEAS-2B cells cultured in 100 mm dishes were treated with or without Cr(VI) at 0.5 μM for 3, 6 and 8 weeks. Cells growth and morphology changes were monitored and recorded under microscope throughout the experimental period. Photos are representative of 3-5 sets assays.
Since Cr(VI) exposure reduces E-cadherin expression, enhances vimentin expression, and alters the cell morphology from cobblestone shape into fibroblastoid morphology, we wondered if this might also accompany with enhanced cell migration and invasion. Figure 3 (C) and supplementary Fig. 1 (A-C) showed the cell migration patterns. Unlike controls, Cr(VI)-treated cell showed enhanced tendency of cell scattering and occupied more spaces. These cells also grew slower, migrated randomly toward different directions, lost cell-cell contact, and finally formed a loose cell mass (Suppl. Fig. 1A, B). However, trans-well migration assay and scratch wound closure assay did not show enhanced cell migration in Cr(VI)-treated cells over the control cells (data not shown). Second, to evaluate if these cells might invade matrigel, equal number of cells were seeded into matrigel chamber, and kept growing for 24, 48 and 72 hours to examine the invasion. The results indicated that both cells had very little invasion in matrigel assay at 24, 48 hours, while Cr(VI)-treated cell showed much more invasion by 72 hours (Fig. 4 A, B). Third, owning the invading cells are usually accompanied with enhanced matrix enzyme activity, such as increased MMP9 expression (Nawrocki-Raby et al., 2003), we further analyzed if Cr(VI)-treated cell might show enhanced expression of MMP9. As expected, Western blot assay confirmed that Cr(VI)-treated cell expressed much higher level of MMP9 protein over the control cells (Fig. 4C). Together, these results demonstrate that Cr(VI) promotes BEAS-2B cells fibroblastoid morphological change and invasion, a typical process described as epithelial-mesenchymal transition (EMT).
Figure 4. Chromium induces BEAS-2B cell invasion.
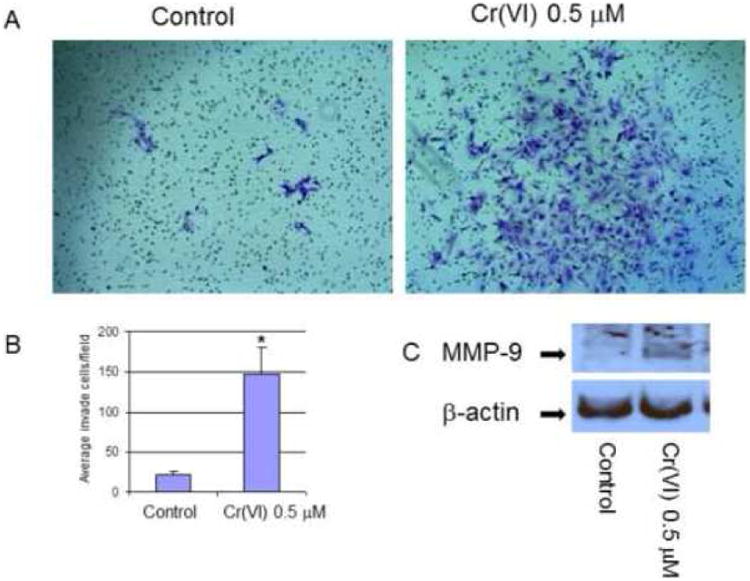
For invasion assay, cells were treated with or without Cr(VI) at 0.5 μM for 6 weeks. 1 × 105 cells from each group were seeded on the top chamber of 24-well plate culture inserts coated with 20 μl of matrigel in duplicate. Cells were cultured for additional 72 hours, invaded cells on the bottom of insert were stained, photographed, and counted (A, B). Quantitative results were compared with control cells without Cr(VI) treatment, *P<0.01 when compared with control (B). For MMP-9 expression assay (C), cytosolic proteins were extracted from cells that were treated as mentioned above, 20 μg of proteins were separated on 10% SDS-polyacrylamide gel to detect MMP-9 expression, anti-β-actin antibody was probed to monitor protein loading. Blots are representative of three separate experiments with similar results, arrows indicate specific band.
Effects of HDAC inhibition on E-cadherin and vimentin protein expression
Knowing that Cr(VI) represses BEAS-2B cell E-cadherin, enhances vimentin expression, promotes invasion, we intended to find out the underlying molecular mechanisms.
First, we used a HDAC inhibitor, TSA to inhibit HDAC expression, and DNMT inhibitor AZA to block DNA methylation and observed E-cadherin/vimentin expression. The results showed that TSA dramatically reversed Cr(VI)-induced E-cadherin repression and vimentin up-regulatioin (Fig. 5A). This effect was also obvious in TSA control cells and suggested HDAC may be an important mechanism in regulating E-cadherin expression. In contrast, AZA had no effect on Cr(VI)-induced E-cadherin/vimentin expression (Fig. 5B).
Figure 5. HDAC1 inhibition on Cr(VI)-induced E-cadherin and vimentin expression.
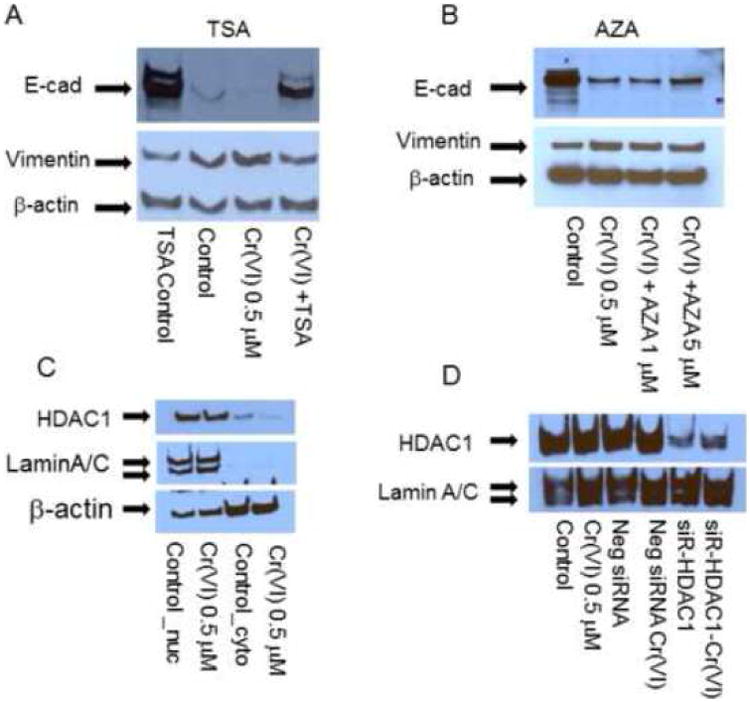
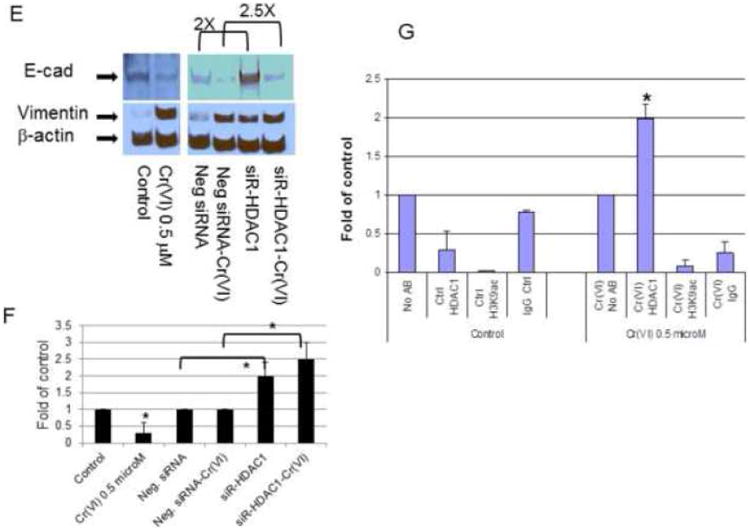
For inhibitor assay, BEAS-2B cells (5 × 105) treated with or without Cr(VI) at 0.5 μM for 6 weeks were seeded in 60 mm dishes. Trichostatin A (TSA, 0.1 μM), 5-aza-2′-deoxycytidine (AZA, 1-5 μM) were added for 24 hours prior to cellular protein extraction to evaluate E-cadherin (E-cad)/vimentin protein expression (A, B). For siRNA transfection assay (C-F), BEAS-2B cells (2.5 × 105) were treated as mentioned above and seeded in 6-well plates. siRNA for HDAC1 and negative control (siR-HDAC1, Neg siRNA) were transfected with Lipofectamine 2000. Nuclear and cytosolic proteins were extracted 48 hours post transfection to evaluate E-cadherin, vimentin protein expression. Blots are representative of three separate experiments with similar results, arrows indicate specific band. Densitometry data are expressed as arbitrary unit and adjusted as fold changes over the control (Fig. 5F), *P<0.05 when compared with their respective controls. ChIP assay and quantitative RT-PCR were performed as described in Materials and Methods. qRT-PCR were performed from input and ChIP material (IP) to detected the relative promoter binding of HDAC1 in E-cadherin promoter, GAPDH served as control. Data represent IP/GAPDH ratio and are adjusted as fold changes over no antibody control (No Ab). Results are mean±SEM from 3 separate experiments, *P<0.01 when compared to the controls (G).
Second, since TAS is a non-specific inhibitor for Class 1 HDAC which includes HDAC1, HDAC2, HDAC3, HDAC8, we therefore used siRNA to knockdown HDAC1, HDAC2, HDAC3 expression and examined E-cadherin expression pattern. The results indicated that Cr(VI) did not affect the overall HDAC1 nuclear protein level (Fig. 5C). siRNA HDAC1 dramatically inhibited HDAC1 expression (Fig. 5D), and enhanced E-cadherin expression in both control and Cr(VI)-treated cells (Fig. 5E), significant difference were noticed when the scanned densitometry data were analyzed (Fig. 5F), P<0.05 when compared with their respective controls. Knocking down HDAC 2, 3 had no effect on E-cadherin expression (suppl. Fig. 2).
Third, to confirm the presence of HDAC1 in E-cadherin gene promoter, we performed CHIP assay to see if there is enhanced HDAC1 binding to E-cadherin promoter in Cr(VI) treated cells. The results (Fig. 5G) clearly showed that binding of HDAC1 in E-cadherin promoter in Cr(VI)-treated cells increased by two folds, however we could not detect H3K9Ac binding to E-cadherin promoter, and H3K9Ac, HDAC1 binding to vimentin promoter (data not shown). Therefore, these results indicated enhanced binding of HDAC1 in E-cadherin promoter and contributed to Cr(VI)-induced E-cadherin repression. Owning AZA had no effect, DNA methylation is unlikely responsible for the loss of E-cadherin expression. The results suggest that HDAC1 plays a critical role in regulating E-cadherin and vimentin gene expression, and this effect is most likely promoter-specific effect in lung epithelial cells.
Catalase reduces Cr(VI)-induced EMT protein expression and invasion
Cr(VI) is characterized by it powerful capability to generate ROS in host cells (Shi et al., 1994), we asked if ROS might be a possible reason for the loss of E-cadherin, enhanced vimentin expression and contribute to EMT and invasion. We used Catalase-stable expressing cell line to see if they might block Cr(VI)-induced E-cadherin suppression and vimentin up-regulation. The results in Fig. 6 showed that catalase-stable expressing BEAS-2B cells effectively attenuated Cr(VI)-induced E-cadherin repression and vimentin up-regulation (Fig. 6A) and reduced cell invasion in matrigel (Fig. 6 B-D). Because catalase-stable cell line itself showed a fibroblast-like morphology (data not shown), we could not differentiate the effect of catalase stable expression and Cr(VI)-induced cell morphological changes. Taken together, these results point out catalase/ROS-mediated effects in Cr(VI)-induced E-cadherin, vimentin protein expression in EMT and invasion processes.
Figure 6. Catalase reduces Cr(VI)-induced E-cadherin suppression, invasion and oncogenic transformation in BEAS-2B cells.
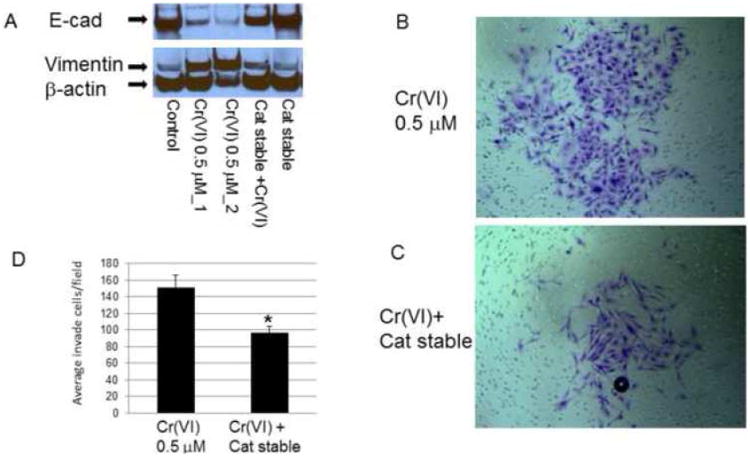
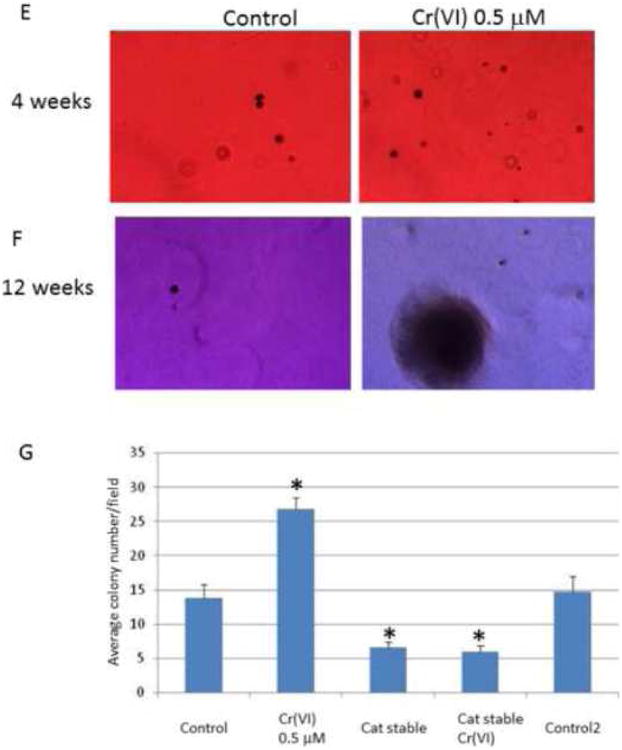
For protein expression assay, BEAS-2B and BEAS-2B-catalase-stably expressing (Cat stable) cells (1 × 106) were treated with or without Cr(VI) for 3 weeks. Cytosolic and nuclear proteins were extracted and separated on 10% SDS-polyacrylamide gel to determine E-cadherin (E-cad) and vimentin expression (A). Blots are representative of three separate experiments with similar results, arrows indicate specific band. For invasion assay (B-D), BEAS-2B and BEAS-2B-catalase-stably expressing (Cat stable) cells were treated with or without Cr(VI) 0.5 μM for 6 weeks and seeded in matrigel chamber as described in Fig 4. For colony formation assay, BEAS-2B and BEAS-2B-catalase-stably expressing (Cat stable) cells (2.5 × 103) (E-G) treated with or without Cr(VI) for 8 weeks, and plated in 0.35% soft agar in duplicate in 10% FBS DMEM media for additional 4-12 weeks to allow colony formation. Colony numbers from 12 weeks group was determined in each well and compared with controls (F). Data are mean±SEM from three separate experiments, *P<0.01 when compared with controls.
Catalase reduces Cr(VI)-induced oncogenic transformation in soft agar colony formation assay
To further understand if chronic and long term incubation of Cr(VI) might induce epithelial cell oncogenic transformation and the role of ROS during this process, we used soft agar assay to investigate if catalase might reduce colony formation, a process that correlates with in vivo oncogenic transformation. Shown in Fig. 6 (E-G) are the representative colony formation in control and Cr(VI)-treated BEAS-2B cells. Fig. 6G is the quantitative data when the number of colony was counted by twelve weeks in different treatment groups. Significantly enhanced colony formation was noticed when the colony number in Cr(VI)-treated groups was compared with controls. In addition, colony number was lower in catalase-stable expressing cells versus control groups (*p<0.01) regardless of Cr(VI) treatment. These results indicate that catalase/ROS play a critical role in Cr(VI)-induced oncogenic transformation, which are also in line with Cr(VI)-induced EMT and invasion processes, and imply a catalaes/ROS-mediated mechanism.
Discussion
The present study describe the primary roles of Cr(VI)-induced morphological change, EMT, invasion and colony formation in lung epithelial cells during oncogenic transformation, and these effects appear to be catalae/ROS-mediated. Accumulating evidence has indicated that chronic inhalation of certain Cr(VI) compounds increase the risk in human lung cancer (Gibb et al., 2000; O'Brien et al., 2003). Cr(VI) compounds also induce inflammatory response and cancer in animal models (Beaver et al., 2009). The mechanism has been attributed to its effect in activating oncogenic pathways and inducing DNA damage (O'Hara et al., 2007; Wise et al., 2008). Different forms of chromium, such as zinc, lead, strontium chromate also induce neoplastic transformation (Wise et al., 2006).
Recently, two groups have reported that mildly cytotoxic Cr(VI) exposure leads to BEAS-2B cells morphological change, partial loss of contact inhibition, increased resistance to Cr(VI)-induced apoptosis and transformation (Xie et al., 2007; Costa et al., 2010). Cr(VI) also interferes cell adhesion (Costa et al., 2010). These initial results provide excellent phenomenal description implicating that Cr(VI) may disturb cell polarity. In this study, we advance these observations by showing that Cr(VI) is able to induce EMT and invasion during oncogenic transformation. Additionally, Catalase can prevent some of these effects. Although we are unable to predict Cr(VI)-induced extensive genomic alternations during oncogenic transformation, the present and previously studies provide clue that Cr(VI) causes extensive gene structure alternation which ultimately result in the formation of tumorigenic cell clones.
E-cadherin is an important epithelial cell adhesion molecule, maintains normal cell:cell adherent junctions, cell polarity and closely associated with cancer cell invasion, metastasis and poor patients prognosis (Umbas et al., 1994; Berx et al., 1995; van Roy and Berx, 2008). Loss of E-cadherin is one hallmark for EMT and triggers the activation of specific downstream signal pathways that facilitate later steps of metastasis (Onder et al., 2008). Morphological change, EMT, invasion and transformation described in this work probably reflected the enormous cell genomic re-arrangement during oncogenic transformation induced by Cr(VI). Recent concept for early metastasis and parallel evolution theory of primary and metastatic tumor have indicated that propensity of cell metastasis is determined early in the neoplastic process, rather than near its end stage and not dependent on the new genetic abnormalities that occur after tumor have been established (Gray, 2003; Hunter, 2004). We reason that at the time when Cr(VI)-treated cells re-organize their genome to become a tumor, or during transformation, cells also acquire EMT and invasive properties. Meanwhile, numerous other cell signaling may also be altered, such as microarray detected gene expression changes and resistant to apoptosis.
In the current work, EMT occurs prior to the time that cells were put onto the soft agar to exam the colony formation. In fact, it is not clear if EMT and oncogenic transformation might be two independent events which occurred parallel to each other; or they are one sequential event that was detected at different time in the same cell population, namely EMT earlier and transformation later. Based on the above discussion, we are prone to the latter hypothesis; however, future studies are required to address these complex issues.
A number of mechanisms have been suggested for CDH1 gene (which encodes E-cadherin protein) repression or silencing. These include activation of transcription factors such as Snail (Batlle et al., 2000), Twist (Yang et al., 2004), Slug (Bolos et al., 2003), ZEB1 (Pena et al., 2005), SIP1 (Comijn et al., 2001), KLF8 (Wang et al., 2007), expression of certain miRNA (Ma et al., 2010), and DNA hypermethylation in its gene promoter (Strathdee, 2002). Several cytokines or growth factors also cause EMT and activation of these transcription factors (Thiery and Sleeman, 2006). In human tumors, somatic mutation, chromosomal deletion, proteolytic cleavage, and silencing of CDH1 promoter are common causes for its repression (Strathdee, 2002). However, metal compound chromium has not been shown to cause E-cadherin repression, EMT and invasion prior to the current study.
Exactly how Cr(VI) represses E-cadherin transcription is awaiting for further clarification, the present work have identified several transcription repressors that are activated by Cr(VI), including Slug, ZEB1 and KLF8. We do not rule out any miRNA (Ma et al., 2010) that might also be induced by Cr(VI), and potentially represses E-cadherin expression. In addition, we show that Cr(VI) is able to enhance HDAC1 binding in E-cadherin gene promoter and possibly keeps the gene in a deacetylated state, all of which may contribute to its transcriptional repression, and their combination may exert maximal effects.
One interesting question is how could Cr(VI) exposure leads to cell reprogram its genome and results in the repression of E-cadherin and meanwhile activation of vimentin, a mesenchymal marker. Eepigenetic alternations induced by Cr(VI) and ROS appear to be critical for this effect, as artificially modify the HDAC1 expression by TSA or siRNA also alter E-cadherin/vimentin expression, and catalase partially blocks these phenotypes. A detailed study on the chromatin structure changes induced by Cr(VI) will definitely help to delineate the molecular mechanisms.
HDAC1 is found in a number of multiprotein complexes and regulates gene transcription, including transcriptional corepressors that lead to hypoacetylation of histones and transrepression of target genes (Lee et al., 2000). It also represses estrogen-related receptor alpha gene transactivation (Matsuyama et al., 2010), and negatively regulates C/EBPdelta-dependent haptoglobin expression in intestinal epithelial cells (Turgeon et al., 2008). A recent study show that Cr(VI) is able to retain HDAC1 binding to chromatin, and form EDTA-reversible, chromatin-HDAC1 crosslinks, which result in Cyp1a1 gene in silent states (Schnekenburger et al., 2007b). Our results indicate E-cadherin repression is also associated with HDAC1 in its gene promoter, and knockdown HDAC1 with siRNA or by its inhibitor TSA both reverse E-cadherin expression, therefore, suggest an unappreciated mechanism in Cr(VI)-induced E-cadherin transcription control.
The current concept that Cr(VI) is mostly an inducible gene repressor, which represses many inducible gene transcriptions but does not affect constitutive gene expression (Borges and Wetterhahn, 1989; McCaffrey et al., 1994; Majumder et al., 2003; Wei et al., 2004). These observations can be explained by its effects on DNA cross-link, alteration of chromatin structure, and disruption of gene expression. Our results reveal that expression of a number of transcriptional factors such as Snail, Twist, β-catenin and NF-κB p65 (data not shown) are repressed, which are in line with these concepts. However, we also note a few genes that their transcriptions are activated, such as Slug, ZEB1, and KLF8. These results therefore provide additional evidence arguing that inhibition of inducible gene expression is probably a promoter specific event in the context of Cr(VI) chronic exposure. In those genes which are not corss-linked by Cr(VI), gene transcription may proceed after proper stimulation. In the current study, vimentin is the only mesenchymal marker induced by Cr(VI). Other markers such as N-cadherin or fibronectin either has no change or even has reduced expression, and this phenomenon has been described as partial EMT (Leroy and Mostov, 2007) and differs from classical EMT, in which all mesenchymal markers change concurrently. Partial EMT is also common for cells that do not acquire full EMT markers (Johnen et al., 2012; Roxanis, 2013), owning the special character of Cr(VI) in cross-linking DNA, we consider this might be a cell type or promoter specific event, as A549 cells do not have the similar responses. Future works are required to explore the mechanisms of EMT in these cells.
One expected result from our observation is that catalase is able to abolish the oncogenic transformation in soft agar analysis. ROS are known to mediate numerous cellular events that contribute to the carcinogenesis (Liou and Storz, 2010). Since Cr(VI) is a major ROS producer and catalase scavenging ROS, therefore, these results indicate ROS-dependent mechanism. Yet, one unexpected result from our study is that Cr(VI) does not potentiate cell migration, despite typical induction of EMT is accompanied with increased cell migration and invasion, the reason is not clear. Since Cr(VI) inhibits cell proliferation and represses inducible gene expression, we hypothesis that Cr(VI) treatment may hamper the cell migration related gene expression and cytoskeletal rearrangements, therefore interferes the cell migratory systems. Further studies are warranted to explore molecular mechanisms related to these discrepancies.
In conclusion, the present work show that Cr(VI) induces EMT with enhanced cell invasion during oncogenic transformation. Catalase reduces Cr(VI)-induced EMT protein expression, invasion, and attenuates oncogenic transformation in human lung epithelial cells. These observations provide insight in chromium-induced carcinogenic mechanism. Future investigation on chromium-induced host cell damage will allow us better understanding its pathogenesis and develop strategy for cancer prevention.
Supplementary Material
Highlight.
We study if Cr(VI) might induce EMT and invasion in epithelial cells.
Cr(VI) induces EMT by altering E-cadherin and vimentin expression.
It also increases cell invasion and promotes oncogenic transformation.
Catalase reduces Cr(VI)-induced EMT, invasion and transformation.
Acknowledgments
This work was supported in part by NIH grants (1R01CA119028).
Footnotes
Conflict of interest: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- Beaver LM, Stemmy EJ, Constant SL, Schwartz A, Little LG, Gigley JP, Chun G, Sugden KD, Ceryak SM, Patierno SR. Lung injury, inflammation and Akt signaling following inhalation of particulate hexavalent chromium. Toxicol Appl Pharmacol. 2009;235:47–56. doi: 10.1016/j.taap.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, van Roy F. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 1995;14:6107–6115. doi: 10.1002/j.1460-2075.1995.tb00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci. 2003;116:499–511. doi: 10.1242/jcs.00224. [DOI] [PubMed] [Google Scholar]
- Borges KM, Wetterhahn KE. Chromium cross-links glutathione and cysteine to DNA. Carcinogenesis. 1989;10:2165–2168. doi: 10.1093/carcin/10.11.2165. [DOI] [PubMed] [Google Scholar]
- Ceryak S, Zingariello C, O'Brien T, Patierno SR. Induction of pro-apoptotic and cell cycle-inhibiting genes in chromium (VI)-treated human lung fibroblasts: lack of effect of ERK. Mol Cell Biochem. 2004;255:139–149. doi: 10.1023/b:mcbi.0000007270.82431.3e. [DOI] [PubMed] [Google Scholar]
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–1278. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- Costa AN, Moreno V, Prieto MJ, Urbano AM, Alpoim MC. Induction of morphological changes in BEAS-2B human bronchial epithelial cells following chronic sub-cytotoxic and mildly cytotoxic hexavalent chromium exposures. Mol Carcinog. 2010;49:582–591. doi: 10.1002/mc.20624. [DOI] [PubMed] [Google Scholar]
- Ding SZ, Fischer W, Kaparakis-Liaskos M, Liechti G, Merrell DS, Grant PA, Ferrero RL, Crowe SE, Haas R, Hatakeyama M, Goldberg JB. Helicobacter pylori-induced histone modification, associated gene expression in gastric epithelial cells, and its implication in pathogenesis. PLoS One. 2010;5:e9875. doi: 10.1371/journal.pone.0009875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding SZ, O'Hara AM, Denning TL, Dirden-Kramer B, Mifflin RC, Reyes VE, Ryan KA, Elliott SN, Izumi T, Boldogh I, Mitra S, Ernst PB, Crowe SE. Helicobacter pylori and H2O2 increase AP endonuclease-1/redox factor-1 expression in human gastric epithelial cells. Gastroenterology. 2004;127:845–858. doi: 10.1053/j.gastro.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Gibb HJ, Lees PS, Pinsky PF, Rooney BC. Lung cancer among workers in chromium chemical production. Am J Ind Med. 2000;38:115–126. doi: 10.1002/1097-0274(200008)38:2<115::aid-ajim1>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Gray JW. Evidence emerges for early metastasis and parallel evolution of primary and metastatic tumors. Cancer Cell. 2003;4:4–6. doi: 10.1016/s1535-6108(03)00167-3. [DOI] [PubMed] [Google Scholar]
- Hunter KW. Host genetics and tumour metastasis. Br J Cancer. 2004;90:752–755. doi: 10.1038/sj.bjc.6601590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnen N, Francart ME, Thelen N, Cloes M, Thiry M. Evidence for a partial epithelial-mesenchymal transition in postnatal stages of rat auditory organ morphogenesis. Histochem Cell Biol. 2012;138:477–488. doi: 10.1007/s00418-012-0969-5. [DOI] [PubMed] [Google Scholar]
- Lee SK, Kim JH, Lee YC, Cheong J, Lee JW. Silencing mediator of retinoic acid and thyroid hormone receptors, as a novel transcriptional corepressor molecule of activating protein-1, nuclear factor-kappaB, and serum response factor. J Biol Chem. 2000;275:12470–12474. doi: 10.1074/jbc.275.17.12470. [DOI] [PubMed] [Google Scholar]
- Leroy P, Mostov KE. Slug is required for cell survival during partial epithelial-mesenchymal transition of HGF-induced tubulogenesis. Mol Biol Cell. 2007;18:1943–1952. doi: 10.1091/mbc.E06-09-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010;44:479–496. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KJ, Jiang J, Swartz HM, Shi X. Low-frequency EPR detection of chromium(V) formation by chromium(VI) reduction in whole live mice. Arch Biochem Biophys. 1994;313:248–252. doi: 10.1006/abbi.1994.1384. [DOI] [PubMed] [Google Scholar]
- Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247–256. doi: 10.1038/ncb2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder S, Ghoshal K, Summers D, Bai S, Datta J, Jacob ST. Chromium(VI) down-regulates heavy metal-induced metallothionein gene transcription by modifying transactivation potential of the key transcription factor, metal-responsive transcription factor 1. J Biol Chem. 2003;278:26216–26226. doi: 10.1074/jbc.M302887200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama R, Takada I, Yokoyama A, Fujiyma-Nakamura S, Tsuji N, Kitagawa H, Fujiki R, Kim M, Kouzu-Fujita M, Yano T, Kato S. Double PHD fingers protein DPF2 recognizes acetylated histones and suppresses the function of estrogen-related receptor alpha through histone deacetylase 1. J Biol Chem. 2010;285:18166–18176. doi: 10.1074/jbc.M109.077024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- McCaffrey J, Wolf CM, Hamilton JW. Effects of the genotoxic carcinogen chromium(VI) on basal and hormone-inducible phosphoenolpyruvate carboxykinase gene expression in vivo: correlation with glucocorticoid- and developmentally regulated expression. Mol Carcinog. 1994;10:189–198. doi: 10.1002/mc.2940100403. [DOI] [PubMed] [Google Scholar]
- Nawrocki-Raby B, Gilles C, Polette M, Martinella-Catusse C, Bonnet N, Puchelle E, Foidart JM, Van Roy F, Birembaut P. E-Cadherin mediates MMP down-regulation in highly invasive bronchial tumor cells. Am J Pathol. 2003;163:653–661. doi: 10.1016/S0002-9440(10)63692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickens KP, Patierno SR, Ceryak S. Chromium genotoxicity: A double-edged sword. Chem Biol Interact. 2010;188:276–288. doi: 10.1016/j.cbi.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien T, Xu J, Patierno SR. Effects of glutathione on chromium-induced DNA crosslinking and DNA polymerase arrest. Mol Cell Biochem. 2001;222:173–182. [PubMed] [Google Scholar]
- O'Brien TJ, Ceryak S, Patierno SR. Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res. 2003;533:3–36. doi: 10.1016/j.mrfmmm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- O'Hara KA, Vaghjiani RJ, Nemec AA, Klei LR, Barchowsky A. Cr(VI)-stimulated STAT3 tyrosine phosphorylation and nuclear translocation in human airway epithelial cells requires Lck. Biochem J. 2007;402:261–269. doi: 10.1042/BJ20061427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- Pena C, Garcia JM, Silva J, Garcia V, Rodriguez R, Alonso I, Millan I, Salas C, de Herreros AG, Munoz A, Bonilla F. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: clinicopathological correlations. Hum Mol Genet. 2005;14:3361–3370. doi: 10.1093/hmg/ddi366. [DOI] [PubMed] [Google Scholar]
- Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- Roxanis I. Occurrence and significance of epithelial-mesenchymal transition in breast cancer. J Clin Pathol. 2013 doi: 10.1136/jclinpath-2012-201348. [DOI] [PubMed] [Google Scholar]
- Schnekenburger M, Peng L, Puga A. HDAC1 bound to the Cyp1a1 promoter blocks histone acetylation associated with Ah receptor-mediated trans-activation. Biochim Biophys Acta. 2007a;1769:569–578. doi: 10.1016/j.bbaexp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnekenburger M, Talaska G, Puga A. Chromium cross-links histone deacetylase 1-DNA methyltransferase 1 complexes to chromatin, inhibiting histone-remodeling marks critical for transcriptional activation. Mol Cell Biol. 2007b;27:7089–7101. doi: 10.1128/MCB.00838-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Dalal NS. Generation of hydroxyl radical by chromate in biologically relevant systems: role of Cr(V) complexes versus tetraperoxochromate(V) Environ Health Perspect. 1994;102(3):231–236. doi: 10.1289/ehp.94102s3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Dong Z, Dalal NS, Gannett PM. Chromate-mediated free radical generation from cysteine, penicillamine, hydrogen peroxide, and lipid hydroperoxides. Biochim Biophys Acta. 1994;1226:65–72. doi: 10.1016/0925-4439(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Snow ET. Effects of chromium on DNA replication in vitro. Environ Health Perspect. 1994;102(3):41–44. doi: 10.1289/ehp.94102s341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns DM, Kennedy LJ, Courtney KD, Giangrande PH, Phieffer LS, Wetterhahn KE. Reduction of chromium(VI) by ascorbate leads to chromium-DNA binding and DNA strand breaks in vitro. Biochemistry. 1995;34:910–919. doi: 10.1021/bi00003a025. [DOI] [PubMed] [Google Scholar]
- Strathdee G. Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin Cancer Biol. 2002;12:373–379. doi: 10.1016/s1044-579x(02)00057-3. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Kondo K, Hirose T, Nakagawa H, Tsuyuguchi M, Hashimoto M, Sano T, Ochiai A, Monden Y. Microsatellite instability and protein expression of the DNA mismatch repair gene, hMLH1, of lung cancer in chromate-exposed workers. Mol Carcinog. 2005a;42:150–158. doi: 10.1002/mc.20073. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Kondo K, Ishikawa S, Uchihara H, Fujino H, Sawada N, Miyoshi T, Sakiyama S, Izumi K, Monden Y. Microscopic analysis of the chromium content in the chromium-induced malignant and premalignant bronchial lesions of the rat. Environ Res. 2005b;99:267–272. doi: 10.1016/j.envres.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Turgeon N, Valiquette C, Blais M, Routhier S, Seidman EG, Asselin C. Regulation of C/EBPdelta-dependent transactivation by histone deacetylases in intestinal epithelial cells. J Cell Biochem. 2008;103:1573–1583. doi: 10.1002/jcb.21544. [DOI] [PubMed] [Google Scholar]
- Umbas R, Isaacs WB, Bringuier PP, Schaafsma HE, Karthaus HF, Oosterhof GO, Debruyne FM, Schalken JA. Decreased E-cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res. 1994;54:3929–3933. [PubMed] [Google Scholar]
- van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65:3756–3788. doi: 10.1007/s00018-008-8281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Son YO, Chang Q, Sun L, Hitron JA, Budhraja A, Zhang Z, Ke Z, Chen F, Luo J, Shi X. NADPH oxidase activation is required in reactive oxygen species generation and cell transformation induced by hexavalent chromium. Toxicological sciences: an official journal of the Society of Toxicology. 2011;123:399–410. doi: 10.1093/toxsci/kfr180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zheng M, Liu G, Xia W, McKeown-Longo PJ, Hung MC, Zhao J. Kruppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 2007;67:7184–7193. doi: 10.1158/0008-5472.CAN-06-4729. [DOI] [PubMed] [Google Scholar]
- Wei YD, Tepperman K, Huang MY, Sartor MA, Puga A. Chromium inhibits transcription from polycyclic aromatic hydrocarbon-inducible promoters by blocking the release of histone deacetylase and preventing the binding of p300 to chromatin. J Biol Chem. 2004;279:4110–4119. doi: 10.1074/jbc.M310800200. [DOI] [PubMed] [Google Scholar]
- Wise SS, Holmes AL, Wise JP., Sr Particulate and soluble hexavalent chromium are cytotoxic and genotoxic to human lung epithelial cells. Mutat Res. 2006;610:2–7. doi: 10.1016/j.mrgentox.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Wise SS, Holmes AL, Wise JP., Sr Hexavalent chromium-induced DNA damage and repair mechanisms. Rev Environ Health. 2008;23:39–57. doi: 10.1515/reveh.2008.23.1.39. [DOI] [PubMed] [Google Scholar]
- Xie H, Holmes AL, Wise SS, Huang S, Peng C, Wise JP., Sr Neoplastic transformation of human bronchial cells by lead chromate particles. Am J Respir Cell Mol Biol. 2007;37:544–552. doi: 10.1165/rcmb.2007-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.