

Abstract
Background
Untreated human immunodeficiency virus (HIV) infection is characterized by progressive depletion of CD4+ T lymphocyte (CD4) count leading to the development of opportunistic diseases (acquired immunodeficiency syndrome (AIDS)), and more recent data suggest that HIV is also associated with an increased risk of serious non-AIDS (SNA) diseases including cardiovascular, renal, and liver diseases and non-AIDS-defining cancers. Although combination antiretroviral treatment (ART) has resulted in a substantial decrease in morbidity and mortality in persons with HIV infection, viral eradication is not feasible with currently available drugs. The optimal time to start ART for asymptomatic HIV infection is controversial and remains one of the key unanswered questions in the clinical management of HIV-infected individuals.
Purpose
In this article, we outline the rationale and methods of the Strategic Timing of AntiRetroviral Treatment (START) study, an ongoing multicenter international trial designed to assess the risks and benefits of initiating ART earlier than is currently practiced. We also describe some of the challenges encountered in the design and implementation of the study and how these challenges were addressed.
Methods
A total of 4000 study participants who are HIV type 1 (HIV-1) infected, ART naïve with CD4 count > 500 cells/μL are to be randomly allocated in a 1:1 ratio to start ART immediately (early ART) or defer treatment until CD4 count is <350 cells/ μL (deferred ART) and followed for a minimum of 3 years. The primary outcome is time to AIDS, SNA, or death. The study had a pilot phase to establish feasibility of accrual, which was set as the enrollment of at least 900 participants in the first year.
Results
Challenges encountered in the design and implementation of the study included the limited amount of data on the risk of a major component of the primary endpoint (SNA) in the study population, changes in treatment guidelines when the pilot phase was well underway, and the complexities of conducting the trial in a geographically wide population with diverse regulatory requirements. With the successful completion of the pilot phase, more than 1000 participants from 100 sites in 23 countries have been enrolled. The study will expand to include 237 sites in 36 countries to reach the target accrual of 4000 participants.
Conclusions
START is addressing one of the most important questions in the clinical management of ART. The randomization provided a platform for the conduct of several substudies aimed at increasing our understanding of HIV disease and the effects of antiretroviral therapy beyond the primary question of the trial. The lessons learned from its design and implementation will hopefully be of use to future publicly funded international trials.
Introduction
Randomized controlled trials comparing different strategies for the clinical management of people with human immunodeficiency virus type 1 (HIV-1) in the current environment of the treatment of HIV pose a multitude of challenges in design and implementation. In this article, we report on our experience in designing and initiating the Strategic Timing of Initiating AntiRetroviral Treatment (START) study, a multicenter international randomized clinical trial evaluating the clinical impact of early versus deferred initiation of antiretroviral therapy in HIV-infected individuals. The study is funded primarily by the Division of Acquired Immunodeficiency Syndrome (DAIDS) of the US National Institute of Allergy and Infectious Diseases (NIAID), with cofunding from several governmental agencies in the United States, Europe, and Australia. Six pharmaceutical companies are donating antiretroviral drugs for the duration of the trial. We summarize the rationale, design, governance, and plan of implementation of the study and describe some of the challenging issues encountered and the approaches we have adopted to overcoming them.
Rationale for a randomized trial of early treatment of HIV
Combination antiretroviral treatment (ART) has resulted in a substantial decrease in morbidity and mortality in persons with HIV infection [1,2]. The optimal time to start ART for asymptomatic HIV infection, however, remains one of the key unanswered strategic questions in the clinical management of HIV-infected individuals. The level of CD4+ T lymphocyte cells (CD4 count) is the primary factor used in deciding when to initiate ART. Some treatment guidelines recommend that ART should be initiated at a CD4 count of 350 cells/μL [3–5]; other guidelines recommend the initiation of ART when the CD4 cell count falls to a level below 500 cells/μL [6,7]. These variations arise because there are no randomized trials assessing the risks and benefits of treatment at a CD4 count > 350 cells/μL. There are a number of reasons supporting deferral of therapy. First, although current ART regimens are very effective in suppressing viral load, increasing CD4 count and enhancing immune function, with greatly improved tolerance and convenience, viral eradication is not an attainable goal with current treatments. This is because viral DNA, which is capable of producing new replication-competent virus upon cellular activation, persists within the host genome of some long-lived cells, including resting memory CD4+ cells, which precludes the elimination of the virus by ART alone for several decades [8]. Thus, daily treatment needs to be taken for life and requires a very high level of adherence in order to maintain efficacy, which is challenging for many patients. A consequence of suboptimal adherence is an increased risk of the development of drug resistance. Second, many drugs still have significant toxicity, which is often cumulative. Given the limited experience with many of the drugs to date (compared with the decades over which they will probably need to be used), it is to be expected that there will be unforeseen toxicities emerging as the drugs begin to approach their first or second decade of use. Third, the drugs are expensive. Because treatment is lifelong, there are serious financial constraints limiting access to treatment, particularly for those in resource-limited settings. Fourth, the excess risk of death associated with untreated HIV may be small at a CD4 count > 500 cells/μL, as suggested by a multicohort collaborative analysis comparing the death rate in untreated HIV-infected people with the general population [9]. Furthermore, the absolute risk of opportunistic diseases that constitute the acquired immunodeficiency syndrome (AIDS) is small at a CD4 count above 350 cells/μL [10,11], and the cost-effectiveness of initiating ART at a CD4 count above 350 cells/μL is uncertain.
On the other hand, more recent data indicate that there is a great potential for a reduction in serious morbidity and mortality due to non-AIDS conditions and in the risk of onward transmission of HIV from earlier initiation of ART. In the last few years, it has been recognized that HIV infection has broader effects beyond opportunistic diseases that may increase mortality even in early stages of the disease [12]. These effects include an excess risk of cardiovascular, renal, and hepatic diseases and malignancies not traditionally associated with a compromised immune system [13–26]. Such serious non-AIDS (SNA) conditions are now thought to be related to the processes of immune activation and inflammation that are known to accompany HIV infection, rather than the immune-compromised state per se [27,28]. In the Strategies for Management of AntiRetroviral Therapy (SMART) study, which compared continuous versus interrupted ART in 5472 participants with baseline CD4 counts > 350 cells/μL, the risk of both AIDS and SNA conditions was higher with interrupted ART compared to continuous ART [13]. This difference in AIDS and SNA conditions was also apparent among the subgroup of participants who were either ART naïve or who had previously taken ART but stopped more than 6 months before randomization (a comparison of immediate ART for those with CD4 > 350 cells/μL versus deferral of ART until the CD4 declined to <250 cells/μL) [29]. The findings from this subgroup, however, lack power because of the small number of participants. Data from observational studies suggest that the risk of AIDS increases as the CD4 count decreases, even at levels above 500 cells/ μL and that, at the same level of CD4 count, the risk of AIDS appears to be lower in patients who have started ART than in those who are ART naïve [10,11]. Data from the SMART study are consistent with these findings. In addition, rates of SNA conditions and death are lower at higher CD4 counts [12,25,30]. More recently, the clinical effect of starting ART at a CD4 count > 350 cells/μL was investigated by two large observational studies using separate multicohort datasets. Findings from the first study suggested that mortality was reduced by about 40% when ART was initiated at a CD4 count between 350 and 500 compared to <350 cells/μL, while mortality was almost halved if ART was initiated at a CD4 count above 500 compared to between 350 and 500 cells/μL [31]. In the second study, however, no evidence of an increase in the risk of AIDS or death was found when ART was started at a CD4 count above 450 compared to between 350 and 450 cells/μL [32].
In addition to the potential individual benefit, earlier treatment has the potential to reduce the risk of HIV transmission through suppression of viral load, a finding with clear import to public health [33–36]. The amount of HIV virus present in the blood and genital secretions is the most important risk factor for onward viral transmission, and ART reduces plasma HIV RNA to very low levels at which HIV transmission is extremely rare, at least for heterosexual transmission [37,38].
On the basis of evidence from large nonrandomized studies and a small subgroup of one randomized trial, the clinical benefit from starting ART at a CD4 count above 500 cells/μL is plausible, although not established. Even when proven, the magnitude of such benefit would need to be reliably estimated in order to quantify the multifaceted risk–benefit trade-offs and cost-effectiveness analyses undertaken to help guide public health policy. This could only be done in a randomized trial and, thus, is the motivation for the START study.
Overview of study design and methods
Design
START is an open-label multicenter international randomized trial comparing serious AIDS and non-AIDS morbidity or mortality between two management strategies for ART-naïve individuals with baseline CD4 counts above 500 cells/μL who are randomized to begin ART immediately (early ART) or to defer ART until the CD4 cell count declines to below 350 cells/μL or AIDS occurs (deferred ART).
A total of 4000 participants are to be randomly allocated over a 3-year period in a 1:1 ratio to the early or deferred ART group and followed up for a minimum of 3 years (Figure 1). The study had two phases: a pilot phase to establish the feasibility of accrual, which was set as the enrollment of at least 900 participants in the first year; and a definitive phase to complete study accrual upon successful conclusion of the pilot phase. Participants allocated to the early ART group are to start treatment immediately. Participants allocated to the deferred ART group are to start treatment when their CD4 count drops to below 350 cells/μL, based on two measurements not more than 1 month apart, or when they develop AIDS.
Figure 1.

Trial schema.
HIV-1: human immunodeficiency virus type 1; ART: antiretroviral treatment; CD4: CD4+ T lymphocyte; AIDS: acquired immunodeficiency syndrome.
The initial ART regimen prescribed to study participants must be chosen from a study-approved list of regimens, which consists of potent drug combinations based on current US Department of Health and Human Services (DHHS) treatment guidelines [6]. As described in Table 1, a study-approved regimen consists of two components. One component is a dual combination of two nucleoside or nucleotide analogue reverse transcriptase inhibitors (NRTIs: tenofovir/emtricitabine, zidovudine/lamivudine, or abacavir/lamivudine). The other component consists of either a non-nucleoside analogue reverse transcriptase inhibitor (NNRTI: efavirenz), or a ritonavir-boosted protease inhibitor (PI: atazanavir, darunavir, lopinavir, or fosamprenavir), or an integrase strand transfer inhibitor (INSTI: raltegravir). The choice of the initial ART regimen from the approved list is left to the clinical judgment of the study clinicians, who are permitted to use any available ART in cases of drug intolerance, toxicity, or for subsequent regimens in case of virological failure. All antiretroviral drugs used in the study must have approval or tentative approval from the US Food and Drug Administration (FDA) or the European Medicines Agency (EMA) and may be obtained either from the study Central Drug Repository (CDR) or through local sources. The large choice of antiretroviral drugs allowed in the study enhances the generalizability of the findings.
Table 1.
Antiretroviral components required for the initial ART regimen
| To construct an antiretroviral regimen, select one component from Column A and one component from Column B | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Column A (NNRTI, PI, or INSTI options) | Column B (dual-NRTI options) | |||||
| NNRTI: efavirenz | or | Ritonavir–boosted PI:atazanavir (1×/day), fosamprenavir (2×/day), fosamprenavir (1×/day), lopinavir (2×/day), lopinavir (1×/day), and darunavir (1×/day) | or | INSTI: raltegravir | + | Dual-NRTI:abacavir/lamivudine, tenofovir/emtricitabine, and zidovudine/lamivudine |
ART: antiretroviral treatment; NNRTI: nonnucleoside analog reverse transcriptase inhibitor; PI: protease inhibitor; INSTI: integrase strand transfer inhibitor; NRTI: nucleoside or nucleotide analog reverse transcriptase inhibitor.
The study is open-label since it is impractical, futile, and even undesirable to blind the treatment assignment. Given the flexibility in the choice of ART regimens and the large number of available drugs, the use of placebo would have been impractical. Regular assessment of the level of HIV RNA viral load, which is impacted by treatment, is routinely used to monitor response to treatment, which would render any attempt to blinding futile. Moreover, the use of placebo, even if it were to be feasible, would create an artificially high pill burden, undermining the ability of the study to assess the risk and benefits of early treatment as would be implemented in practice. The potential for event ascertainment bias due to the lack of blinding is mitigated by the use of objective diagnostic criteria for the clinical events comprising the composite primary endpoint [39,40], and by keeping the study's Endpoint Review Committee (ERC) blinded to treatment allocation. In order to reduce the risk of differential intensity of investigations between the two treatment groups, the same standardized follow-up schedule is used in both groups.
Rationale for the selected treatment strategies
The choice of the two treatment strategies to be compared in START was made with a view to achieving a sufficient difference in exposure to ART between the two groups so that a treatment difference in clinical outcome is plausible. The CD4 cell count of 350 cells/μL below which ART is to be initiated in the deferred ART strategy was based on the treatment guidelines in effect for many regions of the world at the time when the study was first implemented [3–5]. The entry criterion of a CD4 cell count > 500 cells/μL was chosen to ensure a sufficiently large difference in CD4 count between the groups during an average follow-up of 4.5 years, while maintaining CD4 cell counts in both groups at levels where the risk of disease was low. We expected that most individuals would enter the study with CD4 cell count between 500 and 600 cells/μL, with only a few entering having much higher CD4 cell counts. In persons who have never used ART, the CD4 cell count declines by an average of about 60 cells/μL per year [41]; thus, a participant who entered the study with the lowest eligible CD4 cell count would be expected to defer ART by about 2.5 years if randomized to deferring ART. In Appendix A, we describe a simulation study we undertook to derive plausible distributions for CD4 cell counts during follow-up in the two groups under realistic assumptions about the distribution of CD4 cell count at randomization and the evolution of CD4 count both before and after initiation of ART. Some participants in the deferred ART group will initiate ART before reaching an endpoint and before the CD4 count falls below 350 cells/μL either because of the development of HIV-related symptoms that do not constitute an endpoint (as permitted by the protocol) or for another reason. We assumed that only 70% of participants allocated to defer ART would wait until the CD4 cell count drops to <350 cells/μL before initiating ART, and that 20% and 10% would only wait until the CD4 cell count drops to below 400 and 450 cells/μL, respectively. We assumed that 40%, 30%, 20%, and 10% of participants would enter the study with CD4 counts between 501 and 550, between 551 and 600, between 601 and 700, and between 701 and 1000 cells/μL, respectively, which would result in a median CD4 count of 566 (mean of 597) cells/μL. These design assumptions may be conservative if the observed median baseline CD4 count is higher, in which case we would expect longer time to initiation of ART in the deferred ART group and a larger treatment relative difference in the primary outcome. We summarize the results of these simulations below.
Model parameters for the simulation studies were derived from data on 15,274 individuals whose date of HIV seroconversion is well documented as part of 26 cohorts participating in the Concerted Action for HIV SeroConversion and AIDS in Europe (CASCADE) study [42,43]. Figure 2 shows the projected median CD4 count in the two treatment groups during follow-up. The largest treatment difference in CD4 count is expected to occur during the second year and is reduced during subsequent follow-up as more participants in the deferred ART group initiate treatment. The simulation results detailed in Appendix A suggest that the median CD4 count in the early ART group would be higher than in the deferred ART group by 161 and 103 cells/μL 1 and 6 years after randomization, respectively. The decline of the projected median CD4 count after the first year in the early ART group takes account of the possibility that some participants might not adhere to their assigned treatment strategy and interrupt ART. In the CASCADE cohorts, Touloumi et al. [44] estimated that about 20% of those who started ART after 1 year from the date HIV seroconversion interrupted or stopped ART at some point after ART initiation. Thus, if the proportion of participants in the early ART group who stop or interrupt therapy in START is much lower than 20%, our estimate of the treatment difference in CD4 count is likely to be conservative.
Figure 2.

Median projected CD4 cell count during follow-up.
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment.
The Kaplan–Meier plot for the projected time to ART initiation in the Deferred ART group is shown in Figure 3. Median time to ART initiation in the deferred ART group is projected to be 37 months (lower quartile: 17 months and upper quartile: more than 72 months). If all participants in the deferred ART group waited until the CD4 count drops to below 350 cells/μL, the median time to ART initiation would be 41 months (lower quartile: 21 months).
Figure 3.

Kaplan–Meier's plot for projected time from randomization to ART initiation in the deferred ART group under two scenarios about the CD4 count cutoff below which ART is initiated: 350, 400, and 450 cells/μL for 70%, 20%, and 10% of the participants, respectively (deferred ART group); 350 cells/μL for all participants.
ART: antiretroviral treatment; CD4: CD4+ T lymphocyte.
Primary endpoint
The primary outcome for START is time to the development of a nonfatal serious AIDS or non-AIDS condition or death from any cause. The composite primary endpoint has two major components. The first major component is fatal AIDS or a nonfatal but serious AIDS condition. Fatal AIDS is any death whose primary cause is any clinical event satisfying the diagnostic criteria of an AIDS-defining event according to the 1993 Centers for Disease Control (CDC) expanded surveillance definition of AIDS plus additional events associated with immunosuppression in the patient population targeted for enrollment [39,45]. Nonfatal serious AIDS events consist of any AIDS-defining condition but exclude nonfatal esophageal candidiasis and chronic herpes simplex. We refer to this component of the endpoint as AIDS*. The second major component consists of fatal or nonfatal SNA conditions, which we refer to as SNA. Fatal SNA is any death not attributable to AIDS, including deaths due to accidents or violence and those of unknown cause. Nonfatal SNA events consist of (1) cardiovascular disease (myocardial infarction, stroke, coronary artery disease requiring angioplasty, or bypass surgery); (2) end-stage renal disease requiring dialysis or renal transplantation; (3) decompensated liver disease; and (4) all non-AIDS-defining cancers, excluding basal and squamous cell skin cancers [40].
Unlike most trials of the treatment of HIV, which use HIV RNA viral load and CD4 cell count as outcome measures, the primary endpoint in START is clinical. Only clinical endpoint trials, with endpoints including the main serious conditions postulated to be affected by HIV and/or ART, can give reliable estimates of the risk-benefit trade-off of the early versus deferred treatment strategies. While the degree of CD4 increase and viral load suppression after starting ART can be reasonably estimated from existing data, the clinical impact of increases in CD4 count and suppression of HIV RNA by ART while the immune system is still relatively intact is largely unknown, as is the potential for other effects that are not mediated by these markers, such as drug toxicities. The reliable assessment of the clinical impact of starting ART at high CD4 counts compared to waiting until the CD4 count drops to below 350 cells/μL is the main objective of the START study.
START is unique in being the first and, to our knowledge, only HIV treatment trial to use SNA conditions as part of a composite primary endpoint. Based on data from observational studies, the risk of AIDS at CD4 cell counts > 350 cells/μL is small (of the order of 0.5–1.0 per 100 person-years) compared to the risk of SNA, which is expected to be four to five times as large [13]. On the other hand, the anticipated relative risk reduction with the use of ART is much higher for AIDS than for SNA so that the absolute risk reductions are expected to be comparable.
Secondary endpoints
A broad range of clinical secondary outcomes are assessed to allow for a comprehensive evaluation of the benefits and risks of early ART. Some of these outcomes are assessed in all randomized participants and some in subsamples for efficiency. Three key secondary endpoints are the components of the primary composite endpoint (AIDS* and SNA) and all-cause mortality. Other secondary outcomes assessed in all randomized participants include fatal or nonfatal AIDS (including esophageal candidiasis and chronic Herpes simplex); all non-AIDS-defining cancers (including basal and squamous cell skin cancers); bacterial pneumonia; pulmonary embolism; deep vein thrombosis; new-onset diabetes mellitus; coronary artery disease requiring drug treatment; congestive heart failure; peripheral vascular disease; various other adverse events; as well as HIV drug resistance, quality of life, health-care utilization and cost of care, and HIV transmission risk behavior.
Substudies
Studying treatment-naïve individuals with CD4 counts > 500 cells/μL provides a rare opportunity of undertaking substudies to make an unbiased assessment of the impact of ART versus untreated HIV on various outcomes, taking advantage of the randomization and the systematically collected clinical data and stored specimens. It is important, however, that the conduct of substudies does not create a burden on study participants and site personnel such as to compromise the quality of the conduct of the main study. Currently, there are four substudies, which compare early ART and deferred ART in terms of outcomes specific to the question addressed by the substudy.
The neurology substudy will evaluate the impact of early treatment on neurocognitive function in 600 participants from 35 sites. Early ART will be compared to deferred ART in terms of change from baseline in the quantitative neurocognitive performance z score (QNPZ-8), derived from a test battery of eight tests [46–51] administered by trained study staff at baseline; months 4, 8, and 12; and annually thereafter.
The arterial elasticity substudy will coenroll a total of 300 participants at 17 sites to determine whether early ART is superior to deferred ART in increasing large and small artery elasticity measured by trained study staff using a portable radial artery ultrasound device at baseline; months 4, 8, and 12; and annually thereafter. The difference between the two randomized groups in change from baseline in these two measures (averaged over follow-up) will be estimated.
The pulmonary substudy aims to determine whether early ART alters the rate of lung function decline compared to deferral of ART by comparing the two treatment groups in terms of the rate of decline of forced expiratory volume in 1 s (FEV1), which will be measured by a handheld spirometer at baseline and annually in 1000 coenrolled participants.
The bone mineral density (BMD) substudy aims to quantify the relative contributions of HIV and treatment for HIV on rates of hip and spine BMD loss. BMD will be measured at baseline and annually in 400 coenrolled participants.
In each of these four substudies, participants are consented separately for participation before randomization in the main study, so that the substudies' primary comparisons are protected by the randomization.
Since the informed consent process has become increasingly burdensome for participants in recent years, the START study will also include an informed consent substudy. Prior to beginning enrollment, participating clinical sites will be cluster-randomized to use either the standard study consent or a concise consent for all participants. Participant understanding and satisfaction will be assessed using structured questionnaires to be completed on a voluntary basis.
At participating sites, START study participants will also be asked to consent to a genomics substudy and provide a whole blood sample from which DNA will be later extracted to study validated genetic variants that determine the risk of the primary and secondary outcomes assessed in START. Participants may elect to withdraw from this substudy at any time and have their blood sample destroyed.
The START substudies maximize the scientific gains of this unique randomized trial by bringing together a diversity of donors, scientific expertise, and investigators to answer a wide-ranging set of clinical and translational questions of import to the treatment of HIV-infected individuals.
Study population
Table 2 lists the inclusion and exclusion criteria for START. The study has very broad entry criteria; with few exceptions, all previously untreated HIV-infected individuals in reasonably good health and with two recent CD4 cell counts > 500 cells/μL are eligible provided they are willing to adhere to the allocated treatment strategy and data collection schedule. Any prior use of ART, including for prevention of mother-to-child transmission, or any prior immunomodulatory therapy excludes participation in the trial. Individuals are also excluded if they have a history of any AIDS-defining condition; presence of symptoms consistent with HIV disease progression; or a history of myocardial infarction, angioplasty or bypass surgery, stroke, dialysis, renal transplantation, decompensated liver disease, or non-AIDS-defining cancer within 6 months before randomization. For reasons of safety, the study excludes participation of women currently pregnant or breast-feeding. Also excluded are individuals under involuntary incarceration.
Table 2.
START eligibility criteria
| Inclusion criteria | Exclusion criteria |
|---|---|
| Age >18 years | Any previous use of ART or IL-2 |
| Documentation of HIV-1 infection | Diagnosis of any clinical AIDS event |
| Two CD4 cell counts >500 cells/μL at least 2 weeks apart within 60 days before randomization | Presence of HIV disease progression such as oral thrush, unexplained weight loss, or unexplained fever |
| Karnofsky performance score ≥80. Perceived life expectancy of at least 6 months | Cardiovascular event (myocardial infarction, angioplasty coronary artery bypass grafting, and stroke) within 6 months before randomization |
| For women of child-bearing potential, willingness to use contraceptives as described in the product information of the ART drugs they are prescribed | Non-AIDS-defining cancer, excluding basal and squamous cell skin cancer, within 6 months before randomization. Dialysis within 6 months before randomization |
| History of decompensated liver disease | |
| Current imprisonment, or compulsory detention (involuntary incarceration) for treatment of a psychiatric or physical illness |
START: Strategic Timing of AntiRetroviral Treatment; ART: antiretroviral treatment; IL-2: interleukin-2; HIV-1: human immunodeficiency virus type 1; CD4: CD4+ T lymphocyte; AIDS: acquired immunodeficiency syndrome.
The study will enroll participants from diverse populations on five continents without selection for or against prior high risk of the composite primary outcome, for example, high HIV RNA viral load or high risk score for cardiovascular disease. The rationale for the choice of such a heterogeneous population is to enhance the generalizability of the study findings. This will also permit analyses according to baseline risk factors for the primary outcome to be carried out at the completion of the study to explore how ART should most effectively be used in subgroups considering absolute risk reductions and cost-effectiveness.
Not all eligible individuals will be willing to be randomized. The extensive data collected on individuals screened at baseline will help us assess how representatives those who enter the study are compared to the clinic populations from which they were drawn.
Sample size estimation
There were several challenges in the sample size determination in START including estimation of the expected primary event rate during follow-up. Although there are large amounts of data on the rates of AIDS and all-cause mortality by latest CD4 cell count from cohorts of HIV-infected persons, there are insufficient data on these events in ART-naïve individuals in follow-up periods during which CD4 count is >500 cells/μL. Furthermore, only a few cohorts have well-documented causes of death, and there are virtually no data on nonfatal SNA in ART-naïve individuals. Most of the data for SNA come from the SMART [13] and Evaluation of Subcutaneous Proleukin in a Randomized International Trial (ESPRIT) studies [52]. In addition, an association between duration of ART and the risk of myocardial infarction was reported in a multicohort analysis by the Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) study group [53]. In these three studies, however, most participants had been taking ART for several years at the time of enrollment.
Our estimates of the expected event rates in the Early and Deferred ART groups were derived from analyses of event data by latest CD4 from the CASCADE and the UK Collaborative HIV Cohort (CHIC) observational studies [42,54] together with computer simulations of the expected distributions of CD4 cell counts in START. These estimates are described in detail in Appendix B. Because of the uncertainties in some of the assumptions used in deriving the expected event rates, we plan a sample size reestimation before enrollment is completed. The reestimation will be based on the observed event rate pooled across the treatment arms and the hypothesized treatment difference. The observed treatment difference will be unknown to those performing the sample size reestimation.
The sample size has been estimated as 4000 participants to be followed for an average of 4.5 years. The primary analysis will be according to the intention to treat principle, using a stratified two-sided log-rank test with strata defined by geographical region (North America, South America, Europe, Australasia, and Africa) and type 1 error 0.05. A number of assumptions were made in the determination of the sample size. These assumptions are described in detail in Appendix B and are summarized as follows:
Power was set at 90% for the primary endpoint, in part, to ensure that there would be adequate power to address the two major components of the composite endpoint.
Participants will be enrolled over a 3-year period and followed for a minimum of 3 years resulting in a total study duration of 6 years with average follow-up 4.5 years.
The CD4 cell count at entry will be between 501 and 550 cells/μL for 40% of participants, between 551 and 600 cells/μL for 30%, between 601 and 700 cells/μL for 20%, and between 701 and 1000 cells/μL for 10%; with median count of 566 cells/μL.
In the deferred ART group, the average rate of the primary endpoint is 2.8 per 100 person-years over the follow-up period. Nonfatal SNA events will be four times more common than fatal SNA. Approximately 35% of reported nonfatal AIDS* events and 10% of reported nonfatal SNA events will be rejected by the ERC and not count toward the primary endpoint. In SMART, the rates of confirmed or probable nonfatal AIDS and SNA events, as defined in START, were 0.4 and 2.0 per 100 person-years, respectively, in the continuous ART group [13].
Based on the computer simulations described in Appendices A and B, early ART is predicted to reduce the primary endpoint average rate by 28.8% compared to deferred ART. This reduction in the hazard assumes (1) AIDS events represent 23% of the primary events in the deferred arm, and early ART will, on average, reduce this hazard by 43%; and (2) SNA events will represent 77% of events in the deferred ART group, and early ART will, on average, reduce this hazard by 24%. The smaller percentage risk reduction anticipated for SNA events takes into account that some of the deaths will be due to causes that are unrelated to HIV and ART. For example, 18% of deaths in SMART were due to substance abuse, accidents, or violence [13].
The estimated treatment differences take into account likely levels of adherence to the allocated ART strategy. For the deferred ART group, we assumed that 70% of participants would adhere to the deferral CD4 threshold of 350 cells/μL and 30% would initiate ART earlier – 10% before the CD4 cell count declined to 400 cells/μL and 20% while the CD4 count was between 350 and 400 cells/μL. For the early ART group, event rates were estimated from CASCADE participants using the full follow-up time after having started ART; this included some participants who subsequently discontinued ART. Thus, event rate estimates include some allowance for nonadherence.
A loss to follow-up rate of 2.7 per 100 person-years (equivalent to a 15% cumulative lost to follow-up after 6 years) is assumed.
Based on these assumptions, 3822 participants (1911 participants in each treatment group) are required. The number of primary events required is 369.
While many of the underlying assumptions are conservative, for some assumptions there is much uncertainty because of the absence of data (e.g., inadequate or no data on rates of non-AIDS morbidity). Table 3 summarizes study sample size estimates for several scenarios based upon different ratios of the rate of nonfatal SNA events to that of fatal SNA events and the proportion of nonfatal AIDS* events that would be accepted as probable or confirmed by the Endpoint Review Committee. Considering this, a sample size of 4000 participants (2000 in each treatment group) and a target of 370 primary events have been established. This may be modified following sample size reestimation.
Table 3.
Sensitivity of the sample size to assumptions concerning the rate of nonfatal SNA events as a proportion of the rate of fatal serious non-AIDS events and the proportion of events accepted by the Endpoint Review Committee
| Ratio: nonfatal SNA to fatal SNA | % AIDS* events accepted by the ERC | Composite primary event rate per 100 person-years (deferred arm) | Hazard ratio (average over follow-up) | Sample size | Expected number of events |
|---|---|---|---|---|---|
| 3 | 65 | 2.40 | 0.706 | 4,244 | 351 |
| 4 | 65 | 2.81 | 0.712 | 3,822 | 369 |
| 5 | 65 | 3.23 | 0.717 | 3.472 | 383 |
| 3 | 75 | 2.49 | 0.700 | 3,900 | 333 |
| 4 | 75 | 2.91 | 0.707 | 3,545 | 353 |
| 5 | 75 | 3.33 | 0.712 | 3,245 | 367 |
AIDS: acquired immunodeficiency syndrome; SNA: serious non-AIDS.
With the above assumptions, power is 74% to detect the hypothesized 43% reduction in the hazard of AIDS*, and power is 69% to detect the hypothesized 24% reduction in the hazard of SNA.
Study procedures and data collection
Potentially eligible participants will attend screening visits within 2 months before randomization to establish eligibility and collect baseline data. Following screening, eligible participants are randomized in a 1:1 ratio to either early or deferred ART. Randomization is stratified by clinical site. Routine data collection visits (regular study visits) occur at months 1 and 4 after randomization, and every 4 months thereafter. Data collected at study entry and at all follow-up visits include targeted medical history, including smoking status, clinical evaluation, laboratory assessments, and self-reported adherence to ART for those who started ART. Laboratory assessments include, among others, absolute and percent CD4 cell count, HIV RNA viral load, and results of locally performed antiretroviral resistance tests. Plasma and urine specimens are collected prior to randomization and during follow-up, including prior to any ART regimen change, and stored centrally for future analyses.
Additional data collected at study entry and at annual visits include concomitant medications, resting electrocardiogram (ECG), as well as HIV transmission risk behavior, health-care utilization, and quality of life assessments. Prior to randomization, the intended first ART regimen will be collected for all participants. Information on the start of ART, changes to the ART regimen, as well as documentation of important clinical outcomes including components of the composite primary endpoint, certain adverse events, and pregnancy outcomes will be collected during follow-up as they occur.
All participants will be followed to a common closing date when 370 primary endpoints have occurred, currently estimated at the end of 2015.
Study organization and governance
The START study is sponsored by the University of Minnesota and is conducted by the International Network for Strategic Initiatives in Global HIV Trials (INSIGHT) with an organizational structure that reflects the international nature of the collaboration. Scientific and operational oversight for the study is the responsibility of the INSIGHT START study protocol team. The implementation of the protocol is coordinated by INSIGHT staff at the University of Minnesota Coordinating Center (CC) together with four International Coordinating Centers (ICCs) based in Copenhagen, London, Sydney, and Washington. Each ICC is responsible for the management of the study at clinical research sites in multiple countries through one or more Site Coordinating Centers (SCCs). The countries participating in the study and their ICCs are shown in Figure 4. In total, 237 sites from 36 countries are participating in the study; of these, 100 sites from 22 countries enrolled participants in the pilot phase.
Figure 4.
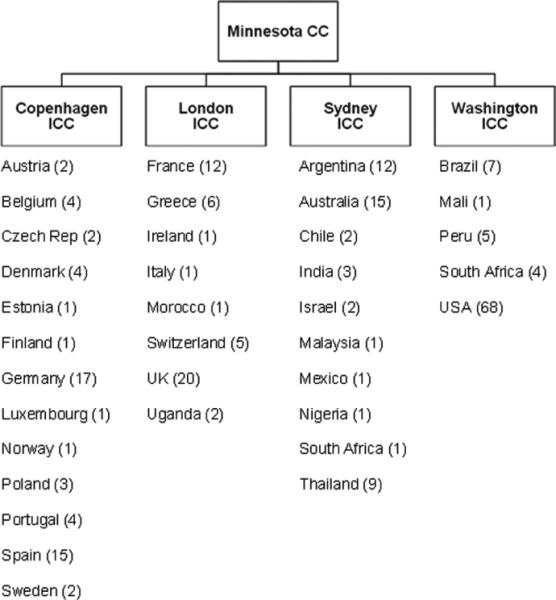
START CCs and participating countries (numbers in parentheses refer to participating sites).
CC: Coordinating Center; ICC: International Coordinating Center.
Clinical event adjudication
A study Endpoint Review Committee (ERC) will review all reported primary or major secondary clinical events to determine the level of diagnostic certainty. Events that are judged as confirmed or probable will be included in the primary analysis. The ERC has established objective criteria for each of the events comprising the primary endpoint and for major secondary endpoints and will carry out the review blinded to the randomized allocation [39,40].
Statistical analysis plan
The primary analysis will be by intent to treat, comparing early and deferred ART in terms of time from randomization to first onset of an event satisfying the primary endpoint definition, using a two-sided log-rank test, Kaplan–Meier plots, and a Cox proportional hazard (PH) model stratified by region. Losses to follow-up will be considered uninformative (i.e., a person who is lost has no greater or lesser chance of experiencing the primary endpoint than a person who is not lost to follow-up and followed for the same period of time since randomization). The same methods will be used to compare the two treatment groups in terms of the major secondary endpoints.
Below we discuss some key issues that needed careful consideration in formulating the data analysis plan. These are adherence to the allocated strategy, assessment of potential ascertainment bias, selection of relevant subgroups within which the treatments are compared in secondary analyses, and the assessment of CD4 count at the time of initiation of treatment in the deferred ART group.
Adherence to the allocated ART management strategy will be assessed by providing a detailed description of ART exposure during follow-up to include time to ART initiation in the deferred ART group, percent of follow-up time spent on ART by treatment group, and the distribution of CD4 count at ART initiation in the deferred ART group.
Because the randomized allocation cannot be masked, it is important to assess the potential for ascertainment bias whereby events are underreported in one treatment group compared to the other. We plan to summarize the quality of follow-up including the frequency of study visit attendance and the timeliness of event reporting and completeness of event documentation by treatment group.
Comparison of early and deferred ART according to subgroups defined by key baseline factors will be mainly limited to the primary and the two major secondary endpoints (AIDS* and SNA). In addition to demographics, CD4 cell count and HIV RNA level at randomization, subgroups will be defined by date of randomization in order to explore the impact of trends in clinical management on the treatment effect, intended ART regimen as specified before randomization (to explore whether the effect of early ART relative to deferred ART is qualitatively different for different treatment regimens), and by risk factors for cardiovascular disease and other SNA conditions to assess the absolute difference in risk for different subgroups defined according to their baseline risk score. The latter includes plans to measure D-dimer, high-sensitivity C-reactive protein, and interlukin-6 at baseline for all study participants. These coagulation and inflammation biomarkers have been shown to be strongly related to all-cause mortality in HIV-infected participants [28,55].
A clear description of the distribution of CD4 count in the deferred ART group at the time of initiation of ART is needed for the interpretation of the study findings. CD4 counts are known to have substantial variability, and a major component of this variability is measurement error (variability of repeated assessments of the same sample). The measured (or observed) CD4 count can be viewed as the sum of the true underlying count and measurement error. The distribution of the measured CD4 cell count at the time of initiation of ART in the deferred ART group is likely to underestimate the level of the true underlying CD4 cell count at which ART is initiated. The reason for this bias is the combination of measurement error, which can be substantial for CD4 cell count, and the CD4 criterion for initiation of ART (only when the measured CD4 count is observed to fall below 350 cells/μL). The bias could be as large as 50 cells/μL as demonstrated in Appendix C. For this reason, we plan to provide summary statistics for the distribution of CD4 cell count, not only at the time of initiation of ART, but also at the regular study visit immediately before that at which the CD4 count was first observed to fall below 350 cells/μL.
Data monitoring
The trial is conducted under the direction of the INSIGHT START study protocol team. Members of the study protocol team – except for the statisticians who prepare the confidential analyses for an independent Data and Safety Monitoring Board (DSMB) – and all participating investigators will be blinded to interim results on clinical outcomes.
The DSMB is convened by NIAID and will review at least annually the general conduct of the study, interim analyses including treatment comparisons in terms of the major clinical outcomes, safety reports, and other data that might impact the design of START, for example, data from other completed trials, and cohorts with similarly defined target populations. The DSMB is requested to recommend early termination or modification of the protocol only if (1) there is proof beyond reasonable doubt that one of the two treatment groups is superior to the other group in terms of the primary outcome and its major components, or (2) a treatment difference is unlikely to be established or refuted by the end of the planned follow-up. As a guideline for (1), the Lan–DeMets spending function analogue of the O'Brien–Fleming boundaries will be used to monitor the primary endpoint comparison [56,57]. The DSMB will be asked not to recommend early termination of the study unless there is evidence of a significant treatment difference based on the spending function boundary for the primary endpoint, and each of the two major components of the primary endpoint – AIDS* and SNA events – are consistent (in the same direction, with Z > 1.5 for each outcome).
At each DSMB review, beginning with the review prior to the end of enrollment when sample size is reestimated, futility analyses will be presented to the DSMB by the unblinded statisticians (those responsible for preparing the DSMB reports) based on conditional and unconditional power. Conditional power incorporates the observed results by treatment group thus far (and uses the originally assumed treatment effect for future data) to calculate the conditional probability of obtaining a significant result by the end of the trial. In contrast, unconditional power does not take into consideration the observed treatment difference. The unblinded statisticians will reestimate what the power was at the beginning of the trial based on the observed event rate in the deferred ART arm, the planned duration of follow-up, and the originally assumed treatment effect. Conditional and unconditional power estimates are used for two different purposes. Conditional power tells us whether we are likely to get a significant result, whereas unconditional power indicates whether a null result would still be meaningful. For example, suppose the unconditional power were only 40%. Even if the true treatment benefit were as originally hypothesized, there would be a 60% chance of missing it. Therefore, a null result would not rule out the originally hypothesized treatment benefit. On the other hand, if unconditional power were high – say 90% – then a null result would effectively rule out the originally hypothesized treatment benefit.
As a guideline, the DSMB is requested to first consider unconditional power. If unconditional power is <70%, the DSMB should then consider conditional power. If conditional power, given the observed data and assuming the originally hypothesized treatment effect thereafter, is <20%, consideration should be given to stopping the trial. We recommend early termination for futility only if both conditional and unconditional power estimates are low, that is, only if a null result is both likely and not meaningful. It is possible that unconditional power is low in the presence of a very large treatment effect of early ART. Hence, there is a need to also consider conditional power. Such a scenario would not be grounds for stopping for futility because conditional power would probably still be high, indicating that a null result is unlikely.
In summary, the DSMB for START is provided with the aforementioned guidelines but will be expected to use their expert and independent judgment concerning early termination. It is recognized that there are a number of considerations in determining whether a trial should be stopped early. For this reason, we proposed guidelines to the DSMB, not rules.
Challenges in study implementation
There were a number of challenges encountered in the implementation of START. Some of these have to do with overcoming difficulties resulting from lack of harmonization of clinical trial regulations across different countries participating in START. These difficulties, which caused a substantial delay in initiating the pilot phase of the study, were described in detail [58] and are summarized below. We also describe how the protocol team addressed the issues raised by changes in treatment guidelines in the United States after enrollment was underway.
Study sponsorship
Development of the protocol began in earnest in the spring of 2007; after 1 year of planning, the trial was very close to initiation. Up to that time, it had been understood that NIAID as the primary funder would also be the study sponsor, as in previous National Institutes of Health (NIH)-funded trials conducted by INSIGHT. In June 2008, however, NIAID informed the study leadership that the European Union (EU) Clinical Trials Directive's requirement for the provision of insurance or indemnity to cover the liability of the sponsor and investigators in the case of injury or death resulting from participation in the study precluded NIH sponsorship of START given the terms of the US Anti-Deficiency Act [59]. Nearly 6 months of dialogue and negotiations ensued between the INSIGHT leadership, NIH, and the University of Minnesota, the institutional recipient of NIAID funds for INSIGHT and START, to achieve consensus. At the end of 2008, the University of Minnesota agreed to serve as the sponsor for START, with the University of Copenhagen as its legal representative in Europe.
Clinical Trial Agreements with six pharmaceutical companies
Following the decision by the University of Minnesota to sponsor START, version 1.0 of the study protocol was completed and sent to participating sites and to the six pharmaceutical companies donating drugs for the study. Negotiations to finalize Clinical Trial Agreements (CTAs) between the University of Minnesota and each pharmaceutical company were vigorously pursued, but took several months to conclude. The main difficulty was agreement around clauses of indemnification. Pharmaceutical companies would not hold harmless the University of Minnesota for product liability or lawsuits resulting from claims concerning unexpected adverse events considered to be related to the drugs. At the end, the University purchased extra insurance to cover this liability, and the last of the six CTAs was signed in November 2009, 7 months after the first participant had been enrolled in the study.
Central Drug Repository
Following the execution of the CTAs, work began on setting up the study's Central Drug Repository (CDR). Almac Clinical Trial Services, with major operations based in Durham, North Carolina, and Craigavon, United Kingdom, was retained to manage distribution of 13 drugs to clinical sites in 22 countries participating in the pilot phase of the study. Establishing the CDR involved many tasks, including projecting drug use in each country; preparing text for drug labels, translating labels, and securing local regulatory approvals of labels; printing multiple-page label booklets based on 29 language variants; acquiring drug and accompanying documentation; applying label booklets to bottles; establishing distribution depots in seven countries; and developing a web-based system for drug orders by sites from the CDR and tracking depot inventories. The lengthy processes for obtaining national approvals of clinical trial drug use, the diversity of national requirements for importation of drug, the complexities of qualified person drug release required in the EU, and the need for additional provision of pharmaceutical documentation resulted in significant delays for some countries to access the study's CDR, which opened for most pilot phase sites between mid-December 2009 and May 2010. Work proceeds on expanding the CDR to supply all newly participating countries and sites in the study.
Designation of the study as an investigational medicinal product trial in Europe
START is comparing two ART management strategies to determine the optimal timing of initiation of ART using FDA- and EMA-approved or tentatively approved antiretroviral drugs. It is not a trial comparing specific drugs, and it is not being carried out under a US Investigational New Drug (IND) application. In spite of this, several (albeit not all) competent authorities in Europe have designated START as a trial of an investigational medicinal product (IMP). As a consequence, procedures for expedited reporting of suspected unexpected serious adverse reactions (SUSARs) to the EU's EudraVigilance had to be established, causing further complications in the conduct of the study.
Impact of changes in HIV treatment guidelines
In 1 December 2009, the DHHS Panel on antiretroviral guidelines released updated guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents [6]. These guidelines recommended initiation of ART in asymptomatic individuals with CD4 cell counts < 500 cells/μL, which was not consistent with the ART management strategy for the deferred ART group in START. Previous DHHS guidelines, which were referenced in the START protocol, had recommended initiation of treatment when the CD4 count is <350 cells/μL and stated that ART might be considered in some patients with CD4+ >350 cells/μL, but that existing data were inadequate to recommend initiation of ART for all patients. The December 2009 DHHS guidelines cite data from a small laboratory-based study and two large observational studies, with particular reference to the North American AIDS Cohort Collaboration on Research and Design (NA-ACCORD) study [28], to support the recommendation for earlier initiation of ART.
The START protocol team was aware of the NA-ACCORD and other studies when version 1.0 of the protocol was written. The results of the NA-ACCORD study were summarized in the START protocol. The findings from NA-ACCORD were discussed with the START DSMB in May 2009. Neither the START protocol team nor the DSMB assessments of the NA-ACCORD data led to any changes in the START protocol. Following the December 2009 update in the US guidelines, however, the START protocol team took a number of actions. First, all investigators were asked whether their clinical uncertainty about the optimal time for ART initiation had changed sufficiently to warrant a protocol revision. The unanimous response was not to change the study design. Second, investigators were reminded about the proper management of participants in the deferred group of START, and additional recommendations were formulated for increased CD4 monitoring as the CD4 count approaches 350 cells/μL. Third, a memorandum was sent to all investigators informing them (1) about the change in the US guidelines, (2) that the protocol was not being modified, and (3) that study participants had to be informed of the new information. Sites were asked to inform study participants that some experts now recommended starting ART in asymptomatic individuals with CD4+ cell counts between 350 and 500 cells/μL and that this recommendation was not based on data from randomized trials. Sites were required to notify their IRBs/RECs of this new information. The INSIGHT Community Advisory Board was centrally involved in this process and, as a result, a statement was issued by community activists and other organizations from around the world questioning the revised guidelines and arguing that people with HIV needed the evidence from a randomized trial like START to make an informed decision about when to start ART [60].
Current status of the trial
The first participant was enrolled on 15 April 2009. Figure 5 illustrates the number of clinical research sites open and participants enrolled over time up to the end of 2010. Study accrual increased steadily in 2010 with the availability of antiretroviral drugs distributed from the CDR. At the end of August 2010, when the study had accrued over 600 participants, DAIDS agreed that feasibility of enrollment had been established and that the study could expand to more sites so that the target accrual of 4000 participants could be achieved by the end of December 2012.
Figure 5.

Cumulative enrollment (left axis) and cumulate number of enrolling sites (right axis) up to December 31, 2010.
At the end of May 2011, a total of 1302 participants had been enrolled. Baseline data are summarized in Table 4. The study population comprises 16% females; 51%, 16%, 16%, 9%, and 8% enrolled from sites in Europe, North America, Australasia, South America, Asia, and Africa, respectively; about two-thirds of the study population acquired HIV through sexual contact with a person of the same sex. Mean age was 37 (standard deviation (SD): 10) years, and the median level of plasma HIV RNA viral load was 4.1 (interquartile range (IQR): 3.6–4.6) log copies/mL. Median CD4 cell count was 635 (IQR: 578–721) cells/μL, which is much higher than the design assumption. Although a higher median CD4 cell count at baseline would be expected to result in a lower event rate, it would also imply a longer time from randomization to ART initiation in the deferred ART group, and thus, the hypothesized treatment difference in the primary outcome is likely to be greater than was anticipated and the net effect would be an increase in power.
Table 4.
Baseline characteristics of 1002 participants enrolled in START as of May 31, 2011
| Demographics | |
|---|---|
| Age (years; mean, SD) | 37 (10) |
| Gender (% female) | 16.2 |
| Race | |
| Asian (%) | 6.3 |
| Black (%) | 17.4 |
| Latino/Hispanic (%) | 11.9 |
| White (%) | 63.5 |
| Other (%) | 1.8 |
| HIV history | |
| Likely mode of HIV infection | |
| Sexual contact with person of same sex (%) | 68.0 |
| Sexual contact with person of opposite sex (%) | 27.2 |
| Injection drug use (%) | 2.4 |
| Blood products (%) | 0.9 |
| Other/unknown (%) | 5.6 |
| Time participant known to be HIV-positive (years; median, IQR) | 1.0 (0.0, 3.0) |
| Laboratory results | |
| CD4+a (cells/mm3; median, IQR) | 635 (578, 721) |
| Nadir CD4+b (cells/mm3; median, IQR) | 529 (460, 609) |
| HIV RNA (log10copies/mL; median, IQR) | 4.1 (3.6, 4.6) |
| Highest HIV RNAb (log10copies/mL; median, IQR) | 4.4 (3.9, 4.9) |
| HIV RNA, undetectable (% with RNA ≤400 copies/mL) | 5.6 |
| Medical history | |
| Hepatitis B (%) | 1.7 |
| Hepatitis C (%) | 5.7 |
| Clinical information | |
| BMI (kg/m2; mean, SD) | 25 (5) |
| BP—systolica (mmHg; mean, SD) | 122 (15) |
| BP—diastolica (mmHg; mean, SD) | 76 (10) |
| CVD risk factors for SNA | |
| Current smoker (%) | 40.5 |
| Diabetes or drug treatment for diabetes (%) | 2.7 |
| Hypertension or use of BP-lowering drugs (%) | 10.3 |
| Hyperlipidemia or use of lipid-lowering drugs (%) | 8.4 |
| History of CVDc (%) | 0.6 |
START: Strategic Timing of AntiRetroviral Treatment; SD: standard deviation; HIV: human immunodeficiency virus; IQR: interquartile range; CD4: CD4+ T lymphocyte; RNA: ribonucleic acid; BMI: body mass index; BP: blood pressure; CVD: cardiovascular disease; SNA: serious non-AIDS.
Average of screening/baseline values.
Documented in participant's record.
History of acute myocardial infarction, stroke or coronary revascularization.
Discussion
The optimal time at which the benefits of treatment of asymptomatic HIV infection outweigh its risks is unknown and has been debated for years [61–76]. There are few controlled trials of ART versus no ART. The Haiti trial [77], and the comparison of early versus deferred ART within a subgroup of the SMART study [29] examined deferral strategies to a CD4 count of 200 and 250, respectively. There are no data from randomized controlled trials at higher CD4 counts. Thus, treatment guidelines have had to rely on weaker forms of evidence (e.g., nonrandomized studies, studies without clinical outcomes and expert opinion). A careful weighing of the potential risks and benefits of initiating ART early by clinicians and patients (something recommended by all guidelines) is difficult if not impossible. Further, treatment guidelines do not take into account costs, and this complicates the development of public health policies. START is designed to address this knowledge gap by evaluating the impact of initiating ART early at a CD4 cell count > 500 cells/μL compared to deferring ART until the CD4 cell count drops to below 350 cells/μL.
Conducting a randomized trial to address the question of when to start ART in HIV-infected but asymptomatic individuals poses significant challenges in design and implementation. There are no reliable surrogate markers of morbidity and mortality that could be used as an outcome measure. High levels of CD4 in HIV-infected persons who have never used ART indicate that the immune system is still relatively intact. However, the clinical impact of increases in CD4 count and suppression of HIV RNA by ART in such persons is unknown, and there may be other effects of HIV and ART, which are not mediated through these pathways. Therefore, CD4 counts and HIV RNA levels cannot be used as surrogate markers for clinical outcomes. The only way to assess the impact of early ART compared to deferred ART is to use a clinical endpoint. Thus, the study needed to be large with extensive follow-up. As in all clinical trials with long follow-up in what has been a fast-moving field, an issue that has to be considered is whether the results from START would still be of relevance at the end of the trial. We consider that this will be the case because there is a far greater stability in treatment and clinical care of people with HIV than in past years. Treatment regimens are now highly successful in maintaining low levels of HIV viral load for many years in the majority of people and, in addition, new drugs are available for the minority of patients who develop drug resistance to the established regimens. The question of when to start ART has been debated for more than 20 years and will remain relevant unless a curative treatment is discovered.
A related issue often raised when designing trials with long follow-up comparing immediate versus deferred treatment is whether changes in clinical management and the introduction of new more potent drugs during the lifetime of the study would confound any treatment effect. The development of new antiretroviral drugs that result in significant improvements in levels of viral load suppression and/or greater increases in CD4 count is possible but, given the high rates of viral suppression achieved in most clinic populations, this seems unlikely. In any case, the use of potentially better drugs in the future by participants in the deferred ART group is one of the consequences of deferring initiation of ART and is part of the strategic question being addressed. Nevertheless, it would be prudent to explore in secondary analyses the contribution of any changes in the treatment environment on the observed effect of early ART compared to deferred ART. In secondary analyses, the START study will compare the early versus deferred ART strategies within subgroups by intended ART regimen (specified prior to randomization) and by calendar time of randomization. These analyses will help to evaluate possible contributions of changes in the ART environment on the treatment difference.
The clinical endpoints that have been used in HIV treatment trials to date are AIDS-defining conditions that are traditionally associated with immunosuppression. The risk of serious AIDS events is very low when the CD4 count is >500 cells/μL (about 6 per 1000 person-years (Appendix B)), and even a large relative reduction in this risk with early treatment would translate into a very modest absolute difference. The primary endpoint in START was chosen as a composite outcome measure of either serious AIDS or SNA conditions. SNA conditions are far more common than AIDS in individuals with high CD4 cell counts [13,78]. The START study is the only HIV treatment trial to our knowledge that has incorporated SNA conditions as part of the primary endpoint. This has posed challenges in estimating an adequate sample size because there are only sparse data from observational studies on the risk of SNA conditions in ART-naïve individuals with CD4 > 500 cells/μL. Data on these outcomes are not routinely collected in cohorts. Our estimates of the anticipated event rates are thus partly based on modeling and computer simulations using data from several large cohorts and from the SMART trial. All models make assumptions some of which are unverifiable, and it is therefore important to monitor these assumptions during follow-up. The study design incorporates a planned sample size reestimation before enrollment is completed. The reestimation will be based on the observed event rate pooled across the treatment arms and the hypothesized treatment difference so that no adjustment for the significance level in the final analysis would be necessary [79,80].
A number of hurdles needed to be overcome in the implementation of the START protocol. To ensure timely enrollment and the generalizability of the findings, the study is being conducted in many countries across six continents in settings with diverse health-care systems and regulatory requirements. The provision of donated antiretroviral drugs to study sites was essential to the success of the study, since many sites were hindered by local treatment guidelines or by lack of funding to access ART in other ways. Providing sites with an adequate supply of drugs required protracted negotiations with six pharmaceutical companies and the establishment of a drug distribution system with a private company. The diversity of the requirements for the conduct of clinical trials across countries and the lack of harmonization even between countries in the same region have all added complexities and costs to the conduct of START. Steps need to be taken to remove the impediments for implementing publicly funded international trials. Some recommended steps are given in Ref. 58.
All clinical trials, particularly those with long follow-up in a fast-moving area, need to respond in a timely manner to changes in the clinical environment, which occur during their conduct. Changes in treatment guidelines in some locations where the START study is conducted required such a response, which included timely communication with site investigators, ethics committees and institutional review boards, and community representatives.
In summary, the question of when ART should be initiated in HIV-1-infected but symptom-free individuals is one of the most important unanswered questions in HIV research. START is designed to evaluate the risks and benefits of starting ART earlier than is currently practiced, and its findings will inform treatment guidelines and public health policy worldwide. With the carefully collected clinical outcomes (AIDS and non-AIDS), substudies, and stored specimens for future nested case control studies, START promises to shed important light on the pathogenesis of HIV infection, the effects of HIV and HIV treatments on chronic immune activation and inflammation, and the important role of SNA conditions in those living with HIV.
Acknowledgements
We would like to thank the START participants without whom this work would not be possible.
Funding The START study is primarily funded by the US National Institutes of Health (NIH grant U01-AI068641) from the Division of Acquired Immunodeficiency Syndrome (DAIDS) of the National Institute of Allergy and Infectious Diseases (NIAID) for the main study; the Department of Bioethics at the NIH Clinical Center and five NIH institutes: National Cancer Institute (NCI); National Heart, Lung, and Blood Institute (NHLBI); National Institute of Mental Health (NIMH); National Institute of Neurological Disorders and Stroke (NINDS); and the National Institute of Arthritis and Musculoskeletal disorders (NIAMS). Financial support is also provided by the French Agence Nationale de Recherches sur le SIDA et les Hépatites Virales (ANRS), the German Ministry of Education and Research, the European AIDS Treatment Network (NEAT), the Australian National Health and Medical Research Council, and the UK Medical Research Council and National Institute for Heath Research. Six pharmaceutical companies (Abbott Laboratories, Inc.; Bristol-Myers Squibb, Gilead Sciences, Inc.; GlaxoSmithKline, Inc.; Merck & Co, Inc.; and Tibotec Pharmaceuticals, Ltd.) have donated over 20 antiretroviral drug formulations to the study's CDR.
Appendix A: distribution of CD4 cell count during follow-up
Introduction
In randomized clinical trials, it is important for treatments to be distinguishable from one another. In START, it was therefore important that the defined treatment strategies lead to differences in CD4 cell count over follow-up that would be projected to lead to clinically relevant differences in the primary endpoint between the two groups. We describe a simulation study we undertook to derive plausible distributions for CD4 cell count in the two groups during follow-up under realistic assumptions about the distribution of CD4 cell count at randomization and the evolution of CD4 count both before and after initiation of ART. We considered three scenarios for the deferred strategy—initiation of ART when CD4 cell count drops to below 350, 400, and 450 cells/µL, respectively.
We derived the distribution of CD4 cell count during follow-up in two steps. We first fitted two models for the evolution of CD4 count over time: model 1 (pre-ART model) for time before initiation of ART starting from the date of HIV seroconversion (SC) (which occurs on average about 1 month after infection with HIV); and model 2 (post-ART model) for time starting from the date of ART initiation. The parameters for these two models were estimated using data from the CASCADE study—a collaboration of 26 cohorts of individuals with well-estimated dates of HIV seroconversion [42]. Second, the distribution of CD4 cell count during follow-up in the early and deferred ART groups was derived by simulating trajectories based on models 1 and 2 conditional on the distribution of CD4 count at entry in START. Participants in START enter the study with a CD4 cell count >500 cells/µL, and within this range, we would expect eligible individuals with CD4 count closer to 500 cells/µL to be more likely to consent to participate in the study than those with much higher CD4 count. We therefore conservatively assumed that at baseline, 40%, 30%, 20%, and 10% of participants will have CD4 count between 500 and 549, between 550 and 599, between 600 and 699, and between 700 and 999 cells/µL, respectively. We describe the implementation of these steps and the way they were combined to simulate the CD4 cell count trajectories.
Modeling CD4 trajectory before and after ART initiation
CD4 counts are known to have substantial variability, and a major component of this variability is measurement error (variability of repeated assessments of the same sample). The measured (or observed) CD4 count can be viewed as the sum of the true underlying count and measurement error. While the clinical event rates were assumed to depend on the true CD4 count, initiation of ART in the deferred group is determined by the observed count. Let Xi(t) and Yi(t) be the true and observed square root CD4 for individual i at time t. Then
where {Ei(t); i = 1,–,I} are independent identically distributed random measurement errors assumed to have a normal distribution with mean of 0 and standard deviation σe. Standard mixed linear models (MLMs) assume that Xi(t) is a piecewise polynomial (usually linear or piecewise linear) function of time but with random coefficients varying between individuals. However, these models imply that within individuals, the true CD4 count tracks a simple curve perfectly over the entire follow-up period. Diggle [81] suggested the addition of a zero-mean stochastic process Wi(t) to provide stochastic variation around the within individual deterministic path suggested by the MLM for the true count. Taylor et al. [82] fitted this extended model to repeated observations on CD4 count in the Multicenter AIDS Cohort (MAC) data [83] with Wi(t) an integrated Ornstein–Uhlenbeck process [84], which includes Brownian motion as a special case. They used a fourth root transformation and found that a model with scaled Brownian motion fitted the data best.
We modeled CD4 count in a square root scale – a transformation suggested by Box–Cox. Time was measured in years, from HIV seroconversion for the CD4 trajectory before ART initiation (pre-ART) and from the time of ART initiation for the trajectory after initiation of ART (post-ART).
Pre-ART
Model 1
True square root CD4 follows an underlying linear decline plus a Brownian motion process
where A and B, the true square root CD4 at the time of HIV seroconversion and slope after seroconversion, respectively, are assumed to be jointly Gaussian with E(A) = α; var(A) = σa2; E(B) = β; var(B) = σb2 and corr(A,B) = rab; cov(A,B) = σab = rabσaσb; W(t) is a scaled Brownian motion process independent of A and B with W(0) = 0; var(W(t)) = δt and corr(W(t1),W(t2)) = min(t1/t2/(t1t2)1/2.
Thus, the population mean of Y(t), the observed square root CD4 at time t, is α + βt, with
The model parameters were estimated by maximum likelihood, and for ease of optimization, model parameters with restricted range were appropriately transformed (σa, σb, σe, and δ were log transformed and rab is arctanh transformed). Adequacy of the MLM is tested by setting δ = 0 and referring the change in −2 log-likelihood to the chi-square distribution with 1 degree of freedom.
Data for model 1
We used data available in CASCADE up to December 2007. All CD4 assessments made before the development of any AIDS-defining illness and before ART initiation were used to estimate model parameters. This resulted in 89,176 CD4 measurements from 15,274 individuals. The mean number of measurements per individual was 5.8 (SD: 6.5), median of 4 (IQR: 2–8), and range of 1–77.
Results
The log-likelihood for the full (extended) model, which includes the Brownian motion component, was −41068.41. Table A1 shows the parameter estimates of the extended model and their standard errors. This model suggests that without treatment, the average annual decline is 1.1 in the square root scale. In terms of absolute CD4 count, this translates into a drop of about 55 cells from 625 to 570 over 1 year. Note the negative correlation between the initial CD4 count at HIV seroconversion and the slope so that individuals with higher initial CD4 count would have a steeper decline.
Table A1.
Parameter estimates for CD4 trajectory before ART initiation under the full model
| Square root CD4 | Estimate | 95% Confidence interval |
|---|---|---|
| Fixed effects | ||
| Mean at SC (A) α | 23.75 | 23.64–23.85 |
| Mean slope (B) β | −1.10 | −1.13 to −1.06 |
| Random effects | ||
| SD(A) σa | 5.37 | 5.28–5.47 |
| SD(B) σb | 0.50 | 0.44–0.57 |
| Corr(A,B) rab | −0.92 | −0.97 to −0.75 |
| Brownian motion | ||
| Variance at 1 year δ | 6.89 | 6.62–7.16 |
| Measurement error | ||
| SD(E) σe | 2.26 | 2.24–2.28 |
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; SD: standard deviation.
For comparison, the log-likelihood for the reduced (standard mixed linear) model was −244149.34. The parameter estimates for the reduced model are given in Table A2.
Table A2.
Parameter estimates for CD4 trajectory before ART initiation under the reduced model
| Square root CD4 | Estimate | 95% Confidence interval |
|---|---|---|
| Fixed effects | ||
| Mean at SC (A) α | 24.14 | 24.03 24.25 |
| Mean slope (B) β | −1.35 | −1.38 to −1.32 |
| Random effects | ||
| SD(A) σa | 5.82 | 5.73 5.91 |
| SD(B) σb | 1.23 | 1.20–1.26 |
| Corr(A,B) rab | −0.38 | −0.41 to −0.36 |
| Measurement error | ||
| SD(E) σe | 2.78 | 2.76 2.79 |
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; SD: standard deviation.
Testing δ = 0
Difference in −2 log-likelihood between the full and reduced model was 6161.86, p < 10−300, providing very strong evidence for the lack of fit of the MLM compared to the full model with a Brownian motion component. Figure A1 shows the standardized residuals for the two models, plotted against CD4 count, and demonstrates the poor fit of the standard MLM.
Figure A1.

Standardized residuals for the standard mixed linear model and the extended model for square root CD4 before initiation of ART (15,274 participants in CASCADE).
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; CASCADE: Concerted Action for HIV SeroConversion and AIDS in Europe.
Predicting individual-specific `true' trajectory
Each individual's true root CD4 at HIV seroconversion and underlying slope are predicted by the best linear unbiased predictors (BLUPs) defined as the conditional mean of X(t) given the individual's observed CD4 counts. This is derived from the conditional mean of A and B, and for any given time t, W(t) is predicted from its conditional mean given the individual's observed CD4 counts. Individual-specific predicted values of A, B, and W(t) are then used to obtain predicted true square root CD4 for that individual through the model equation
Figures A2 and A3 display the distribution of the true square root CD4 at HIV seroconversion (A) and the within individual deterministic slope (B). While the standard MLM predicts that about 14% of individuals would have a positive slope (mean slope of −1.34 with SD of 1.23), this proportion in the full model is only 1%.
Figure A2.

Distribution of “true” square root CD4 at seroconversion (15,274 participants in CASCADE).
CD4: CD4+ T lymphocyte; CASCADE: Concerted Action for HIV SeroCon-version and AIDS in Europe.
Figure A3.

Distribution of the slope of square root CD4 before ART initiation (15,274 participants in CASCADE).
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; CASCADE: Concerted Action for HIV SeroConversion and AIDS in Europe.
Post-ART
Model 2
True square root CD4 follows an underlying path, which is piecewise linear with slope changing 1 year after initiation of ART plus a Brownian motion process. The slope change point at 1 year was chosen by maximizing the likelihood function over a range of values
where A, B, and C are the true square root CD4 at the time of ART initiation, slope during the first year after starting ART, and slope after the first year, respectively. A, B, and C are assumed to be jointly Gaussian with
where W(t) is a Brownian motion process independent of A, B, and C with W(0) = 0; var(W(t)) = δt; and corr(W(t1),W(t2)) = min(t1,t2)/(t1t2)1/2.
Adequacy of the MLM is tested by setting δ = 0 and referring the change in −2 log-likelihood to the chi-square distribution with 1 degree of freedom.
Data for model 2
All CD4 assessments made after starting ART in individuals in CASCADE starting combination ART (cART) when ART-naïve after December 1996 were used to estimate the model parameters. Follow-up is censored at 6 years after ART initiation. There were 65,137 CD4 cell count measurements from 5147 individuals. Mean number of measurements was 12.7 (SD 9.6) with median of 11 (IQR: 5–18) and range of 1–106.
Results
The log-likelihood for the full and reduced (MLM) models was −168110.81 and −170974.66, respectively. Parameter estimates for the full and reduced models are shown in Tables A3 and A4. Under the full model, the maximum likelihood estimate of corr(A,C) = −1 so that the slope after 1 year is a linear function of the baseline value (square root CD4 at ART initiation). Writing C − γ = −ψ(A − α), the model parameters are reduced to α, β, γ, σa, σb, rab, ψ, δ, σe, with
We note the strong negative correlation between baseline CD4 count (A) and both initial gradient of the rise during the first year (B) and subsequent smaller rise after the first year (C). Thus, although both early and later gradients are positive on average, there are more likely to be smaller (and for later gradient even negative) for those starting ART at very high CD4 cell count. This is probably a reflection of the fact that in the CASCADE study, individuals who started ART at high CD4 count were more likely to stop or interrupt treatment than those who started at a lower CD4 count.
Table A3.
Parameter estimates for the CD4 trajectory after ART initiation under the full model
| Square root CD4 | Estimate | 95% Confidence interval |
|---|---|---|
| Fixed effects | ||
| Mean baseline (A) α | 19.69 | 19.52–19.86 |
| Mean slope 1 (B) β | 2.93 | 2.80–3.07 |
| Mean slope 2 (C) γ | 0.10 | 0.04–0.16 |
| Random effects | ||
| SD(A) σa | 5.71 | 5.59–5.84 |
| SD(B) σb | 2.06 | 1.85–2.30 |
| Corr(A,B) rab | −0.44 | −0.50 to −0.37 |
| SD(C)/SD(A) ψ | 0.10 | 0.09–0.11 |
| Brownian motion | ||
| Variance at 1 year δ | 7.83 | 7.52–8.15 |
| Measurement error | ||
| SD(E) σe | 2.19 | 2.17–2.21 |
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; SD: standard deviation.
Table A4.
Parameter estimates for CD4 trajectory after ART initiation under the reduced mixed linear model
| Square root CD4 | Estimate | 95% Confidence interval |
|---|---|---|
| Fixed effects | ||
| Mean at SC α | 19.81 | 19.64–19.98 |
| Mean slope 1 β | 2.88 | 2.74–3.03 |
| Mean slope 2 γ | 0.09 | 0.03–0.14 |
| Random effects | ||
| SD(A) σa | 5.70 | 5.57–5.84 |
| SD(B) σb | 3.90 | 3.77–4.03 |
| SD(C) σc | 1.45 | 1.40–1.50 |
| Corr(A,B) rab | −0.24 | −0.28 to −0.21 |
| Corr(A,C) rac | −0.41 | −0.45 to −0.38 |
| Corr(B,C) rbc | −0.02 | −0.06 to −0.03 |
| Measurement error | ||
| SD(E) σe | 2.66 | 2.64–2.67 |
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; SD: standard deviation.
We also note that the two models are no longer nested. However, the model with Brownian motion has greater log-likelihood (−168110.81) and fewer parameters (nine parameters) compared to the mixed linear model with 10 parameters and log likelihood (−170974.66).
Simulation of CD4 count during follow-up
We simulated CD4 trajectories for 50,000 participants with 10,000 on no treatment (No ART) throughout follow-up of 6 years, 10,000 following the early ART strategy where ART is initiated at entry into the study, and 30,000 following three deferral strategies with 10,000 participants in each group. Deferral strategy 1 (Def1) would start ART as soon as CD4 count is observed to fall below 350 cells/μL and is confirmed 1 month later. Def2 and Def3 would start ART when the observed CD4 count had fallen below 400 and below 450, respectively—also confirmed 1 month later. The purpose of introducing Def2 and Def3 is to take account of the proportion of participants in the deferred ART group who would initiate ART at a CD4 count >350 cells/μL either because of diagnosis of an AIDS-defining illness as is allowed in the study protocol or for any other reason by considering a weighted average of key design parameters (changes in CD4 cell count and primary event rates) in Def1, Def2, and Def3. We also considered two different scenarios for the frequency of regular study visits (four monthly or three monthly) to assess the impact of the frequency of measurements of CD4 cell counts. We present the results assuming four monthly visits (the three monthly frequency of visits resulted in a very small reduction in time to ART initiation in the deferred ART group). The observed CD4 counts at baseline were generated so that 40% would have baseline count ≥500 and <549, 30% have 550–599, 20% have 600–699, and 10% have 700–999. For each participant, the true CD4 cell count was drawn from the conditional distribution of X(t0) in model 1 given the participant's observed count, where t0 is time since HIV seroconversion at enrollment. We estimated t0 by the mean time since HIV seroconversion for the categories of observed CD4: 500–499, 550–599, 600–648, 650–699, 700–799, and 800–999, provided by the CASCADE data used in fitting model 1. The distribution of time since HIV seroconversion by observed CD4 count is given in Table A5. For observed CD4 count between 500 and 999, mean time since HIV seroconversion ranges between 2.92 and 3.28 years and the median between 1.93 and 2.43 years.
Table A5.
Distribution of time since HIV seroconversion by observed CD4 count
| CD4 category | N | Mean | SD | Median | IQR |
|---|---|---|---|---|---|
| 500–549 | 7097 | 3.28 | 2.95 | 2.43 | 1.13–4.54 |
| 550–599 | 5855 | 3.24 | 2.95 | 2.34 | 1.10–4.46 |
| 600–649 | 5165 | 3.14 | 2.96 | 2.17 | 1.05–4.30 |
| 650–699 | 4152 | 2.99 | 2.85 | 2.08 | 0.97–4.15 |
| 700–799 | 6229 | 3.07 | 2.87 | 2.17 | 0.99–4.29 |
| 800–999 | 6652 | 2.92 | 2.87 | 1.93 | 0.87–4.02 |
HIV: human immunodeficiency virus; SD: standard deviation; IQR: interquartile range.
The distributions of true and observed CD4 cell counts at baseline are summarized in Table A6. At study entry, true CD4 count has mean 578 and standard deviation 134 cells/μL. Because of measurement error and the entry criterion (CD4 count >500), the mean of the observed count is higher than the mean of the true count by about 20 cells/μL.
Table A6.
Distribution of CD4 cell count at baseline
| Cells per microliter | True CD4 count | Observed CD4 count |
|---|---|---|
| <200 | <0.1% | 0.0% |
| 200–349 | 1.7% | 0.0% |
| 350–499 | 28.1% | 0.0% |
| 500–649 | 44.9% | 79.9% |
| ≥650 | 25.2% | 28.1% |
| Mean (SD) | 577.5 (133.7) | 597.2 (101.6) |
| Median (IQR) | 561 (485–651) | 566 (531–624) |
CD4: CD4+ T lymphocyte; SD: standard deviation; IQR: interquartile range.
Baseline true and observed CD4 provided initial values to simulate CD4 counts at every month during follow-up (months 1–72) using model 1 for the No ART group throughout follow-up and for Def1–Def3 until the time of ART initiation. Model 2 was used for the early ART group throughout follow-up and for Def1–Def3 after ART initiation. The month of ART initiation was defined for each participant in the deferred groups as the second of two consecutive months (the first of which coincided with a regular study visit) at which CD4 count was less than the CD4 cutoff defining the deferral strategy. Figure A4 shows the Kaplan–Meier plots for time to ART initiation according to the deferral strategy. For Def1, where ART is initiated when CD4 count drops to <350 cells/μL, median time to ART initiation was 41 months with lower quartile 21 months and upper quartile >72 months. For Def2 and Def3, where ART is initiated when CD4 count drops to <400 and to <450 cells/μL, the median (IQR) was 29 (13–61) and 21 (9–45) months, respectively.
Figure A4.

Kaplan–Meier plots for time to initiation of ART.
ART: antiretroviral treatment; CD4: CD4+ T lymphocyte.
The mean and standard deviation of true CD4 count at annual visits for the four treatment strategies are listed in Table A7. Figure A5 shows the median of the true CD4 count during follow-up. The largest difference between early ART and any of the three deferral strategies occurs during the second year of follow-up at months 18, 15, and 12 for Def1, Def2, and Def3, respectively, and is reduced after this time as more patients initiate treatment. One year after randomization, median true CD4 count in early ART was higher than in the No ART, Def1, Def2, and Def3 strategies by 175, 171, 165, and 156 cells/μL, respectively. At year 6, the mean differences were 337, 123, 84, and 60 cells/μL, respectively..
Table A7.
Mean (standard deviation) for true CD4 cell count during follow-up
| Year | Early ART | Def1a | Def2b | Def3c | No ART |
|---|---|---|---|---|---|
| 1 | 700 (216) | 532 (166) | 538 (162) | 546 (159) | 525 (172) |
| 2 | 686 (251) | 515 (178) | 534 (177) | 559 (178) | 478 (196) |
| 3 | 675 (283) | 509 (192) | 535 (198) | 564 (205) | 432 (216) |
| 4 | 665 (311) | 509 (209) | 540 (220) | 572 (234) | 390 (230) |
| 5 | 656 (334) | 515 (228) | 547 (243) | 580 (262) | 352 (239) |
| 6 | 648 (357) | 524 (250) | 557 (268) | 587 (287) | 317 (246) |
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; Def: deferral strategy; No ART: no treatment.
Defer ART until CD4 <350 cells/μL.
Defer ART until CD4 <400 cells/μL.
Defer ART until CD4 <450 cells/μL.
Figure A5.

Median “true” CD4 count during follow-up according to ART deferral strategy.
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment.
Appendix B: sample size determination
Introduction
START is the first HIV treatment trial to incorporate SNA events as part of the primary endpoint, and one of the challenges we faced in the design is that there are no data from observational studies to provide any guidance on the rate of these events in the target populations for START as these events are not systematically collected in cohorts. More generally, there is little information on morbidity and mortality in patients starting ART at a CD4 count > 500 cells/μL. Our assumptions about the event rates were arrived at in several steps. First, we fitted two models for the evolution of CD4 count over time before and after ART initiation and estimated the parameters of these models as described in Appendix A. Second, we estimated the `true' CD4 counts corresponding to the observed (measured) CD4 counts used in the estimation by their BLUPs. If Y(t) is the observed CD4 count at time t and X(t) is the corresponding `true' (unobservable) count, the BLUP of Y(t) is the conditional mean of X(t) given the observed value Y(t). Third, we estimated the relationship between latest (`true' and observed) CD4 count and rates of serious AIDS (including AIDS-related death), death from any cause, and non-AIDS-related death using data from CASCADE [42] (and UK CHIC [10] for serious AIDS or death by latest observed CD4 only). This relationship was estimated separately before and after ART initiation. The rate of SNA events was assumed to be a constant multiple of the rate of non-AIDS-related death, regardless of the level of current CD4 count. We describe the implementation of the third step (steps 1 and 2 are detailed in Appendix A) and the way these steps were combined to estimate the primary event rate during follow-up. We assumed that the event rates are determined by current `true' CD4 count, and that the relationship between current CD4 count and the event rates after ART initiation is the same in the early and deferred treatment groups.
Event rates by latest CD4 count
CASCADE is a collaboration of 26 cohorts of HIV-infected individuals in whom the time of HIV sero-conversion is well estimated. The data contain clinical information including CD4 count, vital status, and cause of death. For the purpose of assessing the rate of clinical events (serious AIDS, all-cause mortality and non-AIDS-related death) by latest CD4 count, only CD4 counts that were measured while the participant was ART naïve or after initiation of ART if the participant had started cART after 1996 and was ART naïve at the time of starting cART were used. Separate analyses were performed for the two periods before (pre-ART) and after ART initiation (post-ART). Each participant's follow-up was divided into time spans with each span starting at the date of a CD4 assessment and ending at the earliest of the date of the next CD4 assessment, 6 months after the initial CD4 assessment, at the time of last follow-up, or the development of the clinical event under study. The time spans were grouped, according to the CD4 count at the start of the span, into the following four categories: <200, 200–350, 350–500, and ≥500. The rates within CD4 categories were calculated by aggregating the number of events and person-years within each category. The estimated rates are given in Table B1.
Table B1.
Event rates by observed latest CD4 count
| ART status | Outcome | CD4 cat | Mean CD4 | Events | Person-years | Rate (per 100 person-years) |
|---|---|---|---|---|---|---|
| Pre-ART | Death | <200 | 131.1 | 147 | 1570.8 | 9.36 |
| 200–349 | 287.4 | 47 | 5085.9 | 0.92 | ||
| 350–499 | 424.3 | 50 | 8237.0 | 0.61 | ||
| ≥500 | 742.9 | 57 | 15291.7 | 0.37 | ||
| Serious AIDS | <200 | 130.1 | 372 | 1617.7 | 23.00 | |
| 200–349 | 287.4 | 172 | 5135.6 | 3.35 | ||
| 350–499 | 424.2 | 111 | 8240.1 | 1.35 | ||
| ≥500 | 742.9 | 95 | 15257.0 | 0.62 | ||
| Non-AIDS death | <200 | 131.1 | 62 | 1570.8 | 3.95 | |
| 200–349 | 287.4 | 38 | 5085.9 | 0.75 | ||
| 350–499 | 424.3 | 46 | 8237.0 | 0.56 | ||
| ≥500 | 742.9 | 47 | 15291.7 | 0.31 | ||
| Post-ART | Death | <200 | 119.3 | 57 | 1327.2 | 4.29 |
| 200–349 | 281.7 | 22 | 2624.0 | 0.84 | ||
| 350–499 | 423.6 | 15 | 3532.3 | 0.42 | ||
| ≥500 | 768.9 | 25 | 8424.9 | 0.30 | ||
| Serious AIDS | <200 | 126.7 | 74 | 907.1 | 8.16 | |
| 200–349 | 282.8 | 38 | 2284.1 | 1.66 | ||
| 350–499 | 423.9 | 18 | 3228.4 | 0.56 | ||
| ≥500 | 771.6 | 21 | 7964.3 | 0.26 | ||
| Non-AIDS death | <200 | 119.3 | 32 | 1327.2 | 2.41 | |
| 200–349 | 281.7 | 20 | 2624.0 | 0.76 | ||
| 350–499 | 423.6 | 15 | 3532.3 | 0.42 | ||
| ≥500 | 768.9 | 23 | 8424.9 | 0.27 |
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment.
For event rates by latest `true' CD4 counts, the same methods as for observed CD4 count were used. The `true' counts were calculated from the models used for the CD4 trajectory described in Appendix A. The results are presented in Table B2.
Table B2.
Event rates by true latest CD4 count
| ART status | Outcome | CD4 cat | Mean CD4 | Events | Person-years | Rate (per 100 person-years) |
|---|---|---|---|---|---|---|
| Pre-ART | Death | <200 | 138.0 | 134 | 1336.3 | 10.03 |
| 200–349 | 291.4 | 54 | 4799.6 | 1.13 | ||
| 350–499 | 426.3 | 45 | 8971.0 | 0.50 | ||
| ≥500 | 711.8 | 68 | 15078.5 | 0.45 | ||
| Serious AIDS | <200 | 138.0 | 339 | 1376.2 | 24.36 | |
| 200–349 | 291.3 | 201 | 4854.3 | 4.14 | ||
| 350–499 | 426.3 | 116 | 8976.4 | 1.29 | ||
| ≥500 | 711.8 | 94 | 15042.9 | 0.62 | ||
| Non-AIDS death | <200 | 138.0 | 54 | 1336.3 | 4.04 | |
| 200–349 | 291.4 | 41 | 4799.6 | 0.85 | ||
| 350–499 | 426.3 | 40 | 8971.0 | 0.45 | ||
| ≥500 | 711.8 | 58 | 15078.5 | 0.38 | ||
| Post-ART | Death | <200 | 124.3 | 53 | 1181.7 | 4.49 |
| 200–349 | 285.4 | 21 | 2534.9 | 0.83 | ||
| 350–499 | 425.0 | 22 | 3752.4 | 0.59 | ||
| ≥500 | 743.9 | 23 | 8439.5 | 0.27 | ||
| Serious AIDS | <200 | 131.5 | 70 | 774.6 | 9.04 | |
| 200–349 | 287.0 | 38 | 2175.7 | 1.75 | ||
| 350–499 | 425.3 | 23 | 3435.3 | 0.67 | ||
| ≥500 | 746.1 | 20 | 7998.3 | 0.25 | ||
| Non-AIDS death | <200 | 124.3 | 29 | 1181.7 | 2.45 | |
| 200–349 | 285.4 | 18 | 2534.9 | 0.71 | ||
| 350–499 | 425.0 | 21 | 3752.4 | 0.56 | ||
| ≥500 | 743.9 | 22 | 8439.5 | 0.26 |
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment.
Expected event rates in START
As described in Appendix A, we simulated CD4 trajectories for 50,000 participants with 10,000 on No ART throughout follow-up of 6 years, 10,000 following the early ART strategy where ART is initiated at entry into the study, and 30,000 following three deferral strategies with 10,000 participants in each group. Def1 would start ART as soon as CD4 count is observed to fall below 350 cells/μL and is confirmed 1 moth later. Def2 and Def3 would start ART when the observed CD4 count had fallen below 400 and 450, respectively – also confirmed 1 month later. The purpose of introducing Def2 and Def3 is to take account of the proportion of participants in the deferred ART group who would initiate ART at a CD4 count > 350 cells/μL either because of diagnosis of an AIDS-defining illness as is allowed in the study protocol or for any other reason by considering a weighted average of key design parameters (changes on CD4 cell count and primary event rates) in Def1, Def2, and Def3. Two different scenarios were considered for the frequency of study visits (four monthly or three monthly).
To calculate the hazards for the clinical events (serious AIDS, all-cause death, and non-AIDS death) at every month of follow-up, we imputed the event rates at any `true' CD4 count assuming that within each CD4 category (<200, 200–349, 350–499, or ≥500), the log rate is piecewise linear in true CD4 and that the rates given in Table B2 apply to the time-averaged CD4 in the category. For each participant and at every month during follow-up, the hazard of the composite primary endpoint was calculated under the following scenarios:
The ratio of the rate of a nonfatal serious non-OD event relative to the rate of non-AIDS death was 3, 4, or 5.
The proportion of serious AIDS events accepted after review by the Endpoint Review Committee (ERC) was 65%, 70%, or 75%.
The proportion of nonfatal serious non-AIDS events accepted after review by the ERC was 80% or 90%.
The ranges considered in the above scenarios are consistent with what was observed in the SMART, ESPRIT, and Subcutaneous Recombinant, Human Interleukin-2 in HIV-infected Patients with Low CD4+ Counts Under Active Antiretroviral Therapy (SILCAAT) studies and reported by the INSIGHT Endpoint Review Committee [13,39,40].
The primary endpoint for START is a composite of two major components – serious AIDS (including AIDS-related death) and SNA (including non-AIDS related death). A small number of participants would be expected to develop events in both components. The proportion of these participants among all participants reaching the primary endpoint was set to be either 5% or 10%.
A weighted average of the cumulative probability of reaching the primary endpoint (the cumulative distribution function (CDF)) in Def1, Def2, and Def3, with weights of 0.7, 0.2, and 0.1, respectively, was used to estimate the distribution of the primary outcome and its components in the deferred group of START. Tables B3, B4 and B5 show the predicted average rate of the primary endpoint and two of its major components.
Table B3.
Average rate by year of follow-up for the composite primary endpoint from any cause according to the frequency of regular visits
| Year | Three monthly follow-up |
Four monthly follow-up |
||
|---|---|---|---|---|
| Rate per 100 person-years | Hazard ratio | Rate per 100 person-years | Hazard ratio | |
| 1 | 2.576 | 0.724 | 2.597 | 0.719 |
| 2 | 2.759 | 0.635 | 2.816 | 0.622 |
| 3 | 2.856 | 0.682 | 2.898 | 0.675 |
| 4 | 2.967 | 0.743 | 2.998 | 0.732 |
| 5 | 3.063 | 0.803 | 3.100 | 0.797 |
| 6 | 3.169 | 0.860 | 3.179 | 0.867 |
Table B4.
Average rate by year of follow-up for serious AIDS according to the frequency of regular visits
| Year | Three monthly follow-up |
Four monthly follow-up |
||
|---|---|---|---|---|
| Rate per 100 person-years | Hazard ratio | Rate per 100 person-years | Hazard ratio | |
| 1 | 0.631 | 0.460 | 0.645 | 0.448 |
| 2 | 0.666 | 0.436 | 0.701 | 0.407 |
| 3 | 0.683 | 0.540 | 0.709 | 0.516 |
| 4 | 0.723 | 0.678 | 0.742 | 0.638 |
| 5 | 0.779 | 0.807 | 0.793 | 0.779 |
| 6 | 0.857 | 0.915 | 0.850 | 0.928 |
AIDS: acquired immunodeficiency syndrome.
Table B5.
Average rate by year of follow-up for serious non-AIDS according to the frequency of regular visits
| Year | Three monthly follow-up |
Four monthly follow-up |
||
|---|---|---|---|---|
| Rate per 100 person-years | Hazard ratio | Rate per 100 person-years | Hazard ratio | |
| 1 | 2.076 | 0.805 | 2.084 | 0.802 |
| 2 | 2.236 | 0.695 | 2.261 | 0.688 |
| 3 | 2.329 | 0.726 | 2.347 | 0.725 |
| 4 | 2.422 | 0.771 | 2.433 | 0.767 |
| 5 | 2.498 | 0.818 | 2.517 | 0.816 |
| 6 | 2.579 | 0.866 | 2.582 | 0.874 |
AIDS: acquired immunodeficiency syndrome.
The cumulative hazard and the CDF of time to the primary endpoint were calculated for each participant at each month of follow-up. The CDF was then averaged across participants at each month of follow-up for each of the five groups. Figure B1 displays the CDF by group under the scenario of 4-month follow-up, rate of nonfatal serious non-OD events was four times the rate of non-AIDS death, 65% of serious AIDS events and 90% of SNA events accepted by ERC, and 5% of participants experiencing both AIDS and SNA.
Figure B1.

Cumulative probability of reaching the primary endpoint according to three different strategies for deferring ART compared to immediate ART and no ART.
ART: antiretroviral treatment; CD4: CD4+ T lymphocyte.
Figure B2 displays the expected CDF in the two treatment groups in START under the previous scenario. This was used to calculate the sample size under the following further assumptions:
Enrollment over 3 years and minimum follow-up 3 years.
Cumulative loss to follow-up of 15% at 6 years.
Two-sided α = 0.05.
Power = 0.90.
Figure B2.

Cumulative probability of reaching the primary endpoint by treatment group compared to no ART.
ART: antiretroviral treatment.
Appendix C: CD4 count at ART initiation in the deferred group of START
Background and methods
The intention of the START protocol is to initiate treatment in the deferred ART group as soon as CD4 count drops below 350 cells/μL so that CD4 count at ART initiation would be below, but close to, 350. As the decision to initiate ART is based on the measured (observed) CD4 cell count, which is measured with error, the observed CD4 count at ART initiation would be expected to underestimate the true count. We explore the extent of this bias using data from the UK CHIC study [54] as well as the simulated data used in estimating the sample size for START. According to the START protocol, participants in the deferred ART group are to initiate ART as soon as CD4 cell count is observed to fall below 350 cells/μL and the measurement is confirmed within 4 weeks. Using data derived from a subset of participants in the UK CHIC study with first CD4 count > 500 while they were ART naïve, we describe the distribution of the measured CD4 cell count at the time when it first dropped below 350 as well as at the first time two consecutive counts were observed to be below 350. We then use the models and simulations results described in Appendix A for the trajectories of true and observed CD4 cell counts of participants in the deferred ART group to estimate the bias in CD4 cell count measured at the time of ART initiation.
Data from the UK CHIC study
Table C1 shows the median value of CD4 count around the time when CD4 drops to below 350. The median of the second of two consecutive values < 350 is 280. These data are based on CD4 counts measured a median of 91 days apart (from last value > 350 to first value < 350); max 183. These findings will therefore not be strictly relevant to the situation where patients are being closely monitored as they approach CD4 350.
Table C1.
Analysis of reaching CD4 count of 350 threshold in UK CHIC study
| Definition of level reaching <350 | Which CD4 count? | Number of patients | Median CD4 count (per mm3) |
|---|---|---|---|
| Two consecutive values <350 while ART naïve | First of the two values <350 | 1118 | 298 |
| Second of the two values <350 | 1118 | 280 | |
| Value after two consecutive values <350 | 1081 | 286 | |
| Second value after two consecutive values <350 | 1037 | 290 | |
| One value <350 while remaining ART naïve | First value <350 | 1656 | 304 |
| Value after first value <350 | 1603 | 350 | |
| Second value <350 | 1534 | 344 |
CD4: CD4+ T lymphocyte; UK CHIC: U.K. Collaborative HIV Cohort; ART: antiretroviral treatment.
Interpretation of what the median value is likely to be in START, where participants will be monitored more regularly as they approach 350, and also the confirmatory value will be done within a short period, is not straightforward and depends on whether the variability in the measured CD4 count reflects variability in the true underlying CD4 count or not. If much of the variability in CD4 count reflects real underlying changes, then there may be real increases in underlying CD4 count after first falling below 350.
On the other hand, if a substantial part of the variability in measured (observed) CD4 counts reflects measurement error, the apparent increase in observed CD4 after first falling below the threshold is not real and is due to the inherent bias caused by measurement error and the deferral strategy based on observed counts (regression to the mean).
Bias in measured CD4 at ART initiation as an estimate of the true count
Measurement error is equally likely to be positive or negative and is zero on average. Consequently, the observed value of CD4 is equally likely to be above or below the true value and on average equals the true value (i.e., an unbiased estimate of the true count). However, this would no longer be the case if the measured CD4 is restricted to values falling below a threshold (350). In this case, the measurement error would more likely be negative than positive, and the observed CD4 would be biased downward. The magnitude of such a bias depends on the amount of variability (e.g., SD) of the measurement error. The larger the variability in the measurement error the larger is the bias. This could explain the apparent increase in the median CD4 count following the value first observed to be below 350 (from 304 to 350) in the UK CHIC data. The alternative explanation (if we were to assume little measurement error) is that there is a real increase in the underlying true CD4 count following a fall below the threshold.
Simulated data – methods
The simulations that were performed to estimate the CD4 trajectories in the deferred ART group are described in Appendix A and are used here to explore the impact of various scenarios of the implementation of the protocol vis-à-vis ART initiation in the deferred group on the distribution of CD4 at ART initiation. The scenarios considered are as follows:
Frequency of regular visits: four monthly or three monthly (only the former is relevant to START).
Frequency of additional CD4 assessments during the month following a CD4 < 350: once, twice, or four times.
ART initiated after two counts < 350 within 1 month: whether these two counts are consecutive or not.
Simulated data – results
About two-thirds of participants in the deferred group are predicted to meet the CD4 criteria for starting ART during the study. This varies between 62% (4-monthly regular visits, one additional visit after one month following CD4 < 350) and 70% (3-monthly regular visits, four additional visits in the month following any CD4 < 350 at a regular visit, and confirmatory count not consecutive). Those who start ART would do so after an average time (from randomization), which varies between 25 and 28 months (4-monthly regular visits) and between 23 and 27 months (3-monthly regular visits).
Table C2 gives median and IQR of CD4 at ART initiation for the 10 different scenarios. Median observed CD4 count varies between 278 and 291 for 4-monthly regular visits and between 282 and 295 for 3-monthly regular visits. The median true CD4 count is consistently higher than the observed value demonstrating the downward bias in the observed count due to the variability in measurement error. For example, for 4-monthly regular visits, four additional visits, and confirmatory count not necessarily consecutive, the median observed CD4 at ART initiation is biased downward by 53 (345–291) cells/μL.
Table C2.
CD4 count at ART initiation at second observed count <350a (simulated data)
| Frequency of regular visits | CD4 frequency during month after first CD4 <350 | Consecutive? | % Start ART | Median count (IQR) |
|
|---|---|---|---|---|---|
| Observed CD4 | True CD4 | ||||
| Four monthly | Once | — | 62 | 278 (232–315) | 311 (270–354) |
| Twice | Yes | 62 | 278 (232–315) | 313 (273–355) | |
| No | 66 | 286 (243–320) | 331 (290–371) | ||
| Four times | Yes | 65 | 284 (241–318) | 325 (287–363) | |
| No | 68 | 291 (250–323) | 345 (304–386) | ||
| Three monthly | Once | — | 63 | 282 (236–318) | 320 (281–362) |
| Twice | Yes | 64 | 283 (238–318) | 323 (283–364) | |
| No | 68 | 290 (248–322) | 340 (302–380) | ||
| Four times | Yes | 66 | 287 (245–321) | 334 (297–372) | |
| No | 70 | 295 (255–324) | 356 (317–395) | ||
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; IQR: interquartile range.
Occurring on average between 25 and 28 months from randomization in those initiating ART during follow-up (approximately two-thirds of participants).
Table C3 shows that the median of the first of the two values < 350 at ART initiation is only slightly higher than the confirmatory value, which is not surprising given the short time between the two assessments.
Table C3.
CD4 count at first observed count <350a (simulated data)
| Frequency of regular visits | CD4 frequency during month after first CD4 <350 | Consecutive? | % Start ART | Median count (IQR) |
|
|---|---|---|---|---|---|
| Observed CD4 | True CD4 | ||||
| Four monthly | Once | — | 62 | 283 (237–319) | 328 (280–362) |
| Twice | Yes | 62 | 281 (235–316) | 318 (277–360) | |
| No | 66 | 289 (246–322) | 336 (296–378) | ||
| Four times | Yes | 65 | 285 (240–319) | 326 (290–365) | |
| No | 68 | 293 (252–325) | 350 (307–391) | ||
| Three monthly | Once | — | 63 | 285 (242–319) | 329 (291–370) |
| Twice | Yes | 64 | 285 (242–319) | 328 (289–368) | |
| No | 68 | 292 (251–323) | 347 (309–387) | ||
| Four times | Yes | 66 | 290 (247–321) | 337 (301–373) | |
| No | 70 | 297 (257–326) | 361 (321–402) | ||
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; IQR: interquartile range.
Occurring on average between 10 days and 1 month before ART initiation in those initiating ART during follow-up (approximately two-thirds of participants).
Table C4 gives the distribution of CD4 at the assessment immediately before (3–4 months before) the first of the two counts triggering ART initiation. The median of the observed value at this assessment ranges between 421 and 455 (4-monthly regular visits) and between 418 and 452 (3-monthly regular visits). Comparison with the true value shows that the observed value is biased upward, not downward as in Tables C2 and C3, because of the requirement that it should be greater than or equal to the threshold of 350.
Table C4.
CD4 count at the assessment immediately before first observed count <350a (simulated data)
| Frequency of regular visits | CD4 frequency during month after first CD4 <350 | Consecutive? | % Start ART | Median count (IQR) |
|
|---|---|---|---|---|---|
| Observed CD4 | True CD4 | ||||
| Four monthly | Once | — | 62 | 433 (387–501) | 378 (335–425) |
| Twice | Yes | 62 | 421 (371–496) | 374 (331–422) | |
| No | 66 | 445 (393–514) | 396 (354–442) | ||
| Four times | Yes | 65 | 425 (380–494) | 372 (334–419) | |
| No | 68 | 455 (399–522) | 410 (366–455) | ||
| Three monthly | Once | Yes | 63 | 429 (387–493) | 376 (335–418) |
| Twice | Yes | 64 | 418 (371–489) | 374 (333–419) | |
| No | 68 | 442 (393–511) | 395 (356–439) | ||
| Four times | Yes | 66 | 424 (381–490) | 375 (338–417) | |
| No | 70 | 452 (399–523) | 410 (370–455) | ||
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; IQR: interquartile range.
Occurring on average between 3 and 5 months before ART initiation in those initiating ART during follow-up (approximately two-thirds of participants).
The distribution of CD4 count at the visit following the confirmatory value (3–4 months after the confirmatory value), if ART had not been started, is summarized in Table C5. Because there is no threshold condition, the observed count is very close to the true count (i.e., there is no bias resulting from measurement error since there is no selection). For 4-monthly regular visits and four additional visits, the median value is 330 (observed) and 332 (true).
Table C5.
CD4 count at the assessment immediately after second observed count <350a (simulated data)
| Frequency of regular visits | CD4 frequency during month after first CD4 <350 | Consecutive? | % Start ART | Median count (IQR) |
|
|---|---|---|---|---|---|
| Observed CD4 | True CD4 | ||||
| Four monthly | Once | — | 59 | 299 (229–378) | 299 (249–355) |
| Twice | Yes | 59 | 300 (229–380) | 300 (248–356) | |
| No | 63 | 317 (241–400) | 317 (366–373) | ||
| Four times | Yes | 62 | 310 (238–393) | 310 (260–365) | |
| No | 66 | 330 (254–414) | 332 (278–388) | ||
| Three monthly | Once | Yes | 62 | 312 (244–391) | 312 (267–362) |
| Twice | Yes | 62 | 312 (244–391) | 314 (266–366) | |
| No | 66 | 329 (261–410) | 330 (284–380) | ||
| Four times | Yes | 64 | 324 (255–401) | 324 (278–372) | |
| No | 69 | 345 (274–424) | 346 (297–396) | ||
CD4: CD4+ T lymphocyte; ART: antiretroviral treatment; IQR: interquartile range.
CD4 count about 3.3 months after second measured CD4 <350 if ART was not initiated.
Finally, for comparison with UK CHIC data, the median of the first simulated observed count < 350 (not confirmed) and measured at a 3-monthly regular visit was 285 (IQR: 242–319) (Table C2). The corresponding median from the UK CHIC analysis was 304 (Table C1).
Conclusions
There is substantial variability in measured (observed) CD4 count including measurement error.
Median measured CD4 at ART initiation is likely to be much lower than 350 cells/μL. Simulated data suggest a value of about 290 for weekly assessments to confirm a drop to <350.
However, the measured value is downwardly biased by about 50 cells. The true value is about 345 for weekly confirmatory assessments (about 330 for confirmatory assessments every 2 weeks).
This is supported by a high value of median CD4 at the assessment following the second of two values < 350.
Addendum
Since submission of this manuscript the results of a large trial (HPTN 052), which studied 1763 HIV sero-discordant couples in whom the HIV positive partner had a CD4 count between 350 and 550 cells/μL, were released [85]. This trial compared immediate initiation of ART (Early ART) versus delaying ART until the CD4 count is between 200 and 250 cells/μL (Delayed ART) and showed a large reduction (96%) in the risk of transmission of HIV to the HIV negative partner and a significant reduction in the risk of clinical disease progression events (predominantly in extra-pulmonary tuberculosis). These results provide the first randomized evidence that ART can be used to reduce the transmission of HIV. They also reaffirm the findings from the Haiti trial [78] that ART should be initiated before CD4 count decline to a level below 200–250 cells/μL and underscores the importance of the question addressed by START.
Footnotes
Protocol Team: James D Neaton (INSIGHT PI), Abdel G Babiker (cochair), Sean Emery (cochair), Fred M Gordin (cochair), Jens D Lundgren (cochair), Brian K Agan, Jose R Arribas, John Baxter, William Burman, David A Cooper, Richard T Davey Jr, Guy De La Rosa, Eileen T Denning, Greg Dore, Ezekiel Emanuel, Alexia Exarchos, Gerd Fätkenheuer, Linda Fischer, Hansjakob Furrer, Daniela C Gey, Birgit Grund, Sally Hodder, Bruno Hoen, Margaret A Johnson, Karin L Klingman, Alan Landay, Gregg Larson, Bruno Ledergerber, Eric Lafebvre, Sandra N Lehrman, Ana Martinez, Marisol Martinez-Tristani, Sue Meger, Ronald T Mitsuyasu, Amanda Mocroft, Jean-Michel Molina, David Munroe, Michael Norton, Ines M Otegui, Nick Paton, Sarah L Pett, Andrew Phillips, Deenan Pillay, Michael Proschan, Claire Rappoport, Peter Reiss, Jurgen Rockstroh, James F Rooney, Michael J Ross, Mauro Schechter, Siegfried Schwarze, Daniel Seekins, Shweta Sharma, Wendy Snowden, Jeff Tryon, Michael J Vjecha, Scott A Wegner, William Woodward.
Substudy Cochairs: Jason V Baker, Daniel Duprez (arterial elasticity); Andrew Carr, Jennifer Hoy (bone mineral density); Matthew Dolan, Amalio Telenti (genomics); Christine Grady (informed consent); Edwina Wright, Bruce Brew, Richard W Price, Kevin Robertson (neurology); Ken M Kunisaki, John Connett, Dennis Niewoehner (pulmonary function).
Endpoint Review Committee: Alan Lifson (chair), Waldo Belloso, Richard Davey, Daniel Duprez, Jose M Gatell, Jennifer Hoy, Court Pedersen, George Perez, Richard Price, Ronald Prineas, John Worley.
Community Advisory Board: Claire Rappoport, Peer D Aagaard, Simon Collins, Nathan Geffen, David H-U Haerry, Joseph Hall, Michael Meulbroek, David Munroe, Dwight Peavey, Siegfried Schwarze, Mirta Valdez, Jo Watson.
Division of AIDS, NIAID: Ellen DeCarlo, Beverly Alston-Smith.
Statistics and Data Management Center: Alain DuChene, Michelle George, Merrie Harrison, Kathy-Huppler Hullsiek, Eric Krum, Carol Miller, Ray Nelson, Kien Quan, Siu-Fun Quan, Mollie P Roediger, Terri Schultz, Gregory Thompson.
International Coordinating Centers: Copenhagen HIV Programme, University of Copenhagen, Denmark – Daniela Grey, Karoline Jensen, Ellen Larsen, Heidi Juncher, Per Jansson, Birgitte G Jensen, Bitten Aagaard, Mary Pearson, Marie L Jakobsen, Ruth Pedersen, Ravi Shastry, Jesper Grarup; Medical Research Council Clinical Trials Unit, London, UK – Fleur Hudson, Alejandro Arenas-Pinto, Nafisah Braimah, Sue Fleck, Michelle Gabriel, Anne Hoppe, Brooke Jackson, Filipo Paccianini, Adrian Palfreeman, Nicki Smith; The Kirby Institute, University of New South Wales, Sydney, Australia – Cate Carey, Megan Evans, Sally Hough, Simone Jacoby, David Courtney-Rodgers, Lara Cassar, Nisha Seneviratne, Alexis Shambrook, Rose Robson, Joe Levitt; Veterans Affairs Medical Center, Washington, DC, USA – Elizabeth Finley, Barbara Standridge, Adriana Sánchez, Melissa Douglas, Douglas Thomas, Laura Mol, Haidee Elvis, Melissa Turner and Janet Royal.
Site Coordinating Centers and Clinical Site Investigators for the Pilot Phase: Argentina – Waldo Belloso, Ines Otegui, Isabel Lanusse, Alejandra Moricz, Marisa Garofalo, Lucas Roby; Marcelo Losso, Claudia Checa Cabot (Hospital General de Agudos JM Ramos Mejia); Marisa del Lujan Sanchez (Hospital Italiano de Buenos Aires); Gustavo Lopardo, Emiliano Bissio (FUNCEI); Hector Laplume, Lucia Daciuk (Hospital Nacional Profesor Alejandro Posadas); Sergio Lupo, Luciana Peroni (CAICI); Arnaldo Casiro, Graciela Guaragna (Hospital General de Agudos); Daniel David, Ana Alfonsina Crinejo (Hospital Rawson). Australia – Emanuel Vlahakis, Isabel Prone (Taylor Square Private Clinic); David Cooper, Kate Sinn (St. Vincent's Hospital); Jennifer Hoy, Jessica Costa (The Alfred Hospital); Tim Read, Julie Silvers (Melbourne Sexual Health Centre); Norman Roth, Helen Lau (Prahran Market Clinic); David Baker, Robyn Vale (East Sydney Doctors). Belgium – Nathan Clumeck, Kabamba Kabeya (Centre Hospitalier Universitaire St. Pierre); Eric Florence, Pascale Borguet (Institute of Tropical Medicine). Brazil – Tâmara NL Souza, Carina Yoshida; Luiz Pereira Jr, Ana L Toscano (Instituto de Infectología Emilio Ribas); Mauro Schechter, Monica de Souza (Hospital Escola Sao Francisco de Assis). Chile – Marcelo Wolff, Gladys Allendes (Fundacion Arriaran). Denmak – Lars Mathiesen, Philippa Collins (Hvidovre University Hospital); Jan Gerstoft, Lene P Jensen (Rigshospitalet). Finland – Matti Ristola, Outi Debnam (Helsinki University Central Hospital). France – Jean P Aboulker, Catherine Capitant; Jean-Michel Molina, Laurence Schnell-Niedbalski (Hopital Saint-Louis); Francois Boue, Imad Kansau (Hopital Antoine Beclere); Bruno Hoen, Catherine Bourdeaux (CHU de Besancon, Hopital Saint Jacques); Renaud Verdon, Pascale Goubin (CHU Cote de Nacre); Jerome Pacanowski, Benedicte Lefebvre (Hopital Saint-Antoine); Yasdan Yasdanpanah, Thomas Huleux (Hopital Gustave Dron); Laurence Weiss, Asmaa Boudribila (Hopital Europeen Georges Pompidou); Christine Katlama, Yasmine Dudoit (Groupe Hospitalier Pitie-Salpetriere); Cécile Goujard, Martine Mole (Hopital de Bicetre); Yves Levy, Chrystel Chesnel (Hopital Henri Mondor); Jean-Paul Viard, Aline Maignan (Hopital Hotel Dieu). Germany – Klaus Tillmann, Dejan Adzic; Christoph Stephan, Kathleen Mantzsch (Johann Wolfgang Göthe University Hospital); Martin Hartmann, Carolin Zschoche (Universitaetsklinikum Heidelberg); Hartwig Klinker, Susanne Wiebecke (University of Wuerzburg, Medizinische Poliklinik); Gerd Faetkenheuer, Eleonore Rund (Klinik I für Innere Medizin der Universitat zu Köln); Jurgen Rockstroh, Brigitta Spaeth (Medizinische Universitaetsklinik, Bonn); Johannes R Bogner, Ilse Ott (Klinikum Innenstadt, Infektionsambulanz und Tagesklinik CRS); Andreas Plettenberg, Nicole Hundrieser (ifi-Studien u.Projekte); Stefan Esser, Heidi Wiehler (Klinik fur Dermatologie, Venerologie, Allergologie); Stefan Reuter, Cecilie Feind (Universitaetsklinikum Duesseldorf); Martin Hower, Claudia Bachmann (Infectious Diseases and Pneumology Klinikum Dortmund gGmbH); Bernd Salzberger, Erika Jaeger (University Clinic Regensburg); Jan van Lunzen, Nadine Zerche (University Medical Centre Hamburg-Eppendorf); Heiko Jessen, Carmen Zedlack (Gemeinschaftspraxis Jessen-Jessen-Stein); Thomas Harrer, Ellen Harrer (Universitaetsklinikum Erlangen); Keikawus Arasteh, Torsten Meier (EPIMED-Gesellschaft fuer epidemiologische und klinische Forschung in der Medizin mbH); Matthias Stoll, Kirsten Hoeper (Medizinische Hochschule Hannover); Christian Hoffmann, Susanne Heesch (ICH Study Center). Greece – Angelos Hatzakis, Giota Touloumi, Vicky Gioukari; Athanasios Skoutelis, Ioannis Baraboutis (Evangelismos General Hospital); Antonios Papadopoulos, Anastasia Antoniadou (Attikon University General Hospital); Andreas Katsambas, Vassilios Paparizos (Syngros Hospital). Israel – Eynat Kedem, Hadar Lender; Shimon Pollack (Rambam Medical Center). Italy – Giuseppe Tambussi, Vega Rusconi (Ospedale San Raffaele). Mali – Sophia Siddiqui, Susan Orsega, Christian Yoder; Sounkalo Dao, Mamadou Cisse (Serefo/Cesac Mali). Morocco – Hakima Himmiche; Kamal Marhoum El Filali, Ikbale Erradey (University Hospital Centre Ibn Rochd). Peru – Alberto La Rosa, Juan Vega, Romina Chinchay; David Iglesias (Asociación Civil Impacta Salud y Educación); Fernando Mendo, Monica Del Portal (Hospital Nacional Edgardo Rebagliati Martins); Raul Salazar (Hospital Nacional Guillermo Almenara Irigoyen). Poland – Andrzej Horban, Elzbieta Bakowska (Wojewodzki Szpital Zakazny). South Africa – Robin Wood, Christie Heiberg; Catherine Orrell, Elizabeth Fielder (Desmond Tutu HIV Foundation). Spain – Paco Lopéz, Begoña Portas; Bonaventura Clotet, Patricia Cobarsi (Hospital Universitari Germans Trias I Pujol); Jose Maria Gatell, Ana González (Hospital Clinico de Barcelona CRS); Jose Arribas, Maria Luisa Montes Ramirez (Hospital La Paz CRS); Rafael Rubio, Mariano Matarranz del Amo (H Doce de Octubre); Jesus Sanz Sanz, Ignacio de los Santo Gil (Hospital La Princesa). Switzerland – Anna Barbara Christen; Bernard Hirschel, Olivia Zehfus (Unite VIH/SIDA Geneva); Hansjakob Furrer, Susanne Zgraggen (Poliklinik fur Infektiologie); Nicolas Müller, Susan Hochstrasser (University Hospital Zurich). Thailand – Thidarat Jupimai, Kanlaya Charoentonpuban, Sasiwimol Ubolyam, Wanida Thiansanguankul, Kobkeaw Laohajinda; Kiat Ruxrungtham, Anchalee Avihingsanon (Chulalongkorn University Hospital); Ploenchan Chetchotisakd, Manassinee Horsakulthai (Khon Kaen University, Srinagarind Hospital). United Kingdom – Margaret Johnson, Anne Carroll (Royal Free Hospital); Brian Gazzard, Christopher Higgs (Chelsea and Westminster Hospital); Martin Fisher, Nicky Perry (Brighton and Sussex University Hospitals NHS Trust); Alan Winston, Ken Legg (Imperial College Healthcare NHS Trust); Martin Wiselka, Sharon Holling (Leicester Royal Infirmary). United States – Wafaa El-Sadr, Luis A Fuentes (Harlem Family Center/Columbia University); David Mushatt, Connie Scott (Tulane University Medical Center); Richard M Novak, Gabriel Culbert (University of Illinois at Chicago); Ann Labriola, Douglas Thomas (VA Medical Center); Melissa Murphy, Norma Martinez (The Research and Education Group, Portland); Norman Markowitz, Jonathan Clemmer (Henry Ford Hospital CRS); Rodger MacArthur, Martha Farrough (Wayne State University CRS); William Burman, Laurie Luna (Denver Public Health); Daniel Nixon, Vinnie Mitchell (Virginia Commonwealth University Medical); Edward Telzak, Megan Tuohy (Bronx-Lebanon Hospital Center CRS); Gerald Friedland, Laurie Andrews (Yale University School of Medicine); Calvin Cohen, Julia Green (Community Research Initiative of New England); William Keith Henry, Edie Gunderson (Minneapolis Medical Research Foundation, Hennepin County Medical Center); Sally Hodder, Nancy Reilly (New Jersey Medical School Adult Clinical Research Center); Edwin DeJesus, Janiza Veloz (Orlando Immunology Center); Charurut Somboonwit, Mable Chow (Hillsborough County Health Department, University of South Florida); Ellen Marie Tedaldi, Christiane Geisler (Temple University); Stephen Weis, Isabel Vecino (University of North Texas Health Science Center); Timothy Wilkin, Valery Hughes (Cornell University); Amy Weintrob, David Wallace (Walter Reed Army Medical Center); Anuradha Ganesan, Irma Barahona (National Naval Medical Center); Jason Maguire, Susan M Banks (Naval Medical Center Portsmouth); Jason Okulicz, Terry Sjoberg (Brooke Army Medical Center); Nancy Crum-Cianflone (Naval Medical Center San Diego); Richard Davey, Jessica Springer (National Institutes of Health Clinical Center); Brian Agan, Michelle Linfesty, Sara Echols.
Pharmaceutical Companies: Abbott, Bristol-Myers Squibb, Gilead, GlaxoSmithKline, Merck, Tibotec.
Drug Distribution Center: Dorothy Coyle, Julie Eckstrand (ALMAC Clinical Trials Services).
Specimen Repository: Marie Hoover.
Wake Forest ECG Reading Center: Elsayed Soliman, Zhu Ming Zhang.
Conflict of interest Six pharmaceutical companies (Abbott Laboratories, Inc.; Bristol-Myers Squibb, Gilead Sciences, Inc.; GlaxoSmithKline, Inc.; Merck & Co, Inc.; and Tibotec Pharmaceuticals, Ltd.) have donated over 20 antiretroviral drug formulations to the study's CDR. The University of Minnesota, the sponsor of START, receives royalties from the use of abacavir, one of the HIV medicines that can be used in START.
References
- 1.Bhaskaran K, Hamouda O, Sannes M, et al. Changes in the risk of death after HIV seroconversion compared with mortality in the general population. JAMA. 2008;300(2):5–9. doi: 10.1001/jama.300.1.51. [DOI] [PubMed] [Google Scholar]
- 2.Mocroft A, Ledergerber B, Katlama C, et al. Decline in the AIDS and death rates in the EuroSIDA study: An observational study. Lancet. 2003;362:22–29. doi: 10.1016/s0140-6736(03)13802-0. [DOI] [PubMed] [Google Scholar]
- 3.European AIDS Clinical Society (EACS) Guidelines for the clinical management and treatment of HIV infected adults in Europe. Version 5. Dec, 2009. [(accessed 22 March 2012)]. Available at: http://www.europeanaidsclinicalsociety.org/images/stories/EACS-Pdf/version-5-november2009-eacs-guidelines-cologne.pdf. [Google Scholar]
- 4.Gazzard BG, on behalf of the BHIVA Treatment Guidelines Writing Group British HIV Association guidelines for the treatment of HIV-1-infected adults with antiretroviral therapy. HIV Med. 2008;9:563–608. doi: 10.1111/j.1468-1293.2008.00636.x. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organisation [(accessed 1 October 2010)];Antiretroviral therapy for HIV infection in adults and adolescents – Recommendations for a public health approach 2010 revision. Available at: http://whqlibdoc.who.int/publications/2010/9789241599764_eng.pdf. [PubMed]
- 6.Panel on Antiretroviral Guidelines for Adults and Adolescents Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. [(accessed 1 October 2010];Department of Health and Human Services. 2009 Dec 1;:1–161. Available at: http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf.
- 7.Thompson MA, Aberg JA, Cahn P, et al. Antiretroviral treatment of adult HIV infection: 2010 Recommendations of the International AIDS Society – USA Panel. JAMA. 2010;304(3):321–33. doi: 10.1001/jama.2010.1004. [DOI] [PubMed] [Google Scholar]
- 8.Richman DD, Margolis DM, Delaney M, et al. The challenge of finding a cure for HIV infection. Science. 2009;323:1304–07. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- 9.Study Group on Death Rates at High CD4 Count in Antiretroviral Naive Patients Death rates in HIV-positive antiretroviral-naive patients with CD4 count greater than 350 cells per microL in Europe and North America: A pooled cohort observational study. Lancet. 2010;376:340–45. doi: 10.1016/S0140-6736(10)60932-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The UK Collaborative HIV Cohort (CHIC) Study Steering Committee Rate of AIDS diseases or death in HIV infected antiretroviral therapy-naïve individuals with high CD4 count. AIDS. 2007;21:1717–21. doi: 10.1097/QAD.0b013e32827038bf. [DOI] [PubMed] [Google Scholar]
- 11.Guiguet M, Porter K, Phillips A, Costagliola D, Babiker AG. Clinical progression rates by CD4 cell category before and after the initiation of combination antiretroviral therapy (cART) Open AIDS J. 2008;2:3–9. doi: 10.2174/1874613600802010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phillips AN, Neaton J, Lundgren J. The role of HIV in serious diseases other than AIDS. AIDS. 2008;22:2409–18. doi: 10.1097/QAD.0b013e3283174636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The Strategies for Management of Antiretroviral Therapy (SMART) Study Group CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. 2006;355:2283–96. doi: 10.1056/NEJMoa062360. [DOI] [PubMed] [Google Scholar]
- 14.Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with HIV disease. J Clin Endocrinol Metab. 2007;92:2506–12. doi: 10.1210/jc.2006-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phillips AN, Carr A, Neuhaus J, et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: Exploratory analyses from the SMART trial. Antivir Ther. 2007;13:177–87. doi: 10.1177/135965350801300215. [DOI] [PubMed] [Google Scholar]
- 16.The Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) Study Group HIV-induced immunodeficiency and mortality from AIDS-defining and non-AIDS-defining malignancies. AIDS. 2008;22:2143–53. doi: 10.1097/QAD.0b013e3283112b77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silverberg MJ, Neuhaus J, Bower M, et al. Risk of cancers during interrupted antiretroviral therapy in the SMART study. AIDS. 2007;21(14):1957–63. doi: 10.1097/QAD.0b013e3282ed6338. [DOI] [PubMed] [Google Scholar]
- 18.Patel P, Hanson DL, Sullivan PS, et al. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992–2003. Ann Intern Med. 2008;148:728–36. doi: 10.7326/0003-4819-148-10-200805200-00005. [DOI] [PubMed] [Google Scholar]
- 19.Clifford GM, Polesel J, Rickenbach M, et al. Cancer risk in the Swiss HIV Cohort Study: Associations with immunodeficiency, smoking, and highly active antiretroviral therapy. J Natl Cancer Inst. 2005;97(6):425–32. doi: 10.1093/jnci/dji072. [DOI] [PubMed] [Google Scholar]
- 20.Engels EA, Brock MV, Chen J, et al. Elevated incidence of lung cancer among HIV-infected individuals. J Clin Oncol. 2006;24(9):1383–88. doi: 10.1200/JCO.2005.03.4413. [DOI] [PubMed] [Google Scholar]
- 21.Kirk GD, Merlo C, O'Driscoll P, et al. HIV infection is associated with an increased risk of lung cancer, independent of smoking. Clin Infect Dis. 2007;45(1):103–10. doi: 10.1086/518606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaturvedia AK, Pfeifferb RM, Changa L, et al. Elevated risk of lung cancer among people with HIV. AIDS. 2007;21:207–13. doi: 10.1097/QAD.0b013e3280118fca. [DOI] [PubMed] [Google Scholar]
- 23.McGinnis KA, Fultz SL, Skanderson M, et al. Hepato-cellular carcinoma and non-Hodgkin's lymphoma: The roles of HIV, hepatitis C infection, and alcohol abuse. J Clin Oncol. 2006;24(31):5005–09. doi: 10.1200/JCO.2006.05.7984. [DOI] [PubMed] [Google Scholar]
- 24.Mocroft A, Soriano V, Rockstroh J, et al. Is there evidence for an increase in the death rate from liver-related disease in patients with HIV? AIDS. 2005;19:2117–25. doi: 10.1097/01.aids.0000194799.43799.ea. [DOI] [PubMed] [Google Scholar]
- 25.The Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) Study Group Liver-related deaths in persons infected with the human immunodeficiency virus. Arch Intern Med. 2006;166:1632–41. doi: 10.1001/archinte.166.15.1632. [DOI] [PubMed] [Google Scholar]
- 26.Atta MG, Gallant JE, Rahman MH, et al. Antiretroviral therapy in the treatment of HIV-associated nephropathy. Nephrol Dial Transplant. 2006;21(10):2809–13. doi: 10.1093/ndt/gfl337. [DOI] [PubMed] [Google Scholar]
- 27.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nature Med. 2006;12(12):1365–71. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 28.Kuller LH, Tracy R, Belloso W, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5(10):e203. doi: 10.1371/journal.pmed.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.The Strategies for Management of Antiretroviral Therapy (SMART) Study Group Clinical outcomes in antiretroviral naïve patients and those not taking antiretroviral therapy at entry to SMART. J Infect Dis. 2008;197(8):1133–44. doi: 10.1086/586713. [DOI] [PubMed] [Google Scholar]
- 30.The Strategies for Management of Antiretroviral Therapy (SMART) Study Group Inferior clinical outcome of the CD4+ count-guided antiretroviral treatment interruption strategy in SMART: Role of CD4+ counts and HIV-RNA levels during follow-up. J Infect Dis. 2008;197:1145–55. doi: 10.1086/529523. [DOI] [PubMed] [Google Scholar]
- 31.Kitahata MM, Gange SJ, Abraham AG, et al. Effect of early versus deferred antiretroviral therapy for HIV on survival. N Engl J Med. 2009;360:1815–26. doi: 10.1056/NEJMoa0807252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.When to Start Consortium Timing of initiation of antiretroviral therapy in AIDS-free HIV-1-infected patients: A collaborative analysis of 18 HIV cohort studies. Lancet. 2009;373:1352–63. doi: 10.1016/S0140-6736(09)60612-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fideli ÜS, Allen SA, Musonda R, et al. Virologic and immunologic determinants of heterosexual transmission of human immunodeficiency virus type 1 in Africa. AIDS Res Hum Retroviruses. 2001;17(10):901–10. doi: 10.1089/088922201750290023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tovanabutra S, Robison V, Wongtrakul J, et al. Male viral load and heterosexual transmission of HIV-1 sub-type E in northern Thailand. J Acquir Immune Defic Syndr. 2002;29:275–83. doi: 10.1097/00126334-200203010-00008. [DOI] [PubMed] [Google Scholar]
- 35.Quinn TC, Wawer MJ, Sewankambo N, et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. N Engl J Med. 2000;342:921–29. doi: 10.1056/NEJM200003303421303. [DOI] [PubMed] [Google Scholar]
- 36.Donnell D, Baeten JM, Kiarie J, et al. Heterosexual HIV-1 transmission after initiation of antiretroviral therapy: A prospective cohort analysis. Lancet. 2010;375(9731):2092–98. doi: 10.1016/S0140-6736(10)60705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vergidis PI, Falagas ME, Hamer DH. Meta-analytical studies on the epidemiology, prevention, and treatment of human immunodeficiency virus infection. Infect Dis Clin North Am. 2009;23:295–308. doi: 10.1016/j.idc.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 38.Attia S, Egger M, Müllera M, Zwahlen M, Low N. Sexual transmission of HIV according to viral load and antiretroviral therapy: Systematic review and meta-analysis. AIDS. 2009;23(11):1397–404. doi: 10.1097/QAD.0b013e32832b7dca. [DOI] [PubMed] [Google Scholar]
- 39.Lifson AR, Rhame FS, Belloso WH, et al. Reporting and evaluation of HIV-related clinical endpoints in two multicenter international clinical trials. HIV Clin Trials. 2006;7(3):125–41. doi: 10.1310/7MER-XFA7-1762-E2WR. [DOI] [PubMed] [Google Scholar]
- 40.Lifson AR, INSIGHT Endpoint Review Committee Development of diagnostic criteria for serious non-AIDS in HIV clinical trials. HIV Clin Trials. 2010;11(4):205–19. doi: 10.1310/hct1104-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolbers M, Babiker AG, Sabin C, et al. Pretreatment CD4 cell slope and progression to AIDS or death in HIV-infected patients initiating antiretroviral therapy – The CASCADE collaboration: A collaboration of 23 cohort studies. PLoS Med. 2010;7(2):e1000239. doi: 10.1371/journal.pmed.1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.CASCADE (Concerted Action on SeroConversion to AIDS and Death in Europe) Collaboration [(accessed 26 July 2010)];CASCADE: Participating cohorts. 2009 Available at: http://www.cascade-collaboration.org.
- 43.CASCADE (Concerted Action on SeroConversion to AIDS and Death in Europe) Collaboration Changes in the uptake of antiretroviral therapy and survival in people with known duration of HIV infection in Europe: Results from CASCADE. HIV Med. 2000;1(4):224–31. doi: 10.1046/j.1468-1293.2000.00033.x. [DOI] [PubMed] [Google Scholar]
- 44.Touloumi G, Pantazis N, Antoniou A, et al. Highly active antiretroviral therapy interruption: Predictors and virological and immunologic consequences. J Acquir Immune Defic Syndr. 2006;42(5):554–61. doi: 10.1097/01.qai.0000230321.85911.db. [DOI] [PubMed] [Google Scholar]
- 45.Centers for Disease Control and Prevention 1993 revised classification system for HIV infection and expanded surveillance definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41:1–19. [PubMed] [Google Scholar]
- 46.Trites RL. Lafayette Grooved Pegboard Task. Instruction/Owner's Manual. Lafayette Instrument Company; Lafayette, IN: 1989. [Google Scholar]
- 47.D'Elia LF, Satz P, Uchiyama CL, et al. Color Trails Test. Psychological Assessment Resources Inc; Odessa, FL: 1996. [Google Scholar]
- 48.The Psychological Corporation . Wechsler Adult Intelligence Scale-Revised. The Psychological Corporation; San Antonio, TX: 1981. [Google Scholar]
- 49.Benedict RH, Schretlen D, Groninger L, Brandt J. Hopkins verbal learning test – Revised: Normative data and analysis of inter-from and test-retest reliability. Clin Neuropsychol. 1998;12(1):43–55. [Google Scholar]
- 50.Reitan RM. Manual for Administration of Neuropsychological Test Batteries for Adults and Children. Indiana University Press; Bloomington, IN: 1969. [Google Scholar]
- 51.Spreen O, Strauss E. A Compendium of Neuropsychological Tests: Administration, Norms, and Commentary. 2nd edn Oxford University Press; New York: 1998. [Google Scholar]
- 52.The INSIGHT-ESPRIT Study Group and SILCAAT Scientific Committee Interleukin-2 therapy in patients with HIV infection. N Engl J Med. 2009;361:1548–59. doi: 10.1056/NEJMoa0903175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.The Data Collection on Adverse Events of Anti-HIV Drugs (D:A:D) Study Group Combination antiretroviral therapy and the risk of myocardial infarction. N Engl J Med. 2003;349:1993–2003. doi: 10.1056/NEJMoa030218. [DOI] [PubMed] [Google Scholar]
- 54.The UK Collaborative HIV Cohort (CHIC) Study [(accessed 26 July 2010)];Participating sites. Available at: http://www.ukchic.org.uk.
- 55.Boulware D, Huppler Hullsiek K, Puronen C, et al. Higher levels of CRP, D-dimer, IL-6, and hyaluronic acid before initiation of antiretroviral therapy (ART) are associated with increased risk of AIDS or death. J Infect Dis. 2011;203:1637–1646. doi: 10.1093/infdis/jir134. PMCID: PMC3096784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O'Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979;35:549–56. [PubMed] [Google Scholar]
- 57.Lan KG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika. 1983;70:659–63. [Google Scholar]
- 58.Neaton JD, Babiker AG, Bohnhorst M, et al. Regulatory impediments jeopardizing the conduct of clinical trials in Europe funded by the National Institutes of Health. [(accessed 21 March 2012)];Clin Trials. 2010 7:705–18. doi: 10.1177/1740774510376547. Also available at: http://www.law.cornell.edu/uscode/text/31/1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.US Anti-Deficiency Act of 2005. USC §1341(a)(1)(A) 2000 Antideficiency act reports. [Google Scholar]
- 60.INSIGHT Community Advisory Board [(accessed 1 December 2010)];Community statement on the START trial and the change in the US DHHS treatment guidelines. 2010 May 5; Available at: http://i-base.info/home/community-statement-supporting-start-trial.
- 61.Ho DD. Time to hit HIV, early and hard. N Engl J Med. 1995;333:450–51. doi: 10.1056/NEJM199508173330710. [DOI] [PubMed] [Google Scholar]
- 62.Walker BD, Basgoz N. Treat HIV-1 infection like other infections – Treat it. JAMA. 1998;280:91–93. doi: 10.1001/jama.280.1.91. [DOI] [PubMed] [Google Scholar]
- 63.Burman WJ, Reves RR, Cohn DL. The case for conservative management of early HIV disease. JAMA. 1998;280:93–95. doi: 10.1001/jama.280.1.93. [DOI] [PubMed] [Google Scholar]
- 64.Henry K. The case for more cautious, patient-focused antiretroviral therapy. Ann Intern Med. 2000;132(4):306–11. doi: 10.7326/0003-4819-132-4-200002150-00009. [DOI] [PubMed] [Google Scholar]
- 65.Harrington M, Carpenter CC. Hit HIV-1 hard, but only when necessary. Lancet. 2000;355(9221):2147–52. doi: 10.1016/S0140-6736(00)02388-6. [DOI] [PubMed] [Google Scholar]
- 66.Thorner A, Rosenberg E. Early versus delayed antiretroviral therapy in patients with HIV infection: A review of the current guidelines from an immunological perspective. Drugs. 2003;63(13):1325–37. doi: 10.2165/00003495-200363130-00001. [DOI] [PubMed] [Google Scholar]
- 67.Lane HC, Neaton JD. When to start therapy for HIV infection: A swinging pendulum in search of data. Ann Intern Med. 2003;138:680–81. doi: 10.7326/0003-4819-138-8-200304150-00018. [DOI] [PubMed] [Google Scholar]
- 68.Holmberg SD, Palella FJ, Lichtenstein KA, Havlir DV. The case for earlier treatment of HIV infection. Clin Infect Dis. 2004;39:1699–704. doi: 10.1086/425743. [DOI] [PubMed] [Google Scholar]
- 69.Schechter M. Therapy for early HIV infection: How far back should the pendulum swing? J Infect Dis. 2004;190:1043–45. doi: 10.1086/422852. [DOI] [PubMed] [Google Scholar]
- 70.Wang C, Masho SW, Nixon DE. When to start antiretroviral therapy. Curr HIV/AIDS Rep. 2006;3(2):66–73. doi: 10.1007/s11904-006-0020-3. [DOI] [PubMed] [Google Scholar]
- 71.Phillips AN, Gazzard BG, Clumeck N, et al. When should antiretroviral therapy for HIV be started? BMJ. 2007;334:76–78. doi: 10.1136/bmj.39064.406389.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gallant JE. When to start antiretroviral therapy: A swinging pendulum? Top HIV Med. 2008;16(2):82–88. [PubMed] [Google Scholar]
- 73.Wilkin TJ, Gulick RM. When to start antiretroviral therapy? Clin Infect Dis. 2008;47:1580–86. doi: 10.1086/593311. (Comment in Clin Infect Dis 2009; 48(8): 1162; author reply 1162-63.) [DOI] [PubMed] [Google Scholar]
- 74.Sabin CA, Phillips AN. Should HIV therapy be started at a CD4 cell count above 350 cells/microl in asymptomatic HIV-1-infected patients? Curr Opin Infect Dis. 2009;22(2):191–97. doi: 10.1097/qco.0b013e328326cd34. [DOI] [PubMed] [Google Scholar]
- 75.Jain V, Deeks SG. When to start antiretroviral therapy. Curr HIV/AIDS Rep. 2010;7(2):60–68. doi: 10.1007/s11904-010-0044-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gatell JM. When and why to start antiretroviral therapy? J Antimicrob Chemother. 2010;65(3):383–85. doi: 10.1093/jac/dkp487. [DOI] [PubMed] [Google Scholar]
- 77.Severe P, Juste MJ, Ambroise A, et al. Early versus standard antiretroviral therapy for HIV-infected adults in Haiti. N Engl J Med. 2010;363:257–65. doi: 10.1056/NEJMoa0910370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lau B, Gange S, Moore RD. Risk of non-AIDS related mortality may exceed risk of AIDS-related mortality among individuals enrolling into care with CD4+ counts greater than 200 cells/mm3. J Acquir Immune Defic Syndr. 2007;44(2):179–87. doi: 10.1097/01.qai.0000247229.68246.c5. [DOI] [PubMed] [Google Scholar]
- 79.Gould AL. Interim analyses for monitoring clinical trials that do not affect the type I error rate. Stat Med. 1992;11:55–66. doi: 10.1002/sim.4780110107. [DOI] [PubMed] [Google Scholar]
- 80.Proschan MA, Liu Q, Hunsberger S. Practical midcourse sample size modification in clinical trials. Control Clin Trials. 2003;24:4–15. doi: 10.1016/s0197-2456(02)00240-4. [DOI] [PubMed] [Google Scholar]
- 81.Diggle PJ. An approach to the analysis of repeated measurements. Biometrics. 1988;44:959–71. [PubMed] [Google Scholar]
- 82.Taylor JMG, Cumberland WG, Sy JP. A stochastic model for analysis of longitudinal AIDS data. J Am Stat Assoc. 1994;89:727–36. [Google Scholar]
- 83.Multicenter AIDS Cohort Cohort description. Available at: http://www.statepi.jhsph.edu/macs/macs.html.
- 84.Blackwell PG. Encyclopedia of Biostatistics. John Wiley & Sons, Ltd; Ornstein-Uhlenbeck process. Published Online: 15 July 2005; doi: 10.1002/0470011815.b2a07038. [Google Scholar]
- 85.Cohen MS, Chen YQ, McCauley M, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med. 2011;365:493–505. doi: 10.1056/NEJMoa1105243. [DOI] [PMC free article] [PubMed] [Google Scholar]