

Abstract
Substituted pyrimidine inhibitors of the Clk and Dyrk kinases have been developed, exploring structure-activity relationships around four different chemotypes. The most potent compounds have low-nanomolar inhibitory activity against Clk1, Clk2, Clk4, Dyrk1A and Dyrk1B. Kinome scans with 442 kinases using agents representing three of the chemotypes show these inhibitors to be highly selective for the Clk and Dyrk families. Further off-target pharmacological evaluation with ML315, the most selective agent, supports this conclusion.
Keywords: Clk, Dyrk, Kinase inhibitor, Splicing, Pyrimidine
The cdc2-like kinases (Clks) are CMGC group (cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAP kinases), glycogen synthase kinases (GSK) and CDK-like kinases) dual-specificity kinases, capable of autophosphorylation at tyrosine residues while phosphorylating substrates at serine and threonine residues.1 There are four isoforms, Clk1-Clk4, which impact mRNA splicing by phosphorylating the serine- and arginine-rich (SR) family of splicing proteins.2–4 The SR proteins guide spliceosome assembly by establishing mRNA splice sites, and the distribution and activity of SR proteins is dependent upon their phosphorylation states.5 The ability to selectively alter specific spliceosome operations using small molecules could offer a means of intervening in disorders resulting from aberrant mRNA splicing.6,7 Recent studies have implicated Clks and SR proteins in insulin-induced alternative splicing of PKCβII mRNA,8 HIV-1 gene expression,9 alternative splicing of adenovirus E1A,10 and tissue factor pre-mRNA splicing in endothelial cells related to procoagulant activity.11 The role of Clk2 in glucose metabolism pathways and hepatic gluconeogenesis has also been described.12 Earlier studies established Clk3 expression in mouse spermatozoa13 and associated Clk kinases with induction of neuronal differentiation in PC12 cells.14
The first selective small-molecule inhibitor of the Clk kinases, benzothiazole TG003 (Figure 1), was reported in 2004 by Hagiwara and coworkers.15 TG003 is a potent inhibitor of Clk1 (20 nM) and Clk4 (15 nM), exhibits modest activity against Clk2 (200 nM), and is inactive against Clk3. Hagiwara’s work demonstrated that alternative pre-mRNA splicing could be altered, both in vitro and in vivo, with the use of a Clk inhibitor. This splicing regulation was linked to the suppression of SR protein phosphorylation. Chronogen, Inc. later patented a number of quinolines as low-micromolar Clk1 inhibitors.16 KH-CB19 was disclosed in 2011 as a specific and potent inhibitor of Clk1 (20 nM) and Dyrk1A (55 nM).17 The compound, inspired by the β-carboline alkaloid bauerine C, also exhibited moderate activity against Clk3 (530 nM) and was noted by the authors to be a potent inhibitor of Clk4, though no IC50 value was reported. Another natural-product-inspired Clk inhibitor, Leucettine L41, was also reported in 2011.18 This compound, modeled on the marine alkaloid leucettamine B, is highly active against Clk1 (15 nM), Dyrk1A (40 nM), and Dyrk2 (35 nM), while moderately active against Clk3 (4.5 μM).
Figure 1.
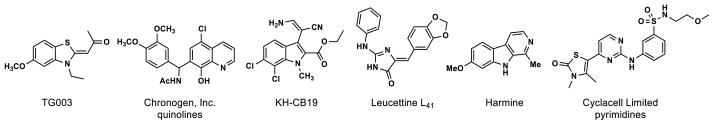
Reported Clk and Dyrk kinase inhibitors
Like the Clk kinases, dual-specificity tyrosine phosphorylation-regulated kinases (Dyrks) are part of the CMGC group, capable of autophosphorylation at tyrosine residues while phosphorylating substrates at serine and threonine residues.19 Seven mammalian isoforms have been described: Dyrk1A, Dyrk1B (Mirk), Dyrk1C, Dyrk2, Dyrk3 (REDK), Dyrk4A, and Dyrk4B.20 Dyrk1A, the best-characterized isoform,21 plays an important role in neuronal development and neurite formation,22 with balanced expression levels crucial to proper growth and function.23 Found on the Down Syndrome (DS) critical region of chromosome 21,24 overexpression of Dyrk1A in mouse models has been linked to changes in neurodevelopment consistent with the DS phenotype.25,26 Improper expression of Dyrk1A has also been implicated in neurodegeneration in both Alzheimer’s disease and Pick disease.27,28 Furthermore, a connection between Dyrk1A and pre-mRNA splicing has been established through its phosphorylation of spliceosomal protein SF3bI, part of the U2 small nuclear ribonucleoprotein particle (snRNP).29 Underexpression of Dyrk1A brought on by gene mutation or truncation is also problematic.30,31 Mounting evidence suggests that some members of the Dyrk family have cell-protective functions.19,32,33 Dyrk1B34 may serve as a checkpoint kinase connected to the Hedgehog signaling pathway, providing cancer cells impaired by chemotherapeutics with the opportunity to repair themselves.35 Dyrk1B overexpression has been detected in numerous cancers, suggesting this kinase may be an important target for small-molecule intervention.36,37
Very few potent and selective Dyrk inhibitors have been described. Harmine, a plant-derived β-carboline alkaloid, (Figure 1) is an ATP-competitive Dyrk1A (33–80 nM) and Dyrk1B (166 nM) inhibitor.22,38 TG003 has also recently been demonstrated as a potent inhibitor of Dyrk1A (12 nM) and Dyrk1B (130 nM), in addition to its Clk1 and Clk4 activity (vide supra).39 A Cyclacell Limited patent covers a number of thiazolone-substituted aminopyrimidines that inhibit Dyrk1A with potencies <100 nM.40
In an earlier report,39 substituted quinazoline modulators of Lamin A splicing discovered in a high-throughput phenotypic screen at the NIH Chemical Genomics Center (NCGC) were disclosed (PubChem AID 1487). Literature precedent establishing quinazolines as ATP-competitive kinase inhibitors led to the hypothesis that the compound might be acting as a Clk inhibitor, which was validated with a commercial kinase profile.41 Activity against the related CMGC group Dyrk kinases was also discovered. To identify additional Clk4 inhibitors, a screen of NCGC libraries was begun using bioluminescent luciferase-based Kinase Glo qHTS assays monitoring ATP consumption or ADP formation.42 Structure-activity relationship (SAR) studies around the quinazoline core were also carried out. These efforts culminated in the identification of potent ATP-competitive inhibitors of both the Clk and Dyrk kinases (Figure 2). Compound 1 was developed first as a potent inhibitor of Clk1, Clk4 and Dyrk1A.39 Further optimization led to Clk4-selective inhibitor 2.43
Figure 2.
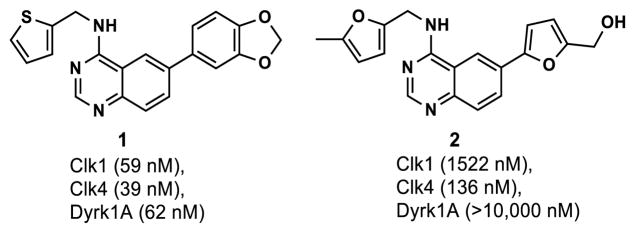
Quinazoline Clk/Dyrk kinase inhibitors
Continuing in the effort to identify Clk and Dyrk inhibitors with selectivity profiles different from those of the quinazolines shown in Figure 2, we now describe SAR studies with several pyrimidine chemotypes that were also identified as described above. This work has resulted in the development of a chemical probe, ML315.44 Thus, a screen against Clk4 was carried out using an NCGC library of 375 pyrimidines having three different core structures; the most active compound from each series is shown in Figure 3. Series 2 was chosen for follow-up because the most potent compound against Clk4 (3, 63 nM) was a member of this series. Pyrimidine 3 was the only compound of the 375 screened with an IC50 <100 nM.
Figure 3.

Three series of pyrimidine Clk4 inhibitors
The absence of clear SAR patterns among Series-2 compounds in the initial screening set prompted us to employ the Topliss scheme45 for selecting substitution patterns on the benzylamine (R1) of the pyrimidines synthesized in a follow-up matrix library. Aromatics that were present in the most active compounds in the initial screen were maintained at R2. Figure 4 shows the results of this effort, and while these compounds were not especially potent, an interesting selectivity pattern did emerge: when the pyrimidine was substituted with both a chlorinated benzylamine at R1 and an oxygenated benzene at R2, the compound tended to be active against Clk4 but inactive against Dyrk1A (see A2, A4, C2, G2, and G4).
Figure 4.

IC50 values (μM) against Clk4/Dyrk1A for matrix library pyrimidines (Kinase Glo (Promega) assay)
Synthesis involved microwave-assisted nucleophilic aromatic substitution (SNAr) of 2,4-dichloropyrimidine with five functionalized benzyl amines, providing regioisomeric aminopyrimidines 4 and 5 in ratios of 2–3:1 and 81–99% yield (Scheme 1, Eq. 1).46 The regioisomers were easily separated on scale by silica gel chromatography. Subsequent microwave-assisted Suzuki cross-coupling reactions of intermediate 4 with the requisite arylboronic acids afforded the desired compounds 6, which were purified by reverse-phase preparative HPLC.
Scheme 1.
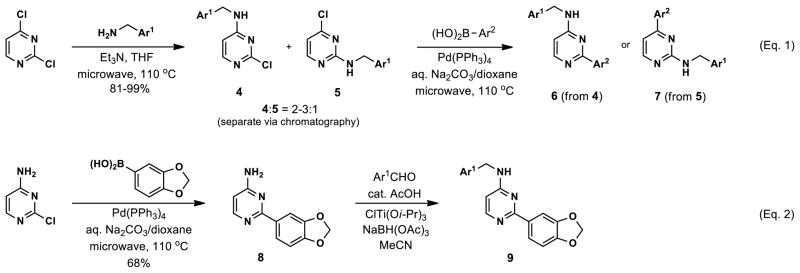
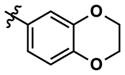
Synthesis of substituted aminopyrimidines
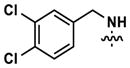
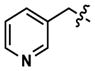
Merging structural features of both compound 3 and the Clk4-selective compounds of Figure 4, we synthesized halogen-substituted pyridine analogs in an effort to maintain the potency of 3, while introducing Clk4 selectivity. Other modifications including the incorporation of a quinoline and a pyrimidine at Ar1, as well as a pyridine scan, were carried out. Compound synthesis followed the route shown in Scheme 1, Eq. 2, which allowed a common intermediate (8), made by Suzuki cross-coupling between commercially-available 4-amino-2-chloropyrimidine and 3,4-(methylenedioxy)phenylboronic acid, to be used in parallel reductive aminations.47 Final compounds 9 were purified by reverse-phase preparative HPLC. No improvements in Clk4-selectivity were observed and the poor potencies of these compounds prompted us to forego further analog synthesis involving modification of the pyridine. Additional analogs containing halogenated benzylamines at Ar1 were also synthesized and tested, but again no improvements were observed in either potency or selectivity. Details of these studies may be found in the Supporting Information.
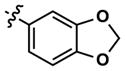
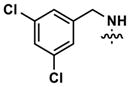
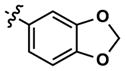
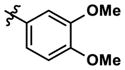
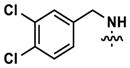
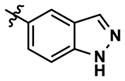
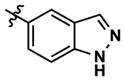
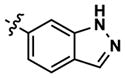
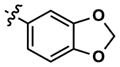
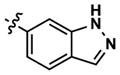
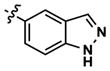
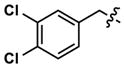
At this point, we turned our attention to the series of compounds shown in Table 1, determined to examine the effect of changing the relative positions of the core nitrogen atoms to produce pyrimidines having a guanidyl arrangement of nitrogens rather than the amidinyl arrangement found in all analogs generated to this point. Compound synthesis was carried out largely according to Scheme 1, Eq. 1 and a variation of Eq. 2 utilizing the isomeric 2-amino-4-chloropyrimidine as starting material. Anilines 26–29 were synthesized via a Suzuki cross-coupling/nucleophilic aromatic substitution sequence from 2,4- dichloropyrimidine. Details are provided in the Supporting Information. Significant improvements in potency were immediately realized (qHTS assay results are shown in parentheses in Tables 1 and 2). Comparing the amidinyl compounds (Figure 4) to their corresponding guanidinyl isomers (Table 1), a striking improvement in potency was observed. Among the guanidyl analogs, clear patterns in SAR were also seen: compounds incorporating the benzodioxolane moiety at R2 were the most potent Clk4 inhibitors (14 and 15, 40–50 nM), followed by compounds incorporating the p-methoxybenzene (10 and 11, 210–290 nM), and then m-methoxybenzene moieties (12 and 13, 3.7–5.8 μM). This trend was not observed in the amidinyl series, where the corresponding analogs all had similar potencies (13–41 μM). Within the guanidyl set, compounds substituted with 3,4-dichlorobenzyl amine at R1 performed slightly better than their 4-chlorobenzylamine-substituted counterparts (compare 10–15). The 3,4- and 3,5-dichlorobenzylamines performed similarly (compare 15 and 17), while a notable 10-fold selectivity for Clk4 over Dyrk1A was observed for the 3,4-dibromobenzylamine analog 16. Introducing the 3-picolyl substituent into the guanidyl series once again produced a potent, but not selective, inhibitor (19) of Clk4.
Table 1.
IC50 valuesa (nM) for pyrimidines having a guanidyl core
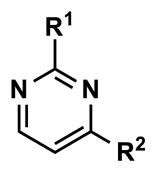
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | Clk1 | Clk2 | Clk3 | Clk4a | Dyrk1Aa | Dyrk1B |
| 10 |
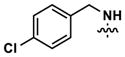
|
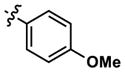
|
277 | 1092 | -- | 319 (290) | 558 (2070) | 871 |
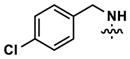
| 11 |

|
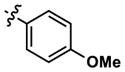
|
76 | 409 | -- | 92 (210) | 203 (1160) | 411 |
| 12 |
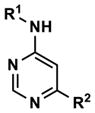
|
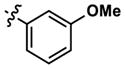
|
>10,000 | >10,000 | -- | 6806 (5820) | 6361 (7330) | 6782 |
| 13 |
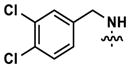
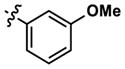
|
|
5459 | >10,000 | -- | 3858 (3680) | 4654 (5820) | >10,000 |
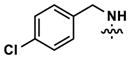
| 14 |
|
|
(50) | (260) | ||||
| 15 |
|
|
8 | 20 | 3634 | 14 (40) | 26 (160) | 38 |
| 16 |
|
|
24 | 30 | 3232 | 18 (80) | 79 (820) | 112 |
| 17 |
|
|
2 | 23 | >10,000 | 4 (50) | 4 (50) | 7 |
| 18 |
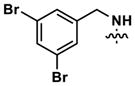
|
|
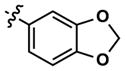
13 | 41 | -- | 9 (40) | 11 (70) | 18 |
| 19 |
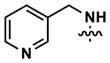
|
|
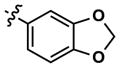
11 | 60 | 1473 | 16 (20) | 7 (40) | 12 |
| 20 |
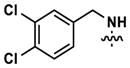
|
|
16 | 39 | >10,000 | 13 (120) | 18 (210) | 55 |
| 21 |
|
|
>10,000 | -- | -- | >10,000 (26,020) | -- (26,020) | -- |
| 22 |
|
|
41 | 190 | 2934 | 30 (30) | 129 (330) | 125 |
| 23 |
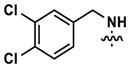
|
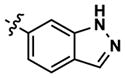
|
183 | 429 | -- | 123 (5820) | 169 (1640) | 599 |
| 24 |

|
|
73 | 462 | 2663 | 60 (30) | 61 (150) | 93 |
| 25 |
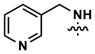
|
|
(40) | (210) | ||||
| 26 |
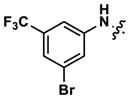
|
|
102 | 345 | -- | 86 (130) | 107 (520) | 137 |
| 27 |
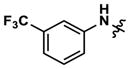
|
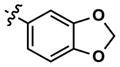
|
34 | 52 | >10,000 | 42 (70) | 10 (70) | 18 |
| 28 |
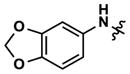
|
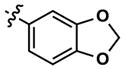
|
180 | 259 | -- | 75 (120) | 25 (130) | 42 |
| 29 |
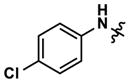
|
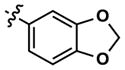
|
1262 | >10,000 | -- | 5450 (370) | 273 (520) | 884 |
IC50 values in parentheses determined using Kinase Glo (Promega) qHTS assay. The remaining IC50 values determined by Reaction Biology (www.reactionbiology.com). Compounds were tested in 10-dose IC50 mode with 3-fold serial dilution starting at 10 μM. Reactions were carried out at 10 μM ATP.
Table 2.
IC50 valuesa (nM) for Series 1–3 compounds substituted with optimized groups at R1 and R2
| Cmpnd | Scaffold | R1 | R2 | Clk1 | Clk2 | Clk3 | Clk4 | Dyrk1A | Dyrk1B |
|---|---|---|---|---|---|---|---|---|---|
| 30 |
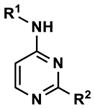
|
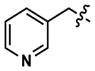
|
|
1598 | 1903 | -- | 1294 (290) | 1434 (2070) | 829 |
| 31 |
|
|
964 | 665 | -- | 574 (150) | 349 (650) | 179 | |
| 32 |
|
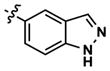
|
1783 | 238 | -- | 241 (290) | 2352 (4630) | 514 | |
|
| |||||||||
| 33 |
|
|
|
2370 | 8791 | -- | 9284 (5820) | 4579 (26,020) | 8862 |
| 34 |
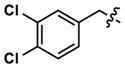
|
|
626 | 1557 | -- | 526 (580) | 3454 (4120) | >10,000 | |
| 35 |

|
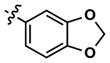
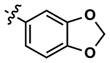
|
68 | 231 | >10,000 | 68 (130) | 282 (2600) | 1156 | |
| 36 |
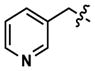
|

|
4805 | >10,000 | -- | 7535 (9230) | >10,000 (26,020) | >10,000 | |
|
| |||||||||
| 37 |
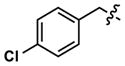
|
|

|
(2320) | (9230) | ||||
| 38 |
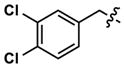
|
|
(4630) | (13,040) | |||||
| 39 |

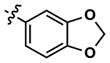
|
|
(1640) | (3280) | |||||
| 40 |
|
|
(6540) | (16,420) | |||||
IC50 values in parentheses determined using Kinase Glo (Promega) qHTS assay. The remaining IC50 values determined by Reaction Biology (www.reactionbiology.com). Compounds were tested in 10-dose IC50 mode with 3-fold serial dilution starting at 10 μM. Reactions were carried out at 10 μM ATP.
Altering the benzodioxolane moiety at R2 was also explored to examine the effects of several important structural variations. Expanding the five-membered ring to the corresponding benzodioxane (20) resulted in losses in both potency and selectivity in the qHTS assay, while opening the ring to produce veratrole-substituted analog 21, resulted in a complete loss of activity. Nitrogen-containing heterocyclic replacements were also introduced to explore hydrogen bonding properties in this region of the inhibitor. When the benzimidazole moiety proved challenging to incorporate, isomeric indazoles were substituted. With 3,4-dichlorobenzylamine at R1, the 1H-indazol-5-yl isomer (22) produced a potent and selective (>10-fold) inhibitor of Clk4, whereas the 1H-indazol-6-yl isomer (23) appeared to be significantly less potent in the bioluminescent qHTS assay. With the 3-picolyl substituent at R1, both indazole isomers 24 and 25 were potent Clk4 inhibitors, but of moderate selectivity (5-fold in the bioluminescent qHTS assays). Several compounds replacing the benzylamines at R1 with substituted anilines were also synthesized. Anilines 26–28 were found to be potent against Clk4, but lacked selectivity versus Dyrk1A. Notably, 4-chloroaniline-substituted 29, the analog of 4-chlorobenzylamine-substituted 14, displayed a sharp drop in potency against Clk4, demonstrating the importance of the one-carbon linker between the guanidyl pyrimidine core and the attached aromatic at R1.
Revisiting the three pyrimidine scaffolds from Figure 3, substituents that provided the best potencies and selectivities in the guanidyl series above were introduced at R1 and R2. Compound synthesis is described in the Supporting Information. The results are shown in Table 2. Though most of the compounds did not perform as well as their corresponding Table-1 isomers, one compound (35) did exhibit acceptable potency (130 nM) and a very good 20-fold selectivity for Clk4 over Dyrk1A. Compound 32 was also notable for its 16-fold selectivity for Clk4 versus Dyrk1A.
Most of the compounds from Tables 1 and 2 were also sent for Reaction Biology Clk and Dyrk isoform selectivity profiles to determine their activity against Clk1-Clk4 and Dyrk1A-Dyrk1B. As may be seen from the tables, the data obtained from these profiles was largely in agreement with the results from the initial qHTS bioluminescent assays (shown in parentheses). The greatest differences were noted for compounds 23 and 29; however, the trends associated with these compounds still held true. It is also worth noting that the degree of isoform selectivity was diminished in the Reaction Biology panel relative to the qHTS assay results, showing the difficulty associated with developing highly isoform-selective Clk and Dyrk inhibitors in the absence of protein structural data for rational design.
To examine selectivity within the kinome, a DiscoverRX® KINOMEscan® analysis of 442 kinases was performed using three inhibitors representing different structural types (22, 32 and 35).
This assay, run at 10 μM, gauges the degree to which a compound competitively displaces an active-site ligand anchored to a bead from a kinase linked to a DNA tag using quantitative PCR. Dendrograms displaying the results of the analysis are shown in Figure 5. It may be seen from the figure that compounds 32 and 35 were very selective for the Clk and Dyrk kinases, while 22 was somewhat more promiscuous. These results comport with the greater potency observed for 22 in the Clk/Dyrk assays. Ricerca LeadProfiling® was also used to evaluate the off-target pharmacology of 35 in a 67-assay screen. Targets for which >50% inhibition or stimulation (at 10 μM) was observed included adrenergic α2A, dopamine transporter (DAT), and norepinephrine transporter (NET). A complete listing of targets and results for the screen may be found in the Supporting Information. The small number of potent off-target hits in the Ricerca screen is consistent with the limited activity observed in the KINOMEscan® (Figure 5), providing additional evidence that 35 is a highly selective Clk/Dyrk inhibitor.
Figure 5.

Dendrogram depiction of DiscoverRX® KINOMEscan® kinase binding selectivity analysis. Larger spheres represent <10% control at 10 μM and smaller spheres represent <30% control at 10 μM (% control refers to the percentage of kinase remaining bound to the anchored active-site ligand in the presence of a competitive inhibitor). Activity for 35: <10% = Clk1, Clk2, Clk4, Dyrk1A, PRKCE; <30% = CSNK1E, MAP3K1. Activity for 32: <10% = BIKE, Clk2, Dyrk1A; <30% = AAK1, Clk1, Clk4, GAK, LATS1, PIM1, PIP5K1A, PIP5K2B, RIOK3, SRPK3. Activity for 22: <10% = BIKE, CIT, Clk1, Clk2, Clk4, CSNK1D, CSNK1E, CSNK1G2, DRAK, Dyrk1A, Dyrk1B, Erk8, Flt3, HIPK3, KIT, MYLK4, PIP5K2C, PRKCE, PRKCH; <30% = BMPR2, Clk3, CSNK1A1, DDR2, Dyrk2, Erk5, GSK3B, HIPK4, HUNK, IGF1R, IKK-alpha, IRAK3, MAP3K1, PIP5K1A, PIP5K2B, RIOK1, RIOK2, RIOK3, ROCK1, SRPK3. (Dendrograms generated using the online tool at: http://tripod.nih.gov/?p=226)
Compound 35 was further subjected to in vitro analysis of its pharmacokinetic properties. Details may be found in the Supporting Information. Though the solubility of 35 in both phosphate buffered saline (PBS) and the assay media is limited, it is 17–25 times greater than the IC50 value of the compound, indicating that potency is not limited by solubility. The compound is stable in both mouse and human plasma, moderately cell permeable, and although its hepatic microsome stability is low, this should not affect the utility of the compound as a probe for cell-based studies.
Compound 17 was the most potent among the compounds tested and was also part of the guanidyl family of compounds for which the majority of SAR data was generated. It was therefore chosen as an appropriate representative for homology modeling studies. From a Clk4 homology model constructed using a Clk1 crystal structure (PDB ID: 1Z57, 87% sequence identity) as a template with the modeling program SYBYL48 from Tripos, the docking pose shown in Figure 6a was predicted using the Docking Module of SYBYL. In this arrangement, compound 17 forms several hydrogen bonds with amino acid residues Leu244, Lys191, and Asp325 in the hinge region of the enzyme. The importance of the hydrogen bond between the oxygen of the benzodioxole and Leu244 may be seen when it is compared to analogs in which the dioxole ring is replaced by either a p-methoxy (10/11) or m-methoxy (12/13) substituent. In the m-methoxy analogs, the oxygen atom is not in proximity to Leu244, while in the p-methoxy cases, the methyl group would be expected to rotate out of the plane of the ortho C-H groups, positioning the oxygen lone pairs away from the Leu244 residue and disrupting hydrogen bond formation. These observations are reflected in the steep drops in inhibitory activity for compounds 10–13. The same effect is observed for veratrole-substituted analog 21 for similar reasons. Ring-expanded benzodioxane analog 20, on the other hand, is still a potent Clk4 inhibitor as the two methylene carbons are held rigidly in the ring, placing the lone pairs on the para oxygen atom at angles conducive to hydrogen bond formation with Leu244. Comparing compounds having the guanidyl core to compounds having the amidinyl core, the importance of the guanidyl nitrogen as a hydrogen bond acceptor with Lys191 may also be seen. In the amidinyl series, a hydrogen bond with Lys191 is absent and the compound is significantly less potent. The surface representation in Figure 6b suggests that the aromatic ring of the benzylamine moiety sits in a hydrophobic pocket where van der Waals interactions between the halogenated benzylamine and the pocket are important for binding.
Figure 6.
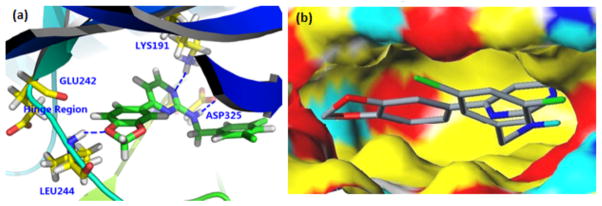
Docking pose for compound 17 in Clk4 homology model: (a) Important hydrogen bond interactions with the hinge region of the enzyme; (b) surface representation showing hydrophobic pocket. (Graphics prepared using Pymol.)
In conclusion, a new series of aryl-substituted aminopyrimidines with activity against the Clk and Dyrk families of kinases has been described. Four substitution patterns around the central pyrimidine were explored, and a number of new compounds were discovered with activities <100 nM against combinations of Clk1, Clk2, Clk4, Dyrk1A and Dyrk1B. The most potent agents have activities <10 nM. Three compounds with different substitution patterns were subjected to DiscoverRX® KINOMEscan® analysis, revealing different levels of selectivity within the kinome between the chemotypes. The off-target pharmacology and in vitro pharmacokinetic properties of the most selective of these agents, 35, were further evaluated and support the idea that this compound is a selective Clk/Dyrk inhibitor with adequate solubility, stability, and cell permeability to allow it to be used in cell-based biological studies. Compound 35 (ML315), therefore, represents a complementary addition to the very small collection of existing Clk/Dyrk inhibitors (Table 3). Its biochemical profile, when compared to other inhibitors, should make it a useful biochemical tool, particularly if used in parallel with other inhibitors to dissect Clk/Dyrk biology.
Table 3.
IC50 values (nM) against Clk and Dyrk kinases for small-molecule inhibitors
| Compound | Clk1 | Clk2 | Clk3 | Clk4 | Dyrk1A | Dyrk1B |
|---|---|---|---|---|---|---|
| 35 (ML315) | 68 | 231 | >10,000 | 68 | 282 | 1156 |
| 139 | 59 | 1902 | 6936 | 39 | 62 | 697 |
| 243 | 1522 | 1648 | >10,000 | 136 | >10,000 | 4420 |
| TG00339 | 19 | 95 | 3000 | 30 | 12 | 130 |
| KH-CB1917 | 20 | NR* | 530 | NR* | 55 | NR* |
| Leucettine L4118 | 15 | NR* | 4500 | NR* | 40 | NR* |
NR = not reported
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (grants U54 HG005031, RO3 MH084827-01, and U54 MH084681-01) and the Division of Preclinical Innovation, National Center for Advancing Translational Sciences of the National Institutes of Health. The dendrograms depicting kinase binding selectivity were generated using the online tool at: http://tripod.nih.gov/?p=226. We thank Benjamin Neuenswander and Patrick Porubsky for compound purification and curation. We thank Arianna Mangravita-Novo and Layton H. Smith of the Sanford-Burnham Medical Research Institute at Lake Nona for solubility and PK profiling measurements.
Footnotes
Supplementary data may be found online at _____
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and alllegal disclaimers that apply to the journal pertain.
References and notes
- 1.Nayler O, Stamm S, Ullrich A. Biochem J. 1997;326:693. doi: 10.1042/bj3260693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prasad J, Manley JL. Mol Cell Biol. 2003;23:4139. doi: 10.1128/MCB.23.12.4139-4149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duncan PI, Stojdl DF, Marius RM, Scheit KH, Bell JC. Exp Cell Res. 1998;241:300. doi: 10.1006/excr.1998.4083. [DOI] [PubMed] [Google Scholar]
- 4.Bullock AN, Das S, Debreczeni JÉ, Rellos P, Fedorov O, Niesen FH, Guo K, Papagrigoriou E, Amos AL, Cho S, Turk BE, Ghosh G, Knapp S. Structure. 2009;17:352. doi: 10.1016/j.str.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long JC, Caceres JF. Biochem J. 2009;417:15. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Blanco MA, Baraniak AP, Lasda EL. Nat Biotech. 2004;22:535. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- 7.Hagiwara M. BBA-Proteins Proteom. 2005;1754:324. [Google Scholar]
- 8.Jiang K, Patel NA, Watson JE, Apostolatos H, Kleiman E, Hanson O, Hagiwara M, Cooper DR. Endocrinology. 2009;150:2087. doi: 10.1210/en.2008-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong R, Balachandran A, Mao AYQ, Dobson W, Gray-Owen S, Cochrane A. Retrovirology. 2011;8:47. doi: 10.1186/1742-4690-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yomoda J-i, Muraki M, Kataoka N, Hosoya T, Suzuki M, Hagiwara M, Kimura H. Genes Cells. 2008;13:233. doi: 10.1111/j.1365-2443.2008.01163.x. [DOI] [PubMed] [Google Scholar]
- 11.Eisenreich A, Bogdanov VY, Zakrzewicz A, Pries A, Antoniak S, Poller W, Schultheiss HP, Rauch U. Circ Res. 2009;104:589. doi: 10.1161/CIRCRESAHA.108.183905. [DOI] [PubMed] [Google Scholar]
- 12.Rodgers JT, Haas W, Gygi SP, Puigserver P. Cell Metab. 2010;11:23. doi: 10.1016/j.cmet.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menegay H, Moeslein F, Landreth G. Exp Cell Res. 1999;253:463. doi: 10.1006/excr.1999.4655. [DOI] [PubMed] [Google Scholar]
- 14.Myers MP, Murphy MB, Landreth G. Mol Cell Biol. 1994;14:6954. doi: 10.1128/mcb.14.10.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, Sumi K, Yomoda J-i, Murray MV, Kimura H, Furuichi K, Shibuya H, Krainer AR, Suzuki M, Hagiwara M. J Biol Chem. 2004;279:24246. doi: 10.1074/jbc.M314298200. [DOI] [PubMed] [Google Scholar]
- 16.Hekimi S, McBride K, Hihi AK, Kianicka I, Wang Y, Hayes SL, Guimond MP, Sévigny G, Dumas D, Smith J. WO 2008/014602 A1. Quinoline Derivatives. 2008 Feb 7;
- 17.Fedorov O, Huber K, Eisenreich A, Filippakopoulos P, King O, Bullock AN, Szklarczyk D, Jensen LJ, Fabbro D, Trappe J, Rauch U, Bracher F, Knapp S. Chem Biol. 2011;18:67. doi: 10.1016/j.chembiol.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Debdab M, Carreaux F, Renault S, Soundararajan M, Fedorov O, Filippakopoulos P, Lozach O, Babault L, Tahtouh T, Baratte B, Ogawa Y, Hagiwara M, Eisenreich A, Rauch U, Knapp S, Meijer L, Bazureau JP. J Med Chem. 2011;54:4172. doi: 10.1021/jm200274d. [DOI] [PubMed] [Google Scholar]
- 19.Aranda S, Laguna A, de la Luna S. FASEB J. 2011;25:449. doi: 10.1096/fj.10-165837. [DOI] [PubMed] [Google Scholar]
- 20.Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost H-G. J Biol Chem. 1998;273:25893. doi: 10.1074/jbc.273.40.25893. [DOI] [PubMed] [Google Scholar]
- 21.Becker W, Sippl W. FEBS J. 2011;278:246. doi: 10.1111/j.1742-4658.2010.07956.x. [DOI] [PubMed] [Google Scholar]
- 22.Göckler N, Jofre G, Papadopoulos C, Soppa U, Tejedor FJ, Becker W. FEBS J. 2009;276:6324. doi: 10.1111/j.1742-4658.2009.07346.x. [DOI] [PubMed] [Google Scholar]
- 23.Tejedor FJ, Hämmerle B. FEBS J. 2011;278:223. doi: 10.1111/j.1742-4658.2010.07954.x. [DOI] [PubMed] [Google Scholar]
- 24.Guimera J, Casas C, Estivill X, Pritchard M. Genomics. 1999;57:407. doi: 10.1006/geno.1999.5775. [DOI] [PubMed] [Google Scholar]
- 25.Yabut O, Domogauer J, D’Arcangelo G. J Neurosci. 2010;30:4004. doi: 10.1523/JNEUROSCI.4711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park J, Song WJ, Chung KC. Cell Mol Life Sci. 2009;66:3235. doi: 10.1007/s00018-009-0123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrer I, Barrachina M, Puig B, Martínez de Lagrán M, Martí E, Avila J, Dierssen M. Neurobiol Dis. 2005;20:392. doi: 10.1016/j.nbd.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 28.Wegiel J, Gong CX, Hwang YW. FEBS J. 2011;278:236. doi: 10.1111/j.1742-4658.2010.07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Graaf K, Czajkowska H, Rottmann S, Packman LC, Lilischkis R, Luscher B, Becker W. BMC Biochem. 2006;7:7. doi: 10.1186/1471-2091-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fotaki V, Dierssen M, Alcántara S, Martínez S, Martí E, Casas C, Visa J, Soriano E, Estivill X, Arbonés ML. Mol Cell Biol. 2002;22:6636. doi: 10.1128/MCB.22.18.6636-6647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Møller RS, Kübart S, Hoeltzenbein M, Heye B, Vogel I, Hansen CP, Menzel C, Ullmann R, Tommerup N, Ropers HH, Tümer Z, Kalscheuer VM. Am J Hum Gen. 2008;82:1165. doi: 10.1016/j.ajhg.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida K. Biochem Pharmacol. 2008;76:1389. doi: 10.1016/j.bcp.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 33.Guo X, Williams JG, Schug TT, Li X. J Biol Chem. 2010;285:13223. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leder S, Weber Y, Altafaj X, Estivill X, Joost HG, Becker W. Biochem Biophys Res Commun. 1999;254:474. doi: 10.1006/bbrc.1998.9967. [DOI] [PubMed] [Google Scholar]
- 35.Lauth M, Bergstrom A, Shimokawa T, Tostar U, Jin Q, Fendrich V, Guerra C, Barbacid M, Toftgard R. Nat Struct Mol Biol. 2010;17:718. doi: 10.1038/nsmb.1833. [DOI] [PubMed] [Google Scholar]
- 36.Friedman E. J Cell Biochem. 2007;102:274. doi: 10.1002/jcb.21451. [DOI] [PubMed] [Google Scholar]
- 37.Mercer SE, Friedman E. Cell Biochem Biophys. 2006;45:303. doi: 10.1385/CBB:45:3:303. [DOI] [PubMed] [Google Scholar]
- 38.Adayev T, Wegiel J, Hwang YW. Arch Biochem Biophys. 2011;507:212. doi: 10.1016/j.abb.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mott BT, Tanega C, Shen M, Maloney DJ, Shinn P, Leister W, Marugan JJ, Inglese J, Austin CP, Misteli T, Auld DS, Thomas CJ. Bioorg Med Chem Lett. 2009;19:6700. doi: 10.1016/j.bmcl.2009.09.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S, Wood G, Duncan KW, Meades C, Gibson D, McLachlan JC, Perry A, Blake D, Zheleva DI, Fischer PM. Pyrimidin-4-yl-3,4-thione Compounds and Their Use in Therapy. WO 2005/042525 A1. 2005 May 12;
- 41.http://www.reactionbiology.com/pages/kinase.htm.
- 42.Tanega C, Shen M, Mott BT, Thomas CJ, MacArthur R, Inglese J, Auld DS. Assay Drug Dev Technol. 2009;7:606. doi: 10.1089/adt.2009.0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenthal AS, Tanega C, Shen M, Mott BT, Bougie JM, Nguyen DT, Misteli T, Auld DS, Maloney DJ, Thomas CJ. Bioorg Med Chem Lett. 2011;21:3152. doi: 10.1016/j.bmcl.2011.02.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.An MLPCN probe report describing ML315 will soon be published on the NCBI bookshelf: http://www.ncbi.nlm.nih.gov/books/NBK47352/
- 45.Topliss JG. J Med Chem. 1972;15:1006. doi: 10.1021/jm00280a002. [DOI] [PubMed] [Google Scholar]
- 46.Garner J, Hill T, Odell L, Keller P, Morgan J, McCluskey A. Aust J Chem. 2004;57:1079. [Google Scholar]
- 47.Gutierrez CD, Bavetsias V, McDonald E. Tetrahedron Lett. 2005;46:3595. [Google Scholar]
- 48.SYBYL 8.0. Tripos Associates; St. Louis, MO: 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.