

Abstract
Diphenoxylate, a well-known opioid agonist and anti-diarrhoeal agent, was recently found to block Kv1.3 potassium channels, which have been proposed as potential therapeutic targets for a range of autoimmune diseases. The molecular basis for this Kv1.3 blockade was assessed by the selective removal of functional groups from the structure of diphenoxylate as well as a number of other structural variations. Removal of the nitrile functional group and replacement of the C-4 piperidinyl substituents resulted in several compounds with submicromolar IC50 values.
Autoimmune disorders are characterized by an organism mistakenly mounting an immune response against itself. Examples of these disorders include multiple sclerosis (MS) psoriasis, type-1 diabetes, Crohn’s Disease, and rheumatoid arthritis1. The aberrant immune response is mediated by autoantibodies and self-reactive lymphocytes. The lymphocytes involved are T cells and once activated they proliferate and cause tissue damage. One particular type of T cells known as effector memory (TEM) cells have been linked to autoimmune disorders2–4.
Human T cells express two types of K+ channels, the voltage-gated Kv1.3 and the calcium-activated KCa3.1 channel5, 6. Both Kv1.3 and KCa3.1 play a role in regulating membrane potential and calcium signalling during the activation of T cells5. Calcium influx, which is crucial to the process, is only possible if the T cells are able to maintain a negative membrane potential through a counterbalancing potassium efflux via Kv1.3 and/or KCa3.1 channels5, 7, 8.
Blockade of Kv1.3 or KCa3.1 channels is a possible method for treating autoimmune and inflammatory diseases by suppressing T-cell proliferation and modulating their activities9. Importantly, naïve and central memory T cells (TCM) upregulate KCa3.1 channels upon activation leaving the number of Kv1.3 channels largely unchanged. The opposite occurs in TEM cells which upregulate Kv1.3 channels when activated5. Blockade of the Kv1.3 channel therefore provides an opportunity for intervention by therapeutic agents, leaving naïve and TCM cells free to address other immunogenic threats (e.g. infections).
A number of studies have shown that blockade of Kv1.3 potassium channels results in functional inhibition of T cell activation/proliferation and cytokine secretion5, 8, 10. In one study, the potent Kv1.3 blocking peptide ShK and a series of related analogues were able to treat both adoptive transfer and chronic relapsing experimental autoimmune encephalomyelitis (EAE) in rats11. Small molecule blockers of Kv1.3 channels have also been investigated12. The most potent of these compounds is PAP-1 (IC50 2 nM) which has been shown to suppress delayed type hypersensitivity (DTH) and allergic contact dermatitis (ACD) in Lewis rats when dosed orally, by i.p. injection or topically13, 14.
In searching for new compounds that might have clinical potential as Kv1.3 channel blockers we noticed that diphenoxylate (1) had been shown, in a small clinical trial, to successfully treat psoriasis and other inflammatory skin conditions15, 16. Diphenoxylate also shows structural similarity to a number of Kv1.3 blockers (Figure 1) and when assessed it was found to block Kv1.3 channels with an IC50 of 5 μM12. With a view to optimising the Kv1.3 blockade shown by diphenoxylate, we examined which elements of the chemical structure are required for biological activity.
Figure 1.
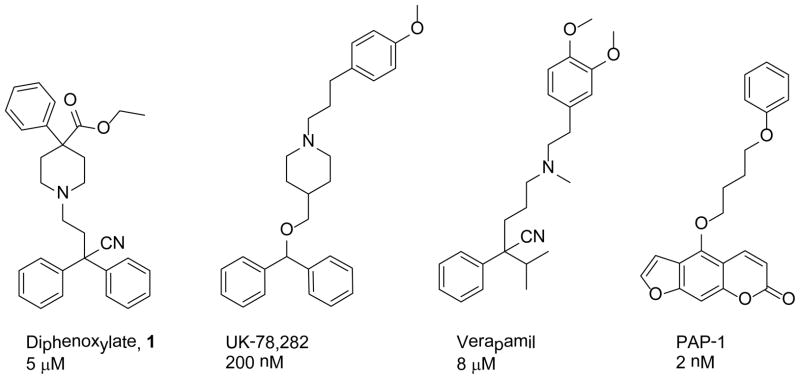
Structures and Kv1.3 blocking activity of diphenoxylate (1), UK-78,282, verapamil and PAP-1.
As our principle objective was to delineate the pharmacophoric elements of diphenoxylate with respect to Kv1.3 blockade, analogues were prepared where one or more of the functional elements of 1 were removed or altered. The first reports of diphenoxylate synthesis date back to Janssen et al. in 195917 and that synthesis provided the basis for the work described here. As exemplified in the synthesis of the methyl ester 2, it was found that the alkylation of the key piperidine precursor (3) with diphenylbromopropionitrile (4) could be accelerated and the yield improved by microwave heating in acetonitrile (ACN) in the presence of N,N-diisopropylethylamine (DIPEA). This synthetic step (Scheme 1) formed the basis for all analogues described here, with variations derived from commercial or synthesized building blocks. Full synthesis details are provided as supplementary material.
Scheme 1.
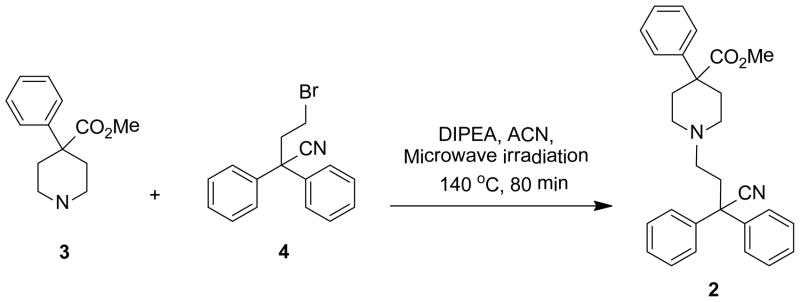
Synthesis of diphenoxylate analogue 2
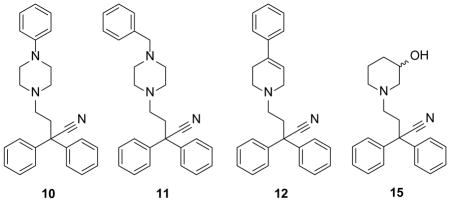
Other compounds prepared in this way included 5, 6 and 7 in which the ester, piperidine 4-phenyl substituent and the nitrile were replaced by a hydrogen atom, respectively or 8 where both piperidine 4-substituents were removed. In compound 9 the 2-biphenylbutyronitrile portion is replaced by a simple phenylpropionyl group. Other analogues that were prepared included replacements of the piperidine ring such as piperazine (10, 11) and tetrahydropyridine (12). Replacement of the ester with a hydroxyl group (13) was undertaken as the 4-phenylpiperidin-4-ol group is a well-known fragment in established drugs. The activity of 13 discussed later, prompted the removal of the 4-phenyl ring to produce 14 and the related piperidine-3-ol analogue (15). To complement our SAR exploration, the truncated analogues 16 and 17 were also produced. A further series of analogues based on compound 14 were synthesized to produce 18, 19, 20 and 21.
Compounds were assessed for their ability to block Kv1.3 channels using manual whole-cell patch-clamp as previously described14, 18. Briefly, L929 cells stably expressing Kv1.3 channels were subjected to depolarizing step pulses from −80 mV to +40 mV to elicit Kv1.3 currents. Compounds were manually perfused and in most cases 3–5 different concentrations were tested at least 2–3 times. All compounds were washed out again to differentiate true pharmacological effects from unspecific current “run-down”. IC50 values were determined by fitting the reduction of area under the current curve after reaching equilibrium block to the Hill equation. Full details are provided as supplementary material.
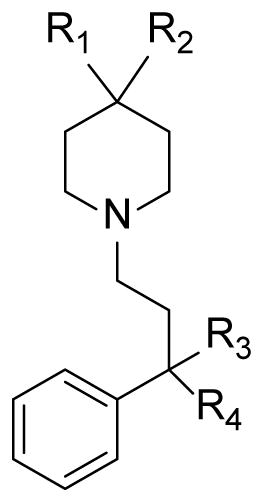
The compounds tested demonstrated a range of potencies, clearly showing the specific contributions of the diphenoxylate substructures. In the simplest analogue, replacement of the ethyl ester with a methyl ester (2) resulted in a marginal improvement in activity (Table 1). Removing the ethyl ester from diphenoxylate altogether in 5, also slightly improved blocking potency. In contrast, removal of the phenyl ring from the piperidine (6) resulted in a substantial decrease in activity (~50 fold). Compound 7 which lacked the cyano group was 6-fold more active than diphenoxylate itself.
Table 1.
Kv1.3 blockade
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | Kv1.3 (IC50 μM) |
| Diphenoxylate, 1 | -C6H5 | -C=OOC2H5 | -CN | -C6H5 | 5.012 |
| 2 | -C6H5 | -C=OOCH3 | -CN | -C6H5 | 2.52 ± 0.15 |
| 5 | -C6H5 | -H | -CN | -C6H5 | 2.61 ± 0.11 |
| 6 | -H | -C=OOC2H5 | -CN | -C6H5 | 230 ± 7.5 |
| 7 | -C6H5 | -C=OOC2H5 | -H | -C6H5 | 0.90 ± 0.10 |
| 8 | -H | -H | -CN | -C6H5 | 20.5 ± 1.6 |
| 9 | -C6H5 | -C=OOC2H5 | -H | -H | 0.80 ± 0.10 |
| 10a | 1.86 ± 0.03 | ||||
| 11a | 1.27 ± 0.10 | ||||
| 12a | 4.20 ± 0.80 | ||||
| 13 | -C6H5 | -OH | -CN | -C6H5 | 1.70 ± 0.20 |
| 14 | -H | -OH | -CN | -C6H5 | 0.75 ± 0.05 |
| 15a | > 100 | ||||
| 16 | -H | -OH | -H | -C6H5 | ~75.0 |
| 17 | -H | -OH | -H | -H | > 100 |
| 18 | -H | -CH3 | -CN | -C6H5 | 5.80 ± 0.3 |
| 19 | -H | -OCH3 | -CN | -C6H5 | > 100 |
| 20 | -H | -NH2 | -CN | -C6H5 | > 100 |
| 21 | -H | -NHBoc | -CN | -C6H5 | ~25.0 |
Of the more extensively pruned compounds, compound 8 was 4-fold less potent than diphenoxylate demonstrating that while the ester is not needed, further removal of the phenyl ring was not well tolerated suggesting that this aromatic group is required for activity (Table 1). Pruning of the diphenylmethane/cyano moiety of diphenoxylate had a significant effect on activity. Removal of both the cyano group and one phenyl ring from this group (9) resulted in a similar improvement in potency relative to compound 7. Compound 9 has been flagged as an interesting substance for future optimization.
Having systematically removed groups from the structure of diphenoxylate, we investigated
Diphenoxylate has already been shown to be able to treat inflammatory skin conditions including the autoimmune disorder psoriasis15, 16 and this clinical observation, though unproven in a large clinical trial, is consistent with modest Kv1.3 channel blockade12. Efforts to produce a suitable topical formulation of diphenoxylate would seem unlikely due to the side-effect potential of this narcotic agent.
Exploitation of diphenoxylate as a lead compound for developing Kv1.3 channel blockers presents a number of challenges12. Diphenoxylate has moderate potency and a relatively high molecular weight and lipophilicity. Compounds with high logP values are often found to be able to block potassium channels and the need to demonstrate a clear SAR for diphenoxylate was imperative. From a structural perspective we saw partial similarities to the known Kv1.3 blockers UK 78,282, verapamil and PAP-1 (Figure 1). These included the basic aliphatic nitrogen, diphenylmethyl moiety, cyano group and the location of aromatic rings at both ends of the molecules. We began by pruning these functional groups on diphenoxylate to explore their effect on Kv1.3 blockade. In some cases these groups were replaced by other functional groups of varying sizes and properties.
The principle outcomes were the demonstration that Kv1.3 blockade could be maintained and even enhanced by the removal of one or more of diphenoxylate’s key functional groups. For example, removal of the R4-phenyl ring and the nitrile was tolerated such that compound 9 (IC50 0.8 μM) had improved potency. The replacement of the phenyl and carboxylic ester substituents by a single hydroxyl group in compound 14 also showed improved activity (IC50 0.75 μM). The combination of these modifications however, yielded a poorly active compound 17 (IC50 > 100 μM). This contradicted the overall pharmacophore that we had tentatively proposed12 for Kv1.3 inhibitors. The inner cavity, which constitutes the binding site for most small molecule Kv1.3 blockers is somewhat large19 and it seems probable that the compounds bind in various ways such that a single pharmacophore will not describe the SAR.
From a drug discovery perspective, the retention of blockade with both reduced molecular size and lipophilicity is encouraging. While PAP-1 shows good potency it has no ionizable functional groups and a logP value of 4.0314 resulting in a relatively low oral availability of only 25% and solubility issues for formulation20. Both compounds 14 and 9 have reduced ClogD7.4 values relative to diphenoxylate as well as reduced molecular weights (320.4 and 351.5, respectively). Diphenoxylate is a poorly soluble substance due to its relatively high lipophilicity (ClogP7.4 4.27). The lipophilicity of compound 9 was reduced (ClogD7.4 3.47) however, the removal of groups to generate compound 14 resulted in a substantially lower ClogD7.4 value of 1.46.
The concept of lipophilic ligand efficiency (LipE) has recently emerged as an influential descriptor to assist lead optimization21 and is calculated by subtracting the pIC in its IC50. Also in contrast to 9, the removal of a cyano group plus a phenyl ring (17) also resulted in no activity. The difference in SAR between diphenoxylate and 14 suggests that these two compounds are likely to have two distinct binding modes and/or locations within the Kv1.3 channel. It is possible that these compounds may also be binding to a different state of the channel (i.e. open or closed) which may account for their dissimilar SAR.
Another important aspect of investigating the SAR of diphenoxylate is to place focus on the ester group which is linked to its activity at opioid receptors. The ester on diphenoxylate is metabolized in vivo to the carboxylic acid (diphenoxin) which is the active opioid agent22. Any future work would need to monitor mu opioid activity and avoiding an ester would need to be considered. Compound 14 circumvents this problem, however any optimization of 9 would need to bear this in mind.
This study has identified two new series of Kv1.3 blockers derived from the anti-diarrhoeal compound diphenoxylate. Successive deletion of functional groups was able to improve activity although the SAR was not consistent between the compound classes. Removal of the ester, cyano and an aromatic ring were tolerated and in many cases improved activity. These deletions also reduced both MW and lipophilicity presenting compounds worthy of further investigation. There is a need for Kv1.3 blockers with improved selectivity and biopharmaceutical properties, and this study provides a starting point for further investigations.
Experimental
Electrophysiology
L929 cells stably expressing Kv1.3 channels were used for all electrophysiology experiments. All experiments were conducted in the whole-cell configuration of the patch-clamp technique with a holding potential of −80 mV unless otherwise stated. Pipette resistances averaged 2.0 MΩ, and series resistance compensation of 80% was employed when currents exceeded 2 nA. Kv1.3 currents were elicited by repeated 200-ms or 500-ms pulses from −80 mV to 40 mV, applied at intervals of 30 or 60 s. Kv1.3 currents were recorded in normal Ringer solution with a Ca2+-free pipette solution containing (in mM): 145 KF, 10 HEPES, 10 EGTA, 2 MgCl2, pH 7.2, 300 mOsm. IC50-values and Hill coefficient were determined by fitting the Hill equation to the reduction of area under the current curve. All compounds tested were >95% purity as determined by RP-HPLC.
Chemistry
Alkylation Reactions
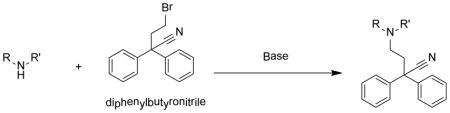
General method for the preparation of diphenoxylate analogues
Method A
To a solution of alkyl halide (1–2 eq) in ACN (or DMF) was added amine (1–2 eq) with diisopropylamine (2–3 eq) and refluxed overnight under N2. Solvent was concentrated in vacuo yielding a crude residue, which was dissolved in EtOAc and washed with saturated NaHCO3, H2O, saturated NaCl, dried with anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was either reacted directly or purified.
Method B
To a solution of alkyl halide (1–2 eq) in ACN (or DMF) was added amine (1–2 eq) with diisopropylamine (2–3 eq). The resulting mixture was placed in the microwave reactor (90W) and reaction commenced for 80 mins at 140°C. Solvent was concentrated in vacuo yielding a crude residue, which was dissolved in EtOAc and washed with saturated NaHCO3, H2O, saturated NaCl, dried with anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was either reacted directly or purified.
Methyl 1-(3-cyano-3,3-diphenylpropyl)-4-phenylpiperidine-4-carboxylate (2)23
According to method A, 4-bromo-2,2-diphenylbutanenitrile (270 mg, 0.90 mmol) was treated with methyl 4-phenylpiperidine-4-carboxylate (120 mg, 0.55 mmol). The yellow oil was purified via RP-HPLC to afford a cream solid (149 mg, 62 %). Mp: 184–186 °C (lit. 180–181 °C). 1H NMR (300 MHz, CDCl3) δ 7.50 – 7.31 (m, 15H, Ar H), 3.75 (s, 3H, COOCH3), 2.72-2.62 (m, 4H, 4 × CHpip), 2.48 (t, J = 14.2 Hz, 2H, NCH2CH2C), 2.22 (t, J = 12.8 Hz, 2H, NCH2CH2C), 1.98 – 1.86 (m, 4H, 4 × CHpip). 13C NMR (600 MHz, CDCl3) δ 175.2, 140.9, 139.8, 138.6, 128.1, 127.1, 126.8, 126.2, 125.0, 120.9, 54.7, 51.4, 50.2, 48.7, 47.8, 36.3, 32.4. Mass Spectrum (ESI): m/z 439.5 [M+H]+.
2,2-diphenyl-4-(4-phenylpiperidin-1-yl)butanenitrile (5)24
According to method B, 4-bromo-2,2-diphenylbutanenitrile (120 mg, 400 μmol) was treated with 4-phenylpiperidine (90 mg, 560 μmol). The crude product was purified by column chromatography using a gradient solvent system (100% hexane – 33% EtOAc in hexane) to afford a yellow oil (82 mg, 54%). 1H NMR (400 MHz, CDCl3) δ 7.46 – 7.30 (m, 12H, Ar H), 7.26 – 7.19 (m, 3H, Ar H), 3.06 (ad, J = 10.8 Hz, 2H, 2 × CHpip), 2.76-2.65 (m, 2H, NCH2CH2C), 2.58 – 2.45 (m, 3H, NCH2CH2C, CH), 2.19 – 2.06 (m, 2H, 2 × CHpip), 1.89 – 1.77 (m, 4H, 4 × CHpip). 13C NMR (101 MHz, CDCl3) δ 146.07, 140.01, 128.96, 128.44, 127.96, 126.84, 126.80, 126.21, 122.08, 55.11, 54.61, 50.10, 42.52, 36.68, 33.27. Mass Spectrum (ESI): m/z 381.2 [M+H]+. (HR-ESI) Found: [M+H]+, 381.2311. C27H28N2 requires [M+H]+, 381.2325.
Ethyl piperidine-4-carboxylate (22)25
Isonipecotic acid (1.29g, 10.0 mmol) was dissolved in absolute ethanol (50 ml). The solution was cooled to 0°C and thionyl chloride (2.91 ml, 40.0 mmol) added dropwise. The solution was then stirred and refluxed for 48 h. The solvent was removed in vacuo yielding yellow oil, which was dissolved in EtOAc and washed with 10% NaOH. The organic layer was dried with anhydrous Na2SO4, filtered and concentrated in vacuo to afford a clear oil (1.48 g, 94%). 1H NMR (400 MHz, CDCl3) δ 4.12 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.07 (dt, J = 12.6, 3.6 Hz, 2H, 2 × CHpip), 2.61 (td, J = 12.3, 2.7 Hz, 2H, 2 × CHpip), 2.38 (tt, J = 11.3, 3.9 Hz, 1H, CH), 1.94 – 1.78 (m, 2H, 2 × CHpip), 1.59 (adtd, J = 13.4, 11.4, 4.0 Hz, 2H, 2 × CHpip), 1.24 (t, J = 7.1 Hz, 3H, OCH2CH3). Mass Spectrum (ESI): m/z 158.2 [M+H]+.
Ethyl 1-(3-cyano-3,3-diphenylpropyl)piperidine-4-carboxylate (6)
According to method B, 4-bromo-2,2-diphenylbutanenitrile (50 mg, 167 μmol) was treated with ethyl piperidine-4-carboxylate (22) (31 mg, 200 μmol). The crude product was purified by column chromatography using a gradient solvent system (100% hexane – 33% EtOAc in hexane) to afford orange oil (38 mg, 61%). 1H NMR (400 MHz, CDCl3) δ 7.48 – 7.23 (m, 10H, Ar H), 4.14 (q, J = 7.1 Hz, 2H, OCH2CH3), 2.86 (ad, J = 11.5 Hz, 2H, 2 × CHpip), 2.68 – 2.57 (m, 2H, NCH2CH2C), 2.50 – 2.40 (m, 2H, NCH2CH2C), 2.35 – 2.22 (m, 1H, CH), 2.09 – 1.97 (m, 2H, 2 × CHpip), 1.94 – 1.84 (m, 2H, 2 × CHpip), 1.82 – 1.68 (m, 2H, 2 × CHpip), 1.26 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ 174.94, 140.01, 128.92, 127.93, 126.78, 122.07, 60.31, 54.96, 53.22, 50.03, 41.01, 36.72, 28.17, 14.20. Mass Spectrum (ESI): m/z 377.5 [M+H]+. (HR-ESI) Found: [M+H]+, 377.2232. C24H28N2O2 requires [M+H]+, 377.2224.
Ethyl 4-phenylpiperidine-4-carboxylate (23)26
4-phenylpiperidine-4-carboxylic acid (1.00 g, 3.27 mmol) was added to a stirred solution of ACN (20ml) with ethyl iodide (523 μl, 6.55 mmol) and DIPEA (1.71 ml, 9.82 mmol) and stirred at room temperature overnight. ACN was then removed in vacuo, added to a 1:1 mixture of TFA/DCM (20 ml) and stirred for 30 mins. TFA/DCM was then removed in vacuo. The resulting residue was then dissolved in DCM and washed with 2M NaOH, saturated NaCl, H2O, dried with anhydrous Na2SO4 and filtered. The solvent was then concentrated in vacuo to afford a creamy white solid (653 mg, 86%). 1H NMR (300 MHz, CDCl3) δ 7.40 – 7.24 (m, Ar H), 4.21 (q, 2H, J = 7.0 Hz, OCH2CH3), 3.13 (dt, 2H, J = 3.5 Hz, 2 × CHpip), 2.82 (td, J = 2.0 Hz, 2 × CHpip), 2.57 (ad, J = 14 Hz, 2H, 2 × CHpip), 1.87 (td, 2H, J = 4.0 Hz, 2 × CHpip), 1.20 (t, 3H, J = 4 Hz, OCH2CH3). Mass Spectrum (ESI): m/z 234.3 [M+H]+.
Ethyl 1-(3,3-diphenylpropyl)-4-phenylpiperidine-4-carboxylate (7)27
According to method B, (3-bromopropane-1,1-diyl)dibenzene (127 mg, 463 μmol) was treated with ethyl 4-phenylpiperidine-4-carboxylate (23) (120 mg, 514 μmol). The crude product was purified by column chromatography using a gradient solvent system (100% hexane – 66% EtOAc in hexane) to afford a crude oil (174 mg. 79%). 29 mg of this crude sample was then further purified using preparative TLC using a solvent system of 5% MeOH in CHCl3 to afford clear gel (24 mg, 65%). 1H NMR (400 MHz, CDCl3) δ 7.32 – 7.16 (m, 15H, Ar H), 4.05 (q, 2H, J = 7.0 Hz, OCH2CH3), 3.95 – 3.85 (m, 1H, CH), 2.76 (ad, J = 11.2 Hz, 2H, 2 × CHpip), 2.48 (ad, J = 13.1 Hz, 2H, NCH2CH2CH), 2.23 – 2.16 (m, 4H, NCH2CH2CH, CH, 2 × CHpip), 2.10 – 1.99 (m, 2H, 2 × CHpip), 1.96 – 1.83 (m, 2H, 2 × CHpip), 1.07 (t, J = 7.1 Hz, 3H. OCH2CH3). 13C NMR (101 MHz, CDCl3) δ 174.43, 144.82, 128.54, 128.51, 128.46, 127.88, 127.86, 126.17, 125.82, 61.02, 60.81, 57.06, 51.56, 49.27, 49.24, 47.37, 38.27, 33.85, 32.83, 14.07. Mass Spectrum (ESI): m/z 428.5 [M+H]+. (HR-ESI) Found: [M+H]+, 428.2563. C29H33NO2 requires [M+H]+, 428.2584.
2,2-diphenyl-4-(piperidin-1-yl)butanenitrile (8)23
A mixture of diphenylacetonitrile (2.00 g, 10.35 mmol) and anhydrous sodium hydride (1.52 g, 50.67 mmol) in anhydrous DMF (30 ml) was stirred at room temperature for 30 min. 1-(2-chloroethyl)piperidine (2.85 g, 19.43 mmol) in anhydrous DMF (10 ml) was added and solution refluxed at 90 °C for 24 h. The solution was concentrated in vacuo, taken up in water, made basic with potassium carbonate, then extracted into ethyl acetate and concentrated in vacuo. 100 mg of the crude product was purified by RP-HPLC to afford 72 mg of a white solid. Mp: 72 – 74 °C. 1H NMR (300 MHz, CDCl3) δ 7.47 – 7.35 (m, 10H, Ar H), 3.62 (t, J = 12.2 Hz, 2H, NCH2CH2C), 3.22-3.14 (m, 4H, 4 × CHpip), 2.66 (t, J = 11.2 Hz, 2H, NCH2CH2C), 2.28 – 2.01 (m, 4H, 4 × CHpip), 1.46 (m, 2H, NCH2CH2CH2). 13C NMR (600 MHz, CDCl3) δ 138.0, 129.4, 128.6, 126.4, 121.2, 53.8, 49.9, 33.0, 30.9, 22.7, 21.9. Mass Spectrum (ESI): m/z 305.4 [M+H]+.
Ethyl 4-phenyl-1-(3-phenylpropyl)piperidine-4-carboxylate (9)28
According to method B, 4-bromo-2,2-diphenylbutanenitrile (89 μl, 585 μmol) was treated with ethyl 4-phenylpiperidine-4-carboxylate (23) (150 mg, 643 μmol). The crude product was purified by column chromatography using a gradient solvent system (100% DCM – 5% MeOH in DCM) to afford a yellow oil (163 mg, 80%). 1H NMR (400 MHz, CDCl3) δ 7.44 – 7.17 (m, 10H, Ar H), 4.15 (q, 2H, J = 7.0 Hz, OCH2CH3), 2.78 (ad, J = 11.5 Hz, 2H, 2 × CHpip), 2.61 – 2.55 (m, 4H, NCH2CH2CH2, 2 × CHpip), 2.39 – 2.29 (m, 2H, NCH2CH2CH2), 2.21 – 2.08 (m, 2H, 2 × CHpip), 2.05 – 1.92 (m, 2H, 2 × CHpip), 1.86 – 1.75 (m, 2H, NCH2CH2CH2), 1.20 (t, J = 7.1 Hz, 3H. OCH2CH3). 13C NMR (101 MHz, CDCl3) δ 174.46, 143.02, 142.17, 128.49, 128.40, 128.31, 126.93, 125.82, 125.76, 60.79, 58.19, 51.52, 49.33, 33.91, 33.80, 28.73, 14.08. Mass Spectrum (ESI): m/z 352.3 [M+H]+. (HR-ESI) Found: [M+H]+, 352.2254. C23H29NO2 requires [M+H]+, 352.2271.
2,2-diphenyl-4-(4-phenylpiperazin-1-yl)butanenitrile (10)29
According to method B, 4-bromo-2,2-diphenylbutanenitrile (100 mg, 333 μmol) was treated with 1-phenylpiperazine (36 μl, 233 μmol). The crude product was purified by column chromatography using a gradient solvent system (100% hexane – 33% EtOAc in hexane) to afford a yellow oil (62 mg, 49%). 1H NMR (400 MHz, CDCl3) δ 7.41 – 7.16 (m, 12H, Ar H), 6.82 (add, J = 8.8, 0.9 Hz, 2H, Ar H), 6.79 – 6.73 (m, 1H, Ar H), 3.12 – 3.06 (m, 4H, 2 × Piperazine CH2), 2.61 – 2.54 (m, 2H, NCH2CH2C), 2.54 – 2.48 (m, 4H, 2 × Piperazine CH2), 2.47 – 2.40 (m, 2H, NCH2CH2C). 13C NMR (101 MHz, CDCl3) δ 151.25, 140.02, 129.11, 128.97, 128.00, 126.82, 122.10, 119.73, 116.06, 54.74, 53.38, 50.04, 49.06, 36.64. Mass Spectrum (ESI): m/z 382.5 [M+H]+. (HR-ESI) Found: [M+H]+, 382.2284. C26H27N3 requires [M+H]+, 382.2278.
4-(4-benzylpiperazin-1-yl)-2,2 diphenylbutanenitrile (11)30
According to method B, 4-bromo-2,2-diphenylbutanenitrile (100 mg, 333 μmol) was treated with 1-benzylpiperazine (81 μl, 470 μmol). The crude product was purified by column chromatography using a gradient solvent system (100% hexane – 33% EtOAc in hexane) to afford a pale yellow oil (82 mg, 62%). 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.09 (m, 15H, Ar H), 3.47 (s, 2H, Benzyl CH2), 2.62 – 2.31 (m, 12H). 13C NMR (101 MHz, CDCl3) δ 139.87, 129.32, 128.96, 128.32, 127.99, 127.31, 126.78, 122.03, 62.74, 54.54, 52.99, 52.57, 49.98, 36.26. Mass Spectrum (ESI): m/z 396.6 [M+H]+. (HR-ESI) Found: [M+H]+, 396.2441. C27H29N3 requires [M+H]+, 396.2434.
4-phenyl-1,2,3,6-tetrahydropyridine (24)31
4-phenylpiperidin-4-ol (100 mg, 564 μmol) was dissolved in 9M HCl and stirred at 80°C for 24 hours. The solution was then cooled to 0°C and NaOH added and stirred till the solution was basic (pH > 8). EtOAc was then added and organic layer washed with saturated NaHCO3, saturated NaCl, H2O, dried with anhydrous Na2SO4, filtered and concentrated in vacuo to yield a clear oil (61 mg, 68%). 1H NMR (400 MHz, CDCl3) δ 7.43 – 7.38 (m, 2H, Ar H), 7.37 – 7.32 (m, 2H, Ar H), 7.29 – 7.23 (m, 1H, Ar H), 6.18 – 6.14 (m, 1H, CH), 3.55 (dd, J = 5.9, 2.8 Hz, 2H, PhC=CHCH2), 3.13 (t, J = 5.7 Hz, 2H, PhCCH2CH2), 2.48 (ddq, J = 5.6, 4.4, 2.7 Hz, 2H, PhCCH2). Mass Spectrum (ESI): m/z 160.2 [M+H]+.
2,2-diphenyl-4-(4-phenyl-5,6-dihydropyridin-1(2H)-yl)butanenitrile (12)
According to method B, 4-bromo-2,2-diphenylbutanenitrile (65 mg, 217 μmol) was treated with 4-phenyl-1,2,3,6-tetrahydropyridine (24) (35 mg, 217 μmol). The crude product was purified by column chromatography using a gradient solvent system (100% DCM – 2% MeOH in DCM) to afford a yellow oil (72 mg, 49%). 1H NMR (400 MHz, CDCl3) δ 7.47 – 7.21 (m, 15H, Ar H), 6.07 – 6.03 (m, 1H, CH), 3.22 (ad, J = 2.4 Hz, 2H, PhC=CHCH2), 2.83 – 2.72 (m, 4H, PhCCH2CH2, NCH2CH2C), 2.71 – 2.55 (m, 4H, PhCCH2, NCH2CH2C). 13C NMR (101 MHz, CDCl3) δ 140.76, 140.00, 135.16, 128.99, 128.29, 127.99, 127.06, 126.78, 124.97, 122.07, 121.48, 54.54, 53.45, 50.63, 50.07, 37.07, 28.10. Mass Spectrum (ESI): m/z 379.1 [M+H]+. (HR-ESI) Found: [M+H]+, 379.2186. C27H26N2 requires [M+H]+, 379.2169.
4-(4-hydroxy-4-phenylpiperidin-1-yl)-2,2-diphenylbutanenitrile (13)32
According to method A, 4-bromo-2,2-diphenylbutanenitrile (2.00 g, 6.66 mmol) was treated with 4-phenylpiperidin-4-ol (1.18 g, 6.66 mmol). 100 mg of the crude product was purified by RP-HPLC to afford 66mg of a beige solid. Mp: 218–220 °C (lit. 221–223 °C). 1H NMR (300 MHz, CDCl3) δ 7.45 - 7.26 (m, 15H, Ar H), 3.56 (t, 2H, J = 11.4 Hz, NCH2CH2C), 3.27 (t, 2H, J = 11.0 Hz, NCH2CH2C), 3.11-3.00 (m, 4H, 4 × CHpip), 2.91-2.79 (m, 4H, 4 × CHpip). 13C NMR (600 MHz, CDCl3) δ 145.3, 134.0, 129.5, 128.9, 128.7, 128.1, 126.4, 124.2, 121.1, 69.2, 54.2, 49.8, 49.4, 35.4, 33.3. Mass Spectrum (ESI): m/z 397.5 [M+H]+.
4-(4-hydroxypiperidin-1-yl)-2,2-diphenylbutanenitrile (14)
4-bromo-2,2-diphenylbutanenitrile (2.14 g, 7.12 mmol), potassium iodide (1.17 g, 7.12 mmol) and DIPEA (4.13 ml, 23.7 mmol) were added to 30 ml of DMF and stirred at RT for 10 mins. 4-hydroxypiperidine (800 mg, 7.91 mmol) was then added to the reaction which was heated overnight at 90°C. DMF was then removed in vacuo and residue dissolved in DCM (40 ml) which was washed with saturated NaHCO3, H2O, saturated NaCl, dried with anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography using a gradient solvent system (100% DCM – 10% MeOH in DCM) to afford yellow oil (1.84 g, 73% yield). This was then recrystallized in a 60:40 mixture of H2O and MeOH to afford a clear solid (824 mg, 33%). 1H NMR (400 MHz, CDCl3) δ 7.54 – 7.49 (m, 4H, Ar H), 7.44 – 7.39 (m, 4H, Ar H), 7.38 – 7.32 (m, 2H, Ar H), 4.26 (s, 1H, OH), 3.46 – 3.24 (m, 2H, 2 × CHpip), 3.23 – 3.15 (m, 3H, NCH2CH2C, CH), 3.11 – 3.02 (m, 2H, NCH2CH2C), 2.56 (s, 2H, 2 × CHpip), 1.90 (ad, J = 13.3 Hz, 2H, 2 × CHpip), 1.63 (as, 2H, 2 × CHpip). Mass Spectrum (ESI): m/z 321.2 [M+H]+.
4-(3-hydroxypiperidin-1-yl)-2,2-diphenylbutanenitrile (15)
According to method B, 4-bromo-2,2-diphenylbutanenitrile (100 mg, 333 μmol) was treated with 3-hydroxypiperidine (37 mg, 366 μmol). The crude product was purified by column chromatography using a gradient solvent system (100% DCM – 10% MeOH in DCM) to yield a yellow oil which was then further purified using preparative TLC using a solvent system of 10% MeOH in DCM to afford yellow oil (45 mg, 42%). 1H NMR (400 MHz, CDCl3) δ 7.45 – 7.12 (m, 10H, Ar H), 3.79 – 3.65 (m, 1H, OHCH), 2.57 – 2.36 (m, 6H, 2 × CHpip, NCH2CH2C, NCH2CH2C), 2.20 – 2.06 (m, 2H, 2 × CHpip), 1.76 – 1.64 (m, 1H, 1 × CHpip), 1.53 – 1.36 (m, 3H, 3 × CHpip). 13C NMR (101 MHz, CDCl3) δ 140.08, 128.94, 127.97, 126.81, 122.32, 66.09, 60.53, 54.68, 53.85, 49.97, 36.59, 31.53, 21.32. Mass Spectrum (ESI): m/z 321.2 [M+H]+. (HR-ESI) Found: [M+H]+, 321.1969. C21H24N2O requires [M+H]+, 321.1961.
1-(3,3-diphenylpropyl)piperidin-4-ol (16)33
According to the method B, (3-bromopropane-1,1-diyl)dibenzene (118 mg, 430 μmol) was treated with 4-hydroxypiperidine (100mg, 478 μmol). The crude product was purified by column chromatography using a gradient solvent system (100% hexane – 66% EtOAc in hexane) to afford a white solid (97 mg. 58%). Mp: 118–120 °C. 1H NMR (400 MHz, CDCl3) δ 7.23 – 7.14 (m, 8H, Ar H), 7.13 – 7.06 (m, 2H, Ar H), 3.95 – 3.84 (m, 1H, OHCH), 3.61 (as, 1H, diphenyl-CH), 2.76 – 2.59 (m, 2H, 2 × CHpip), 2.28 – 2.15 (m, 4H, NCH2CH2C, NCH2CH2C), 2.11 – 1.95 (m, 2H, 2 × CHpip), 1.82 (ad, J = 9.3 Hz, 2H, 2 × CHpip), 1.57 – 1.48 (m, 2H, 2 × CHpip). 13C NMR (101 MHz, CDCl3) δ 144.78, 128.45, 127.84, 126.17, 67.89, 56.75, 51.05, 49.19, 34.40, 32.90. Mass Spectrum (ESI): m/z 296.5 [M+H]+. (HR-ESI) Found: [M+H]+, 296.2021. C20H25NO requires [M+H]+, 296.2009.
1-(3-phenylpropyl)piperidin-4-ol (17)
According to method B, (3-bromopropyl)benzene (92 μl, 719 μmol) was treated with 4-hydroxypiperidine (80 mg, 790 μmol). The crude product was purified by column chromatography using a gradient solvent system (100% Hexane – 100% CHCl3 – 10% MeOH in CHCl3). Fractions containing the product were then reduced in vacuo, CHCl3 added, filtered and subsequently concentrated in vacuo again to afford a yellow oil (91 mg, 58%). 1H NMR (400 MHz, CDCl3) δ 7.37 – 7.12 (m, 5H, Ar H), 3.84 – 3.63 (m, 1H, CH), 2.81 (bs, 2H, 2 × CHpip), 2.74 – 2.58 (m, 2H, NCH2CH2CH2), 2.51 – 2.34 (m, 2H, NCH2CH2CH2), 2.33 – 2.12 (m, 2H, 2 × CHpip), 1.91 (ad, J = 26.3 Hz, 4H, NCH2CH2CH2, 2 × CHpip), 1.63 (ad, J = 8.5 Hz, 2H, 2 × CHpip). 13C NMR (101 MHz, CDCl3) δ 141.91, 128.38, 128.34, 125.83, 67.47, 57.82, 50.87, 34.11, 33.69, 28.49. Mass Spectrum (ESI): m/z 220.2 [M+H]+. (HR-ESI) Found: [M+H]+, 220.1693. C14H21NO requires [M+H]+, 220.1696.
4-(4-methylpiperidin-1-yl)-2,2-diphenylbutanenitrile (18)34
According to method A, 4-bromo-2,2-diphenylbutanenitrile (1.00 g, 3.33 mmol) was treated with 4-methylpiperidine (394 μl, 3.33 mmol). 100 mg of the crude product was purified by RP-HPLC to afford 48 mg of a yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.48 – 7.27 (m, 10H, Ar H), 3.49 – 3.32 (m, 4H, 4 × CHpip), 2.98 (t, J = 12.8 Hz, 2H, NCH2CH2C), 2.96 (t, J = 11.2 Hz, 2H, NCH2CH2C), 1.86 (m, 1H, NCH2CH2CH), 1.84 – 1.70 (m, 4H, 4 × CHpip), 1.26 (s, 3H, CH3). 13C NMR (600 MHz, CDCl3) δ 138.7, 137.6, 132.4, 130.1, 121.3, 51.6, 42.6, 38.3, 37.4, 35.6, 30.9, 26.6. Mass Spectrum (ESI): m/z 319.7 [M+H]+.
4-(4-methoxypiperidin-1-yl)-2,2-diphenylbutanenitrile (19)
4-(4-hydroxypiperidin-1-yl)-2,2-diphenylbutanenitrile (14) (40 mg, 125 μmol) was dissolved in DMF (10 ml) containing NaH (9 mg, 375 μmol) and stirred for room temperature for 30 mins. MeI (9 μl, 144 μmol) was then added dropwise and reaction stirred under N2 at room temperature overnight. DMF was then removed in vacuo and residue dissolved in DCM (40 ml) and washed with saturated NaHCO3, H2O, saturated NaCl, dried with anhydrous Na2SO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography using a gradient solvent system (100% DCM – 5% MeOH in DCM) to afford yellow oil (36 mg, 86%). 1H NMR (400 MHz, CDCl3) δ 7.36 – 7.19 (m, 10H, Ar H), 3.25 (s, 3H, OCH3), 3.17 – 3.09 (m, 1H, CH), 2.76 – 2.29 (m, 6H, 2 × CHpip, NCH2CH2C, NCH2CH2C), 2.23 – 1.98 (m, 2H, 2 × CHpip), 1.93 – 1.76 (m, 2H, 2 × CHpip), 1.52 (as, 2H, 2 × CHpip). 13C NMR (101 MHz, CDCl3) δ 139.94, 128.98, 128.96, 128.04, 126.77, 53.16, 50.51, 47.50, 41.06, 37.10, 32.05. Mass Spectrum (ESI): m/z 335.2 [M+H]+. (HR-ESI) Found: [M+H]+, 335.2125. C22H26N2O requires [M+H]+, 335.2118.
4-(4-aminopiperidin-1-yl)-2,2-diphenylbutanenitrile (20)
Tert-butyl 1-(3-cyano-3,3-diphenylpropyl)piperidin-4-ylcarbamate (21) (250 mg, 596 μl) added to 1:1 mixture of TFA/DCM (10 ml) and stirred for 30 mins. TFA/DCM was then removed in vacuo. The resulting residue was then dissolved in DCM and washed with 2M NaOH, saturated NaCl, H2O, dried with anhydrous Na2SO4 and filtered. The solvent was then concentrated in vacuo to afford a clear oil (155 mg, 91%). 1H NMR (400 MHz, CDCl3) δ 7.42 – 7.26 (m, 10H, Ar H), 2.88 – 2.77 (m, 2H, 2 × CHpip), 2.68 – 2.57 (m, 3H, 2 × CHpip, CH), 2.47 – 2.38 (m, 2H, NCH2CH2C), 2.08 – 1.96 (m, 2H, NCH2CH2C), 1.85 – 1.73 (m, 2H, 2 × CHpip), 1.41 – 1.30 (m, 2H, 2 × CHpip). 13C NMR (101 MHz, CDCl3) δ 140.05, 128.91, 127.92, 126.79, 122.09, 54.71, 52.75, 50.04, 48.61, 36.86, 35.85. Mass Spectrum (ESI): m/z 320.3 [M+H]+. (HR-ESI) Found: [M+H]+, 320.2135. C21H25N3 requires [M+H]+, 320.2121.
Tert-butyl piperidin-4-ylcarbamate (25)35
Tert-butyl 1-benzylpiperidin-4-ylcarbamate (500 mg, 1.72 mmol) was dissolved in methanol (10 ml) and stirred. 36% aqueous HCl (169 μl, 1.72 mmol) was added followed by Pd(OH)2/C (525 mg, 689 μmol). Oxygen was then removed under reduced pressure and H2 added to the system (via a balloon) and stirred overnight. The reaction mixture was then filtered through celite and fitrate reduced in vacuo. DCM was then added and washed with 2M NaOH, H2O, saturated NaCl, dried with anhydrous Na2SO4, filtered and concentrated in vacuo to yield a yellow oil (321 mg, 94%). 1H NMR (300 MHz, MeOH) δ 3.63 – 3.46 (m, 1H, CH), 3.34, (s, NH), 3.23 (ad, J = 12.9 Hz, 2H, 2 × CHpip), 2.97 – 2.71 (m, 2H, 2 × CHpip), 2.08 – 1.09 (m, 2H, 2 × CHpip), 1.61 – 1.40 (m, 11H, 2 × CHpip, 3 × Boc CH3). Mass Spectrum (ESI): m/z 201.2 [M+H]+.
Tert-butyl 1-(3-cyano-3,3-diphenylpropyl)piperidin-4-ylcarbamate (21)
According to method A without the use of DIPEA, 4-bromo-2,2-diphenylbutanenitrile (320 mg, 1.07 mmol) was treated with tert-butyl piperidin-4-ylcarbamate (25) (256 mg, 1.28 mmol) and K2CO3 (442 mg, 3.20 mmol). The crude product was purified by column chromatography using a gradient solvent system (100% DCM – 10% MeOH in DCM) to afford a yellow oil (252 mg, 56%). 1H NMR (400 MHz, CDCl3) δ 7.41 – 7.11 (m, 10H, Ar H), 3.36 (dd, J = 20.6, 9.8 Hz, 1H, CH), 2.70 (ad, J = 11.8 Hz, 2H, 2 × CHpip), 2.62 – 2.41 (m, 2H, NCH2CH2C), 2.41 – 2.27 (m, 2H, NCH2CH2C), 1.99 (dd, J = 16.1, 6.3 Hz, 2H, 2 × CHpip), 1.82 (ad, J = 11.4 Hz, 2H, 2 × CHpip), 1.46 – 1.20 (m, 11H, 2 × CHpip, 3 × Boc CH3). 13C NMR (101 MHz, CDCl3) δ 155.16, 140.02, 128.92, 127.94, 126.79, 122.08, 79.27, 54.65, 52.55, 49.99, 47.59, 36.86, 32.49, 28.41. Mass Spectrum (ESI): m/z 420.2 [M+H]+.
References
- 1.Biswas S, Manikandan J, Pushparaj PN. Bioinformation. 2011;6:153. doi: 10.6026/97320630006153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prinz JC. J Eur Acad Derm Vener. 2003;17:257. doi: 10.1046/j.1468-3083.2003.00720.x. [DOI] [PubMed] [Google Scholar]
- 3.Bowcock AM, Krueger JG. Nat Rev Immunol. 2005;5:699. doi: 10.1038/nri1689. [DOI] [PubMed] [Google Scholar]
- 4.Sallusto F, Lenig D, Förster RLM, Lanzavecchia A. Nature. 1999;401:708. [Google Scholar]
- 5.Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. Trends Pharmacol Sci. 2004;25:280. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wulff H, Beeton C, Chandy KG. Curr Opin Drug Discov Dev. 2003;6:640. [PubMed] [Google Scholar]
- 7.Cahalan MD, Wulff H, Chandy KG. J Clin Immunol. 2001;21:235. doi: 10.1023/a:1010958907271. [DOI] [PubMed] [Google Scholar]
- 8.Lin CS, Boltz RC, Blake JT, Nguyen M, Talento A, Fischer PA, Springer MS, Sigal NH, Slaughter RS, Garcia ML. J Exp Med. 1993;177:637. doi: 10.1084/jem.177.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wulff H, Pennington M. Curr Opin Drug Discov Dev. 2007;10:438. [PubMed] [Google Scholar]
- 10.DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Nature. 1984;307:465. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- 11.Matheu MP, Beeton C, Garcia A, Chi V, Rangaraju S, Safrina O, Monaghan K, Uemura MI, Li D, Pal S, de la Maza LM, Monuki E, Flugel A, Pennington MW, Parker I, Chandy KG, Cahalan MD. Immunity. 2008;29:602. doi: 10.1016/j.immuni.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen W, Howard BL, Neale DS, Thompson PE, White PJ, Wulff H, Manallack DT. Curr Med Chem. 2010;17:2882. doi: 10.2174/092986710792065072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azam P, Sankaranarayanan A, Homerick D, Griffey S, Wulff H. J Invest Dermatol. 2007;127:1419. doi: 10.1038/sj.jid.5700717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitz A, Sankaranarayanan A, Azam P, Schmidt-Lassen K, Homerik D, Hansel W, Wulff H. Mol Pharmacol. 2005;68:1254. doi: 10.1124/mol.105.015669. [DOI] [PubMed] [Google Scholar]
- 15.Lanier EW. US 4,383,000. 1983
- 16.Lanier EW. Arch Dermatol. 1985;121:1486. [PubMed] [Google Scholar]
- 17.Janssen P, Jageneau A, Huggens J. J Med Pharm Chem. 1959:299. doi: 10.1021/jm50005a001. [DOI] [PubMed] [Google Scholar]
- 18.Harvey AJ, Baell JB, Toovey N, Homerick D, Wulff H. J Med Chem. 2006;49:1433. doi: 10.1021/jm050839v. [DOI] [PubMed] [Google Scholar]
- 19.Bruhova I, Zhorov BS. BMC Struct Biol. 2007;7:5. doi: 10.1186/1472-6807-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW, Kolski-Andreaco A, Wei E, Grino A, Counts DR, Wang PH, LeeHealey CJ, BSA, Sankaranarayanan A, Homerick D, Roeck WW, Tehranzadeh J, Stanhope KL, Zimin P, Havel PJ, Griffey S, Knaus HG, Nepom GT, Gutman GA, Calabresi PA, Chandy KG. Proc Natl Acad Sci USA. 2006;103:17414. doi: 10.1073/pnas.0605136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leeson PD, Springthorpe B. Nat Rev Drug Discov. 2007;6:881. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 22.Sweetman S. Martindale: the complete drug reference. 36. London: Pharmaceutical Press; 2009. [Google Scholar]
- 23.Adelstein GW. US 3,917,615. 1975
- 24.Yen CH. US 4,053,477. 1977
- 25.Diouf O, Depreux P, Chavatte P, Poupaert JH. Eur J Med Chem. 2000;35:699. doi: 10.1016/s0223-5234(00)00163-x. [DOI] [PubMed] [Google Scholar]
- 26.Nagarathnam D, Wetzel JM, Miao SW, Marzabadi MR, Chiu G, Wong WC, Hong X, Fang J, Forray C, Branchek TA, Heydorn WE, Chang RS, Broten T, Schorn TW, Gluchowski C. J Med Chem. 1998;41:5320. doi: 10.1021/jm980506g. [DOI] [PubMed] [Google Scholar]
- 27.GB 1037380. Orgamol. 1966
- 28.Janssen PAJ, Eddy NB. J Med Pharmaceut Ch. 1960;2:31. doi: 10.1021/jm50008a003. [DOI] [PubMed] [Google Scholar]
- 29.Daly JW, Camerini-Otero C, Shapiro CA, Ma J, Ziffer H, Vélez L, Harper JL. Drug Develop Res. 2006;67:842. [Google Scholar]
- 30.Myaji H, Sugimoto O, Okazaki T, Kyota H, Segawa M. JP 07291945. 1995
- 31.Kim E, Park CS, Han T, Bae MH, Chong W, Lee CH, Shin YA, Ahn BN, Kim MK, Shin CY, Son MH, Kim JK, Moon HS, Shim HJ, Kim EJ, Kim SH, Lim JI. Bioorg Med Chem Lett. 2008;18:4993. doi: 10.1016/j.bmcl.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 32.Adelstein GW, Yen CH, Dajani EZ, Bianchi RG. J Med Chem. 1976;19:1221. doi: 10.1021/jm00232a010. [DOI] [PubMed] [Google Scholar]
- 33.Storey TDJ, Arstad E, Guilbert B, Gaeta A. WO 2008075040. 2007
- 34.Dupre DJ, Elks J, Hems BA, Speyer KN, Evans RM. J Chem Soc. 1949:500. [PubMed] [Google Scholar]
- 35.Mase T, Houpis IN, Akao A, Dorziotis I, Emerson K, Hoang T, Iida T, Itoh T, Kamei K, Kato S, Kato Y, Kawasaki M, Lang F, Lee J, Lynch J, Maligres P, Molina A, Nemoto T, Okada S, Reamer R, Song JZ, Tschaen D, Wada T, Zewge D, Volante RP, Reider PJ, Tomimoto K. J Org Chem. 2001;66:6775. doi: 10.1021/jo0157425. [DOI] [PubMed] [Google Scholar]