

Abstract
Sexually transmitted carcinogenic Human Papillomavirus (HPV) infections are extraordinarily prevalent worldwide. However, most incident HPV infections clear within a few years, whereas a small minority persists to invasive cancer. Recent studies indicate that detection of methylated viral DNA may distinguish women with cervical intraepithelial neoplasia grade 2+ (CIN2+) from those with a carcinogenic HPV type infection that shows no evidence of CIN2+. Several studies have reported a positive association between methylation of CpG sites in the L1 gene and CIN2+, while there are inconclusive results regarding methylation of CpG sites in the Upstream Regulatory Region (URR). In this review, we summarize the current state of knowledge on HPV DNA methylation in cervical carcinogenesis, and discuss the merits of different methods used to measure HPV DNA methylation. To follow the promising leads, we suggest future studies to validate the use of methylated carcinogenic HPV DNA as a predictive and/or diagnostic biomarker for risk of cervical cancer among HPV-positive women.
Keywords: human papillomavirus, methylation, cervical cancer, biomarker, epigenetics
I. Introduction
The development of cervical cancer is linked to persistent infection with at least one of 13 types (i.e. HPV16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68) of human papillomavirus (HPV) from the alphapapillomavirus genus (hereafter HPV refers to these 13 types) (1). HPV infections are very common, yet cervical cancer and its immediate precursor lesion, cervical intraepithelial neoplasia grade 3 (CIN3, precancer) are relatively uncommon. Approximately 90% of incident HPV infections become undetectable using standard test methods within a few years (2), whereas persistent infections are significantly associated with progression to CIN3 lesions (3, 4), of which approximately 30% will progress to invasive cancer over three decades (5). Although current vaccines for HPV16/18 hold great promise, the predominant mechanism for cervical cancer prevention for the foreseeable future will continue to be screening and treating women with precancerous lesions. The transition between clearance and progression represents the critical dichotomy between benign HPV infection and substantial cancer risk that requires clinical attention (6).
The new cervical cancer screening guidelines in the United States recommend HPV co-testing among women 30 years and older (7). Although more sensitive than cytology, HPV testing has modest specificity and positive predictive value for detection of precancer, and cannot distinguish infections that will resolve from those that will progress (6). Thus, an important question is how to triage HPV-positive women. This public health need warrants further research on the mechanisms of, and the development and validation of novel biomarkers associated with, HPV-induced transformation and progression to precancer.
HPV viral methylation has emerged as a novel biomarker that may help distinguish benign HPV infections from those that progress to precancer (8-28). Detection of epigenetic changes, specifically if related to development of cervical precancer, may serve as a predictive or diagnostic biomarker for risk of cervical cancer amongst HPV-positive women. In this review, we describe molecular methods used to detect CpG methylation, and summarize the current state of knowledge on HPV DNA methylation in cervical carcinogenesis. We also identify important gaps in our knowledge required to decide how detection of HPV viral genome methylation could be used to prevent cervical cancer.
II. Background
IIa. HPV and the Viral Life Cycle
HPV is a double-stranded, circular DNA virus approximately 8,000 base pairs in size. The distribution of CpG sites is uneven throughout the viral genome (Figure 1). All oncogenic HPV types code for six early genes (E6, E7, E1, E2, E4, and E5) involved in viral gene expression and replication, and two late genes (L2 and L1) involved in capsid formation (3). The L1 protein self-assembles into viral-like particles and is the active component in the currently licensed HPV vaccines. The upstream regulatory region (URR) located between the L1 and E6 genes, contains the E6 promoter and an enhancer region with cis-responsive elements that regulate viral gene expression, replication, and packaging into viral particles (Figure 1) (29, 30). The primary HPV oncogenes, E6 and E7, interact with a large number of cellular targets, including the cellular tumor suppressor proteins p53 and pRb, which are central regulators of apoptosis and cell cycle, respectively (for reviews see (31-34)). During productive infection, E6 and E7 are expressed at relatively low levels, in part due to transcriptional repression by E2. During the carcinogenic process, transcription of E6 and E7 is deregulated, leading to their over-expression (33). This deregulation may be mediated by the integration of HPV DNA into the host genome, often resulting in disruption of the E2 gene with increased E6 and E7 transcripts spliced into host sequences, causing increased HPV oncogene expression (35, 36). However, HPV integration is not a necessary step in malignant transformation, and other mechanisms, such as alterations of the E2 binding sites in the URR or altered expression of E2, may be implicated (14, 16, 37). Based on these molecular mechanisms of HPV oncogenesis, a number of biomarkers have been developed including those associated with HPV oncogene activity (i.e., E6 and E7 mRNA expression) and with cell cycle deregulation (38, 39). The most extensively studied biomarker is p16INK4a, a cyclin-dependent kinase inhibitor that correlates with increased expression of the oncogenic E7 protein (40-45).
Figure 1. Organization of the HPV16 Genome, Upstream Regulatory Region (URR) and Location of 112 CpG Sites.
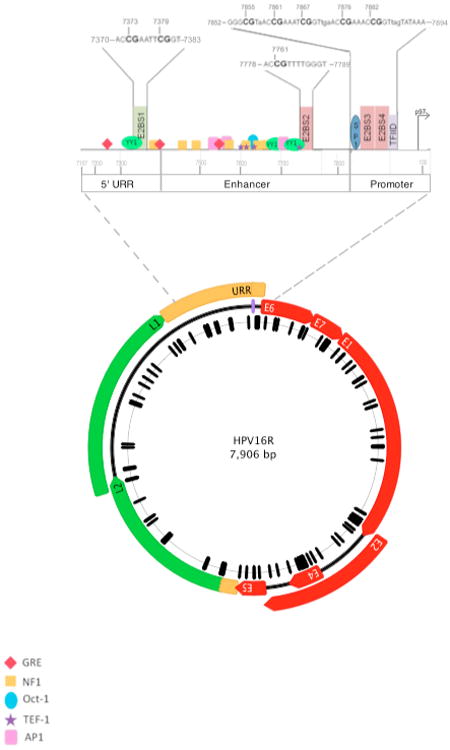
The 7,906 base pair, circular reference genome for HPV16R (NC_001526) is illustrated. Genes expressed early in the viral life cycle (early genes) are shown in red and are prefixed with an “E” in white, corresponding to their names. Genes expressed late in the viral life cycle (late genes) are shown in green and are prefixed with an “L” in black. Two non-coding regions are displayed in yellow, labeled the upstream regulatory region (URR) between the stop codon of L1 and the start codon of E6, and the non-coding region (NCR) located between the stop codon of E5 and the start codon of L2. A purple line at position 7906/1 denotes the first position of the genome. The black hash marks correspond to the 112 CpG sites within the HPV16R genome and are displayed relative to their genomic position. This figure was made using Geneious Pro 5.6.3 (71).
An enlargement of the URR is shown above the genome and illustrates the topology of the CpG sites, four E2 binding sites and other DNA-protein binding motifs. The nucleotide sequence of each E2 binding site is shown in capital letters with CpG sites indicated in bold and the nucleotide position is shown for the cytosine based on the HPV16R sequence. The color of each binding sites corresponds to whether binding of E2 inhibits (red) or stimulates (green) E6 and E7 transcription. Transcription factor sites are depicted as indicated: YY1 transcription factor YY1, binds to E2 in the 5′ URR to stimulate transcription (66, 72). NF1, Nuclear Factor 1, GRE, glucocorticoid response elements, AP1, Activator Protein 1, and TEF-1, Transcription Enhancer Factor-1, are transcriptional activators (73). SP1, Specificity Factor 1 and TFIID, Transcriptional Factor IID are transcriptional activators that can be sterically hindered by E2 binding (67). CpG site 31 overlaps the SP1 biding site and TFIID binds to the TATA box to initiate transcription.
IIb. CpG Methylation of DNA from Oncogenic HPV
In humans, DNA methylation is facilitated by a family of DNA methyltransferases that catalyze the addition of a methyl group to cytosines at the 5′ position of a CpG dinucleotide pair and is typically detected by bisulfite modification of DNA (methods discussed below; Figure 2A, 2B) (46, 47). The methyl group can alter chromatin conformation and DNA topology resulting in displacement of transcription factors and alterations in expression (48, 49). Methylation of CpG-rich stretches of human DNA located in promoter regions of genes, termed “CpG Islands,” are essential for normal biological processes (50). Disruption of CpG island methylation has been documented in malignant cellular transformation (51).
Figure 2. Chemistry and Analysis of DNA CpG Methylation.
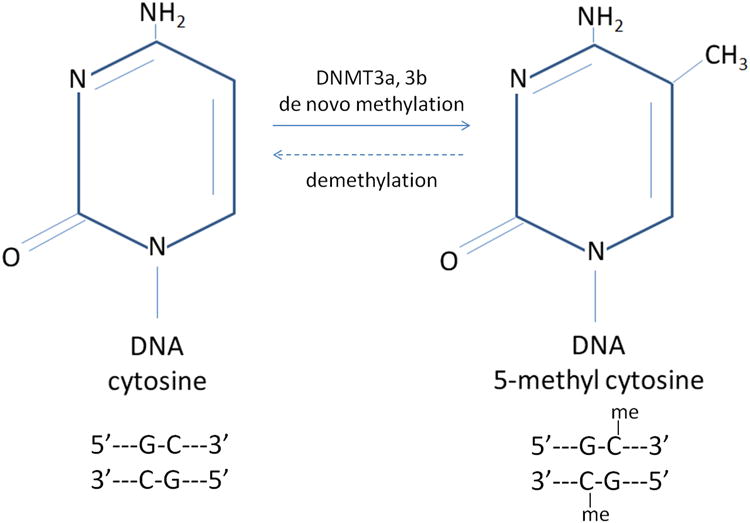
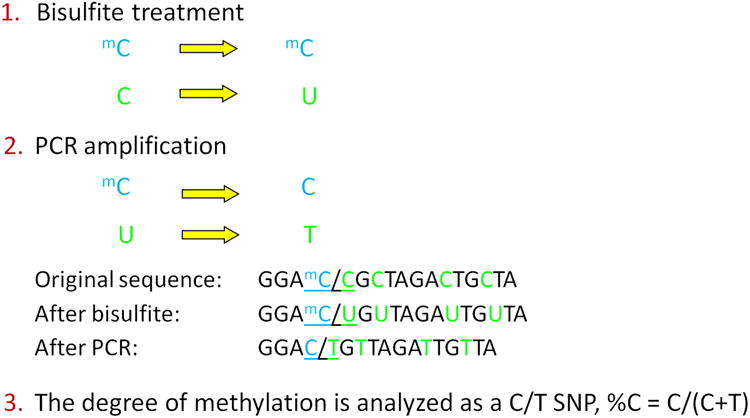
(Panel A) Specific cytosines within the double-stranded DNA helix can be methylated by a family of DNA Methyltransferases (DNMT3a, 3b) to form 5-methyl cytosine residues. Removal of methyl groups (“demethylation”) occurs via the same family of enzymes. (Panel B) Bisulfite conversion of cytosines and downstream analysis of CpG site methylation. Bisulfite treatment of DNA deaminates unmethylated cytosine residues (labeled as “C”) on single stranded DNA molecules, converting them to uracils (labeled “U”), while 5-methyl cytosines are protected from conversion (labeled, “mC”) (step 1). Polymerase Chain Reaction (PCR) amplification of bisulfite modified DNA, converts uracils to thymidines (labeled, “T”) by DNA polymerase (step 2). The ratio of C/C+T indicates the proportion of methylated cytosines in the assayed sample (step 3).
Although there are no classical CpG islands within the HPV genome, regions of high density and conservation of CpG sites (52) suggest the potential for a functional role (Figure 3). The molecular basis and covalent alterations of methylation at individual CpG sites is poorly understood. Targeted methylation of CpG sites may represent a mechanism by which HPV switches from a productive infection to one leading to transformation (16). Alternatively, methylation of HPV DNA may serve as a host defense mechanism for silencing viral replication and transcription.
Figure 3. CpG Sites in Aligned Carcinogenic HPV Genomes.
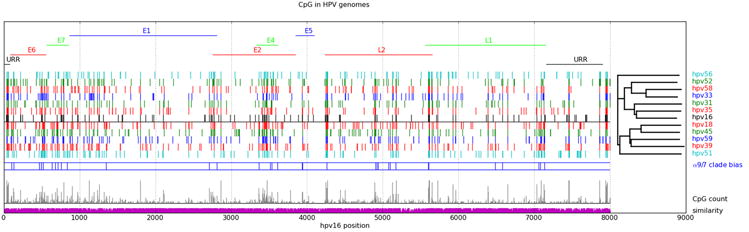
Carcinogenic HPV genotypes obtained from PAVE (http://pave.niaid.nih.gov) were aligned using MUSCLE (http://www.drive5.com/muscle/). Gene features and numbering are referenced to HPV16R. CpG sites for each genome are identified together with the phylogenetic relationships of the alignment. Phylogenetic and molecular evolutionary analyses were conducted using MEGA version 5 (74). Single representatives of alpha-6 (hpv56) and alpha-5 (hpv51) flank the alpha-9 and and alpha-7 clades above and below the horizontal line. Alpha 9/7 clade bias is indicated where a chi-square indicates a significant difference (p<0.05) for the presence of CpG in the two clades. CpG count is the (arbitrarily scaled) count of CpG sites at each position, while similarity is the maximal nucleotide frequency at the alignment position.
Recent studies have shown that methylation of HPV viral DNA may distinguish the presence of CIN2+ from an acute and clearing HPV infection (Table 1) (8-27). Similar associations between HPV methylation and other HPV-associated anogenital and head and neck cancers have also been reported (53-56).
Table 1. Published Studies of Human Papillomavirus DNA Methylation.
| First Author (Year) | Method | Specimen Type | Sample Size (Cases/Controls) | Outcomes | Location and No. of CpGs |
|---|---|---|---|---|---|
| Methylation of CpG Sites within the URR Associated with Asymptomatic and Low-grade Infections | |||||
| Badal (2003) | MS-restriction digestion and bisulfite sequencing | Exfoliated cervical cells and FFPE biopsy samples | 56/25 | NILM, CIN1-3. SCC | URR (11) |
| Hublarova (2009) | MS-restriction digestion | Exfoliated cervical cells and fresh frozen biopsies | 120/218 | NILM, CIN1-CIN3, ICC | URR (6) |
| Piyathilake (2011) | Pyrosequencing | Exfoliated cervical cells | 30/45 | ≤CIN1, CIN2+ | URR (6) |
| Xi (2011) | Bisulfite Sequencing | Exfoliated cervical cells | 94/117 | < CIN2/3, CIN2/3 | URR (11) |
| Mazumder (2011) | MS-restriction digestion | Fresh frozen biopsies | 205/34 | CIN1-3, ICC | URR (NS) |
| Methylation of CpG Sites within the URR, particularly in E2 Binding sites, is Associated with High-grade Lesions and Cancer | |||||
| Bhattacharjee (2006) | MS-restriction digestion and bisulfite sequencing | Exfoliated cervical cells and fresh frozen biopsies | 57/15 | NILM, SCC | URR (11) |
| Hong (2008) | Pyrosequencing | Exfoliated cervical cells | 14/56 | NILM, CIN1-3, ICC | URR (8) |
| Ding (2009) | Bisulfite Sequencing | Cervical scrapes/biopsy tissues | 36/17 | LSIL, HSIL, SCC | URR (15) |
| Vinokurova & von Knebel Doeberitz (2011) | LCM, Bisulfite Sequencing, COBRA | FFPE tissue sections | 40/3 | NILM, LSIL, HSIL | URR (16) |
| Snellenberg (2011) | Methylation Independent PCR, Luminex xMap | FFPE biopsy samples, exfoliated cervical cells | 48/17 | NILM, CIN3, SCC | E2 binding sites, URR (3) |
| Methylation of L1 is Associated with High-grade Lesions and Cancer | |||||
| Kalantari (2004) | Bisulfite Sequencing | Not specified | 64/51 | NILM, CIN1-3, ICC | 3′ L1 – URR (19) |
| Brandsma (2009) | Bisulfite Sequencing | Cervical cells (PreservCyt) | 9/4 | NILM, ASC-US, LSIL, CIN1-3 | Genome (113) |
| Fernandez (2009) | Bisulfite sequencing and MSP | Not specified | 70/17 | NILM, CIN1-3, SCC | Genome (110; BS.) L2/L1 (BS; MSP) |
| Kalantari (2009) | LCM, bisulfite sequencing | FFPE biopsy samples | 5/2 | NILM, ICC | 3′ end of L1 – URR (19) |
| Kalantari (2010) | Bisulfite sequencing | Exfoliated cervical cells | 21/0 | LSIL, HSIL | 3′ L1 (4) |
| Sun (2011) | Epityper and Pyrosequencing | Exfoliated cervical cells | 39/46 | NILM, LSIL/CIN1, CIN2/3, ICC | L1 – URR (32) |
| Mirabello (2012) | Pyrosequencing | Exfoliated cervical cells | 65/34 | Cleared HPV, persistent HPV, CIN3 | Genome (67) |
| Mirabello (2012) | Pyrosequencing | Exfoliated cervical cells | 181/95 | Cleared HPV, persistent HPV, CIN2+ | Genome (66) |
| Methylation of Other HPV Types is Associated with High-grade Lesions and Cancer | |||||
| Badal (2004) | Bisulfite Sequencing | Not specified | 6/5 | NILM, ICC | L1 – URR (39) |
| Turan (2006) | Bisulfite sequencing | Cervical smears/tumor biopsies | 13/11 | NILM, ASC-US, LSIL, HSIL, SCC | L1 – URR (31) |
| Fernandez (2009) | Bisulfite sequencing, MSP for E2 | Not specified | 29/10 | NILM, CIN1-3, SCC | Genome (168) |
| Wentzensen (2012) | Pyrosequencing | Exfoliated cervical cells | HPV18: 40/42 | <CIN2/ASCUS, CIN3 | Genome (106) |
| HPV31: 45/43 | <CIN2/ASCUS, CIN3 | Genome (80) | |||
| HPV45: 12/11 | <CIN2/ASCUS, CIN3 | Genome (105) | |||
Abbreviations: ASC-US, Atypical squamous cells of undetermined significance; BS, Bisulfite sequencing; CIN, Cervical intraepithelial neoplasia; COBRA, Combined bisulfite restriction analysis; E2, E2 gene of human papillomavirus; FFPE, Formalin-fixed, paraffin-embedded; HSIL, High-grade intraepithelial lesion; HPV; Human papillomavirus; ICC, Invasive cervical cancer; L1, L1 gene of human papillomavirus; LCM, Laser-capture microdissection; LSIL, Low-grade intraepithelial lesion; MS, Methylation sensitive; MSP, Methylation specific PCR; NILM, Negative for intraepithelial lesion or malignancy; PCR, Polymerase chain reaction; SCC, Squamous cervical cancer; URR, Upstream regulatory region
III. DNA Methylation Assays (Table 2)
Table 2. Methods Used to Study Human Papillomavirus Methylation.
| Method | Description | Strengths | Limitations |
|---|---|---|---|
| Non-Bisulfite Based Assays | |||
| Methylation-sensitive restriction digestion | Pair of restriction methylation sensitive and insensitive isochizomer endonucleases. Identification of methylation status by Southern blotting or PCR. | Non-bisulfite method Rapid and cost- effective, highly sensitive. | Incomplete restriction digestion. Limited availability of restriction sites. Qualitative. |
| Bisulfite Based Assays | |||
| Quantitative Methylation- Specific PCR | Methylation-specific probes allow for quantitative determination of methylation patterns and prevalence within a mixed pool of PCR products. Methylation can also be detected by methylation-specific primer design (Methylation-Specific PCR). | High specificity and sensitivity, has the ability to detect one copy of methylated DNA among several thousand. High throughput, quantitative. | Detects methylation status of several CpGs simultaneously over a short genomic region. |
| Sequencing of Individual Clones | Cloning bisulfite-treated PCR products into plasmid vectors and sequencing individual clones. | Provides methylation information for single DNA molecules, allows for determination of variable methylation patterns. | Slow, methylation status may be over or underrepresented as only a select few clones are chosen. Qualitative. |
| Pyrosequencing | Sequencing PCR products for C/T polymorphisms, based on light-based detection of nucleotide incorporation. | Rapid, high-throughput, for quantitative assessment of CpG site methylation, allows for assessments of DNA methylome. | Provides an average value of percent methylation. |
| EpiTYPER | Base specific cleavage of transcribed RNA from PCR products yields distinct patterns for the methylated and non-methylated DNA, measured by mass spectrometry. | High-throughput, quantitative assessment of CpG site methylation. | Provides an average value of percent methylation. |
| Combined Bisulfite Restriction Analysis (COBRA) | PCR amplification of bisulfite treated DNA followed by methylation dependent restriction digestion of methylation-specific sites. | Provides semi-quantitative information regarding the methylation status at a specific region. | Dependent on the availability of informative restriction sites. |
| Luminex® xMAP™ | Bead-based detection assay. Involves the hybridization and fluorescence detection of probes specific for the “C” of methylated or the “T” of unmethylated CpG sites coupled to microspheres. | High-throughput, more sensitive than bisulfite sequencing. | Detects only methylation frequencies. |
| Next-Generation Assays | |||
| Illumina | Multiplex, massively parallel next generation sequencing of PCR products for C/T polymorphisms. | Analysis of the methylation status of individual HPV DNA molecules. | Limited applicable statistical methods to handle the high-density, single CpG loci resolution data. |
| Pacific Biosciences | Detects nucleotide binding to the polymerase in real time and is sensitive to base modifications, such as methylated CpG sites in the DNA template. | High-throughput, no need for bisulfite modification of DNA. | Not adapted to studies of HPV methylation, high error rate, and technically complex. |
IIIa. Non-Bisulfite Based Assays
Earlier studies of HPV methylation used methylation-sensitive restriction digestion assays that involved pairs of methylation-sensitive and methylation-insensitive restriction endonucleases (or isochizomers), for example HpaII and MspI, respectively (10-12, 21). Following digestion, methylation status can be determined by Southern blotting or polymerase chain reaction (PCR). This method is simple, relatively inexpensive, and does not require bisulfite treatment of DNA, which is prone to DNA degradation and incomplete conversion (methods discussed below). However, limitations include the large amount of DNA required to perform the assay and the limited availability of methylcytosine-sensitive restriction sites in the HPV genome (26). This approach lacks the ability to survey multiple sites and is not quantitative.
IIIb. Bisulfite-Based Assays
Sodium bisulfite deaminates cytosine residues on single-stranded DNA molecules and converts them to uracils, while 5-methyl cytosines remain protected from conversion. When bisulfite-modified DNA is subjected to PCR, the uracil residues are converted to thymidines by DNA polymerase in the amplified products (57). The ratio of C/C+T indicates the proportion of methylated cytosines at each C in the assayed sample (Figure 2B). The methylation status of bisulfite-treated DNA can be determined by several methods.
Methylation-specific PCR (MSP) utilizes primers designed to distinguish methylated from unmethylated DNA, taking advantage of the sequence differences generated from bisulfite modification (58). This technique is sensitive and specific; however, it does not provide quantitative information and does not allow for determination of methylation status at the nucleotide level (57). Quantitative MSP (Q-MSP) is a rapid, high-throughput technique based on the fluorescent analysis of PCR products. Individual probes specific for each methylation-dependent sequence (i.e., C or T) at CpG sites allow for a fluorescent-based quantitative assay for methylation patterns (59).
Analysis of PCR products by direct sequencing can be used to study HPV DNA methylation at individual CpG sites, indicated by C/T nucleotides. Many studies have relied on sequencing of cloned PCR products from bisulfite-treated DNA, and sequencing of several individual clones to estimate percent methylation (13, 16, 17, 19-22, 24, 25, 54-56). Although this technique can provide information on multiple CpG sites in a region, it is labor intensive, insensitive to low levels of methylation, and inadequate for the analysis of large numbers of samples (60). The introduction of next-generation, single molecule sequencing makes cloning and sequencing obsolete (see below).
One of the newer high-throughput techniques, pyrosequencing, involves the photon-based detection of released inorganic phosphate (PPi) during nucleotide incorporation (61, 62). Recent studies have successfully designed pyrosequencing assays for the study of HPV DNA methylation (8, 23, 26-28). Pyrosequencing allows for quantitative assessment of methylation status at multiple CpG sites within relatively short reads (∼30 nucleotides) and provides an average estimate of methylation levels at a specific site by providing percent incorporation of C (methylated) vs. T (unmethylated) in bisulfite-treated DNA (60). Other methods include the EpiTYPER assay, which involves subsequent base-specific cleavage of RNA (i.e., cleaves at uracils and cytosines) generated from the PCR products by T7 polymerase and mass spectrometry (8, 26), Combined Bisulfite Restriction Analysis (COBRA;(16)), and the Luminex® xMap™ system, a bead-based assay that involves the hybridization and fluorescent detection of C/T at the C position of the CpG site using specific probes (14).
IIIc. Next-Generation Assays
Next-generation sequencing techniques can be used to analyze the methylation status of single CpG sites in individual HPV DNA molecules and to assess the relative heterogeneity of HPV DNA methylation in cervical cell samples by sequencing a particular amplicon at greater depth (i.e., thousands of single molecules from a single bisulfite-treated sample). Technologies such as the Illumina HiSeq 2000 enable multiplex, parallel sequencing of bisulfite-treated and amplified HPV DNA fragments, and are more accurate than pyrosequencing in deciphering homopolymeric (i.e., stretches of identical nucleotides) regions of the DNA template (63). Even more novel “third generation” sequencing technologies, such as PacBio (Pacific Biosciences, Menlo Park, CA), are being adapted for methylation analyses (64).
IV. HPV Methylation
Overall, studies have suggested an association between methylation of CpG sites in the HPV genome and detection of cervical precancer and cancer. The positive relationship between methylation in the L1 gene and CIN2+ seems to be relatively consistent in most studies (8, 9, 19, 20, 24-26, 28); however, the association between methylation in the URR and CIN2+ is less clear. Some studies have found decreased methylation of CpG sites in the URR in samples with CIN2+ (10-13, 15, 23), yet others report an association between increased methylation of CpG sites in the URR and CIN2+ (14, 16, 21, 22). Data for HPV16 and other HPV types are reviewed below.
IVa. HPV16 Methylation
IVa1. Increased Methylation of L1 Associated with CIN2, CIN3, and Cancer
Kalantari and colleagues studied CpG methylation in fragments containing the 3′ end of the L1 open reading frame (ORF) through the URR in 115 clinical samples (17). They found increased methylation in cervical carcinomas (n=47), particularly in the L1 gene, while methylation was relatively reduced in low grade CIN (n=17) and cytologically-normal samples (n=51) (17). This finding was confirmed in two follow-up studies, one using laser capture microdissection to evaluate 3 cancer samples (19) and the other, a pilot study of L1 DNA methylation and chromosomal integration in a very small set of abnormal Pap smear samples (18). Based on these studies and others at extracervical sites (54, 55), the authors speculated that L1 genes may be hypermethylated as a result of integration into the cellular DNA (18).
The first study to map the whole HPV16 genome was conducted by Brandsma and colleagues (24). Using bisulfite sequencing, they mapped 113 CpG sites from a small collection of 13 cervical (non-malignant) samples at different stages of progression (24). Their results indicated a pattern of increased methylation, particularly in the E5, L2, and L1 region with increasing disease severity. Fernandez et al., mapped 110 CpG sites in the HPV16 genome using bisulfite sequencing, and detected hypermethylation of L1 and L2 with increased lesion severity in samples from a small group of women with briefly described cervical diagnoses of normal cytology (n=10), CIN1 (n=17), CIN2/3 (43), cervical cancer (n=17), and a set of cervical cancer cell lines (20). In a more recent publication using pyrosequencing, Sun et al., examined 32 sites in the L1 ORF and URR and found an association between increased DNA methylation and CIN3+ exclusively in the L1 region in 85 samples (26). Using pyrosequencing, Mirabello et al., investigated the association between HPV16 DNA methylation and cervical disease with serial samples from a prospective cohort study, covering all gene regions and 67 CpG sites in the HPV16 genome (8). In pre-diagnostic HPV16 samples (taken before onset of disease or HPV clearance), a CpG site in L2 (position 4261) showed increased methylation associated with development of incident CIN3 (n=20). In diagnostic samples (collected at the time of diagnosis), increased methylation at CpG sites in the E2, L2, and L1 region was associated with risk of CIN3 (n=30) compared to women who cleared their HPV16 infections (n=34). An independent replication study of a larger number of samples (n = 273) including CIN2 and cancer cases, conducted by Mirabello and colleagues (28), confirmed higher methylation in L2, L1 and E2-E4 regions among cases with CIN2+. This study also demonstrated that viral load did not affect methylation levels and that women older than the median age of 28 years tended to have higher methylation levels compared to women <28 years. The analysis of serial CIN3 samples prior to CIN3 diagnosis suggested that most regions of the HPV16 genome increased methylation over time, particularly in the L1 and L2 genes.
IVa2. Studies Investigating Methylation in the URR
The URR is a region of interest in HPV16 methylation studies since viral transcription is regulated by binding of the HPV E2 protein to four conserved binding sites (E2BS, 5′-ACCN6CGT-3′, (65)) throughout the URR (Figure 1). E2 has the ability to both stimulate and repress oncogene transcription; depending on which site it binds (66, 67). Because they contain conserved CpG sites, E2BSs are of particular interest; however, the distinct functional roles of these and other transcription factor binding sites in the URR make simple mechanistic interpretations difficult.
The following studies suggest that decreased methylation of the HPV16 URR is associated with increased severity of cervical neoplasia. One of the earliest reports on HPV16 DNA methylation conducted by Badal et al., (11) analyzed the methylation status of the enhancer and promoter region of the URR in samples from women with normal Pap smears (n=25), CIN1-CIN3 (n=23), and cancers (n=33). They found an overall greater proportion of methylation in samples from women with normal cytology compared to women with CIN1-CIN3 and cancer. In a small subset of 15 samples, they mapped methylation at 11 CpG sites in the URR enhancer/promoter region using bisulfite Sanger sequencing of PCR products and found that, in addition to the normal samples showing methylation at all 11 sites, 1 CIN3 and 2 cancer samples showed complete promoter and enhancer methylation, while additional cancer samples showed methylation in the promoter region, but not the enhancer. Taken together, these results weakened their main finding, indicating that methylation in the enhancer and promoter region of the URR was heterogenous in a subset of CINs and cancers. A study conducted by Hublarova and colleagues (12) evaluated methylation status in a group of samples comprised of normal cytology (n=21), CIN1 (n=8), CIN2/3 (n=89), and cancers (n=23) using restriction digestion and PCR. They observed higher methylation frequencies of the enhancer and promoter region of the URR in normal cytological specimens (81%) and decreased methylation in CIN3s and cancers (31.5% and 43.4%, respectively). Methylation within the URR was also associated with reduced E6 expression. Although their results indicated a trend toward decreased methylation in the URR with severity of cervical neoplasia, methylation was present in nearly half of the cancers. Mazumder et al., (15) showed that methylation of the URR gradually decreased from CIN to cervical cancer, irrespective of whether the DNA was in an episomal or integrated state in over 200 cancers and 34 CIN1-CIN3 samples. It is difficult to interpret these findings however, as the group of CINs was heterogeneous, the major difference in methylation status was seen between cancers of different stages, and their methylation assays were not quantitative.
More recent studies have used bisulfite sequencing assays to determine the methylation status of CpG sites within the URR. Piyathilake et al., (23) studied HPV16 methylation of the URR promoter region and one CpG site (7862) in the enhancer region in 45 women with ≤ CIN1 and 30 women with CIN2+. After adjusting for demographic and lifestyle risk factors for cervical cancer, they found that increased methylation of CpG sites in the promoter and URR enhancer site 7862 was associated with a reduced likelihood of being diagnosed with CIN2+ (23). A recent paper by Xi et al. (13) studied the relationship between the number of methylated CpG sites in the URR (i.e., 0, 1, 2-3, ≥4) using bisulfite sequencing of cloned molecules from 211 samples. Their findings indicated a lower overall frequency of methylation of ≥ 4 of the 11 CpG sites within the URR in the 94 women with CIN2/3 compared to the 107 women without CIN2/3. The likelihood of being diagnosed with CIN2/3 was significantly inversely related to having ≥ 4 methylated CpGs in the URR region, regardless of co-variates such as age, race, and HPV16 variant. Compared with the other mentioned studies, the outcome in this analysis was CIN2/3, and only two cancers were included.
In contrast to the above reports, other studies using different methods have indicated that increased methylation, particularly in CpG sites overlapping E2BSs, is associated with increased severity of CIN. One of the first studies to look specifically at methylation of all 16 CpGs within the URR was conducted by Bhattacharjee & Sengupta, (21) using methylation-sensitive restriction enzyme digestion in 57 invasive cervical cancer and 15 cytologically-normal samples and bisulfite Sanger sequencing of PCR products in a subset of cancers. Their results indicated that methylation of the promoter region in the URR, which contains two E2 binding sites, was increased in cancer cases compared with cytologically-normal controls. This association was particularly significant for site 58 which overlaps with E2BS4 (Figure 1). Hong and colleagues studied methylation of the HPV16 URR in a small group of samples, focusing specifically on CpG sites within the promoter and enhancer regions (27). Using pyrosequencing of bisulfite-treated DNA, they observed that the proportion of methylated samples was highest among cancer cases (n=22, 85% methylated), followed by specimens with normal cytology (n=10, 71% methylated) and CIN3s (n=6, 46% methylated). Methylation levels were lowest among cases with CIN1/2 (n=5, 29% methylated). Ding and colleagues investigated the methylation patterns of 15 CpG sites within the URR in a small set of clinical specimens ranging from low-grade cytology (LSIL; n=17), high-grade cytology (HSIL; n=21) and squamous cell carcinoma (n=15) (22). Using bisulfite sequencing of 5 cloned fragments per sample, they found low frequencies of methylation at CpG sites in the 5′ URR in samples with LSIL, whereas in HSIL specimens, more frequent methylation was observed in the enhancer and the 5′ URR region. Cervical cancer samples showed the highest frequency of methylation, with differences in the promoter region being the most pronounced. A recent study by Vinokurova & von Knebel Doeberitz (16) analyzed the methylation status of the HPV16 URR in distinct stages of epithelial differentiation (i.e., basal, intermediate, and superficial layers of the epithelium) in microdissected tissue from 3 samples. Using a combination of cloning and sequencing of bisulfite-treated DNA and a modified version of the COBRA assay (Table 2) (68), they reported complete methylation of all 16 CpGs in the URR in cytologically normal, so called “latent” samples. This finding was consistent for all three epithelial cell layers. In low-grade lesions with productive HPV infection (characterized by expression of L1), the promoter region was only methylated in cells from the superficial layer of the epithelium, while the enhancer region was only methylated in cells from the basal and intermediate cell layers. The 5′URR was unmethylated in all layers. In HPV-transformed cells from CIN3 lesions, the basal and intermediate layers displayed methylation in the 5′ URR and enhancer regions only, with consistent methylation present in two CpG sites overlapping an E2 binding site (E2BS1, Figure 1). Through a series of functional studies, they reported that methylation of CpG sites within the E2BS1 was associated with increased E6 promoter activity and that the methylated E2BS1 may recruit additional, uncharacterized protein complexes that regulate transcription in transforming infections. Finally, Snellenberg and colleagues focused specifically on E2BS methylation using the Luminex xMap system (14). They determined the methylation status of three E2BSs (excluding E2BS2 in the enhancer region) in 29 cervical squamous cell carcinomas, 38 CIN3s, and 17 cytologically-normal cervical scrapes. Methylation frequencies of all three E2BSs were significantly higher in cancer cases compared to both CIN3s and controls. Moreover, all cancer cases showed methylation of at least one E2BS, compared to 58% of CIN3s, and 24% of controls.
V. Methylation of Other Carcinogenic HPV Types
One of the first studies on HPV18, conducted by Badal et al., used bisulfite sequencing of cloned molecules to investigate methylation of the L1-URR region in 5 cytologically-normal and 6 cervical cancer samples (10). They observed increased methylation in L1 amongst both cancers and normal samples. Methylation in the URR was less consistent, with hypomethylation observed in the enhancer region and increased methylation in the promoter region in normal samples compared with cancer cases. Turan and colleagues studied HPV18 DNA methylation in a total of 11 samples with no cytologic abnormalities, 8 with low-grade cytologic changes, 4 with high-grade cervical changes, and 14 cancers using bisulfite sequencing of the 3′ region of the L1 gene and URR (25). They found low levels of methylation distributed throughout the URR amongst all samples. Methylation levels were low in cytologically-normal samples, with only sporadic methylation observed in the L1 region, while two high-grade lesions and most cancer samples showed hypermethylation in L1. A report by Fernandez et al., analyzed all 168 CpG sites in the HPV18 genome using cell lines and bisulfite sequencing of cloned fragments from a few samples without evidence of neoplasia and some primary cervical cancers and found increased methylation of HPV 18 L1 in cancers compared with specimens with no detectable disease (20); however, the clinical/epidemiological details of the samples were not presented in detail. Using MSP, they analyzed methylation of the HPV18 E2 gene in a small set of specimens consisting of 6 cytologically-normal infections, 7 CIN1, 9 CIN2/3 and 10 carcinomas and found increasing E2 methylation with disease severity.
In a recent study, Wentzensen and colleagues performed whole genome methylation analysis using pyrosequencing of CpG sites in HPV31, HPV18, and HPV45 (9). Methylation levels in women with CIN3 compared to women with infections with <CIN2 (controls) were analyzed for all three HPV types. Increased methylation in E2, L2, and L1 was observed in specimens from women with CIN3 compared to controls in all three HPV types. While patterns of viral methylation were dominated by high methylation levels in L1, distinct patterns of inter- and intra-gene methylation correlation for alpha-9 types (HPV16 and HPV31) compared to alpha-7 types (HPV18 and 45) were observed. These findings indicate that the association of viral methylation with precancer is a feature of the four most important oncogenic HPV types, which together cause about 90% of all cervical cancers.
VI. Interpreting the HPV DNA Methylation Literature
The finding of increased CpG methylation in the HPV16 and HPV18 L1 ORFs in cancers compared to cells from women without reported lesions is consistent in the literature. However, there are conflicting results with regard to level of methylation of the URR and cervical precancer and cancer. A critical source of discrepancy arises from the variability in the CpG methylation assays used in different studies (Table 2). For example, studies using methylation-sensitive restriction digestion assays and bisulfite cloning and sequencing are only able to obtain qualitative (i.e., yes/no) data on CpG methylation status and have insufficient sensitivity for detecting HPV DNA methylation at low levels (57). Furthermore, methylation-sensitive restriction digestion, as evidenced by Badal et al., has limited sensitivity for detecting methylation at specific CpG sites (11). Several of the studies that used cloning and sequencing of bisulfite-treated DNA were also limited by analyzing a small number of molecules per sample (26).
Case definitions are also very important. Most studies identified for this review analyzed samples representative of the entire HPV-related disease spectrum, from normal cytology to invasive cervical cancer. However, we caution against drawing conclusions when outcomes are differentially ascertained and defined (i.e., histology versus cytology). Moreover, most of the studies had small sample groups for each categorical diagnostic state and many used convenience samples and did not report the criteria used to classify cases.
Sampling differences influence HPV DNA methylation analyses, as many of the reviewed studies are based on exfoliated cervical cell specimens collected for cytology that often contain diverse cell types. Detection of methylated HPV CpG sites may be diluted by the presence of multifocal and heterogeneous populations with different epigenetic patterns. Laser capture microdissection should enable validation of methylation status for women who have heterogeneous disease (i.e., CIN1, CIN3) and multiple type infections to see whether topographic comparisons recapitulate observed case-control differences.
Despite conflicting results, several consistent findings regarding methylation in the HPV16 URR emerged from the published data. In general, most studies showed very low methylation levels at CpG site 7862 in the enhancer region in the HVP16 URR, even among cases that had high methylation at other proximal CpG sites. Notably, this site overlaps an E2 binding site (E2BS2, Figure 1) and is located in between two nucleosomes that form part of the HPV16 origin of replication ((11, 16, 17, 21, 22, 27). Methylation of the promoter region in cervical cancers was also consistent, even in studies where increased methylation in the URR among cervical cancers was not the main finding ((11, 13, 14, 21-23, 27). The HPV16 promoter region contains two E2 binding sites (E2BS3 and E2BS4) that, if modified by methylation, may block E2 from binding and enhance transcription of the E6 and E7 oncogenes (Figure 1). These consistencies highlight the need for future hypothesis-driven studies to analyze the presence and functional relevance of CpG site and region specific methylation in the HPV16 URR.
VII. Suggested HPV DNA Methylation Studies
VIIa. Important Statistical Questions
Carefully-chosen statistical methods can sharpen evaluation of specific scientific or clinical hypotheses in HPV DNA methylation studies. For example, a global statistical test addresses whether overall HPV DNA methylation or methylation of particular HPV gene regions is important. When the goal is identification of individual CpG sites associated with disease, then a site-specific analysis is required. Also when the distribution of methylation level at CpG sites is not normal, a non-parametric test, such as Mann-Whitney test for two groups or Kruskal-Wallis test for more than two groups should be used to identify differentially methylated sites. The high number of CpG sites in the HPV genome, and numerous potential methylation-based data points available to analyze, inevitably raises the question of multiple comparisons-related false discovery; while Bonferroni-corrected significance levels are common, discovery-phase projects warrant more liberal thresholds. Graphic visualizations of correlations between methylation of different CpG sites will be important for informing the development of a panel of biomarkers and for comparing methylation patterns across HPV types (9). Incorporation of genomic information such as HPV ORFs, splice sites, and transcription factor binding sites may lead to hypotheses concerning the functional significance of the DNA methylation changes that can be tested through more fundamental studies. Analyses that incorporate the correlation between adjacent CpG sites, focusing on haplotypes rather than on specific CpG sites, may provide better understanding of the underlying mechanism.
Methylation has the potential to serve as a predictive biomarker if it is present prior to the diagnosis of precancer. This determination requires specific attention to serial changes in methylation levels, as in many molecular studies of time-varying biomarkers. Thus far, preliminary data indicates that methylation at most regions of the HPV16 genome, particularly in the L2 and L1 genes, is higher in CIN3 at the time of diagnosis (8, 28). Nevertheless, the temporal changes in HPV CpG methylation from exposure to persistence to subsequent precancer and cancer require further study. As of now, it is not clear whether methylation of HPV DNA reflects an early event that can be measured as a prognostic biomarker of later CIN2+, or whether it is a late event that will serve only as a diagnostic biomarker. Carefully designed studies need to consider intervals between specimen collections; the analytic challenge will be finding the best way to examine changes in methylation levels over time, including appropriate case/control definitions, potential covariates, and the range and depth of serial data (i.e., how many time points) needed to understand the natural history of HPV DNA methylation.
VIIb. DNA Methylation Analyses in Other HPV Types and Assay Design
If the observation of increased CpG methylation in the L1 ORFs among precancers and cancers remains consistent across most carcinogenic HPV types, it should be possible to develop a multiplex assay that measures CpG methylation at sites that are highly discriminatory for CIN3 versus transient infections. Such an assay will need to identify and then pool individual CpG sites into one test. This type of assay will be crucial for the triage of HPV-positive women, so that one test could be used regardless of the type of HPV infection. Selecting too many sites could increase the complexity and cost of the assay, and may increase the risk of false positive results. Nevertheless, optimal discrimination may only be achieved when multiple, less correlated sites are measured for a specific viral type. Additional studies to address the architecture of HPV methylation and the correlation amongst CpG sites across different HPV types will be needed to optimize an assay. Furthermore, it will be important to have some redundancy of coverage, to account for genomic variants, viral integration, and/or assay complexity.
VIIc. Using Methylation Data to Determine HPV Type Attribution in Cervical Lesions
Determining the HPV type responsible for a lesion has historically been a challenge when multiple HPV genotypes are detected. Multiple lesions may be caused by different types or a single type may be responsible for a precancerous lesion, but may be surrounded by transient infections of other HPV types (69). Wentzensen and colleagues evaluated the methylation levels in cases and controls by infection status with multiple versus single (18, 31, and 45) carcinogenic HPV types (9) . Single-type infections were distinguished by higher methylation levels compared with multiple-type infections, where causal attribution was less clear.
VIId. Host and Viral Factors Influencing HPV DNA Methylation
Only a few studies have analyzed whether demographic and lifestyle factors affect HPV DNA methylation and/or modify the association between HPV DNA methylation and carcinogenesis (8, 13, 15, 22, 23, 28). One study found that oral contraceptive use was associated with lower methylation of the URR (22) while, Piyathilake et al., found that reported healthy-eating was associated with increased methylation in the enhancer region of the URR (23). One large study suggested that older women had higher methylation levels and increased risk compared to younger women (28). In summary, it appears that the association between cervical neoplasia and HPV DNA methylation is not easily explained by demographic and other risk factors. More definitive studies are needed with larger sample sizes, particularly to analyze the relationship between demographics, co-factors, HPV methylation and cervical cancer development.
Although the primary focus of this review is on viral methylation in cervical cells and HPV progression to precancer/cancer, it is important to note that epigenetic changes occur in both the host and viral genome of cells during neoplastic transformation (for a review see (70)).
VIII. Conclusions
Based on future studies expanding the oncogenic types and the association of CpG methylation with precancer, it is likely that HPV viral methylation will have a role as a candidate biomarker for translation to clinical use. Since HPV methylation can be measured from the same specimen used for an HPV test, the most likely scenario will be as a triage test for women screening HPV-positive. The methylation assay could guide decisions about immediate intervention and future follow-up and identify women who need to undergo further evaluation.
Millions of women are still at risk of cervical cancer and an enormous public health challenge remains, particularly for women infected prior to the vaccine, and for those oncogenic types not included in the current vaccines. The future demands additional scientific breakthroughs in managing oncogenic cervical infections to prevent cervical cancer.
Acknowledgments
This research was supported [in part] by the Intramural Research Program of the National Institutes of Health (NIH) and the National Cancer Institute (5U01CA078527 to RDB)
Footnotes
Disclosure of Potential Conflicts of Interest: There are no financial disclosures.
References
- 1.Cogliano V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F. Carcinogenicity of human papillomaviruses. Lancet Oncology. 2005;6(4):204. doi: 10.1016/s1470-2045(05)70086-3. [DOI] [PubMed] [Google Scholar]
- 2.Ho GYF, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. New England Journal of Medicine. 1998;338(7):423–8. doi: 10.1056/NEJM199802123380703. [DOI] [PubMed] [Google Scholar]
- 3.Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370(9590):890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- 4.Bodily J, Laimins LA. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol. 2011;19(1):33–9. doi: 10.1016/j.tim.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCredie MR, Sharples KJ, Paul C, Baranyai J, Medley G, Jones RW, et al. Natural history of cervical neoplasia and risk of invasive cancer in women with cervical intraepithelial neoplasia 3: a retrospective cohort study. Lancet Oncol. 2008;9(5):425–34. doi: 10.1016/S1470-2045(08)70103-7. [DOI] [PubMed] [Google Scholar]
- 6.Schiffman M, Wentzensen N, Wacholder S, Kinney W, Gage JC, Castle PE. Human papillomavirus testing in the prevention of cervical cancer. J Natl Cancer Inst. 2011;103(5):368–83. doi: 10.1093/jnci/djq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saslow D, Solomon D, Lawson HW, Killackey M, Kulasingam SL, Cain J, et al. American cancer society, american society for colposcopy and cervical pathology, and american society for clinical pathology screening guidelines for the prevention and early detection of cervical cancer. Am J Clin Pathol. 2012;137(4):516–42. doi: 10.1309/AJCPTGD94EVRSJCG. [DOI] [PubMed] [Google Scholar]
- 8.Mirabello L, Sun C, Ghosh A, Rodriguez AC, Schiffman M, Wentzensen N, et al. Methylation of human papillomavirus type 16 genome and risk of cervical precancer in a costa rican population. J Natl Cancer Inst. 2012;104(7):556–65. doi: 10.1093/jnci/djs135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wentzensen N, Sun C, Ghosh A, Kinney W, Mirabello L, Wacholder S, et al. Methylation of HPV18, HPV31, and HPV45 genomes and cervical intraepithelial neoplasia grade 3. JNCI. 2012 Aug 31; doi: 10.1093/jnci/djs425. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Badal S, Badal V, Calleja-Macias IE, Kalantari M, Chuang LS, Li BF, et al. The human papillomavirus-18 genome is efficiently targeted by cellular DNA methylation. Virology. 2004;324(2):483–92. doi: 10.1016/j.virol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Badal V, Chuang LS, Tan EH, Badal S, Villa LL, Wheeler CM, et al. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: genomic hypomethylation correlates with carcinogenic progression. J Virol. 2003;77(11):6227–34. doi: 10.1128/JVI.77.11.6227-6234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hublarova P, Hrstka R, Rotterova P, Rotter L, Coupkova M, Badal V, et al. Prediction of human papillomavirus 16 e6 gene expression and cervical intraepithelial neoplasia progression by methylation status. Int J Gynecol Cancer. 2009;19(3):321–5. doi: 10.1111/IGC.0b013e31819d8a5c. [DOI] [PubMed] [Google Scholar]
- 13.Xi LF, Jiang M, Shen Z, Hulbert A, Zhou XH, Lin YY, et al. Inverse association between methylation of human papillomavirus type 16 DNA and risk of cervical intraepithelial neoplasia grades 2 or 3. PLoS One. 2011;6(8):e23897. doi: 10.1371/journal.pone.0023897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snellenberg S, Schutze DM, Claassen-Kramer D, Meijer CJ, Snijders PJ, Steenbergen RD. Methylation status of the E2 binding sites of HPV16 in cervical lesions determined with the Luminex(R) xMAP system. Virology. 2012;422(2):357–65. doi: 10.1016/j.virol.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Mazumder Indra D, Singh RK, Mitra S, Dutta S, Chakraborty C, Basu PS, et al. Genetic and epigenetic changes of HPV16 in cervical cancer differentially regulate E6/E7 expression and associate with disease progression. Gynecol Oncol. 2011;123(3):597–604. doi: 10.1016/j.ygyno.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 16.Vinokurova S, von Knebel Doeberitz M. Differential methylation of the HPV 16 upstream regulatory region during epithelial differentiation and neoplastic transformation. PLoS One. 2011;6(9):e24451. doi: 10.1371/journal.pone.0024451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalantari M, Calleja-Macias IE, Tewari D, Hagmar B, Lie K, Barrera-Saldana HA, et al. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J Virol. 2004;78(23):12762–72. doi: 10.1128/JVI.78.23.12762-12772.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalantari M, Chase DM, Tewari KS, Bernard HU. Recombination of human papillomavirus-16 and host DNA in exfoliated cervical cells: a pilot study of L1 gene methylation and chromosomal integration as biomarkers of carcinogenic progression. J Med Virol. 2010;82(2):311–20. doi: 10.1002/jmv.21676. [DOI] [PubMed] [Google Scholar]
- 19.Kalantari M, Garcia-Carranca A, Morales-Vazquez CD, Zuna R, Montiel DP, Calleja-Macias IE, et al. Laser capture microdissection of cervical human papillomavirus infections: copy number of the virus in cancerous and normal tissue and heterogeneous DNA methylation. Virology. 2009;390(2):261–7. doi: 10.1016/j.virol.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez AF, Rosales C, Lopez-Nieva P, Grana O, Ballestar E, Ropero S, et al. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 2009;19(3):438–51. doi: 10.1101/gr.083550.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhattacharjee B, Sengupta S. CpG methylation of HPV 16 LCR at E2 binding site proximal to P97 is associated with cervical cancer in presence of intact E2. Virology. 2006;354(2):280–5. doi: 10.1016/j.virol.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 22.Ding DC, Chiang MH, Lai HC, Hsiung CA, Hsieh CY, Chu TY. Methylation of the long control region of HPV16 is related to the severity of cervical neoplasia. Eur J Obstet Gynecol Reprod Biol. 2009;147(2):215–20. doi: 10.1016/j.ejogrb.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 23.Piyathilake CJ, Macaluso M, Alvarez RD, Chen M, Badiga S, Edberg JC, et al. A higher degree of methylation of the HPV 16 E6 gene is associated with a lower likelihood of being diagnosed with cervical intraepithelial neoplasia. Cancer. 2011;117(5):957–63. doi: 10.1002/cncr.25511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandsma JL, Sun Y, Lizardi PM, Tuck DP, Zelterman D, Haines GK, 3rd, et al. Distinct human papillomavirus type 16 methylomes in cervical cells at different stages of premalignancy. Virology. 2009;389(1-2):100–7. doi: 10.1016/j.virol.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turan T, Kalantari M, Calleja-Macias IE, Cubie HA, Cuschieri K, Villa LL, et al. Methylation of the human papillomavirus-18 L1 gene: a biomarker of neoplastic progression? Virology. 2006;349(1):175–83. doi: 10.1016/j.virol.2005.12.033. [DOI] [PubMed] [Google Scholar]
- 26.Sun C, Reimers LL, Burk RD. Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynecol Oncol. 2011;121(1):59–63. doi: 10.1016/j.ygyno.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong D, Ye F, Lu W, Hu Y, Wan X, Chen Y, et al. Methylation status of the long control region of HPV 16 in clinical cervical specimens. Mol Med Report. 2008;1(4):555–60. [PubMed] [Google Scholar]
- 28.Mirabello L, Schiffman M, Ghosh A, Rodriguez A, Vasiljevic N, Wentzensen N, et al. Elevated methylation of HPV16 DNA is associated with the development of high grade cervical intraepithelial neoplasia. Int J Cancer. 2012 Jul 31; doi: 10.1002/ijc.27750. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernard HU. Gene expression of genital human papillomaviruses and considerations on potential antiviral approaches. Antivir Ther. 2002;7(4):219–37. [PubMed] [Google Scholar]
- 30.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2(5):342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 31.McLaughlin-Drubin ME, Meyers J, Munger K. Cancer associated human papillomaviruses. Curr Opin Virol. 2012 doi: 10.1016/j.coviro.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghittoni R, Accardi R, Hasan U, Gheit T, Sylla B, Tommasino M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes. 2010;40(1):1–13. doi: 10.1007/s11262-009-0412-8. [DOI] [PubMed] [Google Scholar]
- 33.Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clinical Science. 2006;110(5):525–41. doi: 10.1042/CS20050369. [DOI] [PubMed] [Google Scholar]
- 34.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550–60. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 35.el Awady MK, Kaplan JB, O'Brien SJ, Burk RD. Molecular analysis of integrated human papillomavirus 16 sequences in the cervical cancer cell line SiHa. Virology. 1987;159(2):389–98. doi: 10.1016/0042-6822(87)90478-8. [DOI] [PubMed] [Google Scholar]
- 36.Wentzensen N, Vinokurova S, Von Knebel Doeberitz M. Systematic review of genomic integration sites of human papillomavirus genomes in epithelial dysplasia and invasive cancer of the female lower genital tract. Cancer Research. 2004;64(11):3878–84. doi: 10.1158/0008-5472.CAN-04-0009. [DOI] [PubMed] [Google Scholar]
- 37.Wentzensen N, von Knebel Doeberitz M. Biomarkers in cervical cancer screening. Dis Markers. 2007;23(4):315–30. doi: 10.1155/2007/678793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reuschenbach M, Clad A, von Knebel Doeberitz C, Wentzensen N, Rahmsdorf J, Schaffrath F, et al. Performance of p16INK4a-cytology, HPV mRNA, and HPV DNA testing to identify high grade cervical dysplasia in women with abnormal screening results. Gynecol Oncol. 2010;119(1):98–105. doi: 10.1016/j.ygyno.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 39.Sahasrabuddhe VV, Luhn P, Wentzensen N. Human papillomavirus and cervical cancer: biomarkers for improved prevention efforts. Future Microbiol. 2011;6(9):1083–98. doi: 10.2217/fmb.11.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klaes R, Friedrich T, Spitkovsky D, Ridder R, Rudy W, Petry U, et al. Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int J Cancer. 2001;92(2):276–84. doi: 10.1002/ijc.1174. [DOI] [PubMed] [Google Scholar]
- 41.Wentzensen N, Bergeron C, Cas F, Eschenbach D, Vinokurova S, von Knebel Doeberitz M. Evaluation of a nuclear score for p16INK4a-stained cervical squamous cells in liquid-based cytology samples. Cancer. 2005;105(6):461–7. doi: 10.1002/cncr.21378. [DOI] [PubMed] [Google Scholar]
- 42.Guo M, Warriage I, Mutyala B, Patel S, Lin E, Gong Y, et al. Evaluation of p16 immunostaining to predict high-grade cervical intraepithelial neoplasia in women with Pap results of atypical squamous cells of undetermined significance. Diagn Cytopathol. 2011;39(7):482–8. doi: 10.1002/dc.21415. [DOI] [PubMed] [Google Scholar]
- 43.Denton KJ, Bergeron C, Klement P, Trunk MJ, Keller T, Ridder R. The sensitivity and specificity of p16(INK4a) cytology vs HPV testing for detecting high-grade cervical disease in the triage of ASC-US and LSIL pap cytology results. Am J Clin Pathol. 2010;134(1):12–21. doi: 10.1309/AJCP3CD9YKYFJDQL. [DOI] [PubMed] [Google Scholar]
- 44.Petry KU, Schmidt D, Scherbring S, Luyten A, Reinecke-Luthge A, Bergeron C, et al. Triaging Pap cytology negative, HPV positive cervical cancer screening results with p16/Ki-67 Dual-stained cytology. Gynecol Oncol. 2011;121(3):505–9. doi: 10.1016/j.ygyno.2011.02.033. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt D, Bergeron C, Denton KJ, Ridder R. p16/ki-67 dual-stain cytology in the triage of ASCUS and LSIL papanicolaou cytology: results from the European equivocal or mildly abnormal Papanicolaou cytology study. Cancer Cytopathol. 2011;119(3):158–66. doi: 10.1002/cncy.20140. [DOI] [PubMed] [Google Scholar]
- 46.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 47.Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. 2011;12(2):206–22. doi: 10.1002/cbic.201000195. [DOI] [PubMed] [Google Scholar]
- 48.Stein RA. DNA methylation profiling: a promising tool and a long road ahead for clinical applications. Int J Clin Pract. 2011;65(12):1212–3. doi: 10.1111/j.1742-1241.2011.02804.x. [DOI] [PubMed] [Google Scholar]
- 49.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–22. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Illingworth RS, Bird AP. CpG islands--‘a rough guide’. FEBS Lett. 2009;583(11):1713–20. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 51.Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17(3):330–9. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 52.Galvan SC, Martinez-Salazar M, Galvan VM, Mendez R, Diaz-Contreras GT, Alvarado-Hermida M, et al. Analysis of CpG methylation sites and CGI among human papillomavirus DNA genomes. BMC Genomics. 2011;12:580. doi: 10.1186/1471-2164-12-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wiley DJ, Huh J, Rao JY, Chang C, Goetz M, Poulter M, et al. Methylation of human papillomavirus genomes in cells of anal epithelia of HIV-infected men. J Acquir Immune Defic Syndr. 2005;39(2):143–51. [PubMed] [Google Scholar]
- 54.Kalantari M, Villa LL, Calleja-Macias IE, Bernard HU. Human papillomavirus-16 and -18 in penile carcinomas: DNA methylation, chromosomal recombination and genomic variation. Int J Cancer. 2008;123(8):1832–40. doi: 10.1002/ijc.23707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Balderas-Loaeza A, Anaya-Saavedra G, Ramirez-Amador VA, Guido-Jimenez MC, Kalantari M, Calleja-Macias IE, et al. Human papillomavirus-16 DNA methylation patterns support a causal association of the virus with oral squamous cell carcinomas. Int J Cancer. 2007;120(10):2165–9. doi: 10.1002/ijc.22563. [DOI] [PubMed] [Google Scholar]
- 56.Park IS, Chang X, Loyo M, Wu G, Chuang A, Kim MS, et al. Characterization of the methylation patterns in human papillomavirus type 16 viral DNA in head and neck cancers. Cancer Prev Res (Phila) 2011;4(2):207–17. doi: 10.1158/1940-6207.CAPR-10-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fraga MF, Esteller M. DNA methylation: a profile of methods and applications. Biotechniques. 2002;33(3):632–4. 6–49. doi: 10.2144/02333rv01. [DOI] [PubMed] [Google Scholar]
- 58.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93(18):9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28(8):E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dupont JM, Tost J, Jammes H, Gut IG. De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal Biochem. 2004;333(1):119–27. doi: 10.1016/j.ab.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 61.Tost J, Gut IG. DNA methylation analysis by pyrosequencing. Nat Protoc. 2007;2(9):2265–75. doi: 10.1038/nprot.2007.314. [DOI] [PubMed] [Google Scholar]
- 62.Colella S, Shen L, Baggerly KA, Issa JP, Krahe R. Sensitive and quantitative universal Pyrosequencing methylation analysis of CpG sites. Biotechniques. 2003;35(1):146–50. doi: 10.2144/03351md01. [DOI] [PubMed] [Google Scholar]
- 63.Schweiger MR, Kerick M, Timmermann B, Isau M. The power of NGS technologies to delineate the genome organization in cancer: from mutations to structural variations and epigenetic alterations. Cancer Metastasis Rev. 2011;30(2):199–210. doi: 10.1007/s10555-011-9278-z. [DOI] [PubMed] [Google Scholar]
- 64.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7(6):461–5. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Androphy EJ, Lowy DR, Schiller JT. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature. 1987;325(6099):70–3. doi: 10.1038/325070a0. [DOI] [PubMed] [Google Scholar]
- 66.Tan SH, Baker CC, Stunkel W, Bernard HU. A transcriptional initiator overlaps with a conserved YY1 binding site in the long control region of human papillomavirus type 16. Virology. 2003;305(2):486–501. doi: 10.1006/viro.2002.1779. [DOI] [PubMed] [Google Scholar]
- 67.Tan SH, Leong LE, Walker PA, Bernard HU. The human papillomavirus type 16 E2 transcription factor binds with low cooperativity to two flanking sites and represses the E6 promoter through displacement of Sp1 and TFIID. J Virol. 1994;68(10):6411–20. doi: 10.1128/jvi.68.10.6411-6420.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25(12):2532–4. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wentzensen N, Schiffman M, Dunn T, Zuna RE, Gold MA, Allen RA, et al. Multiple human papillomavirus genotype infections in cervical cancer progression in the study to understand cervical cancer early endpoints and determinants. Int J Cancer. 2009;125(9):2151–8. doi: 10.1002/ijc.24528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wentzensen N, Sherman ME, Schiffman M, Wang SS. Utility of methylation markers in cervical cancer early detection: appraisal of the state-of-the-science. Gynecol Oncol. 2009;112(2):293–9. doi: 10.1016/j.ygyno.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Drummond A, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, et al. Geneious v5.6. 2012 Available from http://www.geneious.com.
- 72.O'Connor MJ, Tan SH, Tan CH, Bernard HU. YY1 represses human papillomavirus type 16 transcription by quenching AP-1 activity. J Virol. 1996;70(10):6529–39. doi: 10.1128/jvi.70.10.6529-6539.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoppe-Seyler F, Butz K. Cellular control of human papillomavirus oncogene transcription. Mol Carcinog. 1994;10(3):134–41. doi: 10.1002/mc.2940100304. [DOI] [PubMed] [Google Scholar]
- 74.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]