

Abstract
Strong NF-κB activation requires ligation of both the CD28 co-receptor and TCR. PDK1 acts as a scaffold by binding both PKCθ and CARMA1 and is therefore essential for signaling to NF-κB. Here, we demonstrate the importance of PDK1 threonine (Thr)-513 phosphorylation in regulating the intermolecular organization of PDK1 homodimers. Thr-513 is directly involved in heterotypic PDK1 homodimer formation, in which binding is mediated through the pleckstrin homology (PH) and kinase domains. Upon activation, phosphorylated Thr-513 instead mediates homotypic intermolecular binding through the PH domains. Consequently, cell permeable peptides with a Thr-513 to Ile derivative (PTD-PDK1-Thr-513-Ile) bound the kinase domain and while a Thr-513 to Asp peptide (PTD-PDK1-Thr-513-Asp) bound the PH domain. PTD-PDK1-Thr-513-Ile blocked binding between PDK1 and PKCθ, phosphorylation of PKCθ Thr-538, and activation of both NF-κB and AKT. In contrast, PTD-PDK1-Thr-513-Asp selectively inhibited binding between PDK1 and CARMA1 and blocked TCR/CD28 induced NF-κB activation. Therefore, Thr-513 phosphorylation regulates a critical intermolecular switch governing PDK1 homodimer structure and the capacity to interact with downstream signaling pathway components. Given the pleiotropic functions of PDK1, these data may open the door to the development of immunosuppressive therapies that selectively target the PDK1 to NF-κB pathway in T cell activation.
Keywords: NF-κB, PDK1, PKCθ, CARMA1, T cell
Introduction
Phosphoinositide 3-kinases (PI3Ks) are a family of unique, conserved intracellular lipid kinases that phosphorylate the 3′-hydroxyl group of phosphatidylinositol or phosphoinositides (1). This phosphorylation induces the activation of many intracellular signaling pathways, and thereby mediates cellular functions, such as metabolism, survival, and polarity (2). Activation of the various cell surface receptors induces the activation of PI3K, and this activated PI3K converts the membrane phospholipid phosphatidylinositol-4,5-bisphosphate (PtdIns(4,5)P2) into phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) (2). During this process, phosphoinositide-dependent kinase 1 (PDK1) is activated by PtdIns(3,4,5)P3, and this activated PDK1 mediates multiple signaling pathways that are important for cell metabolism, survival, and activation (3). Further, PDK1 may act as a “master regulator” of the protein kinase A/protein kinase G/protein kinase C (AGC) family of kinases (4). The most well-characterized target of PDK1 is AKT; in fact, PDK1 was initially identified as an AKT kinase (4, 5). AKT activity is regulated via Thr-308 phosphorylation by PDK1 (5, 6) and Ser-473 phosphorylation by mTOR (7, 8).
PDK1 has important roles in activation and function of immune cells (4, 9–13). Previous studies have established that, during coordinated CD28/TCR stimulation of T cells, PDK1 is essential for efficient activation of Protein kinase Cθ (PKCθ) by the PI3K pathway (4) and subsequent assembly of the CARMA1-BCL10-MALT1 (CBM) complex to activate NF-κB (14, 15). PDK1 also plays a crucial role in T cell development, as conditional deletion of PDK1 in double-negative (DN) thymocytes blocks T-cell development at the DN4 stage by impairing pre-TCR induced proliferation (16). However, deleting PDK1 in double-positive (DP) thymocytes does not affect CD4+ single-positive (SP) thymocyte development while CD8SP thymocyte development is largely affected by PDK1 deletion (4).
PDK1 is composed of a kinase and pleckstrin homology (PH) domain. The kinase domain phosphorylates substrate protein and the PH domain binds PtdIns(3,4,5)P3 during PDK1-mediated substrate activation (4). Phosphorylation of PDK1 threonine (Thr)-513 is important for binding to PKCθ and is induced by CD28/TCR stimulation during T-cell activation (4). Initially, the phosphorylation site was found to be autophosphorylated upon binding to the phosphor-lipid complex (17). Previous studies have suggested PDK1 is initially homodimerized, preventing substrate binding (17, 18). However, mutation of PDK1 Thr-513 to Glu (phosphomimic) destabilizes homodimerization and increases binding between PDK1 and AKT, which results in AKT activation through Thr-308 phosphorylation (18).
In this report, we found that PDK1 phosphorylation at Thr-513 induces changes in interdomain binding, thus regulating binding between PDK1 and PKCθ or CARMA1. The phosphorylation destabilized the binding kinase domain and PH domain and increased the binding between PH domains. Moreover, non-phosphomimic Thr-513 peptide binds the PDK1 kinase domain and inhibits binding of PDK1 to PKCθ. In addition, phosphomimic Thr-513 peptide binds to the PDK1 PH domain and inhibits binding of PDK1 to CARMA1 and binding between PH domains, which results in impaired CD28/TCR stimulation and T cell activation. Given the pleiotropic functions of PDK1, these data may open the door to the development of immunosuppressive therapies that selectively target the PDK1 to NF-κB pathway in T cell activation.
Materials and Methods
Cell culture, antibodies, and peptides
HEK293 cells were maintained in DMEM supplemented with 5% FBS. Jurkat T cells were maintained in RPMI 1640 supplemented with 5% FBS. Anti-IκBα and anti-GAPDH antibodies were purchased from SantaCruz. Anti-Myc antibody was purchased from Cell Signaling. Anti-CARMA1 antibody was purchased from Alexis. Anti-HA antibody was purchased from Sigma. Phospho-specific antibody for PKCθ at T538 and anti-PDK1 antibody were purchased from BD Biosciences. Anti-mouse CD3, anti-mouse CD28, anti-human CD3, and anti-human CD28 antibodies were purchased from eBiosciences. Anti-mouse IgG and anti-hamster IgG were purchased from Sigma. FITC-conjugated anti-mouse CD4, PE-conjugated anti-mouse CD69, and PerCP Cy5.5-conjugated anti-mouse CD25 antibodies were purchased from eBiosciences. Peptides, including protein transduction domain (PTD) (DRQIKIWFQNRRMKWKK), PTD-PDK1-Thr-513-Ile (DRQIKIWFQNRRMKWKKEAKNFKIFFVHTP), and PTD-PDK1-Thr-513-Asp (DRQIKIWFQNRRMKWKKEAKNFKDFFVHTP) were synthesized by Anygene.
Plasmids
pCDNA-Myc-PDK1 and pCDNA-HA-PDK1 were constructed by insertion of Myc sequence tagged-Pdk1 open reading frame (ORF) or Ha sequence tagged-Pdk1 ORF into the pCDNA3. pCDNA-Myc-PDK1-PH and pCDNA-HA-PDK1-PH were constructed by insertion of Myc sequence tagged-Pdk1 PH domain sequences or Ha sequence tagged-Pdk1 PH domain sequences into pCDNA3. A QuickChange II XL site-directed mutagenesis kit (Stratagene) was used for mutagenesis of pCDNA-Myc-PDK1, pCDNA-HA-PDK1, pCDNA3-Myc-PDK1-PH, and pCDNA3-HA-PDK1-PH. pEGZ-HA-PKCθ was constructed by insertion of the Pkcq ORF into pEGZ. pCDNA-CARMA1 was constructed by insertion of Carma1 ORF into pCDNA3. Construction of pBIIx and pRenilla were described previously (4).
Flow cytometry
CD4+ T cells were isolated from the spleens and lymph nodes of 6- to 8-week-old C57BL6 mice with the EasySep™ mouse CD4+ T cell enrichment kit (Stem Cell Research). Purified CD4+ T cells were activated with anti-CD3 and anti-CD28 antibodies for indicated times, then stained with indicated antibodies. The stained cells were analyzed on a Guava easyCyte HT (Millipore).
Enzyme-linked immunosorbent assay (ELISA)
Secreted IL-2 was analyzed by IL-2-specific ELISA. Cells were plated at 1 × 104 cells per well in 96-well plates coated with anti–mouse CD3 (5 μg/mL) and anti–mouse CD28 (5 μg/ml) and incubated at 37°C with 5% CO2. Twenty-four hours later, the culture medium was analyzed according to manufacturer protocols (eBioscience)
NF-κB gene luciferase assay
HEK293T cells were plated at 5 × 105 cells per well in a 12-well plate and transfected with NF-κB reporter plasmid (pBIIx), pRenilla, and other plasmids with Lipofectamine 2000 (Invitrogen). Total DNA amounts were normalized to that of empty pCDNA3 vector. After 48 h, cells were lysed in 1× passive lysis buffer. Debris was removed by centrifugation at 14,000 rpm for 5 min at 4°C. Firefly luciferase and Renilla luciferase activity was measured with 20 μl lysate samples. The ‘fold stimulation’ was calculated for each sample by dividing the luciferase activity in the sample (normalized to Renilla luciferase activity) by the activity of a sample containing only empty expression vector. Mouse primary CD4+ T cells or Jurkat T cells were electroporated with NF-κB reporter plasmid (pBIIx), pRenilla, and indicated plasmids with amaxa Nucleofector™ (Lonza). The cells were activated with anti-CD3 and anti-CD28 antibodies.
Co-immunoprecipitation and M450 bead-mediated pull-down assay
To study molecular interactions in HEK293 cells, expression vectors were transfected into HEK293 cells with Lipofectamine 2000 (Invitrogen). Forty hours later, target proteins were immunoprecipitated with anti-HA, anti-PKCθ, anti-CARMA1, anti-PDK1, or anti-Myc antibodies. To study molecular interactions in Jurkat T cells, expression vectors were electroporated into Jurkat T cells with Amaxa Nucleofector (Lonza). The transfected cells were divided into 8 samples. Four samples were treated with peptides for 6 h without stimulation. The remaining 4 samples were pretreated with peptides for 1 h and stimulated with anti-CD3 and anti-CD28 antibodies for 6 h with peptides. Target proteins were immunoprecipitated with anti-HA antibody. Samples were separated by SDS-PAGE and analyzed by immunoblotting with anti-Myc, anti-HA, anti-PKCθ, or anti-PDK1. To study endogenous interactions, 5 × 107 Jurkat or 2 × 105 primary CD4+ T cells were used after stimulation with anti-CD3 and anti-CD28 antibodies. Cell lysates were immunoprecipitated with anti-PDK1 antibody. Samples were separated by SDS-PAGE and analyzes by immunoblotting with anti-PKCθ or anti-PDK1 antibody. For pull-down of peptide binding proteins, chemically activated M450 beads were conjugated with indicated peptides according to manufacturer protocols.
Immunofluorescence microscopy
For fluorescence microscopy, primary CD4+ T cells were incubated with FITC-peptide for 30 min at 37°C. After incubation, T cells were plated on poly L-lysine-coated coverslips for 15 min at 37°C, then fixed with 4% paraformaldehyde for 10 min at room temperature. Cells were incubated with DAPI for nuclear staining. Results were analyzed by Zeiss confocal microscopy.
Results
Binding of the PDK1 PH to kinase domainsis destabilized by Thr-513 phosphorylation
PDK1 Thr-513 phosphorylation is essential for TCR-CD28 stimulation-mediated NF-κB activation (4). This phosphorylation was increased by TCR-CD28 stimulation (Fig. 1A) (4). Many proteins, including PDK1, possess a PH domain. The AKT also has a PH domain and it has been shown that AKT PH domain binds to the kinase domain in inactive conformation, but, two domains are separated during the AKT activation (19). In PDK1, the Thr-513 phosphorylation site is located in the PH domain and it has been known that the phosphorylation site is important for binding of PDK1 to PKCθ. Our binding analysis showed that overexpressed PDK1 PH domain (401–550) bound to the PDK1 kinase domain (1–341) in HEK293 cells (Fig. 1B). Mutation of Thr-513 to Asp (phosphomimic) decreased binding between the PH and kinase domains, but Ile (phosphorylation-disabled) increased binding (Fig. 1B). The PDK1 region responsible for PKCθ binding was analyzed in the ex vivo system by overexpressing each PDK1 deletion fragment in HEK293 cells. The data indicated that PDK1 kinase domain is required for binding to PKCθ and the region overlaps with the PH domain-binding region (Supplemental Fig. 1). Thus, full-length PDK1 Thr-513-Asp binds to PKCθ more strongly than to PDK1 wild-type (WT) because phosphorylation destabilizes binding between kinase domain and PH domain.
FIGURE 1.

Phosphomimic of Thr-513 of PDK1 in PDK1 PH domain destabilizes binding between PDK1 kinase and PH domains. A, Phosphorylation at PDK1 Thr-513 was assessed by immunoblotting with anti-Thr-513 phospho-specific PDK1 antibody after stimulation of mouse primary CD4+ T cells with anti-CD3 and anti-CD28. B, Binding between PDK1 kinase and PH domains in Thr-513 mutant derivatives was analyzed by immunoprecipitation and immunoblotting.
Phosphomimic at PDK1 Thr-513 induced strong homotypic binding of PDK1 PH domains
PDK1 is dimerized and Thr-513 on PDK1 is autophosphorylated via transphosphorylation (17). A recent study showed that a Thr-513 phosphorylation mimic destabilizes homodimerization of PDK1, thus enabling PDK1-substrate binding (18). The previous study employed fluorescence resonance energy transfer (FRET) methods. We used co-immunoprecipitation and immunoblot analyses with tagged PDK1-expressing HEK293 cells. Our data showed that Myc-tagged WT PDK1 co-immunoprecipitated with HA-tagged WT PDK1; in contrast, Myc-tagged WT PDK1 did not co-immunoprecipitate with anti-HA antibody in the absence of HA-tagged WT PDK1 in HEK293 cells (Fig. 2A). Initially, we expected the Thr-513-Asp mutation to abolish binding between tagged PDK1 mutant derivatives. However, Myc-tagged PDK1 Thr-513-Asp still co-immunoprecipitated with HA-tagged PDK1 Thr-513-Asp (Fig. 2A). Because binding of PDK1 PH to kinase domains is destabilized by Thr-513 phosphorylation and PDK1 kinase domain is the substrate-binding site, Thr-513 phosphorylation may lead to binding between PDK1 PH domains. Thus, we tested whether the PDK1 PH domain dimerizes by using differentially tagged PDK1 PH domains expressed in HEK293 cells. Fig. 2B shows that Myc-tagged-PDK1 PH domain co-immunoprecipitated with HA-tagged PDK1 PH domain. In addition, Thr-513-Asp mutation of the PDK1 PH domain dramatically increased binding between tagged domains.
FIGURE 2.
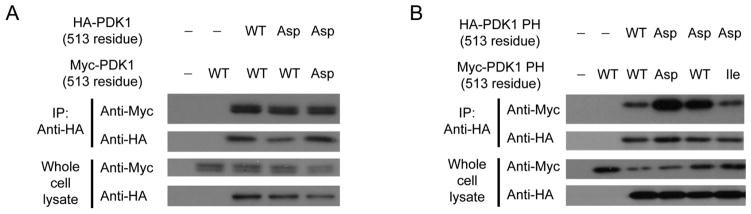
Phosphomimic of Thr-513 of PDK1 in PDK1 PH domain increases PDK1 PH homodimerization. A, Binding between PDK1s carrying mutations at Thr-513 in HEK293 cells were analyzed by immunoprecipitation and immunoblotting. B, Binding between PDK1 PH domains carrying mutations at Thr-513 in HEK293 cells was analyzed by immunoprecipitation and immunoblotting.
Phosphomimic PDK1 Thr-513 region-containing peptide binds to PDK1 PH domain and unphosphomimic PDK1 Thr-513 peptide binds to PDK1 kinase domain
Thr-513 phosphorylation is important for T cell activation because it increases binding between PDK1 and PKCθ, which mediates CBM complex-mediated NF-κB activation (4). In addition, it has been found that Thr-513 is phosphorylated by trans-autophosphorylation (17). Sequence comparison of the Thr-513 region with the phosphorylation sites of PDK1 substrates showed the Thr-513 region is similar to the PDK1 phosphorylation motif (Fig. 3A). To test whether the Thr-513 phosphorylation site can directly regulate binding between PDK1 and PKCθ and binding between PH domains, we designed peptides containing the Thr-513 site and PTD. PTD-PDK1-Thr-513-Ile is composed of 17 PTD amino acids and 6 amino acids before and after the PDK1 Thr-513-Ile substitution; PTD-PDK1-Thr-513-Asp is composed of 17 PTD amino acids and 6 amino acids before and after the PDK1 Thr-513-Asp substitution (Fig. 3B).
FIGURE 3.

PDK1 peptides carrying Thr-513 to Ile and Asp mutations binds to PDK1 kinase domain and PH domain, respectively. A, Sequence alignment between PDK1 target phosphorylation sites and the PDK1 Thr-513 region was performed in ClustalW. B, PTD-PDK1-Thr-513-Asp and PTD-PDK1-Thr-513-Ile were designed as indicated. PTD peptide was used as a control. C and D, Chemically activated M450 beads were conjugated with PTD peptide, PTD-PDK1-Thr-513-Ile, or PTD-PDK1-Thr-513-Asp. The conjugated peptides were incubated with lysate from HEK293 cells transfected with PDK1 kinase or PDK1 PH domain expression plasmids (C) or incubated with lysate from mouse primary CD4+ T cells stimulated with anti-CD3 and anti-CD28 antibodies for 1 hour (D). Binding proteins were analyzed by immunoblotting (C and D).
With the peptides, we tested binding between the peptides and the PDK1 kinase and PH domains. Our pull-down assay showed the PDK1 kinase domain bound to PTD-PDK1-Thr-513-Ile peptide-conjugated M450 beads and the PDK1 PH domain bound to PTD-PDK1-Thr-513-Asp peptide-conjugated M450 beads (Fig. 3C). In addition, PTD-PDK1-Thr-513-Ile and PTD-PDK1-Thr-513-Asp peptide-conjugated M450 beads bound to PDK1 from activated mouse primary CD4+ T cells. This data indicated that the peptide was accessible to the binding region on PDK1 only after T cell activation. In these experiments, the more PDK1 bound to PTD-PDK1-Thr-513-Ile than PTD-PDK1-Thr-513-Asp peptide-conjugated M450 beads (Fig. 3D).
PDK1 Thr-513 region-containing peptides regulate binding of PDK1 to PKCθ or CARMA1
We used co-immunoprecipitation and immunoblot analyses to test whether the designed peptides regulate binding between PDK1 and PKCθ and between PDK1 and CARMA1. PTD-PDK1-Thr-513-Ile inhibited binding between overexpressed PDK1 and PKCθ in HEK293 cells (Fig. 4A). Binding between PDK1 PH domains was inhibited by PTD-PDK1-Thr-513-Asp (Fig. 4B). Moreover, PTD-PDK1-Thr-513-Ile inhibited binding between PDK1 and PKCθ in Jurkat T cells stimulated with anti-CD3 and anti-CD28 antibodies (Fig. 4C). However, PTD-PDK1-Thr-513-Asp did not affect binding between PDK1 and PKCθ, but did inhibit binding between PDK1 and CARMA1 (Fig. 4C). In Jurkat T cells, PTD-PDK1-Thr-513-Ile inhibited binding between PDK1 Thr-513-Ile proteins (Fig. 4D) and PTD-PDK1-Thr-513-Asp inhibited binding between PDK1 Thr-513-Asp proteins with or without stimulation by anti-CD3 and anti-CD28 antibodies (Fig. 4E). We also tested the effect of peptides on binding between WT PDK1 proteins. PTD-PDK1-Thr-513-Ile strongly affected binding between WT PDK1 proteins when Jurkat T cells were not stimulated (Fig. 4F). However, PTD-PDK1-Thr-513-Asp strongly affected binding between WT PDK1 proteins in Jurkat T cells stimulated with anti-CD3 and anti-CD28 antibodies (Fig. 4F).
FIGURE 4.

PDK1 peptides carrying Thr-513 to Ile and Asp mutations inhibits binding of PDK1 to PKCθ and CARMA, respectively. A, The effect of PDK1 peptides on binding between PDK1 and PKCθ in HEK293 cells after overexpression of Myc-PDK1 and HA-PKCθ was analyzed by immunoprecipitation and immunoblotting. B, The effect of PDK1 peptides on binding between PH domains in HEK293 cells after overexpression of Myc-PDK1-PH Thr-513-Asp and HA-PDK1-PH Thr-513-Asp was analyzed by immunoprecipitation and immunoblotting. C, The effect of PDK1 peptides on binding between PDK1 and PKCθ and between PDK1 and CARMA1 in Jurkat T cells after stimulation with anti-CD3 and anti-CD28 antibodies was analyzed by immunoprecipitation and immunoblotting. D, E, and F, The effect of PDK1 peptides on binding between PDK1 Thr-513-Ile proteins (D), between PDK1 Thr-513-Asp proteins (E), and between WT PDK1 proteins (F) after expression of PDK1 Thr-513-Ile, PDK1 Thr-513-Asp, or WT PDK1 in Jurkat T cells with or without stimulation by anti-CD3 and anti-CD28 antibodies was analyzed by immunoprecipitation and immunoblotting as described in Materials and Methods.
TCR-CD28-mediated NF-κB activation is inhibited by PDK1-PH Thr-513-Asp and PDK1-PH Thr-513-Ile
Thr-513 phosphorylation in the PDK1 PH domain is important for binding between the PDK1 kinase domain and PKCθ and between PH domains. The PDK1 PH Thr-513-Ile mutant domain binds to the kinase domain and PDK1 PH Thr-513-Asp mutant domain binds to the PH domain. Thus, PDK1 PH Thr-513-Ile may inhibit binding of PDK1 to PKCθ and PDK1 PH Thr-513-Asp may inhibit homodimerization of PH domains during T-cell activation. Previous reports have shown that PKCθ-mediated CBM complex formation is required for NF-κB activation (20–22). Overexpression of these proteins in HEK293 cells induced strong NF-κB activation, which was enhanced further by PDK1 overexpression. In this system, overexpression of the PDK1 PH domain inhibited NF-κB activation. In addition, overexpression of the PDK1 PH Thr-513-Ile domain or PDK1 PH Thr-513-Asp domain inhibited NF-κB activation (Fig. 5A). The data indicate that binding of PKCθ to PDK1 and PDK1 homotypic dimerization is important for PKCθ and CBM-mediated NF-κB activation. However, this data was obtained from an artificial NF-κB activation system in HEK293 cells, so we determined whether NF-κB activation can be inhibited by PDK1 PH Thr-513-Ile or PDK1 PH Thr-513-Asp expression in mouse primary CD4+ T cells (Fig. 5B) and Jurkat T cells (Fig. 5C). Treatment with anti-CD3 plus anti-CD28 antibodies induced NF-κB activation in Jurkat T cells and mouse primary CD4+ T cells. Under the activating conditions, we introduced an NF-κB reporter plasmid with a PDK1 PH Thr-513-Ile, PDK1 PH Thr-513-Asp, or a PDK1 PH WT expression plasmid into Jurkat T cells or mouse primary CD4+ T cells by electroporation. Our data showed that electroporation-mediated introduction of PDK1 PH WT, PDK1 PH Thr-513-Ile, or PDK1 PH Thr-513-Asp expression plasmids significantly reduced TCR-CD28-mediated NF-κB activation in mouse primary CD4+ T cells (Fig. 5B) and Jurkat T cells (Fig. 5C).
FIGURE 5.

PDK1 PH domain inhibits PDK1-mediated NF-κB activation in T cells. A, The effect of PDK1 PH domain expression on PDK1-PKCθ-CARMA1-Bcl10-Malt1-mediated NF-κB activation was determined by NF-κB-reporter assay. B and C, The effect of PDK1 PH domain expression on TCR-CD28-mediated NF-κB activation in mouse primary CD4+ T cells (B) or Jurkat T cells (C) was determined by NF-κB reporter assay. The Renilla luciferase activity was used as a reference for normalization of gene expression in transiently transfected cells. Results: mean ± SD. Student’s t test: *, p < 0.05; **, p < 0.01.
PDK1 Thr-513 region-containing peptides inhibited NF-κB activation in activated T cells, reducing IL-2 production
Interestingly, PTD-PDK1-Thr-513-Ile inhibited PKCθ phosphorylation and IκBα degradation in activated CD4+ T cells, but PTD-PDK1-Thr-513-Asp inhibited IκBα degradation and not PKCθ phosphorylation (Fig. 6A). In addition, PTD-PDK1-Thr-513-Ile inhibited AKT Thr-308 phosphorylation, but PTD-PDK1-Thr-513-Asp did not (Supplemental Fig. 2). Thus, PTD-PDK1-Thr-513-Asp does not affect the phosphorylation of downstream molecules even though the peptide affects the NF-κB activation during the CD4+ T cell activation. In addition to our investigation on the short-term temporal NF-κB activation through the detection of IκBα degradation, long-term total NF-κB activation levels in mouse primary CD4+ T cells during TCR-CD28 stimulation were determined using NF-κB activation reporter plasmids. PTD-PDK1-Thr-513-Ile significantly inhibited NF-κB activation in CD4+ T cells stimulated with anti-CD3 and anti-CD28 antibodies. PTD-PDK1-Thr-513-Asp also inhibited NF-κB activation, but the inhibitory effect was less substantial (Fig. 6B). In addition, expression of IL-2, the most important NF-κB target during T-cell activation, was also inhibited by PTD-PDK1-Thr-513-Ile and PTD-PDK1-Thr-513-Asp (Fig. 6C). The peptides also reduced the levels of secreted IL-2 protein from activated CD4+ T cells (Fig. 6D). The data indicate that blocking of PDK1 binding to PKCθ by PTD-PDK1-Thr-513-Ile and disruption of PDK1 homotypic dimerization by PTD-PDK1-Thr-513-Asp inhibits TCR-CD28-mediated NF-κB activation (Fig. 6E).
FIGURE 6.

PDK1 peptides carrying Thr-513 to Ile or Asp mutations inhibit TCR-CD28-mediated NF-κB activation in mouse primary CD4+ T cells. A, Effect of PDK1 peptides on phosphorylation of PKCθ and IκBα degradation in mouse primary CD4+ T cells stimulated with anti-CD3 and anti-CD28 antibodies for 30 min. B, The effect of PDK1 peptide (PTD-PDK1-Thr-513-Ile or PTD-PDK1-Thr-513-Asp) on TCR-CD28-mediated NF-κB activation in mouse primary CD4+ T cells was analyzed by NF-κB reporter assay. The Renilla luciferase activity was used as a reference for normalization of gene expression in transiently transfected cells. C and D, The effect of PDK1 peptides on IL-2 production was analyzed by quantitative real-time PCR (C) or ELISA (D). Results: mean ± SD. Student’s t test: *, p < 0.05; **, p < 0.01. E, Schematic model of TCR-CD28-mediated NF-κB activation by PDK1 dimer formation. Thr-513 phosphorylation induces conversion of PDK1 kinase–PH domain binding into PDK1 PH–PH domain binding during T cell activation, which is required for TCR-CD28-mediated NF-κB activation.
Peptides containing the PDK1 Thr-513 region inhibited TCR-mediated T cell activation
To test whether the PTD-tagged peptide is taken up by T cells, PTD-PDK1-Thr-513-Asp and PTD-PDK1-Thr-513-Ile were conjugated with FITC and analyzed by flow cytometry. The FITC-conjugated peptides were transduced to almost all isolated primary CD4+ T cells (Fig. 7A). Fluorescence microscopy confirmed the transduced peptides lay within the CD4+ T cells (Fig. 7B).
FIGURE 7.

Peptides carrying PDK1 Thr-513 influence TCR-CD28-mediated mouse primary CD4+ T-cell activation. A, Uptake of PTD-conjugated peptides was analyzed by flow cytometry with FITC-conjugated peptides. B, Localization of PTD-conjugated peptides was analyzed by fluorescence microscopy with FITC-conjugated peptides. C and D, CD4+ T cells were isolated from C57BL6 mice and activated with anti-CD3 and anti-CD28 antibodies for the indicated times. Cells were stained with fluorochrome-conjugated anti-CD4, anti-CD69, and anti-CD25 antibodies. Surface expression of CD69 (C) and CD25 (D) on CD4+ T cells was analyzed by flow cytometry.
Our data showed that PTD-PDK1-PH Thr-513-Ile and PTD-PDK1-PH Thr-513-Asp inhibited TCR-CD28-mediated NF-κB activation by inhibiting binding between PDK1 and PKCθ and between PDK1 and CARMA1. In addition, PDK1 is linked to T-cell activation because genetic deletion of Pdk1 in CD4+ T cells results in defective NF-κB activation during TCR-CD28-mediated T-cell activation (4). Thus, we tested whether our peptides could inhibit CD4+ T-cell activation in primary mouse CD4+ T cells activated with anti-CD3 and anti-CD28. PTD-PDK1-Thr-513-Ile and PTD-PDK1-Thr-513-Asp inhibited surface expression of CD69 (Fig. 7C and Supplemental Fig. 3) and CD25 (Fig. 7D and Supplemental Fig. 3), and control PTD peptide did not affect expression of activation surface markers. In addition, PTD-PDK1-Thr-513-Ile more strongly inhibited CD4+ T-cell activation than did PTD-PDK1-Thr-513-Asp (Fig. 7C and 7D) like data shown in Fig. 6.
Discussion
TCR-CD28-mediated NF-κB activation is important for CD4+ T cell activation because NF-κB regulates the expression of cytokine genes and the survival of activated T cells (4). Our previous study showed that PDK1 is essential for NF-κB activation and Thr-513 phosphorylation is important for the regulation of the binding between PDK1 and its substrates, PKCθ and CARMA1. However, we did not elucidate the detailed mechanism by which the phosphorylation regulates the binding. Recently, Thr-513 phosphorylation has been suggested to be important for the regulation of PDK1 homodimerization. FRET data demonstrated that PDK1 homodimerizes, and that this homodimerization inhibited PDK1-substrate binding (18). In addition, the PDK1 homodimer is destabilized by phosphomimic mutation at Thr-513, enabling binding between PDK1 and its substrate (17, 18). Our co-immunoprecipitation-mediated binding analysis showed that PDK1 kinase domain binds to PDK1 PH domain during PDK1 homodimerization. In addition, phosphomimic mutation at Thr-513 destabilized binding between the PDK1 kinase and PH domains. Because PDK1 does not form an intramolecular bond between the N- and C-terminal domains (18), we hypothesized that homodimerization occurs via intermolecular binding between the PDK1 kinase and PH domains. We previously demonstrated that a phosphomimic of PDK1 Thr-513 increases binding between PDK1 and PKCθ. Thus, we thought that destabilization of PDK1 homodimer formation through phosphomimic at Thr-513 is important for PDK1-mediated NF-κB activation during T-cell activation. Interestingly, however, Thr-513 phosphomimic still formed a homodimer, but with increased homotypic binding between PH domains.
PDK1 Thr-513 is a transautophosphorylation site (17); thus, Thr-513 is a substrate for PDK1 kinase. Sequence alignment of the Thr-513 region of PDK1 with PDK1 substrate sequences indicates the regions are similar to the substrate motif. In addition, PTD-PDK1-Thr-513-Ile bound to PDK1 kinase domain, but the phosphomimic form, PTD-PDK1-Thr-513-Asp, did not. Instead, PTD-PDK1-Thr-513-Asp bound the PH domain, while PTD-PDK1-Thr-513-Ile did not. Thus, the model developed in our previous study can be modified. Initially, PDK1 dimerizes via binding between the PDK1 kinase and PH domains, but phosphorylation of PDK1 at Thr-513 induces binding between PDK1 PH domains, thus exposing the kinase domain to PKCθ. In addition, PDK1 PH domain homodimerization creates a docking site for CARMA1; the resulting PDK1/PKCθ/CARMA1 complex mediates NF-κB activation during TCR-CD28-mediated T-cell activation.
PTD-PDK1-Thr-513-Asp did not inhibit binding of PDK1 to PKCθ or PKCθ phosphorylation, but did inhibit TCR-CD28-mediated NF-κB activation. This inhibition might be caused by inhibition of homotypic binding between PDK1 PH domains, dramatically reducing binding of PDK1 to CARMA1. This data implies that binding to partners such as CARMA1 requires PDK1 PH domain-mediated dimerization, while AKT phosphorylation and PKCθ phosphorylation do not require PH domain-mediated homotypic binding. In addition, we found that blocking of PDK1 PH domain-mediated dimerization does not affect the AKT activation and AKT overexpression did not affected the PDK1 Thr-513-Asp dimerization. This data may indicate that PH domain binding-mediated PDK1 dimer formation is occurred in cells even though this dimer formation is not essential for AKT activation. Thus, in T-cell activation, PKCθ phosphorylation itself is not an indicator of NF-κB activation because PDK1-mediated signaling complex formation is required for activation. Therefore, PTD-PDK1-Thr-513-Asp can specifically regulate functions of PDK1 in TCR-CD28-mediated NF-κB activation through regulation of binding to CARMA1.
In summary, this report describes a detailed mechanism for PDK1-mediated NF-κB activation in T cells in response to co-engagement of TCR and CD28. Particularly, PDK1 homodimerization without phosphorylation at Thr-513 inhibits binding of PDK1 to PKCθ, but Thr-513 phosphorylation-dependent homodimerization is essential for binding of PDK1 to both PKCθ and CARMA1, required for NF-κB activation during TCR-CD28-mediated T-cell activation. In addition, our data showed that PDK1 Thr-513 is a potential target for development of a regulator of PDK1 function in T-cell activation.
Supplementary Material
Acknowledgments
This work was supported by Future-based Technology Development Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (MEST) (2010-0029942) and by Basic Science Research Program through the NRF funded by MEST (2010-0003187).
Abbreviations used in this paper
- AGC
protein kinase A/protein kinase G/protein kinase C
- CBM
CARMA1-BCL10-MALT1
- FRET
fluorescence resonance energy transfer
- ORF
open reading frame
- PDK1
phosphoinositide-dependent kinase 1
- PH
pleckstrin homology
- PI3Ks
phosphoinositide 3-kinases
- PTD
protein transduction domain
- Thr
threonine
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Koyasu S. The role of PI3K in immune cells. Nature immunology. 2003;4:313–319. doi: 10.1038/ni0403-313. [DOI] [PubMed] [Google Scholar]
- 3.Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR. Essential role of PDK1 in regulating cell size and development in mice. The EMBO journal. 2002;21:3728–3738. doi: 10.1093/emboj/cdf387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park SG, Schulze-Luehrman J, Hayden MS, Hashimoto N, Ogawa W, Kasuga M, Ghosh S. The kinase PDK1 integrates T cell antigen receptor and CD28 coreceptor signaling to induce NF-kappaB and activate T cells. Nature immunology. 2009;10:158–166. doi: 10.1038/ni.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- 6.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 7.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 8.Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- 9.Hagenbeek TJ, Naspetti M, Malergue F, Garcon F, Nunes JA, Cleutjens KB, Trapman J, Krimpenfort P, Spits H. The loss of PTEN allows TCR alphabeta lineage thymocytes to bypass IL-7 and Pre-TCR-mediated signaling. The Journal of experimental medicine. 2004;200:883–894. doi: 10.1084/jem.20040495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly AP, Finlay DK, Hinton HJ, Clarke RG, Fiorini E, Radtke F, Cantrell DA. Notch-induced T cell development requires phosphoinositide-dependent kinase 1. The EMBO journal. 2007;26:3441–3450. doi: 10.1038/sj.emboj.7601761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.So L, Fruman DA. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. The Biochemical journal. 2012;442:465–481. doi: 10.1042/BJ20112092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fayard E, Moncayo G, Hemmings BA, Hollander GA. Phosphatidylinositol 3-kinase signaling in thymocytes: the need for stringent control. Science signaling. 2010;3:re5. doi: 10.1126/scisignal.3135re5. [DOI] [PubMed] [Google Scholar]
- 13.Park SG, Mathur R, Long M, Hosh N, Hao L, Hayden MS, Ghosh S. T regulatory cells maintain intestinal homeostasis by suppressing gammadelta T cells. Immunity. 2010;33:791–803. doi: 10.1016/j.immuni.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hara H, Wada T, Bakal C, Kozieradzki I, Suzuki S, Suzuki N, Nghiem M, Griffiths EK, Krawczyk C, Bauer B, D’Acquisto F, Ghosh S, Yeh WC, Baier G, Rottapel R, Penninger JM. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity. 2003;18:763–775. doi: 10.1016/s1074-7613(03)00148-1. [DOI] [PubMed] [Google Scholar]
- 15.Schulze-Luehrmann J, Ghosh S. Antigen-receptor signaling to nuclear factor kappa B. Immunity. 2006;25:701–715. doi: 10.1016/j.immuni.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Hinton HJ, Alessi DR, Cantrell DA. The serine kinase phosphoinositide-dependent kinase 1 (PDK1) regulates T cell development. Nat Immunol. 2004;5:539–545. doi: 10.1038/ni1062. [DOI] [PubMed] [Google Scholar]
- 17.Gao X, Harris TK. Role of the PH domain in regulating in vitro autophosphorylation events required for reconstitution of PDK1 catalytic activity. Bioorganic chemistry. 2006;34:200–223. doi: 10.1016/j.bioorg.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Masters TA, Calleja V, Armoogum DA, Marsh RJ, Applebee CJ, Laguerre M, Bain AJ, Larijani B. Regulation of 3-phosphoinositide-dependent protein kinase 1 activity by homodimerization in live cells. Science signaling. 2010;3:ra78. doi: 10.1126/scisignal.2000738. [DOI] [PubMed] [Google Scholar]
- 19.Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:e17. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayden MS, Ghosh S. NF-kappaB in immunobiology. Cell research. 2011;21:223–244. doi: 10.1038/cr.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-Garcia ME, Ovechkina YL, Rawlings DJ. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity. 2005;23:561–574. doi: 10.1016/j.immuni.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 22.Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, Chen Y, Lin X. Phosphorylation of CARMA1 plays a critical role in T Cell receptor-mediated NF-kappaB activation. Immunity. 2005;23:575–585. doi: 10.1016/j.immuni.2005.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.