

Abstract
Objective
Angiosarcoma is an aggressive malignancy with endothelial differentiation and notoriously poor prognosis despite aggressive therapy. Limited data are available to guide management decisions. To address this limitation, we present a large retrospective analysis of angiosarcoma patients treated at a single institution over a 25-year period.
Methods
To identify factors that impact angiosarcoma outcomes, we reviewed demographic, tumor and treatment characteristics of angiosarcoma patients evaluated at the University of Wisconsin Hospital between 1987 and 2012.
Results
The cohort included 81 patients diagnosed at age 19–90 yo (median 67). Fifty-five (68%) patients presented with localized disease while 26 (32%) presented with metastases. The primary sites were visceral/deep soft tissue (42%), head and neck/cutaneous (37%), breast (16%) and limbs in the setting of Stewart-Treves (5%). The 5-year overall survival (OS) was 40% with a median of 16 months. By univariate analysis, significant adverse predictors of survival included metastases at presentation, visceral/deep soft tissue tumor location, tumor size > 5 cm, tumor necrosis and the absence of surgical excision. A trend toward prolonged survival was observed with radiation therapy and for chemotherapy in patients with metastases. Age, sex, and prior radiation showed no correlation with survival.
Conclusions
Our large single institution series confirms the poor prognosis of angiosarcoma, supports a central role for surgical excision in management and highlights the need for novel therapies particularly in patients who present with metastatic disease.
Keywords: Angiosarcoma, sarcoma, retrospective study
Angiosarcoma is an aggressive sarcoma with differentiation towards blood or lymphatic endothelium. The tumor can arise at any anatomic location including, most commonly, the scalp, the breast and the extremities.1 The disease accounts for approximately 2% of all soft tissue sarcomas2 and the incidence is rising, probably related to increased use of radiotherapy in the treatment of breast and other cancers.3 Known risk factors for developing angiosarcoma include prior radiation, polyvinyl chloride, arsenic and thorium dioxide exposure, chronic lymphedema, familial syndromes and possibly UV exposure, given the greater frequency of scalp angiosarcoma.1,4 Angiosarcomas are prone to locoregional recurrence, nodal and distant metastases, and have notoriously poor prognoses. The reported rates of advanced/metastatic disease at presentation vary from 16 to 44%, and the overall disease-specific survival is reported as approximately 30–40% in contemporary series.5–10
There are no randomized trials to guide clinical decision-making in the management of angiosarcoma, and prospective studies addressing various treatment options are limited. For localized disease, complete, margin-negative resection is considered to be the treatment of choice whenever possible. The advantage of this approach has been demonstrated in a number of retrospective series.7,10,11 However, even this apparently uncontroversial recommendation remains open to debate in some clinical settings. For example, some authors argue that with the use of multimodal local therapy in scalp and face angiosarcomas, R0 resection offers no advantage over gross tumor resection (“debulking”) that may be associated with fewer complications and improve tolerance of adjuvant therapies.12 Due to relatively high rates of local recurrence even after R0 resection, adjuvant radiation is commonly recommended, as it has been associated with superior survival in some series.6,13 When surgical resection is contraindicated or not possible, chemotherapy is often considered with a goal of either palliation or conversion to resectability. Although no definitive standard of care for systemic therapy has been established, paclitaxel and doxorubicin are among the most active agents.14–16 Targeted therapies are being explored.17–19
As angiosarcoma is a rare tumor and a consensus treatment algorithm is lacking, more evidence is needed to better understand the factors that impact outcomes and the value of various therapies. In this retrospective analysis, we reviewed demographic, tumor and treatment characteristics of 81 patients evaluated at the University of Wisconsin Hospital and Clinic. Factors examined included age, sex, extent and site of disease, association with previous radiotherapy, tumor size, histologic characteristics, and treatments rendered.
PATIENTS AND METHODS
The study was approved by the Institutional Review Board. Patients were identified using a surgical pathology database that spanned the years 1987–2012. Angiosarcoma diagnosis was established originally or confirmed by a senior pathologist with specific interest in soft tissue tumors (G.R.H., B.J.L.). Cases received by the pathology department solely for consultation were excluded from the analysis. Patient records were accessed for age at diagnosis, sex, date of last follow up or death, site of disease and tumor size divided as ≤ 5 cm or > 5 cm. Angiosarcomas were deemed radiation associated if they occurred within or adjacent to prior radiation fields. The architectural pattern and necrosis were assessed by a surgical pathologist blinded to outcomes (D.B.) according to a recently published study by Shon et al20 as follows: vasoformative (> 75% of tumor forming vascular channels with identifiable lumina), non-vasoformative (> 75% of tumor demonstrating architecturally solid epithelioid or spindle cell morphology without vascular channels) or mixed. Operative notes, radiation records (total dose, fractionation) and chemotherapy records (agents, doses, duration of treatment) were reviewed. Subsequent oncology notes, imaging reports, surgical pathology reports and social security death index were reviewed to identify events including local failure, disease progression and death.
The 5-year overall survival (OS) comparisons were performed using the Kaplan-Meier method. Statistical significance was calculated by log rank test. Survival curves were generated using GraphPad Prism software (La Jolla, California).
RESULTS
Patient, Tumor and Treatment Characteristics
The demographic, tumor and treatment characteristics of angiosarcoma patients are presented in Table 1. The cohort consisted of 49 females and 32 males, with median age of 67 years at diagnosis (range, 19–90 years). Fifty-five (68%) patients presented with localized disease while 26 (32%) had metastases most commonly to mesentery/bowel (31%), liver (27%), lungs (19%), and bone (19%). The most common primary tumor site was visceral/deep soft tissue (42%), followed by head and neck/cutaneous (37%) and breast (16%). Four (5%) cases of Stewart-Treves syndrome were identified. Visceral/deep sites included liver (N = 9), bone (N = 7), bladder (N = 3), lung (N = 3), intramuscular (N = 3), bowel (N = 2), and one each of the following: spleen, heart, ovary, omentum, seminal vesicle, adrenal, and aorta (N = 1). Fourteen cases (17%) were considered to be radiation associated, with the majority (8/14, 57%) being related to radiation therapy for breast carcinoma. The median latency period for radiation-associated angiosarcoma was 9 years (range, 4 – 52 years). The size of the primary lesion was available in 64 cases, of which 39 (61%) were 5 cm or less. Four patients with otherwise non-metastatic angiosarcoma presented with lymph node involvement. Three developed metastatic disease within 6 months and expired at 4, 5 and 28 months; one is alive and disease-free at 8 months.
Table 1.
Patient, tumor and treatment characteristics
| N (% total) | ||
|---|---|---|
| Patient Characteristics | Age, Median (range, years) | 67 (19–90) |
| Gender | ||
| male | 32 (40%) | |
| female | 49 (60%) | |
| Localized Disease | 55 (68%) | |
| Metastatic Disease | 26 (32%) | |
| Vital Status | ||
| alive | 28 (35%) | |
| deceased | 53 (65%) | |
| Tumor Characteristics | Primary Site | |
| visceral/deep soft tissue | 34 (42%) | |
| head and neck/cutaneous | 30 (37%) | |
| breast | 13 (16%) | |
| Stewart-Treves | 4 (5%) | |
| Radiation Associated | 14 (17%) | |
| breast | 8 (57%) | |
| bladder | 2 (14%) | |
| liver | 1 (7%) | |
| omentum | 1 (7%) | |
| sternum | 1 (7%) | |
| scalp | 1 (7%) | |
| Primary tumor size: | ||
| Total available | 64 | |
| ≤ 5 cm | 39 (61%) | |
| > 5 cm | 25 (39%) | |
| Histologic pattern | ||
| Total available | 66 | |
| vasoformative | 35 (53%) | |
| epithelioid | 15 (23%) | |
| spindle | 3 (5%) | |
| mixed | 13 (20%) | |
| Necrosis | ||
| Total available | 60 | |
| absent | 36 (60%) | |
| present | 24 (40%) | |
| Treatment | Surgical Excision | 55 (68%) |
| Chemotherapy | 32 (40%) | |
| Radiation therapy | 33 (41%) | |
| Total | 81 |
Sixty-six angiosarcoma cases were available for assessment of the architectural pattern. Vasoformative architecture was observed in 35 (53%) cases, including 13 head and neck/cutaneous, 11 breast, 8 visceral/deep soft tissue and 3 extremity tumors in the setting of Stewart-Treves syndrome. Of the 31 (47%) non-vasoformative angiosarcomas, 15 had epithelioid morphology, 3 had spindle cell morphology and 13 cases were mixed. The majority of cases (20/31) in the non-vasoformative group were visceral/soft tissue based. The majority of assessable radiation associated angiosarcomas (9/13) and all three assessable Stewart-Treves syndrome specimens displayed vasoformative architecture. Representative micrographs are shown in Figure 1. Necrosis was present in 24 (40%) cases, all of which were biopsied/resected prior to initiation of radiation or chemotherapy. Necrosis tended to be more common in non-vasoformative angiosarcomas (14/25, 56%) than vasoformative angiosarcomas (10/35, 29%).
FIGURE 1.
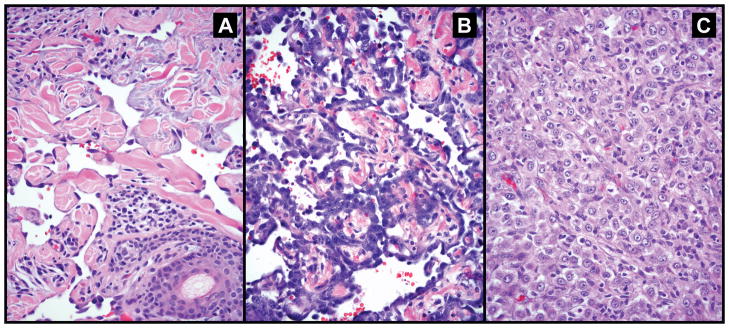
Representative images of the histologic spectrum of angiosarcoma reviewed in this series. (a) vasoformative angiosarcoma showing well-formed irregular anastomosing vascular channels dissecting dermal collagen; the endothelial cells show mild cytologic atypia and lack mitoses; (b) vasoformative angiosarcoma with high grade nuclear features and (c) epithelioid angiosarcoma composed of solid sheet of polygonal cells resembling carcinoma; vascular channels are not seen.
The majority (68%) of patients underwent surgical resection (greater than biopsy) including 44 patients with localized disease and 11 patients with metastatic disease. Nine patients with cutaneous lesions were treated with Mohs micrographic surgery. Radiation therapy was employed in 33 (41%) patients. Radiation therapy was initially employed in the adjuvant setting in 16 patients (48%). In 9 (27%) patients, unresectable local disease prompted definitive radiation therapy. In 7 (21%) patients, radiation was delivered with palliative intent. Only 1 (3%) patient received neoadjuvant radiation therapy. Radiation doses ranged from 8 (single fraction) −80.8 Gy with a median of 56 Gy. Radiation fields standardly covered the involved site with generous margins but did not electively include uninvolved regional lymph nodes. Thirty-two patients (40%) received systemic therapy. The most common indications for systemic therapy were distant metastases (13, 41%) and unresectable local disease (10, 31%). In nine cases (28%), systemic therapy was employed (neo)adjuvantly. Paclitaxel was the most commonly employed agent (N = 21) followed by liposomal doxorubicin (N = 10) and doxorubicin (N = 9).
Patterns of Failure
Of 46 patients with resected, localized disease, 43 were assessable for local control. Sixteen (37%) had documented local failure. Half of these patients experienced distant recurrence either prior to (N = 1), concurrent with (N = 1) or following (N = 6) local recurrence. Local recurrence was detected at a median interval of 6 months after the diagnosis (range, 2–58 months).
Prognostic Factors and Outcomes
Median follow-up time was 13 months (range 0.1 – 264 months). For all patients, the five-year OS was 40% with a median of 16 months (Figure 2), and was not affected by age or sex. The prognostic significance of tumor factors is shown in Figure 3. Metastatic disease at presentation was a critical adverse prognostic factor (median OS of 3 months vs. > 60 months in patients with localized disease). Corresponding 5 year survival rates were 0% and 60%, respectively. The site of the primary lesion significantly influenced survival (median OS of 5 months in patients with visceral/deep soft tissue angiosarcoma vs. > 60 months in patients with non-visceral/deep primaries). Similarly, primary tumor size influenced survival; median OS of patients with tumors ≤ 5 cm and > 5 cm were > 60 months and 10 months, respectively. From a histologic perspective, the presence of necrosis was associated with worse outcomes (median OS of 9 months for tumors with necrosis vs. > 60 months for tumors without necrosis, P = 0.003). Non-vasoformative architectural pattern was associated with inferior outcomes though this did not achieve statistical significance (median OS 33 months for vasoformative vs. 10 months for non-vasoformative tumors, P = 0.32). Similarly, patients with epithelioid angiosarcomas appeared to have an inferior prognosis though this did not achieve statistical significance (median OS 10 months for epithelioid vs. 28 months for non-epithelioid tumors, P = 0.14). Association with prior radiation was not prognostic compared to sporadic angiosarcoma. Although the number of post-radiation angiosarcoma cases is small, we noted significantly better survival in 8 patients with cutaneous tumors of the breast compared to the other 6 radiation-associated angiosarcomas (median OS > 60 months vs. 7 months, P = 0.006).
FIGURE 2.

Five-year overall survival in all patients.
FIGURE 3.

Five-year overall survival in angiosarcoma patients based on (a) localized vs. metastatic disease, (b) visceral/deep soft tissue vs. all other anatomic sites, (c) association with prior radiation, (d) tumor size, (e) vasoformative vs. non-vasoformative architectural pattern and (f) tumor necrosis.
The impact of treatment modalities on angiosarcoma outcomes is shown in Figure 4. Patients undergoing surgery had median OS > 60 months vs. 8 months in those treated non-surgically (P < 0.0001). The positive influence of surgical resection on OS appeared limited to patients initially presenting with localized disease. In 21 patients with cutaneous lesions who underwent Mohs micrographic resection (N = 9) or wide local excision (N = 12), no significant difference in OS was identified between the two surgical techniques (P = 0.89, data not shown). When considering the entire cohort, radiation therapy displayed a trend toward OS benefit; irradiated patients had a median OS of 47 months compared to just 10 months in patients who did not receive radiation therapy (P = 0.067). However, no clear benefit of radiation therapy was observed when patients were segregated by metastatic status at presentation. Furthermore, when only patients presenting with localized disease who underwent surgical resection were considered, radiation therapy was not found to improve OS (P = 0.78, data not shown). The small number of local failures in this subpopulation (N = 16) precluded a statistically meaningful examination of the impact of radiation therapy on local control; median time to local failure exceeded 60 months in both irradiated and unirradiated patients and there was no statistically significant difference between the groups (P = 0.39, data not shown). Similarly, no difference in OS was identified between patients who experienced local failure and those that remained locally controlled (median OS > 60 months for both groups, P = 0.89); this comparison is also hampered by small numbers and a generally favorable prognosis. However, among patients with initially localized disease who underwent oncologic resection, 5 of 16 (31%) irradiated patients failed locally at a median of 10 months (range 6–58 months) while 11 of 27 (41%) unirradiated patients failed locally at a median of 4 months (range 2–41 months). Chemotherapy did not improve outcomes in the entire cohort or in patients presenting with localized disease. However, a trend toward benefit was observed in patients presenting with metastatic disease (median OS of 8 months vs. 1 month without chemotherapy, P = 0.062).
FIGURE 4.

Five-year overall survival in angiosarcoma patients segregated by surgical resection: (a) all patients, (b) localized disease only and (c) metastatic disease only. Five-year overall survival in angiosarcoma patients segregated by radiation therapy: (d) all patients, (e) localized disease only and (f) metastatic disease only. Five-year overall survival in angiosarcoma patients segregated by chemotherapy: (g) all patients, (h) localized disease only and (i) metastatic disease only.
DISCUSSION
Patient and tumor characteristics in our series are generally in agreement with previous studies.5–7,9–11 Angiosarcoma tended to occur in an older patient population, in a variety of anatomic locations, and with a significant percentage of metastatic disease at presentation (32%). In keeping with other reports, the five year OS was 40%. Tumor factors with significant negative impact on outcomes in our series included metastatic disease at presentation, visceral/deep soft tissue location of the tumor, tumor size and the presence of necrosis.
Consistent with other publications, the frequency of radiation-associated angiosarcoma in our series was 17%.10,11 Compared to sporadic angiosarcomas, association with previous radiation did not correlate with survival. However, the median OS of patients with post-radiation angiosarcoma of the breast was significantly better compared to that in other locations and appeared more favorable than in previously reported series by Marchal et al. (15.5 months), Billings et al. (33.5 months), Fury et al. (31 months) or in a recent SEER database review by Mery et al. (35 months).3,7,21,22 Our observations are consistent with those of Brenn et al who suggested more indolent behavior for radiation-induced angiosarcoma of the breast.23 The median interval between radiation and post-radiation angiosarcoma diagnosis in our series was 9 years. One patient presented with cutaneous angiosarcoma four years after initial radiation treatment for prior breast cancer highlighting the need for clinical suspicion even during early radiation therapy follow up.24
Current consensus guidelines from the American Joint Committee on Cancer and College of American Pathologists do not recommend grading of angiosarcoma as conventional grading schemes correlate poorly with prognosis.25 The predictive value of individual histologic parameters such as epithelioid morphology, necrosis, vasoformative architecture and nuclear grade has been examined in the past in a series of post-radiation cutaneous angiosarcomas of the breast, without demonstrating an association with an adverse outcome.22 Subsequently, Deyrup and colleagues, in a series of sporadic cutaneous angiosarcomas, found that epithelioid morphology and necrosis were associated with poorer prognosis.26 More recently, Shon et al. explored more globally the prognostic significance of vasoformative architectural pattern.20 In their series of 98 angiosarcomas from various sites, the authors observed a significant correlation between vasoformative architecture and improved survival. In our series, the vasoformative architectural pattern was associated with a tripling of median OS (33 months compared to 10 months for non-vasoformative architecture). However, this did not achieve statistical significance (P = 0.32). One possible explanation for lack of a statistically significant association is the number of cases available for the assessment of histologic pattern (N = 66). Another potential explanation is a relative overrepresentation of high-grade nuclear features in the vasoformative group of tumors as illustrated in Figure 1B. Furthermore, approximately a third of vasoformative angiosarcomas in our series had necrosis. Thus, in contrast to the study by Shon et al, our data appears to be skewed towards less differentiated tumors, in which case the vasoformative pattern alone may not be prognostic. Tumor necrosis in our series was predictive of worse clinical outcome, which is consistent with the data on cutaneous tumors by Deyrup et al and has also been reported in a previous series of non-selected angiosarcomas from various sites.10 Epithelioid morphology, a feature reported as a negative prognostic factor in several series, was associated with worse survival in our series (median OS 10 months vs. 28 months for non-epithelioid tumors).11 As with vasoformative architecture, however, this did not reach statistical significance (P = 0.14), attributable, at least in part, to the relatively small numbers of epithelioid specimens (N = 15).
Surgery was clearly advantageous when either the entire cohort or patients presenting with localized disease were considered. The utility of Mohs micrographic surgery in the treatment of angiosarcoma has not been examined prospectively, and retrospective studies limited to single cases and small series show variable success.27–29 There is considerable debate regarding the indications for Mohs surgery in soft tissue tumors and particularly angiosarcoma, given the propensity for multifocal growth and notorious difficulty achieving negative margins.13 While our experience is also limited by small numbers, we found no difference in survival between patients who underwent Mohs surgery compared to wide local excision for amenable lesions.
Radiation therapy did show a trend towards benefit in the entire cohort. However, the absence of a survival benefit in patients presenting with localized disease supports a conclusion that the survival benefit of radiation therapy observed in this population reflects, in total or part, the understandable disproportionate use of radiation in patients presenting with (more favorable) localized disease. Small numbers of local failures in the most clinically germane subset – patients presenting with localized disease who underwent oncologic resection – limits the ability to assess the impact of radiation therapy on local control in a statistically meaningful way. Consequently, conclusions on the therapeutic merits of (neo)adjuvant radiation therapy must be offered with considerable circumspection. Nonetheless, the observed absolute reduction in local failure rate (31% vs. 41%) and prolonged time to local failure (median 10 months vs. 4 months) in irradiated patients who presented with localized disease and underwent oncologic resection highlights potential local control benefits of radiation therapy in this rare disease as has been suggested previously and merits rigorous confirmation in larger cohorts.
No angiosarcoma-specific, phase III trial has demonstrated an OS benefit with systemic therapy in this rare disease. However, active agents have been identified. Since the reporting of the ANGIOTAX clinical trial results, the use of paclitaxel in the treatment of angiosarcoma has increased.15 Previous studies have shown increased progression-free survival (PFS) for scalp angiosarcoma treated with paclitaxel as well as activity in metastatic disease.7,14,16,30 In our series, paclitaxel was the most commonly administered chemotherapeutic agent. Moreover, a trend toward an OS benefit was observed in patients with metastases treated with chemotherapy. Again, unfettered conclusions regarding the therapeutic benefits of systemic therapy must be cautiously made. The one month median survival of patients with metastases who did not receive chemotherapy raises the strong possibility that several of these patients had such poor performance status that systemic cytotoxic therapy would not be tolerated. Multi-institutional efforts to prospectively define the role of systemic therapy in the management of this disease are needed.
The impact of biologically targeted agents could not be assessed in this cohort although this is an active area of clinical investigation. Agents targeting the vascular endothelial growth factor (VEGF) pathway have received the greatest attention due to the endothelial differentiation of angiosarcoma. Agulnik and colleagues recently reported results of a phase II trial of bevacizumab monotherapy in patients with unresectable angiosarcoma or epithelioid hemangioendothelioma.17 This VEGF-directed monoclonal antibody resulted in a partial response in 2 of 23 and stable disease in 11 of 23 evaluable angiosarcoma patients. Similarly, the French Sarcoma Group (GSF/GETO) recently reported results of a phase II trial assessing sorafenib in advanced angiosarcoma.18 Sorafenib, a multi-target tyrosine kinase inhibitor with activity against VEGF receptors, was shown to result in a 23% response rate in patients previously treated with cytotoxic chemotherapy. However, no responses were observed in chemotherapy-naïve patients. Clinical trials of VEGF receptor tyrosine kinase inhibitors (sunitinib, sorafenib and pazopanib) in unselected metastatic soft tissue sarcomas are more challenging to interpret with respect to activity in angiosarcoma for several reasons including limited representation of this rare malignancy. However, globally, these studies support modest activity of VEGF antagonism in this disease.31–35
In summary, our series represents a large, contemporary series of angiosarcoma patients from a single institution. Our data confirm the prognostic significance of tumor necrosis and largely support the importance of rigorously assessing angiosarcoma architecture and morphology. This series confirms surgical resection as the main treatment modality when plausible. Although radiation therapy appeared to confer a survival benefit in our patient cohort, further analysis suggests this resulted from a clinically appropriate bias in radiation utilization. Radiation therapy may provide local control benefits but this needs to be confirmed in larger cohorts. Chemotherapy appears to benefit patients with metastatic disease. Collectively, these observations strongly suggest angiosarcoma patients will benefit from multi-disciplinary management by teams with broad expertise in pathologic examination and surgical, radiotherapeutic and systemic management of this rare malignancy. The overall poor outcomes following currently available interventions, particularly for patients presenting with metastatic disease, necessitate an urgent search for novel therapies.
As evidenced by the OS results observed with radiation therapy, this study is constrained by the well-described limitations of retrospective reviews including, most notably, patient and practitioner selection bias. Additionally, the extended timeframe required to identify more than 80 angiosarcoma patients raises the possibility of time-dependent changes in staging and therapy. Nonetheless, large retrospective analyses, like this one, serve a critical need in defining best strategies in pathologic examination and treatment of this rare disease where prospectively defined guidance is largely lacking.
Acknowledgments
FUNDING
This work was supported by the National Institutes of Health [1UL1RR025011].
Footnotes
The authors have declared no conflicts of interest.
References
- 1.Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol. 2010;11:983–991. doi: 10.1016/S1470-2045(10)70023-1. [DOI] [PubMed] [Google Scholar]
- 2.Coindre JM, Terrier P, Guillou L, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer. 2001;91:1914–1926. doi: 10.1002/1097-0142(20010515)91:10<1914::aid-cncr1214>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 3.Mery CM, George S, Bertagnolli MM, Raut CP. Secondary sarcomas after radiotherapy for breast cancer: sustained risk and poor survival. Cancer. 2009;115:4055–4063. doi: 10.1002/cncr.24462. [DOI] [PubMed] [Google Scholar]
- 4.Penel N, Grosjean J, Robin YM, et al. Frequency of certain established risk factors in soft tissue sarcomas in adults: a prospective descriptive study of 658 cases. Sarcoma. 2008;2008:459386. doi: 10.1155/2008/459386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naka N, Ohsawa M, Tomita Y, et al. Angiosarcoma in Japan. A review of 99 cases. Cancer. 1995;75:989–996. doi: 10.1002/1097-0142(19950215)75:4<989::aid-cncr2820750414>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 6.Mark RJ, Poen JC, Tran LM, et al. Angiosarcoma. A report of 67 patients and a review of the literature. Cancer. 1996;77:2400–2406. doi: 10.1002/(SICI)1097-0142(19960601)77:11<2400::AID-CNCR32>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 7.Fury MG, Antonescu CR, Van Zee KJ, et al. A 14-year retrospective review of angiosarcoma: clinical characteristics, prognostic factors, and treatment outcomes with surgery and chemotherapy. Cancer J. 2005;11:241–247. doi: 10.1097/00130404-200505000-00011. [DOI] [PubMed] [Google Scholar]
- 8.McIntosh BC, Narayan D. Head and neck angiosarcomas. J Craniofac Surg. 2005;16:699–703. doi: 10.1097/01.scs.0000159941.70498.b9. [DOI] [PubMed] [Google Scholar]
- 9.Abraham JA, Hornicek FJ, Kaufman AM, et al. Treatment and outcome of 82 patients with angiosarcoma. Ann Surg Oncol. 2007;14:1953–1967. doi: 10.1245/s10434-006-9335-y. [DOI] [PubMed] [Google Scholar]
- 10.Fayette J, Martin E, Piperno-Neumann S, et al. Angiosarcomas, a heterogeneous group of sarcomas with specific behavior depending on primary site: a retrospective study of 161 cases. Ann Oncol. 2007;18:2030–2036. doi: 10.1093/annonc/mdm381. [DOI] [PubMed] [Google Scholar]
- 11.Lahat G, Dhuka AR, Hallevi H, et al. Angiosarcoma: clinical and molecular insights. Ann Surg. 2010;251:1098–1106. doi: 10.1097/SLA.0b013e3181dbb75a. [DOI] [PubMed] [Google Scholar]
- 12.Guadagnolo BA, Zagars GK, Araujo D, et al. Outcomes after definitive treatment for cutaneous angiosarcoma of the face and scalp. Head Neck. 2011;33:661–667. doi: 10.1002/hed.21513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pawlik TM, Paulino AF, McGinn CJ, et al. Cutaneous angiosarcoma of the scalp: a multidisciplinary approach. Cancer. 2003;98:1716–1726. doi: 10.1002/cncr.11667. [DOI] [PubMed] [Google Scholar]
- 14.Fata F, O’Reilly E, Ilson D, et al. Paclitaxel in the treatment of patients with angiosarcoma of the scalp or face. Cancer. 1999;86:2034–2037. [PubMed] [Google Scholar]
- 15.Penel N, Bui BN, Bay JO, et al. Phase II trial of weekly paclitaxel for unresectable angiosarcoma: the ANGIOTAX Study. J Clin Oncol. 2008;26:5269–5274. doi: 10.1200/JCO.2008.17.3146. [DOI] [PubMed] [Google Scholar]
- 16.Italiano A, Cioffi A, Penel N, et al. Comparison of doxorubicin and weekly paclitaxel efficacy in metastatic angiosarcomas. Cancer. 2012;118:3330–3336. doi: 10.1002/cncr.26599. [DOI] [PubMed] [Google Scholar]
- 17.Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2012 Aug 21; doi: 10.1093/annonc/mds237. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 18.Ray-Coquard I, Italiano A, Bompas E, et al. Sorafenib for patients with advanced angiosarcoma: a phase II trial from the French Sarcoma Group (GSF/GETO) Oncologist. 2012;17:260–266. doi: 10.1634/theoncologist.2011-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasenstein JR, Kasmerchak K, Buehler D, et al. Efficacy of Tie2 receptor antagonism in angiosarcoma. Neoplasia. 2012;14:131–140. doi: 10.1593/neo.111770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shon W, Jenkins SM, Ross DT, et al. Angiosarcoma: a study of 98 cases with immunohistochemical evaluation of TLE3, a recently described marker of potential taxane responsiveness. J Cutan Pathol. 2011;38:961–966. doi: 10.1111/j.1600-0560.2011.01790.x. [DOI] [PubMed] [Google Scholar]
- 21.Marchal C, Weber B, de Lafontan B, et al. Nine breast angiosarcomas after conservative treatment for breast carcinoma: a survey from French comprehensive Cancer Centers. Int J Radiat Oncol Biol Phys. 1999;44:113–119. doi: 10.1016/s0360-3016(98)00537-9. [DOI] [PubMed] [Google Scholar]
- 22.Billings SD, McKenney JK, Folpe AL, et al. Cutaneous angiosarcoma following breast-conserving surgery and radiation: an analysis of 27 cases. Am J Surg Pathol. 2004;28:781–788. doi: 10.1097/01.pas.0000126055.33916.0b. [DOI] [PubMed] [Google Scholar]
- 23.Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983–996. [PubMed] [Google Scholar]
- 24.Weaver J, Billings SD. Postradiation cutaneous vascular tumors of the breast: a review. Semin Diagn Pathol. 2009;26:141–149. doi: 10.1053/j.semdp.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Rubin BP, Cooper K, Fletcher CD, et al. Protocol for the examination of specimens from patients with tumors of soft tissue. Arch Pathol Lab Med. 2010;134:e31–39. doi: 10.5858/134.4.e31. [DOI] [PubMed] [Google Scholar]
- 26.Deyrup AT, McKenney JK, Tighiouart M, et al. Sporadic cutaneous angiosarcomas: a proposal for risk stratification based on 69 cases. Am J Surg Pathol. 2008;32:72–77. doi: 10.1097/PAS.0b013e3180f633a3. [DOI] [PubMed] [Google Scholar]
- 27.Goldberg DJ, Kim YA. Angiosarcoma of the scalp treated with Mohs micrographic surgery. J Dermatol Surg Oncol. 1993;19:156–158. doi: 10.1111/j.1524-4725.1993.tb03446.x. [DOI] [PubMed] [Google Scholar]
- 28.Muscarella VA. Angiosarcoma treated by Mohs micrographic surgery. J Dermatol Surg Oncol. 1993;19:1132–1133. doi: 10.1111/j.1524-4725.1993.tb02481.x. [DOI] [PubMed] [Google Scholar]
- 29.Bullen R, Larson PO, Landeck AE, et al. Angiosarcoma of the head and neck managed by a combination of multiple biopsies to determine tumor margin and radiation therapy. Report of three cases and review of the literature. Dermatol Surg. 1998;24:1105–1110. doi: 10.1111/j.1524-4725.1998.tb04083.x. [DOI] [PubMed] [Google Scholar]
- 30.Hirata T, Yonemori K, Ando M, et al. Efficacy of taxane regimens in patients with metastatic angiosarcoma. Eur J Dermatol. 2011;21:539–545. doi: 10.1684/ejd.2011.1403. [DOI] [PubMed] [Google Scholar]
- 31.van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379:1879–1886. doi: 10.1016/S0140-6736(12)60651-5. [DOI] [PubMed] [Google Scholar]
- 32.Sleijfer S, Ray-Coquard I, Papai Z, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organization for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043) J Clin Oncol. 2009;27:3126–3132. doi: 10.1200/JCO.2008.21.3223. [DOI] [PubMed] [Google Scholar]
- 33.von Mehren M, Rankin C, Goldblum JR, et al. Phase 2 Southwest Oncology Group-directed intergroup trial (S0505) of sorafenib in advanced soft tissue sarcomas. Cancer. 2012;118:770–776. doi: 10.1002/cncr.26334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maki RG, D’Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27:3133–3140. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.George S, Merriam P, Maki RG, et al. Multicenter phase II trial of sunitinib in the treatment of nongastrointestinal stromal tumor sarcomas. J Clin Oncol. 2009;27:3154–3160. doi: 10.1200/JCO.2008.20.9890. [DOI] [PMC free article] [PubMed] [Google Scholar]