

Abstract
The gas-phase structures and fragmentation pathways of the singly protonated peptide arginylglycylaspartic acid (RGD) are investigated by means of collision-induced-dissociation (CID) and detailed molecular mechanics and density functional theory (DFT) calculations. It is demonstrated that despite the ionizing proton being strongly sequestered at the guanidine group, protonated RGD can easily be fragmented on charge directed fragmentation pathways. This is due to facile mobilization of the C-terminal or aspartic acid COOH protons thereby generating salt-bridge (SB) stabilized structures. These SB intermediates can directly fragment to generate b2 ions or facilely rearrange to form anhydrides from which both b2 and b2+H2O fragments can be formed. The salt-bridge stabilized and anhydride transition structures (TSs) necessary to form b2 and b2+H2O are much lower in energy than their traditional charge solvated counterparts. These mechanisms provide compelling evidence of the role of SB and anhydride structures in protonated peptide fragmentation which complements and supports our recent findings for tryptic systems (Bythell, B. J.; Suhai, S.; Somogyi, A.; Paizs, B. J. Am. Chem. Soc., 2009, 131, 14057–14065.). In addition to these findings we also report on the mechanisms for the formation of the b1 ion, neutral loss (H2O, NH3, guanidine) fragment ions and the d3 ion.
Keywords: Salt-bridge, proton mobility, anhydride, peptide fragmentation pathways, MALDI, modeling, gas-phase rearrangements
Introduction
Protein identification in the field of proteomics is mainly based on tandem mass spectrometry (MS/MS) of proteolytic peptides.1-3 Most proteomic digestion protocols use trypsin, an enzyme that selectively cleaves after Arg, R and Lys, K (except when followed by Pro, P). Consequently, the vast majority of peptides analyzed in large-scale protein identification studies contain at least one basic amino acid residue at their C-termini. Sequences containing RR or KR can also produce peptides with R at the N-terminus via missed tryptic cleavages or when the next residue is P. Alternatively the use of nontryptic enzymatic digestion of proteins can also lead to peptides with N-terminal R. The peptides generated in these digestions are then usually ionized (by protonation) using either electrospray ionization4 (ESI) or matrix-assisted-laser-desorption/ionization (MALDI)5. ESI predominantly produces doubly or triply charged ions for average-sized peptides, whereas peptide ions produced by MALDI bear only a single ionizing proton (and therefore charge). This difference has serious consequences for the structure and collision-induced-dissociation (CID) chemistry6 of the investigated peptide ions; the singly and multiply protonated forms of the same peptide often produce completely different fragmentation patterns even when using the same experimental conditions (excitation, time-scale, etc.).
This phenomenon has been investigated and partially understood in the framework of the mobile proton model7 of peptide fragmentation. This model states that the energetically most favorable forms of protonated peptides rarely undergo fragmentation reactions directly6,7, but instead require structural changes induced by ion excitation/ activation to facilitate fragmentation. The initial effect of excitation (usually by low-energy CID with inert gas(es)) is to transfer an ionizing proton away from an unreactive, but energetically favorable site of higher gas-phase basicity (Arg, Lys, or His side chain or the N-terminal amino group) to populate the energetically less favored but reactive backbone amide nitrogen sites6. This process in some cases occurs directly, but more often occurs via several intermediate minima. Systems that exhibit facile proton mobilization to the various backbone amide bonds usually result in MS/MS spectra rich in sequence informative b, y, and a ions.8 Conversely, strong sequestration of the ionizing proton(s) by basic amino acid side chains can lead to information-poor MS/MS spectra due to the limited ability to populate amide nitrogen sites.
This suggests that peptides lacking amino acids with side chains of high basicity fragment following minimal activation in either their singly or multiply protonated forms while this is not necessarily the case for peptides that contain Lys or Arg. For example, a peptide having a single Arg residue and no Lys can be difficult to fragment in its singly protonated form while the corresponding doubly charged ion dissociates easily to produce MS/MS spectrum rich in sequence information. This behavior can be explained by assuming that the Arg side chain is likely to remain protonated upon excitation irrespective of the protonation state of the peptide. For the singly charged form of a peptide there is initially no additional mobilized proton available to induce backbone fragmentation. On the other hand for the doubly protonated form, a second ionizing proton is available for facile mobilization to the amide nitrogens. By quantifying the energy needed to induce fragmentation, Wysocki and coworkers7a-c,e showed that proton sequestration by arginine residues is stronger than by lysine residues in line with the basicities of these amino acids.9 In some cases, proton sequestration by arginine can be so strong that charge-remote fragmentation pathways (where the ionizing proton behaves as a spectator and does not take part directly in the dissociation) become more active than charge-directed dissociation channels. A common example being aspartic acid and/or glutamic acid containing peptide ions7b,10 with sequestered ionizing protons. These peptides often produce MS/MS spectra dominated by cleavages C-terminal to these residues due to residue-specific charge-remote chemistries. Such spectra can be difficult to assign because of the limited number of sequence-informative peaks.
The preceding mechanistic considerations suggest that protonated peptide ions lacking easily mobilizable protons are more difficult to fragment and therefore to sequence than peptide ions with mobile protons. This limitation applies more for MALDI-generated ions of tryptic peptides11-15 because of the number of ionizing protons than for those formed by ESI. However, even multiply charged ESI-formed ions of tryptic peptides can be difficult to fragment if enzymatic digestion is incomplete, leaving additional arginine and lysine residues in non-C-terminal positions.
The dissociation chemistry of MALDI-generated peptide ions has been experimentally investigated by numerous groups. Qin and Chait13 studied the fragmentation behavior of more than 200 peptides in a MALDI ion trap (IT) instrument and found that peptide ions with m/z less than 1500 can be effectively fragmented independent of their amino acid composition. The same holds for peptides containing Lys but no Arg up to m/z 3000. On the other hand, Arg containing peptides with m/z greater than 1500 underwent highly preferential fragmentation at the C-termini of Asp and Glu. The fragmentation of peptides containing Arg but no Asp or Glu was much harder to characterize, but these peptides fragmented relatively easily when the positions of the Arg residues were close to the peptide termini.13 In other work, a reference set of MALDI-TOF/TOF spectra of peptides derived from known proteins has been made available to aid new algorithmic approaches to studying the fragmentation behavior of MALDI generated ions.16 In addition, a recent statistical study17 highlighted peptide fragmentation under these conditions. The abundances of y, b, a, immonium, internal ions and their satellite ions generated by small molecule losses were evaluated for a large set of validated spectra. Whilst significant portions of the total ion current could be assigned to one of the above fragment classes, the average unassigned fraction was greater than 20%.
In a recent paper18 we described the proton-driven amide bond cleavage pathways necessary to produce b and y ions from the model singly protonated tryptic peptides, [GnR+H]+ (n = 2 - 4). We demonstrated that three distinct types of proton mobilization pathways can potentially occur: mobilization of Arg side chain guanidinium, amidic, or carboxylic protons. Our calculations also indicated that mobilizing the C-terminal carboxylic acid proton (forming salt-bridge intermediates) leads to the lowest energy transition structures on the amide bond cleavage pathways via two possible mechanisms. This enabled a qualitative rationalization of the experimentally observed amide bond cleavage tendencies which was impossible without considering this means of proton mobilization.
In the present article we investigate the gas-phase dissociation chemistry of the protonated arginylglycylaspartic acid ion, [RGD+H]+, a model system for singly charged protonated peptides containing Arg at the N-terminus. The selection of [RGD+H]+ as a model peptide has several advantages: (1) [RGD+H]+ dissociates to produce many fragments including both backbone cleavages and small molecule (ammonia and water) losses, (2) these fragment types were also observed for many larger peptides containing this sequence (and similar sequences) within the peptide, (3) the mechanisms of formation of some of these fragments have been debated in the MS community for over a decade, (4) a recent IRMPD study19 provided us with useful information on the gas-phase structure of [RGD+H]+, (5) two sources of COOH protons can be investigated, (6) the results of this work can be compared to that of other recent work on similar peptides18,20 and (7) lastly the size of the peptide allows detailed quantum chemical calculations on the fragmentation channels to be performed.
Experimental Section and Computations
All experiments were carried out at Oregon State University. Calculations were performed using the computer clusters of Oregon State University and the German Cancer Research Center. Both the experimental and theoretical techniques are briefly described below.
Mass Spectrometry
Experiments were conducted on an Applied Biosystems 4700 Proteomics Analyzer MALDI-TOF/TOF (Framingham, MA), using ESI on a Finnigan MAT LCQ (San Jose, CA) ion trap (IT) instrument and a 4000 Q Trap LC/MS/MS (Applied Biosystems/MDS Sciex Instruments) triple quadrupole (QQQ) instrument. All chemicals were purchased from Sigma-Aldrich (Milwaukee, WI) and used without further purification. The MALDI matrix was prepared by dissolving α-cyano-4-hydroxycinnamic acid in acetonitrile/water/triflouroacetic acid/monoammonium phosphate (6 mg/ml) 47/47/0.1/6 (v:v) solution at a concentration of 2 mg/mL. The RGD peptide was dissolved in acetonitrile/water/triflouroacetic acid 50/50/0.1 (v:v) solution at a concentration of 100 μg/mL. The sample solutions were then prepared by mixing the matrix/peptide solutions in a 1 to 1 ratio. 0.4 μl of sample solution was applied to each spot on a 196-well target plate and allowed to air dry prior to introduction into the mass spectrometer. The MS/MS spectra consisted of 5000 laser shots per well, twelve replicate wells, with laser fluence constant and at a level low enough to prevent signal saturation. Air was used as the collision gas at 2.0 × 10−8 (“no gas”) & 7.1 × 10−7 mTorr. The [RGD+H]+ ions formed in the MALDI process were selected (200 FWHM m/z) then decelerated to 1keV (laboratory frame) for metastable ion (MI) decomposition or CID.
Samples for the IT experiments were prepared by dissolving RGD in acetonitrile/water/acetic acid 30/70/0.1 (v:v) to form a 2×10−5 mol L−1 solution, which was infused into the ESI source at a rate of 10 μL min−1. The entrance to the sampling capillary was set at −4kV and N2 served as the nebulizing and drying gas (170 °C). CID of [RGD+H]+, was performed by ejecting all ions except [RGD+H]+, and then exciting the latter to fragment in the presence of He buffer gas (10−3 Torr) using a radiofrequency field (0.78 Vp-p)21 under automated gain control. The reproducibility of the relative abundances from multiple, repeated scans was better than ±10%. Additional scans were performed where the excitation level was incrementally increased. These scans began from well below the threshold of product ion formation and continued until all product ions had been observed22. The order of ion appearance energy was thus estimated approximately. An analogous experiment was conducted on the triple quadrupole (QQQ) instrument where the spectrum was recorded as a function of collision energy (with nitrogen as the collision gas). This began well below the precursor fragmentation threshold and incrementally increased providing a fragmentation profile as a function of laboratory collision energy. This offers complementary information to the ion trap experiment.
Calculations
A conformational search engine6,23 devised specifically to deal with protonated peptides was used to scan the potential energy surface (PES) of [RGD+H]+. These calculations began with molecular dynamics (MD) simulations using simulated annealing on various forms of [RGD+H]+ using the Discover program (Biosym Technologies, San Diego, USA), in conjunction with the AMBER force field24 modified in house to allow amide nitrogen and oxygen protonated species. During the MD simulations, structures were regularly saved for further refinement by full geometry-optimization using the same force field. In the next stage of the process, these structures were analyzed by our own conformer-family search program. This program is able to group optimized structures into families based on the similarity of the most important characteristic torsion angles. The most stable species in these families were then fully optimized at the HF/3-21G, B3LYP/6-31G(d), and the B3LYP/6-31+G(d,p) levels. Recent studies have shown that this model chemistry provides good predictions of reaction barrier heights25. The conformer families were regenerated at each level and only structurally nondegenerate conformers are recomputed at the next level to prevent wasting computer time (i.e. only one of N identical structures is recomputed at the next level).
Having scanned the PES, transition structures (TSs) corresponding to the various fragmentation pathways of [RGD+H]+ were then sought. These were calculated at the B3LYP/6-31G(d) and B3LYP/6-31+G(d,p) levels of theory. The resulting TSs were checked using intrinsic reaction coordinate (IRC) calculations to unambiguously define which minima were connected by the TS investigated. Post-reaction complexes and proton-bound dimers were fully optimized at the B3LYP/6-31G(d) and B3LYP/6-31+G(d,p) levels of theory in a manner similar to that used for the various RGD protonation sites and transition structures. Relative energies were calculated by using the B3LYP/6-31+G(d,p) total energies and zero-point energy corrections (ZPE) determined at the B3LYP/6-31G(d) level. Standard enthapies, entropies and free energies were calculated using the rigid-rotor harmonic oscillator (RRHO) approximation. In absolute terms these are likely to be exaggerated due to the presence of too many low-frequency modes; however relative enthapies, entropies and free energies are likely to be much more accurate due to cancellation of errors. Consequently, only relative terms are reported here. It should also be explicitly noted that the entropy values described herein refer to a situation of full thermodynamic equilibrium which is not necessarily the situation in our experiment. It is not our intention to claim this as being the case, merely to illustrate that such “entropic” considerations do not invalidate the conclusions reached in this manuscript. The Gaussian26 suite of programs was used for all ab initio and DFT calculations.
Results and Discussion
(1) Tandem mass spectra (MS/MS) of protonated RGD
The MALDI-TOF/TOF, ESI-IT, and ESI-QQQ (25 eV collision energy) product ion spectra of protonated RGD are summarized in Table 1. These experiments yielded similar N-terminal fragment ions but with large differences in fragment ion abundance. All spectra contain the sequence informative b1, b2 and b2+H2O fragments with significant abundances (the fragmentation pathways of [RGD+H]+are summarized in Figure 1a). The ammonia and guanidine loss fragments27 are present in all spectra too.
Table 1.
Relative abundance in % of the [M+H-NH3]+ “base” (12C) peak area, using peak areas of [RGD+H]+ fragment ions. A typical collision energy of 25 eV is described for the triple quadrupole instrument. For the immonium-type ions the m/z is listed, followed by the relative % abundance in parentheses.
| MI MALDI-TOF/TOF | CID MALDI-TOF/TOF | ESI IT MS2 | ESI QQQ MS2 | |
|---|---|---|---|---|
| [M+H-NH3]+ | 100 | 100 | 100 | 100 |
| b2 | 45 | 74 | 58 | 32 |
| b2 + H2O | 41 | 76 | 28 | 38 |
| b1 | 34 | 65 | 20 | 35 |
| [M+H-59]+ | 24 | 51 | 6 | 16 |
| d3 | − | 44 | 3 | 2 |
| [M+H-H2O]+/ b3 | - | - | 25 | 10 |
| b2 - NH3 | - | - | 17 | 15 |
| Immonium ions | - | 70(476%), 87(176%), 112(81%) |
− | 70(6%), 87(1%), 112(15%) |
Figure 1.

(a) Summary of the fragmentation pathways of [RGD+H]+. (b) Approximate order of appearance-energies of the primary fragmentation channels of [RGD+H]+ from the ion trap experiment. (c) Approximate order of appearance-energies of the primary fragmentation channels of [RGD+H]+ from the triple quadrupole experiment (see Figure S1 (Supporting Information)). (d) Calculated threshold energies (kcal mol−1) and activation entropies at 298K (cal K−1 mol−1) of the pathways forming the major fragment ions of [RGD+H]+.
The CID MALDI-TOF/TOF experiment appears to impart more energy than the IT, QQQ or MI experiment. This is characterized by the shift in the product spectrum to include: large immonium ion peaks, increased overall fragment ion current (relative to the MI experiment, values not shown), and altered relative intensities of the fragment ions (note that the absence of immonium ions in the IT spectra does not necessarily mean that they aren't formed as this instrument discriminates against lower m/z ions). This finding is in keeping with prior work28. The nominally ‘high energy’ d3 ion is observed in the MALDI CID experiment, but not the MALDI MI experiment. Water loss and b2-NH3 peaks are observed in the IT and QQQ experiments, but not in the MALDI spectra. Somewhat surprisingly, a small d3 peak was observed in the IT and QQQ spectra. The ion trap assignment was supported by MS3 of this peak which led to b2, a2 and other N-terminal peaks. The d3 peak was also the major product from MS3 of the water loss peak (data not shown) indicating a mechanism involving consecutive losses of small neutrals must exist (Section 5). This finding is anecdotally supported by the QQQ data (Figure S1, Supporting Information) where an increase in the relative contribution of the d3 ion occurred concurrently with a decrease in the b3 ion relative abundance as the collision energy was increased.
The approximate order of appearance-energies (Figure 1(b)) for the primary fragmentation channels was estimated with the IT instrument by incrementally increasing the parent excitation level (collision energy).22 These scans indicated that the differences in appearance energy were small but often discernable. The scans showed that the b2 ion peak appears at the lowest excitation level and is immediately a large peak. The water and ammonia losses are next, and are not initially large peaks; however the ammonia loss peak becomes much larger as the excitation level is increased. Next are the b1 and b2+H2O ions with the b1 peak larger initially. Finally the 59 loss (guanidine), b2-NH3 and the d3 ion peaks are observed. In these experiments, the relatively long time-scale (~10 ms) of the IT mean kinetic shifts are small29, so the order of the appearance energies is likely to reflect the order of the true threshold energies. In contrast to these findings, the QQQ experiment (Figure S1 (Supporting Information), Figure 1(c)) was unable to distinguish between the onset of the first five peak types (b2, b3/[M+H-H2O]+, [M+H-NH3]+, b1, b2+H2O). This additional data does however clearly support the IT finding that the peaks appear in a very narrow excitation range and the sharp increase in the ammonia loss product with increasing excitation being applied. It should be noted here that these experiments provide only a crude approximation to the quantitative order of appearance energies. Accurate determination of these values requires sophisticated experiments like threshold CID studies in guided ion beam instruments.30 On the other hand, it should be noted that such specialized instrumentation and painstakingly controlled fragmentation conditions are quite different to those practically used in peptide sequencing (unlike those used in this work). However, even the approximate experiments performed in the present work are useful to help judge our computational findings.
(2) Structure and proton mobility of singly protonated RGD
The energetically most favored Arg-containing [M+H]+ structures feature a protonated Arg side chain. Conventionally two major protonation forms of these species are invoked, charge-solvation (CS) and salt-bridge (SB) stabilized structures. In a CS structure the two termini are neutral, whereas in a SB structure the C-terminus or an acidic side chain is deprotonated and another (backbone or amino acid side chain) site is protonated. The relative stability of the CS and SB forms depends strongly on the sequence and size of the peptide. For example, BIRD experiments31 and calculations32 indicate that singly protonated bradykinin forms a SB in the gas phase. The majority of small Arg-containing protonated peptides are thought to have CS structures as the most stable form. A recent IRMPD study indicates that this is the case for protonated Gly-Arg, Val-Arg, Pro-Arg, Lys-Arg, and His-Arg while protonated Arg-Arg has the salt-bridge structure in the gas-phase.33
A selection of the [RGD+H]+ structure families considered in the present paper are depicted in Chart 1 (all energies listed are relative, described using the sum of the electronic energies and zero-point energy contributions. Additionally, the Gibbs free energies and entropies of activation (at 298K) are provided in Table 2 for purposes of comparison). These involve both charge-solvated and salt-bridge structures, the latter with either C-terminal or Asp side chain carboxylate groups. A recent IRMPD and theoretical study19 demonstrated that protonated RGD has a CS structure (structure I in Chart 1, Figure S2) in the gas phase. The energetically most favored SB structure calculated is protonated on the glycine amide oxygen with a deprotonated C-terminus (XI, 8.3 kcal mol−1 relative energy, Chart 1). The energetically most favored SB structure with a deprotonated Asp side chain that features backbone protonation at the N-terminus is less favored at 12.6 cal mol−1 relative energy (structure IV).
Chart 1.

Various structures of protonated RGD: (I) charge-solvated (CS) structure; (II) CS structure with neutral guanidine and protonated N-terminus; (III) CS structure with neutral guanidine and Nprotonated Gly-Asp amide bond; (IV) salt-bridge (SB) structure with deprotonated Asp side chain and protonated N-terminal amino group; (V) SB structure with deprotonated Asp side chain and Nprotonated Gly-Asp amide bond; (VI) SB structure with deprotonated Asp side chain and O-protonated Arg-Gly amide bond; (VII) SB structure with deprotonated Asp side chain and O-protonated Gly-Asp amide bond; (VIII) SB structure with deprotonated C-terminus and protonated N-terminal amino group; (IX) SB structure with deprotonated C-terminus and N-protonated Gly-Asp amide bond; (X) SB structure with deprotonated C-terminus and O-protonated Arg-Gly amide bond; (XI) SB structure with deprotonated C-terminus and O-protonated Gly-Asp amide bond. The relative energies are presented for clarity.
Table 2.
Relative (kcal mol−1) and total (Hartree) energies of selected protonated forms of [RGD + H]+ and the TSs of the various fragmentation pathways. Zero-point energy corrected total energies, Etot+ ZPE are reported at the B3LYP/6-31G+(d,p) theoretical level. The relative energies, Erel (corrected for zero-point energy calculated from B3LYP/6-31G(d) vibrational frequencies) were determined with respect to the global minimum on the PES of [RGD + H]+. Relative Gibbs free energies (ΔG298) at 298K are in kcal/mol, relative entropies (ΔS298) are in cal/mol K.
| Etotal / H | Etotal + ZPE/ H | ΔEel+ ZPE/ kcal mol−1 |
ΔG298/ kcal mol−1 |
ΔS298/ cal K−1 mol−1 |
|
|---|---|---|---|---|---|
| I | −1250.988180 | −1250.592653 | 0.0 | 0.0 | 0.0 |
| II | −1250.966723 | −1250.572800 | 12.5 | 9.6 | 11.4 |
| III | −1250.921509 | −1250.528441 | 40.3 | 39.9 | 0.9 |
| IV | −1250.966245 | −1250.572540 | 12.6 | 11.7 | 3.7 |
| V | −1250.947235 | −1250.555101 | 23.6 | 21.3 | 10.1 |
| VI | −1250.959349 | −1250.565912 | 16.8 | 15.7 | 4.2 |
| VII | −1250.964832 | −1250.571924 | 13.0 | 11.4 | 6.6 |
| VIII | −1250.964982 | −1250.570174 | 14.1 | 13.9 | 0.6 |
| IX | −1250.958198 | −1250.564529 | 17.7 | 16.7 | 4.2 |
| X | −1250.957930 | −1250.565262 | 17.2 | 16.3 | 3.9 |
| XI | −1250.972419 | −1250.579452 | 8.3 | 7.0 | 5.4 |
| XII | −1250.905148 | −1250.514040 | 49.3 | 48.2 | 4.5 |
| XIII | −1250.920132 | −1250.528495 | 40.3 | 39.3 | 3.9 |
| XVI | −1250.918858 | −1250.526632 | 41.4 | 41.8 | −1.5 |
| XVII | −1250.933179 | −1250.539547 | 33.3 | 32.2 | 4.3 |
| XVIII | −1250.927273 | −1250.534842 | 36.3 | 36.3 | 0.2 |
| XIX | −1250.921550 | −1250.529957 | 39.3 | 39.0 | 2.1 |
| XX | −1250.916450 | −1250.523431 | 43.4 | 42.7 | 4.3 |
| XXI | −1250.909141 | −1250.514981 | 48.7 | 48.3 | 1.0 |
| XXII | −1250.940593 | −1250.546724 | 28.8 | 28.6 | 0.5 |
| XXIII | −1250.911452 | −1250.523030 | 43.7 | 42.0 | 9.7 |
| XXIV | −1250.917386 | −1250.524725 | 42.6 | 40.7 | 7.1 |
| XXV | −1250.914905 | −1250.525000 | 42.5 | 41.0 | 8.0 |
| XXVI | −1250.918862 | −1250.527732 | 40.7 | 40.1 | 2.1 |
| XXVII | −1250.899163 | −1250.512393 | 50.4 | 47.7 | 13.9 |
| XXVIII | −1250.913952 | −1250.521069 | 44.9 | 42.8 | 8.3 |
The charge-solvated global minimum structure I, conventionally undergoes two major types of proton transfer reactions upon excitation. The first of these involves mobilization of a guanidinium (Arg side chain) proton leading to a very reactive (good nucleophile, and strong proton withdrawing group) neutral guanidine moiety. One of the energetically most favored such structures features backbone protonation at the N-terminal amino group (structure II, Chart 1) at 12.5 kcal mol−1 relative energy. Alternatively, either the N-terminal or the Asp side chain COOH proton can be mobilized to form the corresponding salt-bridge structures shown in Chart 1. Interconversion between the CS and SB structures occurs via proton transfer reactions. Our recent calculations on the [GnR+H]+ (n = 2 − 4) peptide ions18 indicate that the transition structures for the CS → SB proton transfer reactions are very similar to the corresponding SB structures in terms of geometry and relative energy. Consequently the CS → SB transition does not involve crossing highly energically unfavorable (and therefore potentially rate-limiting) barriers and thus that the SB forms can be populated via low-energy proton transfers.
Several studies have demonstrated that the amide nitrogen protonated species are of critical importance for cleaving amide bonds.6,7,18 The relative energies of the SB structures with G-D amide nitrogen protonation involving C-terminal acid and aspartic acid proton mobilization are comparatively low at 17.7 and 23.6 kcal mol−1, (IX and V) respectively, while the corresponding G-D amide N protonated CS species III is much less favorable at 40.3 kcal mol−1. Additionally, protonation of the R-G nitrogen was found to be highly unstable (see section 3e). All these findings are consistent with our recent data on the tryptic [GnR+H]+ (n = 2 − 4) ions.18
(3) Formation of b2 ions from protonated RGD
Most peptides are sequenced utilizing information on their b and y ions which are predominantly formed on charge-directed fragmentation pathways.6 The structure of b fragment ions has been debated in the literature for some time. The current consensus is that most small b ions are terminated by a five-membered oxazolone34 ring at the C-terminus. The oxazolone isomer of larger bn ions (n ≥ 4) can rearrange by head-to-tail cyclization to form macrocyclic peptide structures.23c,e,35 Alternatively, nearby side chain nucleophiles36 like the guanidine group of Arg can initiate formation of b fragments. For [RGD+H]+ one has to consider the three potential b2 ion isomers shown in Chart 2 and Figure S3 (Supporting Information). Isomer A is the classical oxazolone structure while isomers B and C feature a 9 or 11 membered lactam ring, respectively, formed by Arg side chain initiated amide bond cleavage. Our computations suggest that the energetically most favored isomer is C, the larger lactam ring followed by the classical oxazolone structure at 3.6 kcal mol−1 relative energy. The smaller lactam ring is much less favored energetically than the classical oxazolone structure due to the strained 9-member ring conformation, so will not be discussed in detail here.
Chart 2.
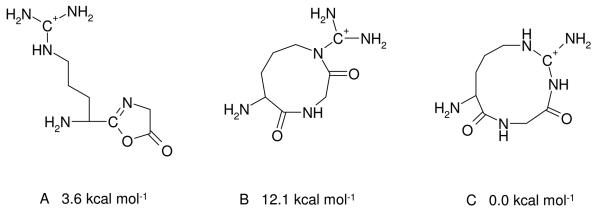
The potential oxazolone (structure A) and the 9-membered (structure B) and 11-membered (structure C) lactam isomers of the b2 ion of RGD. Relative energies of these structures are displayed for clarity.
The oxazolone isomer of the b2 ion can, in principle, be formed via three major categories18 of fragmentation channels; those involving (1) neutral guanidine (classical b2-y1 pathway, section 3a), (2) SB stabilized (sections 3b and 3c), or (3) imine enol structures (section 3d). These reactions involve mobilization of a guanidium, a carboxylic acid, or an amidic proton, respectively. The lactam isomer of the b2 ion can be formed from CS structures via ring formation involving the Arg side chain (section 3e). In the following the related dissociation chemistries will be discussed briefly.
(a) Formation of oxazolone isomer b2 ions on the classical b2-y1 pathway
The classical b2-y1 pathway6,18,23,34 which includes neutral guanidine intermediates (Scheme 1) is initiated by mobilization of one of the protons of the charged guanidinium group to the N-terminus (II, Erel at 12.5 kcal mol−1) followed by proton transfer to the G-D amide nitrogen (III, Erel at 40.3 kcal mol−1). Subsequent cleavage of the protonated G-D amide bond leads to formation of the oxazolone isomer b2 ion (Scheme 1, XII, Figure S4, Table 2) at 49.3 kcal mol−1 threshold energy. On the product-wing of the corresponding TS one finds a post-reaction proton-bound dimer (PBD) of 2-(amino(guanidininobutyl))-5-oxazolone and Asp which can undergo numerous inter-molecular proton transfers under low-energy CID conditions. The dissociation kinetics of such PBDs are mainly determined by the proton affinities of the corresponding monomers: for the classical, charge solvated b2-y1 pathway of [RGD+H]+ the oxazolone fragment will predominantly capture the ionizing proton due to the high proton affinity (PA) of the side chain guanidine group.
Scheme 1.
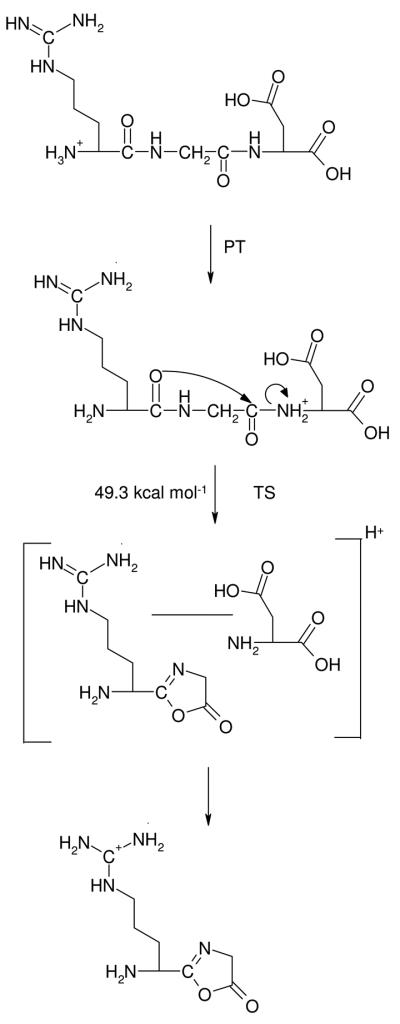
Classical b2-y1 pathway including intermediates with neutral R side chain.
(b) Formation of oxazolone isomer b2 ions via SB intermediates: the b2-y1-SB pathway
The salt-bridge stabilized b2-y1 pathway (b2-y1-SB, Scheme 2) of protonated RGD is initiated by the CS → SB transition which forms SB structures with a proton that has been mobilized to the peptide backbone. Both amide oxygen and nitrogen protonated SB structures can be formed (vide supra), the latter family (IX) are critical intermediates on the amide bond cleavage pathways (Scheme 2). Rotation of the first carbonyl group structure IX produces the reactive configuration from which cleavage of the protonated C-terminal amide bond and simultaneous formation of the oxazolone ring take place through a TS (structure XIII, Figure 2, Table 2) at 40.3 kcal mol−1 relative energy. After leaving the TS, a salt-bridge stabilized proton bound dimer (PBD) of doubly protonated 2-(amino(guanidininobutyl))-5-oxazolone and deprotonated aspartic acid is formed (Figure 2, structure XIV Erel at 22.2 kcal mol−1)). Intermolecular proton transfer in this PBD leads to a CS structure XV (Figure 2, Erel at 29.7 kcal mol−1) that can then dissociate to form the b2 oxazolone isomer and neutral aspartic acid (Scheme 2). This proton transfer is triggered by the high proton affinity of the aspartate anion, [D – H]− (~315 kcal mol−1), which is much greater than that of the oxazolone ring. An alternative salt-bridge stabilized b2-y1 pathway involving the deprotonated Asp side chain instead of the deprotonated C-terminal COOH (Scheme S1, Supporting Information) was also investigated. However, the bond-breaking TS of this pathway (XVI, Figure S5, Supporting Information) is less favored both energetically (Erel at 41.4 kcal mol−1) and entropically (ΔSact = −1.5 cal K−1 mol−1, Table 2, Supporting Information) for this channel.
Scheme 2.
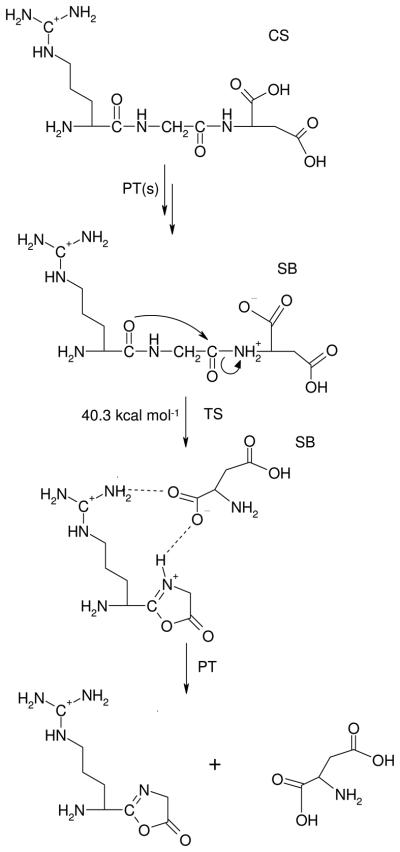
The salt-bridge stabilized b2-y1-SB peptide fragmentation pathway.
Figure 2.
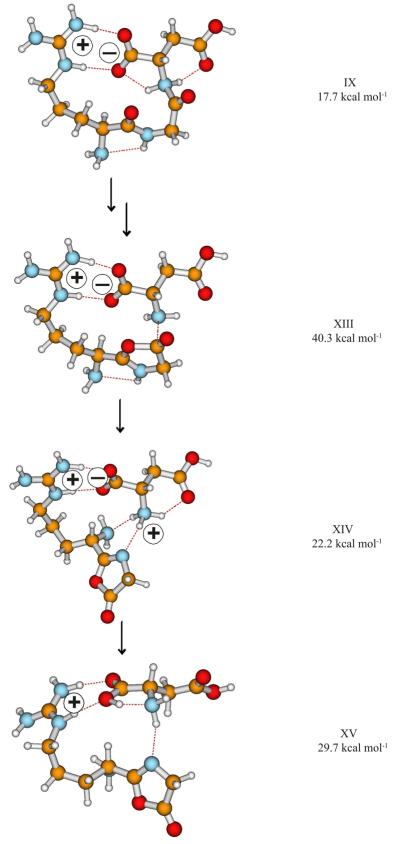
Selected structures on the lowest energy salt-bridge stabilized b2-y1–SB peptide fragmentation pathway of [RGD+H]+. Charge centers are indicated with + & − signs. The relative energies are presented for clarity.
(c) Formation of oxazolone isomer b2 ions via SB intermediates: the b2-y1-Anhydride pathway
Oxazolone isomer b2 ions can be formed from salt-bridge stabilized intermediates on an alternative pathway18 which involves formation of anhydride structures prior to the rate determining cleavage step (Scheme 3). This b2-y1-Anhydride pathway again requires formation of a Gly-Asp amide nitrogen protonated salt-bridge structure, but here the proton has been mobilized from the Asp COOH group (V, 23.6 kcal mol−1). Following the proton transfers to generate this structure; the deprotonated Asp oxygen nucleophilically attacks the Gly carbonyl carbon simultaneously cleaving the Gly-Asp amide bond (Figure 3, structure XVII, TS 33.3 kcal mol−1). This reaction creates a backbone anhydride linkage which following rotations can be facilely cleaved by a second nucleophilic attack on the Gly carbonyl carbon, this time by the Arg carbonyl oxygen (Figure 3, structure XVIII, TS 36.3 kcal mol−1, ΔS298 = 0.2 cal mol−1 K−1). The cleavage of the anhydride C(O)-O bond results in a salt-bridge stabilized PBD of doubly protonated 2-(amino(guanidininobutyl))-5-oxazolone and deprotonated aspartic acid. Again, intermolecular proton transfer in this PBD leads to a CS structure that can dissociate to form the b2 oxazolone isomer and neutral aspartic acid. The equivalent pathway involving the C-terminal COOH proton is possible but this requires 3 kcal mol−1 more energy to initiate (XIX, Figure S6, TS at 39.3 kcal mol−1, ΔSact at 2.1 cal K−1 mol−1). Further discussion of the relevance of this means of proton mobilization as well as salt-bridge and anhydride stuctures to the formation of bn−1+H2O ions is presented in section 4.
Scheme 3.

The most energetically favorable Anhydride b2-y1 peptide fragmentation pathway. This pathway involves the aspartic acid COOH proton being transferred to the G-D amide nitrogen.
Figure 3.

Structures on the most energetically favorable Anhydride b2-y1 peptide fragmentation pathway of [RGD+H]+ (note that the aspartic acid proton is mobilized). SBs are indictaed with + & − signs while the mobilized proton is indicated by small arrows. The relative energies are presented for clarity.
(d) Formation of oxazolone isomer b2 ions via mobilization of the R-G amidic proton
A third means of proton mobilization is also possible, via imine enol structures. Similarly to our previous communication18, the imine enol b2-y1 pathway (Scheme S2, Supporting Information) is also less energetically demanding (TS at 43.4 kcal mol−1, XX, Figure S7, Supporting Information) than the classical b2-y1 pathway but more so than Anhydride b2-y1 pathways. Briefly, this pathway involves mobilization of an amidic proton on the peptide backbone thus generating structures with the neutral – COH=N- moiety (13.4 kcal mol−1). Transfer of the hydroxyl proton followed by nucleophilic attack on the carbonyl group18 cleaves the Gly-Asp amide bond and leads to a proton bound dimer of neutral aspartic acid and the b2 ion protonated at the guanidine group. This dimer then dissociates to form b2 ions and neutral aspartic acid. This mechanism is unlikely to be active for protonated RGD, but could be active for systems lacking acidic residues or an acidic C-terminus37 where the SB amide bond-cleavage pathways cannot occur. These types of systems will be discussed in a subsequent publication.
(e) Formation of lactam isomer b2
The lactam isomer (Chart 2, structure C) of the b2 ion of protonated RGD is energetically more favored than the classical oxazolone structure. This isomer can be formed from CS structures by transferring one of the guanidine protons to the C-terminal amide bond (Scheme S3, Supporting Information) and concerted nucleophilic attack of the neutral guanidine on the carbon center of the protonated amide bond. The corresponding TS (XXI, Erel at 48.7 kcal mol−1, Figure S8, Supporting Information) is energetically much less favored than the SB stabilized (b2-y1-SB) and Anhydride (Anhydride b2-y1) TSs (40.3 and 36.3 kcal mol−1, (XIII & XVIII) respectively). Additionally izomerization reactions in which the b2 lactam isomer (Chart 2, structure C) is formed from the oxazolone form were determined to be energetically unfavorable (>60 kcal mol−1), so no post-bond cleavage b2 ion changes in structure should occur unless significant energy is put into the ion to activate it further. The preceding findings indicate that the oxazolone isomer is the likely b2 ion structure produced from protonated RGD and that it is formed on pathways that involve salt-bridge and anhydride intermediates.
(4) The b2+H2O peptide fragmentation pathway
Formation of the so-called bn−1+H2O ion38 (where n = total number of residues in the protonated peptide, b2+H2O for RGD) involves the loss of the C-terminal amino acid residue to give a product ion which has a carboxyl group at the new C-terminus, i.e. a normal protonated peptide, just one residue shorter ([RG+H]+ in this case). Formation of bn−1+H2O ion is promoted by the presence of basic amino acids in a non-C-terminal position. The mechanism by which this fragment ion type is formed has been debated in the literature for some time.38
Gaskell and coworkers38a proposed a mechanism where the protonated Arg side chain behaves as a spectator and elimination of the C-terminal residue occurs on a charge-remote fragmentation pathway (Scheme S4, Supporting Information). This reaction is initiated by attack of the hydroxyl OH on the carbon of the C-terminal amide bond to form a five-membered ring that is cleaved into three fragments in the next step. Farrugia and O'Hair proposed38e a completely different charge-directed mechanism (Scheme S5, Supporting Information) that involves formation of a salt-bridge intermediate in the first step. Nucleophilic attack by one of the carboxylate oxygens on the carbon of the C-terminal amide bond leads to the same oxazolone derivative as suggested on the pathway of Gaskell and coworkers. This oxazolone then opens up to form an anhydride intermediate that fragments further to form the bn−1+H2O ion.
Substantial experimental, statistical, and theoretical work has been devoted to elucidate the underlying mechanism and chemistry involved in the formation of these ions38; however, no transition structure calculations have been published in support of these proposals. In an attempt to resolve this controversy, we have investigated the above mechanisms using computational and experimental tools. Our calculations led to a new mechanism that involves some of the steps of the pathway proposed by Farrugia and O'Hair38e but important differences too.
Similarly to the most energetically favorable b2-y1 pathway described above, the b2+H2O channel involves salt-bridge intermediates. In fact, the first steps of the two pathways are conceptually identical. Here (Scheme 4) the C-terminal COOH proton is mobilized to the Gly-Asp amide nitrogen (IX, Figure 4). From such salt-bridge stabilized reactive intermediates nucleophilic attack by a carboxylate oxygen on the glycine carbonyl carbon forms an anhydride with simultaneous breaking of the GD amide bond (Scheme 4). This reactions has a low threshold energy at 28.8 kcal mol−1 (XXII). This means that our mechanism forms the anhydride intermediate in a single step from a SB structure which features protonation at the C-terminal amide N. Loss of CO then occurs via another salt-bridge stabilized transition structure at 43.7 kcal mol−1 relative energy (Scheme 4, XXIII, Figure 4). On the product wing of this TS a SB stabilized proton bound dimer of zwitterionic RG and the immonium ion of aspartic acid, Dim, is formed from which an intermolecular proton transfer from the protonated imine group of Dim completes the formation of the new C-terminus of the b2+H2O ion.
Scheme 4.
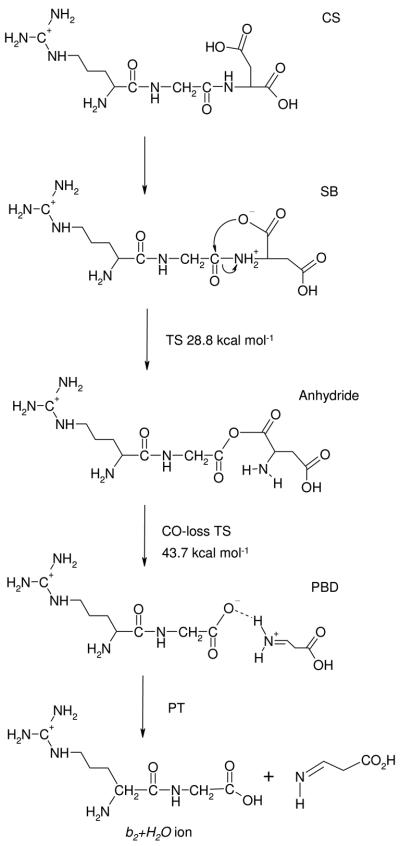
The b2+H2O -forming peptide fragmentation pathway
Figure 4.
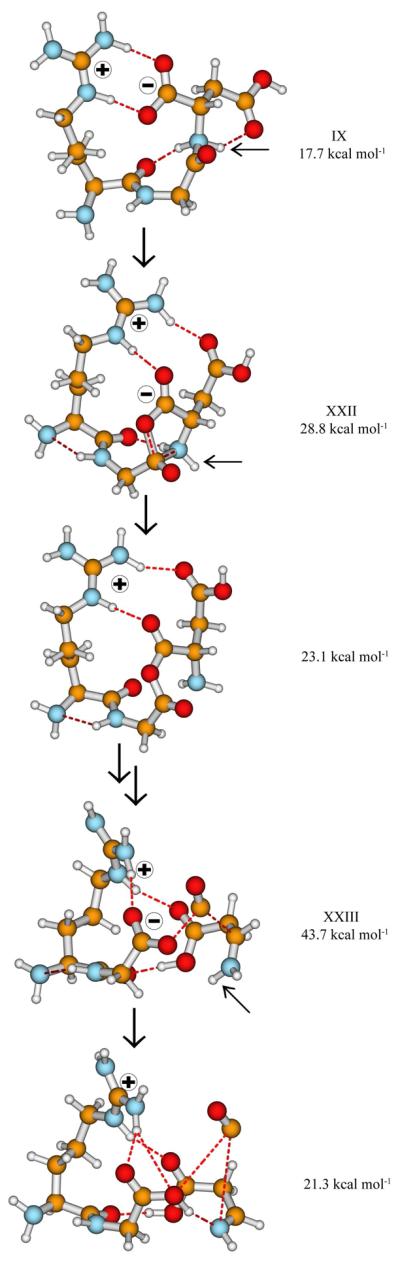
Selected structures on the bn−1+H2O pathway; formation of an anhydride intermediate. Charge centers are indicated with + & − signs while the mobilized proton is noted by small arrows. Relative energies are displayed for clarity.
The b2+H2O-forming anhydride structure is the same type as that proposed previously38e; however, the mechanisms of its formation are quite different. Farrugia and O'Hair proposed forming this anhydride from an oxazolone alcohol via a four-center proton transfer, with concerted amide bond cleavage (Scheme S5, Supporting Information). This oxazolone alcohol is a stable species (~17 kcal mol−1), so was a potential intermediate. However, this mechanism has two problems which make it unlikely to be active: the TS to form the oxazolone alcohol (~31 kcal mol−1) is energetically less favored than our TS to form the anhydride directly, and four-center proton transfer reactions are energetically unfavorable7h,i (>50 kcal mol−1 in this case) so would be rate-limiting for this mechanism.
The mechanism38a proposed by Gaskell and coworkers also suffers from TSs that are energetically much less favored than those proposed here. This charge-remote fragmentation pathway has very limited means of stabilizing the protonated Arg residue as most of the charge donating groups are involved in the charge remote TS. While this situation would potentially improve for larger systems (with more charge solvating groups not part of the TS), this pathway would still be unlikely to be active as the difference in energy was so large (>20kcal mol−1).
It should be noted that the b2-y1-SB (Scheme 2) and b2-y1–Anhydride (TS in Figure S5) pathways as well as the b2+H2O channel (Scheme 4) share the same reactive intermediate with the proton mobilized from the C-terminus to the C-terminal amide nitrogen (structure IX). The branching ratio of these channels is determined by the fate of the SB intermediate. Unfortunately, our modeling does not provide detailed kinetic data on the formation and subsequent reactions of these reactive structures, thus limiting our ability to comment further.
(5) Formation of b1 ions from protonated RGD
Normally, b1 ions are not detected in protonated peptide MS/MS spectra when the N-terminal amino acid has an aliphatic side chain. This is believed to due to the instability of the corresponding acylium ion structure36,39. On the other hand, b1 peaks are often observed when the N-terminal residue is Lys, Arg, His or Met36,40; this experimental finding indicates that these ions have an unusual structure, making the ions stable enough to be detected. Stabilization of most potential b1 ions via a cyclization reaction is not possible due to the lack of a suitable nucleophilic group in the residues side chain. Conversely, Lys, Arg, His and Met side chains can potentially perform this function. Based on this reasoning, cyclization mechanisms have been proposed40. Here we performed calculations to evaluate whether the b1 structures of Arg were stable and whether the proposed mechanism of formation was energetically possible.
Three different b1 structures can be envisaged for arginine; the acylium structure and two lactams with either 6 or 8 membered rings (Chart 3). Our calculations to locate stable acylium structures were unsuccessful, all candidate structures either fell apart (to form CO and the imine of Arg) or cyclized during the course of geometry optimization. The six-membered lactam ring is much more energetically favorable than the 8-membered form (as are the corresponding TSs), so only the former will be described in the following.
Chart 3.
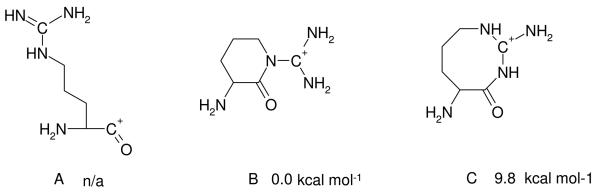
The potential acylium (structure A) and lactam (structures B and C) isomers of the b1 ion of RGD with relative energies displayed.
Formation of the b1 ion can be initiated from CS structures of protonated RGD. Initially, we tried a two-step mechanism (Scheme 5) that involves transfer of the eNH proton of the guadinine group to the N-terminal amide bond and subsequent nucleophilic attack of eN on the carbon of the protonated amide bond. Our calculations however indicate that the corresponding amide N protonated structures are not stable and one of the NH2+ protons always transfers back the arginine side chain during the course of geometry optimization. Subsequently we were successful in trying a mechanism where transfer of the eNH proton, formation of the six-membered lactam ring, and cleavage of the N-terminal amide bond occurs in a concerted manner (for both the 6- and 8-membered rings). The least energetically demanding TS (XXIV, Figure 5, Scheme 5) forms a 6-membered ring at 42.6 kcal mol−1 threshold energy. The reactant wing of this TS was carefully checked using detailed IRC calculations. These indicate that the reaction starts with transfer of the εNH proton which initially approaches the GD amide oxygen. In effect, the proton is shuttled to the carbonyl oxygen, then on to the amide nitrogen. This means that the proton transfer to the RG amide nitrogen is facilitated by the carbonyl oxygen of the glycine residue solvating the charge thereby catalyzing the process.
Scheme 5.

The b1-y2 lactam-forming peptide fragmentation pathway (the numbered steps occur in a concerted manner (see preceding text)).
Figure 5.

The b1-y2 transition structure. The relative energy is presented for clarity.
(5) Pathways leading to elimination of small neutrals from protonated RGD
(a) Loss of ammonia from protonated RGD
Loss of ammonia from the arginine side chain is the largest peak under the majority of spectral conditions. For this pathway to be active, proton transfers to form the NH3 leaving group are necessary.41 This means proton transfer reactions in the protonated guanidine group (Scheme 6) catalyzed by proximal proton accepting groups. Initially we investigated both CS and SB structures with the preformed NH3 moiety (both tautomers in Scheme 6) on the R side chain. These calculations indicated that the CS structures (22-27 kcal mol−1 relative energy) with the preformed NH3 moiety are energetically more favored than the corresponding SB species (>28 kcal mol−1 relative energy). NH3-loss transition structures were then sought from each of these configurations. CS structures were again found to be more favored and the lowest energy TS is at 42.5 kcal mol−1 relative energy (Figure S9, XXV, Supporting Information). The alternate losses of ammonia from the N-terminus were also investigated, but these pathways resulted in TSs of ≥ 50 kcal mol−1 so are not discussed in detail here.
Scheme 6.

The main steps of the ammonia loss pathway: (a) local proton transfer reactions lead to tautomers with the NH3 leaving group (b) loss of ammonia leaves reactive -N=C=NH2+ or -NH+=C=NH moieties behind. Only the arginine side chain is shown for clarity.
(b) Loss of water from protonated RGD
Loss of water occurred only in the IT and QQQ experiments, neither the MI nor the CID MALDI TOF/TOF experiments showed [M+H−H2O]+ ions. Two major pathways can be envisaged for this reaction. The first is a charge-remote channel that represents the major chemistry behind the ‘aspartic acid’ effect.10 This reaction begins with a charge solvated species and involves nucleophilic attack by the aspartic acid carboxyl oxygen on the C-terminal carbonyl group and transfer of the hydroxyl proton from the aspartic acid side chain to the C-terminal OH. The relative energy of the corresponding TS is 40.7 kcal mol−1 (XXVI, Figure 6). On the product side of this TS a cyclic anhydride is formed as the water molecule is expelled. The structure of the [M+H−H2O]+ ion was investigated in MS3 experiments that show combined loss of CO and CO2 but no loss of CO on its own. This result fully supports the anhydride structure of the water-loss fragment. For completeness though, we calculated the alternative oxazolone-forming28,42 mechanisms of water loss and in agreement with our experimental findings the corresponding TS was energetically much less favored (≥50 kcal mol−1) than the one responsible for the formation of the cyclic anhydride.
Figure 6.

The lowest energy water loss transition structure. The relative energy is presented in the figure for clarity.
(c) Formation of the d3 ion
dn ions43 are generally not observed in modern mass spectrometers due to the low-energy collisional activation employed by most current instruments. An occaisional exception for peptides containing acidic residues has been reported by the Gaskell group.44 These ions have also been reported as MS3 products from bn ions with a C-terminal aspartic acid residue.10a,10c We found evidence for both means of d ion formation in this study, however the direct formation was much more energetically favorable. This reaction (Structure XXVII, Figure S10, 50.4 kcal mol−1) involves elimination of three small molecules (H2O, CO and CO2), so it is logical that this process has the most favorable activation entropy of all the reactions studied too (13.9 cal mol−1 K−1). Additionally, consecutive fragmentation to form this ion from the b3 ion is conceptually possible. This requires considerably more energy, but is even more entropically favored so may be a competitive mechanism in instruments where large amounts of energy were put into the ion in a short period of time (consistent with earlier proposals10a,10c).
It should be noted that this mechanism is probably an exception, rather than a general occurrence, so is unlikely to be the same as that which occurs under high-energy CID conditions (collision energy (ELAB) > 5keV)45 or via ultraviolet photodissociation conditions46, where radical species are believed to play a critical role.
(d) Loss of guanidine from [RGD+H]+
Finally, this relatively uncommon ion results from a reaction that requires no proton mobilization. A simple SN2 reaction with a TS at 44.9 kcal mol−1 (XXVIII, Figure S11, Supporting Information) generates the requisite products, namely [PGD+H]+ and neutral guanidine (i.e. the arginine residue is converted into a proline residue). It should be noted however that this TS value should be viewed as a lower bound as density functional methods perform less well for activation barrier height calculations for SN2 reactions47 than for other TS and reaction-path calculations and generally underestimate their barrier heights48.
(6) The competing fragmentation pathways of protonated RGD
Our experiment on the IT where the excitation level was gradually increased to estimate the order of appearance energies indicates b2 < [M+H−H2O]+ ~ [M+H−NH3]+ < b1 ~ b2+H2O < [M+H−59]+ < d3 (Figure 1b). These fragments appear in a narrow excitation range suggesting that the corresponding appearance energies span a narrow range too. Given the very approximate nature of this technique, it was rather surprising that the order of the computed threshold energies (Figure 1c) is in reasonable agreement with the experimentally estimated order. The agreement is less clear for the triple quadrupole experiment (Figure S1, Supporting Information) where the onsets of the first five peak types (b2, b3/[M+H−H2O]+, [M+H−NH3]+, b1, b2+H2O) are indistinguishable. However, this additional data does clearly support the IT finding that the ions appear in a very narrow excitation range though. The computed threshold energies span the 36.3 to 50.4 kcal mol−1 range with activation entropies ranging from 0.2 to 13.9 cal mol−1 K−1 (Figure 1c). Whilst the TSs that form the b and [M+H−H2O]+ ions are separated by 4.4 kcal mol−1, the former is entropically less favorable (Figure 1d). The b3/[M+H−H2O]+, [M+H−NH3]+, b1, and b2+H2O ion peaks appear at similar excitation levels; this is in line with the corresponding computed threshold energies spanning the 40.7 - 43.7 kcal mol−1 range. The calculated activation entropies for the [M+H−NH3]+, b1, and b2+H2O channels are at 7 −10 cal mol−1 K−1 while the water loss is entropically less favored (2.0 cal mol−1 K−1 activation entropy). However, this is compensated by the lowest threshold energy in this group of fragments. The b2+H2O pathway becomes progressively more active as the excitation level rises; this can be rationalized by the high activation entropy (9.7 cal mol−1 K−1 ) of this channel. As discussed above the threshold energy of the [M+H−59]+ pathway is probably underestimated by our computational strategy47,48; this explains why this ion appears experimentally only at higher levels of excitation. d3 has the highest computed threshold energy explaining the late appearance of this fragment in our experiments. Additionally, our calculations indicated that the ammonia loss pathway has several nearly energetically degenerate means of occurring from the guanidine group, which is not the case for the other pathways; this pseudo-degeneracy of fragmentation pathways may help to explain the increased competitiveness of this product type as the incremental excitation initially increases in the ion trap and triple quadrupole instruments.
(7) Conclusions
(1) Our calculations indicate that the formation of b2 and b2+H2O ions of protonated RGD include intermediates, transition structures, and post-reaction complexes stabilized by salt-bridges. Mobilization of a COOH proton leads to SB intermediates which react further to anhydride species which form then b2 and b2+H2O ions.
(2) A new mechanism has been proposed for the long-debated38 formation of bn−1+H2O ions. Our calculations suggest a multi-step mechanism that involves salt-bridge structures with amide N protonation and subsequent anhydride formation. The anhydride then fragments by losing CO as suggested by Farrugia and O'Hair.38e Our modeling also indicates that other mechanisms proposed in the literature38 involve transition structures that are energetically much less favored than those appearing on our SB-stabilized pathway.
(3) An energetically feasible b1-y2 mechanism initiated from charge-solvated structures has been demonstrated for b1 ion formation for protonated peptides with an N-terminal arginine residue. This mechanism operates via transfer of a guanidium proton to the N-terminal amide N which is tightly followed by nucleophilic attack of the newly created neutral guanidine group to cleave the first amide bond.
(4) The ammonia loss reaction is initiated by proton reorganization within the charged guanidine group. Ammonia loss is favored from charge-solvated structures of protonated RGD.
(5) The computed threshold energies and activation entropies reasonably explain the order of appearance energies obtained experimentally in the IT and triple quadrupole instruments by gradually increasing excitation of the parent ion population. This is rather remarkable since the threshold energies span a relatively narrow range.
(6) The interconvertion of charge-solvated and salt-bridge structures plays a key role in the fragmentation of singly protonated arginine containing peptides. The related chemistry is of utmost importance in proteomics studies that are based on sequencing of MALDI generated peptide ions. This and further studies using experimental (MS, ion mobility and IR spectroscopy), theoretical, and statistical studies will help to develop more robust sequencing approaches that will significantly reduce the currently unacceptably high false positive rates of MS-based protein identification.
Supplementary Material
Acknowledgments
The authors thank Yury Vasil'ev and Jeff Morré for running the triple quad experiment. B. B. and D. F. B. thank the NIEHS (ES00210). B. P. and S. S. are grateful for financial support from the Deutsche Forschungsgemeinschaft (SU 244/3-1) and the Landesstiftung Baden-Württemberg (P-LS-Prot/57). The DFT-calculated structures were rendered by Molden to allow their visualization. B.P. acknowledges the Deutsche Forschungsgemeinschaft for a Heisenberg fellowship.
Footnotes
Supporting Information Available
Additional total energies, structures, schemes, figures and a summary table are available in the supporting information. For further details, please refer to the main text.
REFERENCES
- 1.Hunt DF, Yates JR,III, Shabanowitz J, Winston S, Hauer CR. Proc. Natl. Acad. Sci. U.S.A. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aebersold R, Goodlett DR. Chem. Rev. 2001;101:269–296. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 3.Steen H, Mann M. Nature Rev. Mol. Cell Biol. 2004;5:699–711. doi: 10.1038/nrm1468. [DOI] [PubMed] [Google Scholar]
- 4.Yamashita M, Fenn JB. J. Phys. Chem. 1984;88:4451–4459. [Google Scholar]
- 5.Karas M, Hillenkamp F. Anal. Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 6.Paizs B, Suhai S. Mass Spectrom. Rev. 2005;24:508–548. doi: 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- 7.(a) Dongré AR, Jones JL, Somogyi Á, Wysocki VH. J. Am. Chem. Soc. 1996;118:8365–8374. [Google Scholar]; (b) Tsaprailis G, Nair H, Somogyi A, Wysocki VH, Zhong W, Futrell JH, Summerfield SG, Gaskell SJ. J. Am. Chem. Soc. 1999;121:5142–5154. [Google Scholar]; (c) Wysocki VH, Tsaprailis G, Smith LL, Breci LA. J. Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]; (d) Tang X, Boyd RK. Rapid Commun. Mass Spectrom. 1992;6:651. doi: 10.1002/rcm.1290061105. [DOI] [PubMed] [Google Scholar]; (e) Somogyi A, Wysocki VH, Mayer I. J. Am. Soc. Mass Spectrom. 1994;5(8):704. doi: 10.1016/1044-0305(94)80002-2. [DOI] [PubMed] [Google Scholar]; (f) Harrison AG, Yalcin T. Int. J. Mass Spectrom. 1997;165:339. [Google Scholar]; (g) Summerfield SG, Whiting A, Gaskell SJ. Int. J. Mass Spectrom. Ion Proc. 1997;162:149. [Google Scholar]; (h) Csonka IP, Paizs B, Lendvay G, Suhai S. Rapid Commun. Mass Spectrom. 2000;14:417–431. doi: 10.1002/(SICI)1097-0231(20000331)14:6<417::AID-RCM885>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]; (i) Paizs B, Csonka IP, Lendvay G, Suhai S. Rapid Commun. Mass Spectrom. 2001;15:637–650. doi: 10.1002/rcm.279. [DOI] [PubMed] [Google Scholar]; (j) Jorgensen TJD, Gardsvoll H, Ploug M, Roepstorff P. J. Am. Chem. Soc. 2005;127:2785. doi: 10.1021/ja043789c. [DOI] [PubMed] [Google Scholar]; (k) Zehl M, Rand KD, Jensen ON, Jorgensen TJD. J. Am. Chem. Soc. 2008;130:17453–17459. doi: 10.1021/ja805573h. [DOI] [PubMed] [Google Scholar]
- 8.a) Roepstorff P, Fohlmann J. J. Biomed. Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]; b) Biemann K. Biomed. Environ. Mass Spectrom. 1988;16:99. doi: 10.1002/bms.1200160119. [DOI] [PubMed] [Google Scholar]
- 9.a) Harrison AG. Mass Spectrom. Rev. 1997;16:201–217. [Google Scholar]; b) Bleiholder C, Suhai S, Paizs B. J. Am. Soc. Mass Spectrom. 2006;17:1275–1281. doi: 10.1016/j.jasms.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 10.a) Yu W, Vath JE, Huberty MC, Martin SA. Anal. Chem. 1993;65:3015. doi: 10.1021/ac00069a014. [DOI] [PubMed] [Google Scholar]; b) Qin J, Chait BT. J. Am. Chem. Soc. 1995;117:5411–5412. [Google Scholar]; c) Gu C, Tsaprailis G, Breci L, Wysocki VH. Anal. Chem. 2000;72:5804–5813. doi: 10.1021/ac000555c. [DOI] [PubMed] [Google Scholar]; d) Tsaprailis G, Somogyi A, Nikolaev EN, Wysocki VH. Int. J. Mass Spectrom. 2000;196:467–479. [Google Scholar]; e) Paizs B, Suhai S, Hargittai B, Hruby VJ, Somogyi A. Int. J. Mass Spectrom. 2002;219:203. [Google Scholar]
- 11.Spengler B, Kirsch D, Kaufmann R. Rapid Commun. Mass Spectrom. 1991;5:198–202. doi: 10.1002/(SICI)1097-0231(19960731)10:10<1199::AID-RCM643>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 12.Chernushevich IV, Loboda AV, Thomson BA. J. Mass Spectrom. 2001;36:849–865. doi: 10.1002/jms.207. [DOI] [PubMed] [Google Scholar]
- 13.Qin J, Chait BT. Int. J. Mass Spectrom. 1999;190/191:313–320. [Google Scholar]
- 14.Dodds ED, Hagerman PJ, Lebrilla CB. Anal. Chem. 2006;78:8506–8511. doi: 10.1021/ac0614442. [DOI] [PubMed] [Google Scholar]
- 15.Medzihradszky KF, Campbell JM, Baldwin MA, Falick AM, Juhasz P, Vestal ML, Burlingame AL. Anal. Chem. 2000;72:552–558. doi: 10.1021/ac990809y. [DOI] [PubMed] [Google Scholar]
- 16.Falkner JA, Kachman M, Veine DM, Walker A, Strahler JR, Andrews PC. J. Am. Soc. Mass Spectrom. 2007;18:850–855. doi: 10.1016/j.jasms.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Khatun J, Ramkissoon K, Giddings MG. Anal. Chem. 2007;79:3032–3040. doi: 10.1021/ac061455v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bythell BJ, Suhai S, Somogyi A, Paizs B. J. Am. Chem. Soc. 2009;131:14057–14065. doi: 10.1021/ja903883z. [DOI] [PubMed] [Google Scholar]
- 19.Grégoire G, Lemaire J, Ortega JM, Suhai S, Schermann JP, Paizs B, Desfrançois C. manuscript in preparation [Google Scholar]
- 20.(a) Laskin J, Bailey TH, Futrell JH. Int. J. Mass Spectrom. 2004;234:89–99. [Google Scholar]; (b) Laskin J, Bailey TH, Futrell JH. Int. J. Mass Spectrom. 2006;249:462–472. [Google Scholar]
- 21.McLuckey SA, Van Berkel GJ, Goeringer DE, Glish GL. Anal. Chem. 1994;66:689A–696A. doi: 10.1021/ac00086a001. [DOI] [PubMed] [Google Scholar]
- 22.Colorado A, Brodbelt J. J. Am. Soc. Mass Spectrom. 1996;7:1116–1125. doi: 10.1016/S1044-0305(96)00077-3. [DOI] [PubMed] [Google Scholar]
- 23.(a) Paizs B, Suhai S. J. Am. Soc. Mass Spectrom. 2004;15:103. doi: 10.1016/j.jasms.2003.09.010. [DOI] [PubMed] [Google Scholar]; (b) Wyttenbach T, Paizs B, Barran P, Breci LA, Liu D, Suhai S, Wysocki VH, Bowers MT. J. Am. Chem. Soc. 2003;123:13768. doi: 10.1021/ja037204a. [DOI] [PubMed] [Google Scholar]; (c) Harrison AG, Young AB, Bleiholder C, Suhai S, Paizs B. J. Am. Chem. Soc. 2006;128:10364–10365. doi: 10.1021/ja062440h. [DOI] [PubMed] [Google Scholar]; (d) Polfer NC, Oomens J, Suhai S, Paizs B. J. Am. Chem. Soc. 2007;129:5887. doi: 10.1021/ja068014d. [DOI] [PubMed] [Google Scholar]; (e) Bleiholder C, Osburn S, Williams TD, Suhai S, Van Stipdonk M, Harrison AG, Paizs B. J. Am. Chem. Soc. 2008;130:17774. doi: 10.1021/ja805074d. [DOI] [PubMed] [Google Scholar]; (f) Bythell BJ, Knapp-Mohammady M, Paizs B, Harrison AG. J. Am. Soc. Mass Spectrom. 2010 doi: 10.1016/j.jasms.2010.05.006. doi:10.1016/j.jasms.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Case DA, et al. AMBER 99. University of California; San Francisco: See S.I. for full citation. [Google Scholar]
- 25.El Aribi H, Orlova G, Rodriquez CF, Almeida DRP, Hopkinson AC, Siu KWM. J. Phys. Chem. B. 2004;108:18743. [Google Scholar]
- 26.Frisch MJ, et al. Gaussian 03, Revision C.02. See S.I. for full citation. [Google Scholar]
- 27.Papayannopoulos I. Mass Spec. Rev. 1995;14:49–73. [Google Scholar]
- 28.Bythell BJ, Barofsky DF, Pingitore F, Polce MJ, Wang P, Wesdemiotis C, Paizs B. J. Am. Soc. Mass Spectrom. 2007;18:1291–1303. doi: 10.1016/j.jasms.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 29.Lifshitz C. Eur. J. Mass Spectrom. 2002;8:85–98. [Google Scholar]
- 30.Heaton AL, Armentrout PB. J. Am. Chem. Soc. 2008;130:10227–10232. doi: 10.1021/ja801726g. [DOI] [PubMed] [Google Scholar]
- 31.Schnier PD, Price WD, Jockusch RA, Williams ER. J. Am. Chem. Soc. 1996;118:7178–7189. doi: 10.1021/ja9609157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriquez CF, Orlova G, Guo Y, Li X, Siu C-K, Hopkinson AC, Siu KWM. J. Phys. Chem. B. 2006;110:7528–7537. doi: 10.1021/jp046015r. [DOI] [PubMed] [Google Scholar]
- 33.Prell JS, O'Brien JT, Steill JD, Oomens J, Williams ER. J. Am. Chem. Soc. 2009;131:11442–11449. doi: 10.1021/ja901870d. [DOI] [PubMed] [Google Scholar]
- 34.a) Yalcin T, Csizmadia IG, Peterson MR, Harrison AG. J. Am. Soc. Mass. Spectrom. 1996;7:233–242. doi: 10.1016/1044-0305(95)00677-X. [DOI] [PubMed] [Google Scholar]; b) Paizs B, Suhai S. Rapid Commun. Mass Spectrom. 2001;15:2307–2323. doi: 10.1002/rcm.273. [DOI] [PubMed] [Google Scholar]; c) Polfer NC, Oomens J, Suhai S, Paizs B. J. Am. Chem. Soc. 2005;127:17154–17155. doi: 10.1021/ja056553x. [DOI] [PubMed] [Google Scholar]; d) Chen X, Tureček F. J. Am. Soc. Mass. Spectrom. 2005;16:1941–1956. doi: 10.1016/j.jasms.2005.07.023. [DOI] [PubMed] [Google Scholar]; e) Yoon SY, Chamot-Rooke J, Perkins BR, Hilderbrand AE, Poutsma JC, Wysocki VH. J. Am. Chem. Soc. 2008;130:17644–17645. doi: 10.1021/ja8067929. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Oomens J, Young S, Molesworth S, Van Stipdonk M. J. Am. Soc. Mass Spectrom. 2009;20:334–339. doi: 10.1016/j.jasms.2008.10.012. [DOI] [PubMed] [Google Scholar]; g) Bythell BJ, Somogyi Á, Paizs B. J. Am. Soc. Mass Spectrom. 2009 doi: 10.1016/j.jasms.2008.11.021. [DOI] [PubMed] [Google Scholar]; h) Bythell BJ, Erlekam U, Paizs B, Maitre P. Chem. Phys. Chem. 2009;10:883–885. doi: 10.1002/cphc.200800804. [DOI] [PubMed] [Google Scholar]
- 35.(a) Harrison A. J. Am. Soc. Mass Spectrom. 2008;19:1776. doi: 10.1016/j.jasms.2008.06.025. [DOI] [PubMed] [Google Scholar]; (b) Erlekam U, Bythell BJ, Scuderi D, Van Stipdonk M, Paizs B, Maître P. J. Am. Chem. Soc. 2009;131(32):11503–11508. doi: 10.1021/ja903390r. [DOI] [PubMed] [Google Scholar]
- 36.Farrugia JM, O'Hair RAJ, Reid GE. Int. J. Mass Spectrom. 2001;210-211:71–87. [Google Scholar]
- 37.(a) Mouls L, Subra G, Aubagnac J-L, Martinez J, Enjalbal C. J. Mass Spectrom. 2006;41:1470–1483. doi: 10.1002/jms.1118. [DOI] [PubMed] [Google Scholar]; (b) Molesworth S, Leavitt CM, Groenewold GS, Oomens J, Steill JD, Van Stipdonk M. J. Am. Soc. Mass. Spectrom. 2009 doi: 10.1016/j.jasms.2009.06.007. doi:10.1016/j.jasms.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 38.a) Thorne GC, Ballard KD, Gaskell SJ. J. Am. Soc. Mass. Spectrom. 1990;1:249–257. [Google Scholar]; b) Ballard KD, Gaskell SJ. J. Am. Chem. Soc. 1992;114:64–71. [Google Scholar]; c) Vachet RW, Asam MR, Glish GL. J. Am. Chem. Soc. 1996;118:6252–6256. [Google Scholar]; d) Fang S, Takao T, Satomi Y, Mo W, Shimonishi Y. J. Am. Soc. Mass. Spectrom. 2000;11:345–351. doi: 10.1016/s1044-0305(99)00153-1. [DOI] [PubMed] [Google Scholar]; e) Farrugia JM, O'Hair RAJ. Int. J. Mass Spectrom. 2003;222:229–242. [Google Scholar]; f) Hiserodt RD, Brown SM, Swijter DFH, Hawkins N, Mussinan CJ. J. Am. Soc. Mass. Spectrom. 2007;18:1414–1422. doi: 10.1016/j.jasms.2007.04.018. [DOI] [PubMed] [Google Scholar]; g) She Y-M, Krokhin O, Spicer V, Loboda A, Garland G, Ens W, Standing KG, Westmore JB. J. Am. Soc. Mass. Spectrom. 2007;18:1024–1037. doi: 10.1016/j.jasms.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Forbes MW, Jockusch RA, Young AB, Harrison AG. J. Am. Soc. Mass Spectrom. 2007;18:1959–1966. doi: 10.1016/j.jasms.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 39.Tsang CW, Harrison AG. J. Am. Chem. Soc. 1976;98:1301. doi: 10.1021/ja00422a001. [DOI] [PubMed] [Google Scholar]
- 40.a) Yalcin T, Harrison AG. J. Mass Spectrom. 1996;31:1237. doi: 10.1002/(SICI)1096-9888(199611)31:11<1237::AID-JMS416>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; b) Tu YP, Harrison AG. Rapid Commun. Mass Spectrom. 1998;12:849. [Google Scholar]; c) Kulik W, Heerma W. Biomed. Mass Spectrom. 1988;15:419. doi: 10.1002/bms.1200150803. [DOI] [PubMed] [Google Scholar]
- 41.Csonka IP, Paizs B, Suhai S. J. Mass Spectrom. 2004;39:1025–1035. doi: 10.1002/jms.660. [DOI] [PubMed] [Google Scholar]
- 42.(a) Balta B, Aviyente V, Lifshitz C. J. Am. Soc. Mass Spectrom. 2003;14:1192–1203. doi: 10.1016/S1044-0305(03)00479-3. [DOI] [PubMed] [Google Scholar]; (b) Bythell BJ, Dain RP, Curtice SS, Oomens J, Steill JD, Groenewold GS, Paizs B, Van Stipdonk MJ. J. Phys. Chem. A. 2010;114:5076–5082. doi: 10.1021/jp9113046. [DOI] [PubMed] [Google Scholar]
- 43.Johnson RS, Martin SA, Biemann K. Int. J. Mass Spectrom. 1988;86:137–154. [Google Scholar]
- 44.Summerfield SC, Cox KA, Gaskell SJ. J. Am. Soc. Mass. Spectrom. 1997;8:25–31. [Google Scholar]
- 45.Biemann K. In: In: Methods in Enzymology. McCloskey JA, editor. Vol. 193. Academic; San Diego: 1990. pp. 455–479. [DOI] [PubMed] [Google Scholar]
- 46.(a) Cuia W, Thompson MS, Reilly JP. J. Am. Soc. Mass. Spectrom. 2005;16:1384–1398. doi: 10.1016/j.jasms.2005.03.050. [DOI] [PubMed] [Google Scholar]; (b) Yoon H, Chung YJ, Kim MS. J. Am. Soc. Mass. Spectrom. 2008;19:645–655. doi: 10.1016/j.jasms.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 47.Laerdahl JK, Uggerud E. Int. J. Mass Spectrom. 2002;214:277–314. [Google Scholar]
- 48.Gritsenko OV, Ensing B, Schipper PRT, Baerends EJ. J. Phys. Chem. A. 2000;104:8558–8565. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.