

Abstract
Transcription factor NF-E2-related factor-2 (Nrf2) is a key regulator of endogenous anti-oxidant systems shown to play a neuroprotective role in the adult by preserving blood–brain barrier function. The choroid plexus, site for the blood-CSF barrier, has been suggested to be particularly important in maintaining brain barrier function in development. We investigated the expression of Nrf2-and detoxification-system genes in choroid plexus following systemic LPS injections, unilateral cerebral hypoxia-ischemia (HI) as well as the combination of LPS and HI (LPS/HI). Plexuses were collected at different time points after LPS, HI and LPS/HI in 9-day old mice. mRNA levels of Nrf2 and many of its target genes were analyzed by quantitative PCR. Cell death was analyzed by caspase-3 immunostaining and TUNEL. LPS caused down-regulation of the Nrf2-system genes while HI increased expression at earlier time points. LPS exposure prior to HI prevented many of the HI-induced gene increases. None of the insults resulted in any apparent cell death to choroidal epithelium. These data imply that the function of the inducible anti-oxidant system in the choroid plexus is down-regulated by inflammation, even if choroid cells are not structurally damaged. Further, LPS prevented the endogenous antioxidant response following HI, suggesting the possibility that the choroid plexus may be at risk if LPS is united with an insult that increases oxidative stress such as hypoxia-ischemia.
Introduction
Emerging evidence suggests that structural and functional impairment of the blood brain barrier (BBB) contributes to metabolic disorders (Garbuzova-Davis et al 2011). Further, in childhood neurodegenerative lysosomal storage disorders disruption of the BBB is a serious complication (Saha et al 2012). Others have identified inflammation as an important factor in metabolic diseases such as neuronopathic Gaucher (Sun et al 2010) and inhibition of inflammatory mediators augment neurodegeneration in Sandhoff disease mice (Wu and Proia 2004).
The BBB together with the blood–cerebrospinal fluid (CSF) barrier (BCSFB) constitutes cerebral interfaces to maintain homeostasis within the central nervous system: both barriers tightly seal CNS from the changeable milieu of the blood stream. Whereas the BBB is established by specialized endothelial cells of CNS blood vessels, the BCSFB is formed by the epithelial cells of the choroid plexus. The choroid plexus has been suggested to play a particularly important role in the developing brain since it differentiates early, is large in comparison to brain size in the developing animal and many of its transport and enzymatic functions are already present soon after it is formed (Johanson 1995; Johanson et al 2002; Ek et al 2012; Liddelow et al 2012). Between the epithelial cells of the choroid plexus there is a well organized network of tight junctions that constitute a physical barrier already in the embryo (Ek et al 2003; Johansson et al 2006). There are also different transporter proteins present (Strazielle and Ghersi-Egea 2000; Strazielle et al 2004) forming an efficient enzymatic barrier, which prevents the entry of neurotoxic compounds into the CSF and also facilitates export from CSF to blood. The functionality of many of these transport mechanisms is based on a glutathione-dependent detoxification processes involving glutathione-S-transferase (Gst), present in the choroid plexus shortly after it differentiates (Senjo et al 1986; Beiswanger et al 1995). Further, Gst activity has been found at higher levels in the developing plexus than in adult, indicating special importance of this organ for detoxifying mechanisms as well as barrier function in the developing brain (Ghersi-Egea et al 2006).
Free radicals including reactive oxygen species are produced in excess during inflammation as well as during hypoxia-ischemia (HI)/reperfusion and are major mediators of perinatal brain injury (Ferriero 2001). Evidence from clinical studies demonstrates that newborn infants presenting with hypoxic-ischemic encephalopathy (HIE) or post-hemorrhagic ventricular dilatation show an increase in markers of oxidative stress in CSF (Sävman et al 2001; Ogihara et al 2003). Further, higher activity of both super-oxide dismutase (SOD) and glutathione peroxidase (GPx) in CSF correlate with the degree of encephalopathy in infants (Gulcan et al 2005) further implementing a role for an imbalance in redox state in brain pathology. In neonatal rats, we have shown that the anti-oxidant N-acetyl cysteine provides up to 78 % protection from LPS-induced brain damage (Wang et al 2007). Together these studies show that oxidative stress can contribute to perinatal brain injury and suggest that the response of endogenous anti-oxidant systems may play a role in determining neuropathological outcome.
A main regulator of the intracellular anti-oxidant defense is the transcription factor NF-E2-related factor-2 (Nrf2). Activation of Nrf2 has been shown to be neuroprotective in both adult (Innamorato et al 2008) and neonatal brain injury models (Ping et al 2010). Nrf2 also induce protection of brain–blood barrier function after traumatic brain injury by reducing the loss of endothelial cell markers and tight junction proteins (Zhao et al 2007). Systemic LPS down-regulate genes involved in maintenance of barrier function in the choroid plexus of adult animals (Marques et al 2009) and neonatal HI has been reported to cause severe damage to the choroid plexus (Rothstein and Levison 2002). Recently, Nrf2 activation has been reported to protect the BCSFB in vitro from damage caused by H2O2 (Xiang et al 2012), suggesting that Nrf2 may be a regulator of the maintenance of choroid plexus function.
In order to test the hypothesis that inflammation/oxidative stress can alter the gene expression of the endogenous anti-oxidant defense at the blood-CSF interface in the neonate we exposed 9-day old mice to LPS or HI and examined the mRNA expression of Nrf2 and genes related to the Nrf2-system, together with markers involved in apoptosis and barrier mechanisms in the choroid plexus. As LPS is known to sensitize the immature brain to HI via down-regulation of anti-oxidant capacity, we also tested the hypothesis that LPS pre-exposure would change the expression of the anti-oxidant Nrf2-system in response to HI using a well established model of combined LPS/HI (Eklind et al 2001; Wang et al 2009).
Methods
Neonatal LPS injections and hypoxic-ischemic brain injury
All experiments were performed on C57/Bl6 mice housed at Experimental Biomedicine, University of Gothenburg. All experiments were conducted in accordance with Department of Agriculture (Sweden) ethics regarding animal experimentation and approved by the local Animal Ethics Committee, Gothenburg (Licence Number 280/2010). Mice were given a 0.3 mg/kg or 1.0 mg/kg single intraperitoneal injection (i.p.) of LPS (ultra pure E. coli 055:B5, List biological laboratories, Inc) at postnatal day 9 (PND9; day of birth designated PND0). In other groups of mice pups, unilateral HI brain injury was induced at PND9 as previously described (Hedtjärn et al 2002). Briefly, the left carotid artery was permanently ligated under isoflurane anaesthesia and skin was closed using surgical glue (Vetbond, SweVet, Sweden). Animals were left to recover for one hour with the mother and were then introduced to a chamber set at 36 °C with first normal air flowed through it for 10 min, followed by a mixture of normal air and nitrogen so that the oxygen concentration was kept at 10 % for 50 min (HI50). Normal air was then circulated through the chamber for 10 min after which the pups were returned to their mother and left until sacrifice. To induce a combined LPS and HI insult, mice were given a single LPS (1.0 mg/kg, i.p.) or saline injection 14 h before 20 min of HI (LPS/HI and HI20 groups, respectively). It is known from our previous studies that 20 min of HI together with LPS pre-exposure achieves a similar degree of brain injury as detected after prolonged HI (50 min) alone (Eklind et al 2001).
Collection of tissue
No pups died as a result of LPS injections, HI or LPS/HI. At the time of tissue collection, pups were killed with an overdose of pentobarbital and used for histological preparation of brain tissue sections as described below. In separate mice the lateral choroid plexuses were microdissected out for qPCR analysis (see below). In all HI models (HI50, HI20 or LPS/HI), the left (ipsilateral) and right (contralateral) choroid plexuses of left/right lateral ventricles were dissected separately. Following LPS alone treatments, both plexuses in each animal were combined for PCR analysis.
Reverse transcription-quantitative PCR
Choroid plexuses at 2-, 6-, 14-, 24-, 72-h after 0.3 mg/kg of LPS (n=8–16 at each time point from at least 3 mixed litters) as well as saline-injected controls (n=16) were used for qPCR analysis. In another group of animals plexuses were collected at 24 h after 1 mg/kg of LPS (n=8, 3 mixed litters). Following HI50, plexuses were collected at 2-, 6-, 24-h after HI (n=8, 3 mixed litters). Following LPS/HI and HI20 experiments plexus samples were collected at 2 h after HI (n=8, 3 mixed litters). RNA was extracted from tissues with RNeasy Lipid Tissue Mini Kit (Qiagen, Solna, Sweden). Samples were homogenized with Qiasol lysis reagent homogenizer (Qiagen, Solna, Sweden) according to manufacturer’s instructions. Total RNA was measured in a spectrophotometer at 260-nm absorbance. mRNA expression was determined by reverse transcription-quantitative PCR (RT-qPCR). First strand cDNA was synthesized using the Superscript RNase H- reverse transcriptase kit (Invitrogen, CA, USA). Briefly, 200 ng RNA from choroid plexus were mixed with random hexamer primers and dNTP (Roche Molecular Biochemicals, IN, USA) and subjected to the reverse transcription process according to the manufacturer’s instructions. Each PCR (20 μl) contained 2 μl cDNA diluted 1:10, 10 μl Quanti Fast SYBR Green PCR Master Mix (Qiagen, Sweden) and 2 μl PCR primer. The following primers (Qiagen, Solna, Sweden) were used: Mm-Nfe2l2 QuantiTech Primer Assay QT00095270, Mm-Gclc QuantiTech Primer Assay QT00130543, Mm-Gclm QuantiTech Primer Assay QT00174300, Mm-HO-1 QuantiTech Primer Assay QT00159915, Mm-Abcc1 QuantiTech Primer Assay QT00139776, Mm-Gsta1 QuantiTech Primer Assay QT01772883, Mm-Rn QuantiTech Primer Assay 18 s QT1036875, Mm-Gapdh QuantiTech Primer Assay QT01658692, Mm-Ywhaz-1 QuantiTech Primer Assay QT00105350. The amplification protocol comprised an initial 5 min denaturation at 95 °C, followed by 40 cycles of denaturation for 10 s at 95 °C and annealing/extension for 30 s at 60 °C on a LightCycler 480 (Roche, Sweden). Melting curve analysis was performed to ensure that only one PCR product was obtained. For quantification and for estimation amplification efficiency, a standard curve was generated using increasing concentrations of cDNA. The amplification transcripts were quantified with the relative standard curve and normalized against the reference genes Rn 18S, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein (Ywhaz) and glyceraldehyde 3-phosphate dehydrogenase (Gadph).
Caspase-3, MRP1/Abcc1 immunoreactivity and TUNEL labeling
To assess apoptotic activity and to localise efflux transport protein in choroid plexuses, cleaved caspase-3 and MRP1/Abcc1 immunoreactivity was detected on tissue sections. MRP1 has been shown to be specifically localized to the choroid plexus in the rat (Gazzin et al 2008; Ek et al 2010). At 24 h after 1 mg/kg LPS injection, saline injection or HI 50/20, LPS/HI animals were collected and brains prepared for immunocytochemical analysis (n=5 for each group, mixed from 2–3 litters). Pups were transcardially perfused with cold heparinised saline for about 30 s followed by cold Histofix (Histolab, Sweden) for 5 min using a syringe pump. Flow rate was set to the estimated total blood volume (10 % of body weight) per minute. Brains were dissected out and left in Histofix overnight in the fridge, washed in 70 % ethanol, embedded in paraffin and coronal 5 μm sections cut of the hemispheres. For immunohistochemistry, sections were baked in 60 °C oven, paraffin removed by xylene, and sections rehydrated in decreasing concentrations of ethanol. Sections were then gently boiled in citrate buffer (pH 6.0) for 10 min and blocked by incubation in 3 % H2O2 in PBS for 10 min followed by 10 % goat serum for 30 min, before incubation in rabbit anti-active caspase-3 (1:100; BD Pharmingen, Cat#559565) or rabbit anti-MRP1 (1:100; Alexis, Cat#ALX-210-841) overnight in fridge. Sections were then incubated for 2 h with biotinylated secondary antibodies against the appropriate species (1:250; Vector), followed by avidin-peroxide complex (ABC kit PK-6100; Vector) for 1 h, and developed with DAB kit (SK-4100, Vector) including nickel enhancement according to manufacturer’s recommendations.
In addition, DNA strand breaks were detected with deoxynucleotidyl transferease-mediated dUTP nick end labeling (TUNEL). For this, sections were processed as above including citrate buffer treatment, blocked in 3 % bovine serum albumin (30 min) and incubated in TUNEL reaction mixture, made up of 150 U/mL recombinant deoxynucleotidyl transferase (rTdT; Invitrogen, Carlsbad, USA) and 20 nmol/mL biotin-16-dUTP (Roche Applied Science), at 37 °C for 60 min. Visualization was carried out as above with ABC-kit and DAB. Control sections included omission of primary antibodies for immunoreactivity and rTDT for TUNEL labeling.
Statistics
qPCR data collected from LPS injected mice were analyzed with two–way ANOVA followed by Bonferroni’s post-hoc test with a 95 % confident interval. qPCR data from the HI and LPS/HI experiments were determined using paired t-test with a 95 % confident interval. Data from the LPS model are presented as fold change over control±SEM, while data from the HI (HI20/50) and LPS/HI models are presented as fold change between left and right ventricular (left/right) plexus±SEM. Significance levels were set at * p≤0.05, ** p≤0.01, *** p≤0.001. All statistical analyses were performed using GraphPad Prism (GraphPad Software).
Results
Effect of LPS on Nrf2 and Nrf2–gene targets
To examine the effect of systemic inflammation on the Nrf2-system in the choroid plexus, the expression of Nrf2 and its gene targets was studied after systemic LPS administration. Analysis was performed at different time points (2-, 6-, 14-, 24- and 72-h) comparing a saline-injected group of mice (control) with animals injected with 0.3 mg/kg (all time points) or 1.0 mg/kg (24-h time point) of LPS (Fig. 1). The gene expression of Nrf2 and Nrf2 target gene Gclc, was decreased at 24 h after 0.3 mg/kg and 1 mg/kg LPS. For Nrf2 there was a 51 % decrease compared to control after 0.3 mg/kg (p≤0.05) and 62 % decrease after 1 mg/kg (p≤0.05), and for Gclc there was a 56 % decrease after 0.3 mg/kg (p≤0.01) and 57 % after 1 mg/kg (p≤0.05) but either was unaffected at other survival times (Fig. 1a & b). The mRNA expression for Gclm was significantly upregulated by low dose of LPS (0.3 mg/kg) at 2 h (35 % increase compared to control, p≤0.05), while it was down-regulated by 1 mg/kg LPS at 24 h (56 % decrease compared to control, p≤0.05, Fig. 1c). A bi-phasic response was seen for HO-1 transcript after low dose of LPS: mRNA was strongly upregulated at early survival times (2 h: 79 % of increase compared to control, p ≤0.001; 6 h: 65 % increase compared to control, p≤0.01; Fig. 1d) and at the later time point (72 h: 105 % increase compared to control, p≤0.01; Fig. 1d). No effect was seen at other times or after 24 h of 1 mg/kg LPS dose.
Fig. 1.
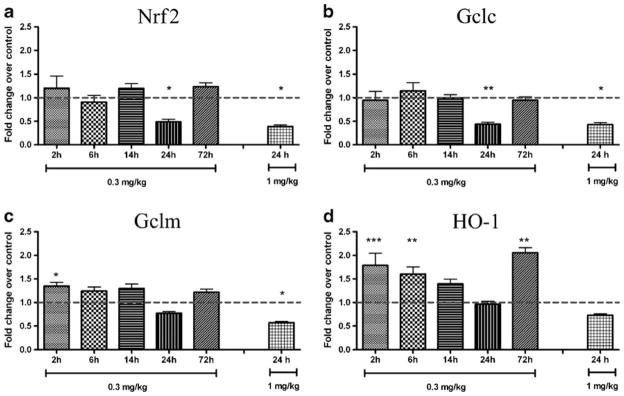
Expression of mRNA of Nrf2 and Nrf2 target genes in choroid plexus after systemic LPS injection. The mRNA expression of Nrf2 and its gene targets were analyzed in choroid plexuses of PND9 mice after i.p. injection of low dose LPS (0.3 mg/kg) at 2–72 h (n=8–16 per group) and at 24 h after high dose LPS (1 mg/kg; n=8). Reverse transcription-quantitative PCR analysis showed a down-regulation of Nrf2 expression at 24 h after injection with both LPS doses (a). Similarly, at 24 h both LPS doses reduced the mRNA of Gclc (b). Gclm was upregulated by low dose of LPS at 2 h while it was down-regulated by high LPS dose at 24 h after treatment (c). A dual phase effect was seen for mRNA of HO-1 after low dose of LPS: mRNA was strongly increased both at early survival times (2 and 6 h) and late time point (72 h). No effect was on HO-1 seen with the higher LPS dose 24 h following the LPS injection (d). Data are shown as fold change over control±SEM, *p≤0.05, **p≤0.01, ***p≤0.001
Effect of HI50 on Nrf2 and Nrf2–gene targets
To investigate the effects of non-infectious injury on the Nrf2-system the mRNA expression of Nrf2 and its target genes were analyzed at 2-, 6-, 24-h following HI50. As has been shown previously (Svedin et al 2007), this insult results in unilateral injury confined to brain regions in the left hemisphere, especially the hippocampus but also cortex, striatum and thalamus whereas the right (contralateral) hemisphere remains undamaged. At 2 h after HI50, Nrf2 mRNA expression in plexuses from the left lateral ventricle was significantly higher compared to expression in plexuses from the right lateral ventricle (50 % higher, p ≤0.01; Fig. 2a) but was not different at 6 and 24 h. Similarly to Nrf2, higher mRNA expressions for Gclc and Gclm were detected at 2 h in plexuses from left ventricle compared to the right (260 % and 130 % higher for Gclc and for Gclm, respectively, p≤0.05; Fig. 2b & c). The expression of Gclc in the plexus from the left ventricle was also significantly higher at 6 h (103 % higher, p<0.05; Fig. 2b). HO-1 mRNA in the left plexus was strongly upregulated at 2 h compared to right plexus (541 % higher, p≤0.01; Fig. 2d) and was also significantly higher 6 h after HI50 (189 % higher, p≤0.01; Fig. 2d).
Fig. 2.
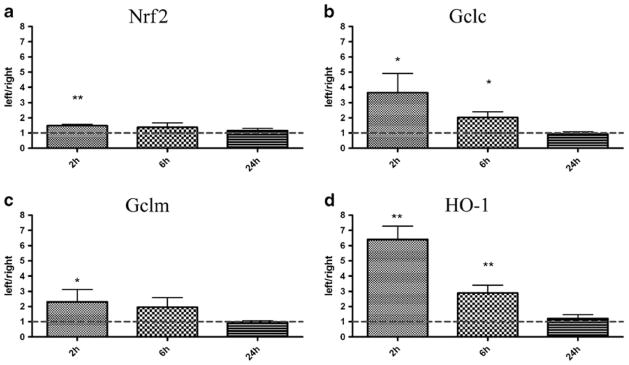
Expression of mRNA for Nrf-2 and Nrf2 target genes in choroid plexuses after 50 min hypoxia-ischemia (HI50). The mRNA expression for Nrf2 and its gene targets were analyzed in choroid plexuses from the ipsilateral (left) and contralateral (right) ventricles at 2-, 6- and 24-h after 50 min HI (HI50) at PND9. Nrf2 gene expression in plexuses from ipsilateral side was significantly higher at 2 h but not at 6- and 24-h after HI in comparison to the plexuses from the contralateral hemisphere (a). All Nrf2 target genes showed similar temporal changes after HI50 with the highest upregulation at 2 h, less upregulation at 6 h and normal levels at 24 h (b–d). Data are shown as fold change between left and right ventricular (left/right) plexus±SEM, *p≤0.05, **p≤0.01
Effect of LPS/HI on Nrf2 and Nrf2–gene targets
To examine whether pre-exposure of LPS influences the expression of antioxidant genes after HI, we investigated the effect of combined LPS/HI. Analysis was performed 2 h after LPS/HI or HI20, the time point with the maximum effect on anti–oxidant responses observed after HI50 alone (Fig. 2). Similar to the results shown following HI50 (Fig. 2a), HI20 treatment increased the Nrf2 mRNA expression in the plexus of the left lateral ventricle (25 % higher, p≤0.05; Fig. 3a) compared to the right, while LPS exposure prior to HI (LPS/HI) abolished this effect (left/right ratio 0.96, p =0.44; Fig. 3a). Gclc mRNA was significantly upregulated in the left plexus 2 h after both HI20 (61 % higher, p≤ 0.05; Fig. 3a) and LPS/HI (78 % higher, p ≤0.01; Fig. 3b). HI20 did not affect the Gclm transcript while there was a down-regulation of the mRNA in the plexus in the left ventricle following LPS/HI (15 % decrease, p≤0.05; Fig. 3c). HO-1 mRNA in the left plexus was upregulated in both HI20 (178 % higher, p ≤0.05; Fig. 3d) and LPS/HI pups (200 % higher, p ≤0.01; Fig. 3d).
Fig. 3.
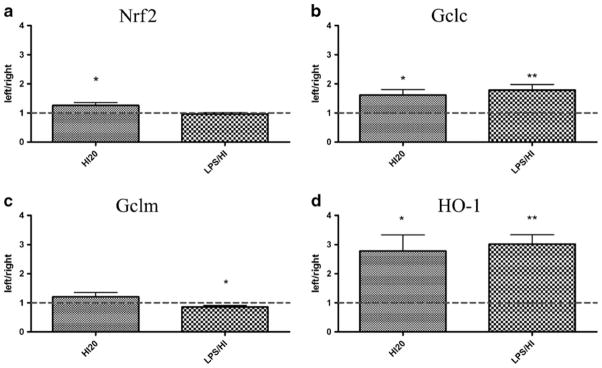
Expression of mRNA for Nrf-2 and Nrf2 target genes in choroid plexuses after LPS/hypoxia-ischemia (LPS/HI). The mRNA expression for Nrf2 and its gene targets were analyzed in choroid plexuses from the ipsilateral (left) and contralateral (right) ventricles at 2 h after saline or LPS administration combined with 20 min HI at PND9 (HI20 and LPS/HI, respectively). Nrf2 gene expression in plexuses from left side was significantly higher after saline exposure but not after LPS (a). Gclm was not altered by saline while it was significantly down-regulated in the left choroid plexus after LPS (c). Gclc (b) and HO-1 (d) genes were upregulated in both saline and LPS injected animals. No significant relevance between two groups. Data are shown as fold change between left and right ventricular (left/right) plexus±SEM, *p≤0.05, **p≤0.01
Effects of inflammation on transport-related genes and efflux protein
To investigate the expression of efflux and detoxification protein in choroid plexus, mRNA levels of MRP1/Abcc1 and Glutathione-S-transferase (Gst) were analyzed after LPS (2–72 h), HI50 (2–24 h), HI20 (2 h) and LPS/HI (2 h). There were no significant changes in MRP1 mRNA expression following LPS (Fig. 4a) or HI50/HI20 (Fig. 4c & e), while LPS/HI induced upregulation of the transcript in the left choroid plexus compared to the right (65 % higher, p≤0.05; Fig. 4e).
Fig. 4.

Transport-related mRNA in choroid plexus. Expression of transport related genes, MRP1/Abcc1 and Glutathione-S-transferase (Gst), in choroid plexus were analyzed in LPS, HI50, HI20, and LPS/HI groups. MRP1 mRNA expression was not altered by either LPS (a) or HI50 (c) but was significantly upregulated in the left plexus after LPS/HI (e). Immunoreactivity for MRP1 in control PND9 choroid plexus was specifically localized to the basolateral membranes of the epithelial cells (g). The staining pattern was not affected by LPS, HI20/50 or LPS/HI, and no staining was detected in other parts of brain (not illustrated). Gst mRNA expression was not significantly affected by LPS (b) while it was strongly increased at 2- and 24-h in HI50 (d) and LPS/HI groups (f). RT-PCR data in LPS group are shown as fold change over control±SEM. RT-PCR data in HI50, HI20, and LPS/HI groups are shown as fold change between left and right ventricular (left/right) plexus±SEM, *p≤0.05, **p≤0.01. Scale bar in D represents 25 μm
The presence of MRP1 protein was also evaluated in tissue sections by immunohistochemistry. In brains from naïve control animals MRP1 protein expression was specifically localized to the choroid plexus and was not detected in other parts of the brain (not illustrated). Control tissue sections showed basolateral localization of this protein in choroidal epithelial cells (Fig. 4g) and this arrangement was not affected in all treatment groups (not illustrated).
The mRNA levels of Gst did not change after LPS (Fig. 4b), while it was higher in left ventricular plexuses both 2 h (640 % higher, p≤0.01) and 24 h after HI50 (690 % higher, p≤0.01; Fig. 4d). HI20 did not affect the mRNA expression of Gst, while there was an increase in expression following LPS/HI in the left plexus compared to the right (262 % higher, p≤0.05; Fig. 4f).
Caspase-3 immunoreactivity and TUNEL reaction
Immunohistochemistry for active caspase-3 was performed to detect apoptosis in choroid plexus after all treatments. In control tissues at PND9 occasional cells in the hippocampus, cortex and thalamus were positive for caspase-3 but the choroid plexus epithelial cells remained devoid of staining (Fig. 5a). No obvious differences in the number of caspase-3 positive cells could be seen after 1 mg/kg LPS injections compared to control tissue (Fig. 5b). In contrast, after HI50 a great number of caspase-3 positive cells were present in brain regions around the left brain ventricle (Fig. 5c). HI20 resulted in some caspase-3 positive cells in hippocampus, while following LPS/HI there were a large number of caspase-3 positive cells in tissues surrounding the lateral ventricles. However, no caspase-3 positive epithelial cells were detected in the choroid plexus following any treatment group. The staining pattern for TUNEL reaction was very similar to caspase-3 immunoreactivity with a large number of labeled brain parenchymal cells in HI50 and LPS/HI animals, some in hippocampus of HI20 animals and very occasional cells in other groups, however, no cells were labeled in choroid plexus in any group (not illustrated).
Fig. 5.

Caspase-3 immunoreactivity after systemic LPS and hypoxia-ischemia (HI). In control tissue (a) as well as 24 h after 1 mg/kg LPS (b), occasional cells were positive for caspase-3 (arrowheads). At 24 h after HI a great number of caspase-3 positive cells were found in hippocampal and thalamic regions surrounding the lateral ventricle in the ipsilateral hemisphere, however, no positive cells were found in the choroid plexus. Lesions were also beginning to appear in tissue at this time (asterix in C). d is higher power micrograph from C showing that none of the choroidal epithelial cells were caspase-3 positive. In HI20 animals, the brain region around the lateral ventricles showed few caspase-3 positive cells (e) with no positive cells in the plexus (f) and in LPS/HI animals extensive caspase-3 labeling was present around ventricles (g) yet no positive cells in plexus (h). Note that all sections were counterstained with acid fuchsin and that no apparent abnormalities were found in choroidal epithelial cells after treatments. This indicates no obvious damage in the choroidal barrier forming cells after either LPS or HI. Scale bar represents 100 μm in A&B, 200 μm in C, E, G and 50 μm in D, F, H
Discussion
In this work we report modulation of the mRNA expression of several components of the Nrf2–dependent antioxidant system in choroid plexus after three different insults in newborn mice. Systemic inflammation, elicited by LPS, induced mixed responses in Nrf2-dependent genes in the choroid plexus, while a prolonged period of HI (50 min) generally resulted in a marked increase in the expression of these transcripts. When LPS preceded HI, several of the HI-induced modulations were prevented. None of the insults were associated with apoptotic cell death or alterations in expression of one of the major efflux transport proteins in the choroid plexus, except for LPS/HI that induced an increased expression of MRP1.
In general, LPS induced a down-regulation of the mRNA expression for Nrf2, Gclc and Gclm at 24 h, an effect that was most pronounced with the higher LPS dose (see Fig. 1). Similarly, Nrf2 regulation has been found to be reduced in liver (Ko et al 2008) and eye (Nagai et al 2009) following LPS-induced endotoxemia, which was related to inflammatory processes. Activation of Nrf2 has been shown to lessen oxidative stress by induction of phase II detoxifying antioxidant enzymes (Calkins et al 2010; Chen et al 2009; Shih et al 2005; Vargas et al 2008). We have previously shown that cell medium from LPS stimulated microglia decreased components of the Nrf2-system in parallel with activation of p38 MAPK in astrocytes (Correa et al 2011). The negative effects on the Nrf2-system were partly reversed by an inhibitor of p38 which protected against oxidative stress. Taken together, our and other data suggest that LPS can have a detrimental effect on several of the Nrf2-dependent antioxidative responses in choroid plexus. There was a dual response of HO-1 in the choroid plexus after LPS with an early upregulation of gene expression at 2 h and 6 h and a second more pronounced effect at 72 h (see Fig. 1d). Our findings are similar to results obtained in adult animals, in which a subseptic dose of LPS induced HO-1 mRNA in the hippocampus and hypothalamus (Maeda et al 2008). HO-1 is also induced in other highly vascularized tissues, such as heart and lung by systemic LPS (Wiesel et al 2000). It is generally accepted that the acute induction of this enzyme is predominantly cytoprotective, however protracted or repeated up-regulation of the HO-1 gene has been suggested to perpetuate cellular dysfunction (Schipper et al 2009). The biphasic effect seen in HO-1 expression after LPS exposure is intriguing and has several similarities with other investigations on systemic inflammation and BBB function (Huber et al 2006). These studies also showed that a peripheral inflammatory stimuli (inflammatory pain) elicites a biphasic responses at the blood–brain barrier with different pathophysiological profiles.
It is unclear from our studies whether the effects noted on the Nrf2 system in the choroid plexus following systemic LPS administration are direct or indirect via peripheral inflammatory factors. The LPS receptor, toll-like receptor (TLR) 4, is present in the choroid plexus and circumventricular organs, and it has been suggested that signals from the periphery are mediated via this route to induce inflammation in the brain parenchyma (Laflamme and Rivest 2001). A recent micro-array analysis revealed that the mouse choroid plexus displays an acute-phase response after systemic LPS, with an up-regulation of genes implicated in immune-mediated cascades and in extracellular matrix remodeling, whereas genes that codes for protein that participate in maintenance of the barrier function were down-regulated (Marques et al 2009). We have recently investigated the mRNA expression of TLRs in the choroid plexus of newborn mice and found that several of the receptors are regulated following peripheral inflammation (Stridh et al, unpublished). Taken together, the studies suggest that circulating LPS is likely to be able to bind to TLR4 at the choroid plexus, resulting in the changes noted in the epithelial plexus cells.
The mRNA expression of Nrf2 and Nrf2–gene targets were increased immediately after HI50/20, but all mRNA changes had returned to normal levels by 24 h (see Figs. 2 and 3). The increase in mRNA for Nrf2 and Nrf2-regulated genes is likely due to an early endogenous response to oxidative stress, which is known to occur during the early reperfusion phase after neonatal HI (Ferriero 2001). Interestingly, when LPS administration preceded HI (LPS/HI), increased mRNA expression for both Nrf2 and Gclm was prevented. Speculatively, such lack of response of components of the endogenous anti-oxidant system may be associated with the increased vulnerability to HI damage following LPS pre-exposure that we have observed earlier (Wang et al 2007).
Apart from functioning as a physical barrier between blood and CSF, the plexus contain active transport mechanisms that restrict compounds from entering the CSF as well as clearing deleterious organic ions from CSF to blood (Ghersi-Egea et al 2006). Together, these mechanisms protect the brain from toxic compounds that may enter or accumulate inside the CSF and brain. MRP1 belongs to the multi-drug resistance related proteins (ABCc family) and actively efflux phase II modified (i.e., glutathione-conjugated) compounds out of cells (Borst et al 2000). Several previous studies in rats have shown that this protein is localized mainly to the epithelial cells of the choroid plexus and may play an important detoxifying role in this organ (Gazzin et al 2008; Ek et al 2010). We could confirm localization of MRP1 to the basolateral membranes of epithelial cells of mice (Fig. 4c). Localization of protein to the blood-facing side of cells implement transport from epithelial cells into the blood, thus, contributing both to barrier function and possibly also removal of toxic compounds from CSF. We did not observe any changes in either the gene or immunohistochemical expression of MRP1 after LPS or HI. However, there was an increase in the mRNA expression of MRP1 following LPS/HI, suggesting a possible effect on transport proteins in this model.
In contrast to MRP1, Gst mRNA was significantly upregulated following both HI50 and LPS/HI (Fig. 4d & f), suggesting that transport mechanisms may be modulated at the glutathione-conjugation step under these conditions. The somewhat discrepant results between the effects on Gst and MRP1 following HI is intriguing, as both Gst and MRP1 have been reported to contain an antioxidant-responsive element (ARE) in the promoter region (Wasserman and Fahl 1997; Kwak et al 2003; Kauffmann et al 2002). However, several other transcription factors appears to be involved in regulation of MRP1 transcription (Meijerman et al 2008; Klaassen and Slitt 2005), while transcriptional regulation of Gsts seems to be mainly Nrf2-dependent (Tirona and Kim 2005). It is thus possible that activation of MRP1 needs activation of additional transcription factors than Nrf2 but that Nrf2 activation is sufficient for Gst gene transcription during HI alone.
We did not find any evidence of caspase-3 activity or any DNA strand breaks in the choroid plexus and the general morphology appeared normal up until 24 h after LPS, HI20/50 and LPS/HI, suggesting that there was no overt damage to this tissue, although surrounding brain tissue showed clear evidence of necrosis and apoptosis (Fig. 5). This is in contrast to a previous study of the choroid plexus after neonatal HI in the rat that showed extensive necrosis of choroid epithelial cells soon after insult (Rothstein and Levison 2002). These differences are difficult to explain but could be due to the different species used, with mouse choroid plexus being less susceptible to HI than the rat. The discrepancy could also be a result of different severity of the overall brain injury in these two models, however, this seems less likely as there was extensive tissue damage in the surrounding brain tissues following both HI and LPS/HI in our study.
Nrf2 is a key regulator of endogenous antioxidant systems (Itoh et al 1999; Johnson et al 2002; Lee et al 2003) and has been shown to play a neuroprotective role in the adult by preserving blood–brain barrier function following traumatic brain injury (Zhao et al 2007). Activation of Nrf2 by isothiocyanates in vitro was recently reported to protect from oxidative damage at the blood-CSF interface by maintaining low permeability of choroid plexus epithelial cells subjected to H2O2 stress (Xiang et al 2012). In the present study, the decreased expression of mRNA for Nrf2 and Nrf2-regulated target genes following LPS implies that the function of the inducible antioxidant system in the choroid plexus is down-regulated by inflammation, even if choroid cells are not structurally damaged. Further, LPS prevented the endogenous antioxidant response following HI, suggesting the possibility that peripheral inflammation may contribute to increased vulnerability of the brain via oxidative mechanisms at the blood–cerebrospinal fluid barrier interface.
Acknowledgments
We would like to thank Simon Klein for his technical help in the preparation of tissues for this study.
Funding This research received financial assistance from the Swedish Medical Research Council (VR 2009–2630; 2009–4067), a Government grant to a researcher in Public Health Service at the Sahlgrenska University Hospital (ALFGBG-142881), European Union grant FP7, (Neurobid, HEALTH-F2-2009-241778), the Leducq foundation (DSRR_P34404), National Institutes of Health (GM 44842), Parkinson-Fonden and Åhlén-stiftelsen and Wilhelm and Martina Lundgren Foundation.
Footnotes
Conflict of interest None.
Presented at the “Brains for Brain Meeting”, Frankfurt, Germany, 9–11 March 2012.
Contributor Information
Barbara D’Angelo, Department of Neuroscience and Physiology, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
C. Joakim Ek, Email: joakim.ek@neuro.gu.se, Department of Neuroscience and Physiology, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Mats Sandberg, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Carina Mallard, Department of Neuroscience and Physiology, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
References
- Beiswanger CM, Diegmann MH, Novak RF, et al. Developmental changes in the cellular distribution of glutathione and glutathi-one S-transferases in the murine nervous system. Neurotoxicol. 1995;16:425–440. [PubMed] [Google Scholar]
- Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst. 2000;92:1295–1302. doi: 10.1093/jnci/92.16.1295. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Vargas MR, Johnson DA, Johnson JA. Astrocyte-specific overexpression of Nrf2 protects striatal neurons from mitochondrial complex II inhibition. Toxicol Sci. 2010;115:557–568. doi: 10.1093/toxsci/kfq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, Vargas MR, Pani AK, et al. Nrf2-mediated neuro-protection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc Natl Acad Sci USA. 2009;106:2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa F, Ljunggren E, Mallard C, Nilsson M, Weber SG, Sandberg M. The Nrf2-inducible antioxidant defense in astrocytes can be both up- and down-regulated by activated microglia: Involvement of p38 MAPK. Glia. 2011;59:785–799. doi: 10.1002/glia.21151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek CJ, Habgood MD, Dziegielewska KM, Saunders NR. Structural characteristics and barrier properties of the choroid plexuses in developing brain of the opossum (Monodelphis Domestica) J Comp Neurol. 2003;460:451–464. doi: 10.1002/cne.10661. [DOI] [PubMed] [Google Scholar]
- Ek CJ, Wong A, Liddelow SA, Johansson PA, Dziegielewska KM, Saunders NR. Efflux mechanisms at the developing brain barriers: ABC-transporters in the fetal and postnatal rat. Toxicol Lett. 2010;197:51–59. doi: 10.1016/j.toxlet.2010.04.025. [DOI] [PubMed] [Google Scholar]
- Ek CJ, Dziegielewska KM, Habgood MD, Saunders NR. Barriers in the developing brain and Neurotoxicology. Neurotoxicol. 2012;33:586–604. doi: 10.1016/j.neuro.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Eklind S, Mallard C, Leverin AL, et al. Bacterial endotoxin sensitizes the immature brain to hypoxic–ischaemic injury. Eur J Neurosci. 2001;13(6):1101–1106. doi: 10.1046/j.0953-816x.2001.01474.x. [DOI] [PubMed] [Google Scholar]
- Ferriero DM. Oxidant mechanisms in neonatal hypoxia-ischemia. Dev Neurosci. 2001;23:198–202. doi: 10.1159/000046143. [DOI] [PubMed] [Google Scholar]
- Garbuzova-Davis S, Louis MK, Haller EM, Derasari HM, Rawls AE, Sanberg PR. Blood–brain barrier impairment in an animal model of MPS III B. PLoS One. 2011;6:e16601. doi: 10.1371/journal.pone.0016601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzin S, Strazielle N, Schmitt C, et al. Differential expression of the multidrug resistance-related proteins ABCb1 and ABCc1 between blood–brain interfaces. J Comp Neurol. 2008;510:497–507. doi: 10.1002/cne.21808. [DOI] [PubMed] [Google Scholar]
- Ghersi-Egea JF, Strazielle N, Murat A, Jouvet A, Buenerd A, Belin MF. Brain protection at the blood-cerebrospinal fluid interface involves a glutathione-dependent metabolic barrier mechanism. J Cereb Blood Flow Metab. 2006;26:1165–1175. doi: 10.1038/sj.jcbfm.9600267. [DOI] [PubMed] [Google Scholar]
- Gulcan H, Ozturk IC, Arslan S. Alterations in antioxidant enzyme activities in cerebrospinal fluid related with severity of hypoxic ischemic encephalopathy in newborns. Biol Neonate. 2005;88:87–91. doi: 10.1159/000084905. [DOI] [PubMed] [Google Scholar]
- Hedtjärn M, Leverin AL, Eriksson K, Blomgren K, Mallard C, Hagberg H. Interleukin-18 involvement in hypoxic-ischemic brain injury. J Neurosci. 2002;22:5910–5919. doi: 10.1523/JNEUROSCI.22-14-05910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber JD, Campos CR, Mark KS, Davis TP. Alterations in blood–brain barrier ICAM-1 expression and brain microglial activation after lambda-carrageenan-induced inflammatory pain. Am J Physiol Heart Circ Physiol. 2006;290:H732–740. doi: 10.1152/ajpheart.00747.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- Itoh S, Yanagishita T, Aoki S, et al. Generation of free radicals and the damage done to the sarcoplasmic reticulum during reperfusion injury following brief ischemia in the canine heart. Jpn Circ J. 1999;63:373–378. doi: 10.1253/jcj.63.373. [DOI] [PubMed] [Google Scholar]
- Johanson CE. Ventricles and cerebrospinal fluid. In: Conn PM, editor. Neuroscience in medicine. J.B Lippincott Company; Philadelphia: 1995. pp. 171–196. [Google Scholar]
- Johanson CE, Jones HC, Stopa EG, Ayala C, Duncan JA, McMillan PN. Enhanced expression of the NA-K-2 Cl cotransporter at different regions of the blood-CSF barrier in the perinatal H-Tx rat. Eur J Pediatr Surg. 2002;12:S47–49. [PubMed] [Google Scholar]
- Johansson PA, Dziegielewska KM, Ek CJ, et al. Blood-CSF barrier function in the rat embryo. Eur J Neurosci. 2006;24:65–76. doi: 10.1111/j.1460-9568.2006.04904.x. [DOI] [PubMed] [Google Scholar]
- Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem. 2002;81:1233–1241. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- Kauffmann HM, Pfannschmidt S, Zoller H, et al. Influence of redox-active compounds and PXR-activators on human MRP1 and MRP2 gene expression. Toxicology. 2002;171:137–146. doi: 10.1016/s0300-483x(01)00570-4. [DOI] [PubMed] [Google Scholar]
- Klaassen CD, Slitt A. Regulation of hepatic transporters by xenobiotic receptors. Curr Drug Metab. 2005;6:309–328. doi: 10.2174/1389200054633826. [DOI] [PubMed] [Google Scholar]
- Ko K, Yang H, Noureddin M, et al. Changes in S-adenosylmethionine and GSH homeostasis during endotoxemia in mice. Lab Invest. 2008;88:1121–1129. doi: 10.1038/labinvest.2008.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1–Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001;15:155–63. doi: 10.1096/fj.00-0339com. [DOI] [PubMed] [Google Scholar]
- Lee JM, Shih AY, Murphy TH, Johnson JA. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem. 2003;278:37948–37956. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Temple S, Møllgård K, et al. Molecular characterisation of transport mechanisms at the developing mouse blood-CSF interface: a transcriptome approach. PLoS One. 2012;7:e33554. doi: 10.1371/journal.pone.0033554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Nakatsuka I, Hayashi Y, Higuchi H, Shimada M, Miyawaki T. Heme oxygenase-1 induction in the brain during lipopolysaccharide-induced acute inflammation. Neuropsychiatr Dis Treat. 2008;4:663–667. doi: 10.2147/ndt.s3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Coppola G, et al. Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J Cereb Blood Flow Metab. 2009;29:921–932. doi: 10.1038/jcbfm.2009.15. [DOI] [PubMed] [Google Scholar]
- Meijerman I, Beijnen JH, Schellens JH. Combined action and regulation of phase II enzymes and multidrug resistance proteins in multidrug resistance in cancer. Cancer Treat Rev. 2008;34:505–520. doi: 10.1016/j.ctrv.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Nagai N, Thimmulappa RK, Cano M, et al. Nrf2 is a critical modulator of the innate immune response in a model of uveitis. Free Radic Biol Med. 2009;47:300–306. doi: 10.1016/j.freeradbiomed.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogihara T, Hirano K, Ogihara H, et al. Non-protein-bound transition metals and hydroxyl radical generation in cerebrospinal fluid of newborn infants with hypoxic ischemic encephalopathy. Pediatr Res. 2003;53:594–599. doi: 10.1203/01.PDR.0000054685.87405.59. [DOI] [PubMed] [Google Scholar]
- Ping Z, Liu W, Kang Z, et al. Sulforaphane protects brains against hypoxic-ischemic injury through induction of Nrf2-dependent phase 2 enzyme. Brain Res. 2010;1343:178–185. doi: 10.1016/j.brainres.2010.04.036. [DOI] [PubMed] [Google Scholar]
- Rothstein RP, Levison SW. Damage to the choroid plexus, ependyma and subependyma as a consequence of perinatal hypoxia/ischemia. Dev Neurosci. 2002;24:426–436. doi: 10.1159/000069052. [DOI] [PubMed] [Google Scholar]
- Saha A, Sarkar C, Singh SP, et al. The blood–brain barrier is disrupted in a mouse model of infantile neuronal ceroid lipofuscinosis: amelioration by resveratrol. Hum Mol Genet. 2012;21:2233–2244. doi: 10.1093/hmg/dds038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sävman K, Nilsson UA, Blennow M, Kjellmer I, Whitelaw A. Non-protein-bound iron is elevated in cerebrospinal fluid from preterm infants with posthemorrhagic ventricular dilatation. Pediatr Res. 2001;49:208–212. doi: 10.1203/00006450-200102000-00013. [DOI] [PubMed] [Google Scholar]
- Schipper HM, Song W, Zukor H, Hascalovici JR, Zeligman D. Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. J Neurochem. 2009;110:469–485. doi: 10.1111/j.1471-4159.2009.06160.x. [DOI] [PubMed] [Google Scholar]
- Senjo M, Ishibashi T, Terashima T, Inoue Y. Successive appearance of glutathione S-transferase-positive cells in developing rat brain: choroid plexus, pia mater, ventricular zone and astrocytes. Neurosci Lett. 1986;66:131–134. doi: 10.1016/0304-3940(86)90178-3. [DOI] [PubMed] [Google Scholar]
- Shih AY, Imbeault S, Barakauskas V, Erb H, Jiang L, Li P, Murphy TH. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J Biol Chem. 2005;280:22925–22936. doi: 10.1074/jbc.M414635200. [DOI] [PubMed] [Google Scholar]
- Strazielle N, Ghersi-Egea JF. Choroid plexus in the central nervous system: biology and physiopathology. J Neuropathol Exp Neurol. 2000;59:561–574. doi: 10.1093/jnen/59.7.561. [DOI] [PubMed] [Google Scholar]
- Strazielle N, Khuth ST, Ghersi-Egea JF. Detoxification systems, passive and specific transport for drugs at the blood-CSF barrier in normal and pathological situations. Adv Drug Deliv Rev. 2004;56:1717–1740. doi: 10.1016/j.addr.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Sun Y, Liou B, Ran H, et al. Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum Mol Genet. 2010;19:1088–1097. doi: 10.1093/hmg/ddp580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedin P, Hagberg H, Sävman K, Zhu C, Mallard C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. J Neurosci. 2007;27:1511–1518. doi: 10.1523/JNEUROSCI.4391-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirona RG, Kim RB. Nuclear receptors and drug disposition gene regulation. J Pharm Sci. 2005;94:1169–1186. doi: 10.1002/jps.20324. [DOI] [PubMed] [Google Scholar]
- Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci. 2008;28:13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Svedin P, Nie C, et al. N-acetylcysteine reduces lipopolysaccharide-sensitized hypoxic-ischemic brain injury. Ann Neurol. 2007;61:263–271. doi: 10.1002/ana.21066. [DOI] [PubMed] [Google Scholar]
- Wang X, Stridh L, Li W, et al. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol. 2009;183:7471–7477. doi: 10.4049/jimmunol.0900762. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci USA. 1997;94:5361–5266. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel P, Foster LC, Pellacani A, et al. Thioredoxin facilitates the induction of heme oxygenase-1 in response to inflammatory mediators. J Biol Chem. 2000;275:24840–24846. doi: 10.1074/jbc.M000835200. [DOI] [PubMed] [Google Scholar]
- Wu YP, Proia RL. Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proc Natl Acad Sci USA. 2004;101:8425–8430. doi: 10.1073/pnas.0400625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J, Alesi GN, Zhou N, Keep RF. Protective effects of isothiocyanates on blood-CSF barrier disruption induced by oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2012;303:R1–7. doi: 10.1152/ajpregu.00518.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Moore AN, Redell JB, Dash PK. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007;27:10240–10248. doi: 10.1523/JNEUROSCI.1683-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]