

Abstract
The MtrCDE multidrug pump, from Neisseria gonorrhoeae, is assembled from the inner and outer membrane proteins MtrD and MtrE, which are connected by the periplasmic membrane fusion protein MtrC. Although it is clear that MtrD delivers drugs to the channel of MtrE, it remains unclear how drug delivery and channel opening are connected. We used a vancomycin sensitivity assay to test for opening of the MtrE channel. Cells expressing MtrE or MtrE-E434K were insensitive to vancomycin; but became moderately and highly sensitive to vancomycin respectively, when coexpressed with MtrC, suggesting that the MtrE channel opening requires MtrC binding and is energy-independent. Cells expressing wild-type MtrD, in an MtrCE background, were vancomycin-insensitive, but moderately sensitive in an MtrCE-E434K background. The mutation of residues involved in proton translocation inactivated MtrD and abolished drug efflux, rendered both MtrE and MtrE-E434K vancomycin-insensitive; imply that the pump–component interactions are preserved, and that the complex is stable in the absence of proton flux, thus sealing the open end of MtrE. Following the energy-dependent dissociation of the tripartite complex, the MtrE channel is able to reseal, while MtrE-E434K is unable to do so, resulting in the vancomycin-sensitive phenotype. Thus, our findings suggest that opening of the OMP via interaction with the MFP is energy-independent, while both drug export and complex dissociation require active proton flux.
Introduction
Gram-negative bacteria utilize homologous tripartite-transport systems to pump cytotoxic compounds, including antibiotics and metals (Pietras et al., 2008; Blair and Piddock, 2009; Misra and Bavro, 2009; Su et al., 2011a) from the cell. These tripartite pumps are composed of inner (IMP) and outer (OMP) membrane proteins, which are connected by a periplasmic membrane fusion protein (MFP). There are 3D structures for all of the individual components: for example, in the case of the AcrABTolC multidrug pump and CusABC copper pump from Escherichia coli, the structure of the IMPs AcrB (Murakami et al., 2002; Yu et al., 2003) and CusA (Long et al., 2010), and of their cognate MFPs, AcrA (Mikolosko et al., 2006) and CusB (Su et al., 2009), and OMPs, TolC (Koronakis et al., 2000) and CusC (Kulathila et al., 2011), have been determined. While the different tripartite pumps use structurally related MFPs and OMPs they use at least three different types of IMPs: predominantly proton-driven transporters, belonging to the RND and MF family, but some also use ABC transporters. For example, the AcrABTolC multidrug pump and CusABC copper pump utilize the RND transporters AcrB (Murakami et al., 2002) and CusA (Long et al., 2010), respectively, while the E. coli EmrABTolC and MacABTolC antibiotic pumps utilizes the MF and ABC transporters EmrB (Lomovskaya and Lewis, 1992) and MacB (Kobayashi et al., 2001) respectively. In addition, the OMPs of the TolC family also participate in Type I secretion systems for protein toxins, such as the HlyBDTolC pump that secretes α-haemolysin (HlyA) (Holland et al., 2005).
IMPs belonging to the RND family, represented by AcrB and CusA, as well as the OMPs, such as TolC and CusC, function as trimeric assemblies. There is evidence that the periplasmic domains of AcrB and TolC contact one another, while AcrA stabilizes this interaction by binding across these proteins (Tikhonova and Zgurskaya, 2004; Touzé et al., 2004; Tamura et al., 2005). Cross-linking studies indicate that the α-helical coiled-coil hairpin of AcrA fits into intra-protomer grooves of the open state of TolC, while its β-/lipoyl domains interact with the periplasmic domain of AcrB, at the interface between adjacent subunits (Lobedanz et al., 2007; Symmons et al., 2009). However, recent studies indicate that AcrA has a propensity to form dimers, which can then assemble into hexamers (Tikhonova et al., 2011; Xu et al., 2011). These findings have led to two different models for the assembly: one in which the AcrA dimers bind to a pair of sites, presumably the intra- and inter-protomer groves, on the surface of TolC and AcrB, strengthening their interaction with one another (Tikhonova et al., 2011); while in the other, the dimers form a hexameric channel, interconnecting AcrB to TolC (Xu et al., 2011). Our previous studies of the MtrCDE pump from Neisseria gonorrhoeae also revealed that the MFP MtrC forms dimers that assemble into a hexameric channel, which has an aperture sufficient to accommodate the MtrE trimer (Janganan et al., 2011a,b). Cross-linking the MtrC dimers in the presence of MtrE stabilized an MtrC : MtrE assembly with a stoichiometry of 6:3. However, we found that the α-helical hairpin domain of MtrC could be cross-linked to the α-helical domain of MtrE, consistent with it binding to intra- and inter-protomer grooves on the surface of trimeric MtrE (Janganan et al., 2011b). Since we also found that trimeric MtrD interacts with trimeric MtrE, albeit weakly, our findings indicate that the MtrCDE pump is assembled with an MtrD : MtrC : MtrE stoichiometry of 3:6:3, in which MtrD and MtrE are held together by MtrC binding across their surfaces. Importantly, recent crystallographic studies of the CusAB complex also revealed an assembly with a stoichiometry of 6:3 (Su et al., 2011b).
Despite the wealth of structural data some fundamental questions on the function of bacterial tripartite multidrug pumps remain unanswered, in particular the role of the membrane fusion proteins in energy transduction between the inner and outer membrane proteins. Previously we isolated a mutant of mtrE, encoding an E434K derivative, which exhibits a higher propensity of opening; however, it only does so in response to the binding of MtrC (Janganan et al., 2011a). Consistent with MtrC binding to the intra- and inter-protomer grooves of MtrE, mutations in these grooves hindered pump function. It was possible to introduce cysteines into the MtrE grooves and the MtrC hairpin that could be cross-linked, locking the pump in the closed-channel state (Janganan et al., 2011b). However, there remains the fundamental question of the role of MtrD in controlling the activity of the pump. Herein, we present evidence that a non-functional MtrD, with charged residues in the trans-membrane (TM) helices mutated to inhibit proton translocation, blocks the MtrE channel because it fails to dissociate from the tripartite complex. This is the first report that the pumping of drugs into the channel of the OMP and its dissociation from the IMP both require energy derived from proton translocation through the IMP.
Results
Full-length MtrC is required to induce opening of the MtrE channel
The MtrE open-state channel can be detected by determining the susceptibility of cells to vancomycin, which only enters cells by passive diffusion very slowly due to its large size, but can gain entry readily through an open OMP (Bavro et al., 2008; Janganan et al., 2011a,b). Cells expressing MtrE or its E434K derivative are insensitive to vancomycin; however, when expressed with MtrC, the cells expressing MtrE become moderately susceptible to vancomycin, while those expressing MtrE E434K become highly susceptible to (discs impregnated with 30 μg of) vancomycin and fail to grow (Fig. 1; Table 1). This behaviour indicates that MtrC induces opening of the MtrE channel and presumably stabilizes the fully open-channel state of MtrE E434K. Previously we established that the purified N-terminal α-helical hairpin domain, incorporating residues 103–183, of MtrC, retains α-helical structure and binds to MtrE and its E434K derivative (Janganan et al., 2011a,b). Indeed, isothermal microcalorimetry (ITC) studies revealed that while NT-MtrC binds to MtrE with a Kd of 9.8 μM, the hairpin binds to MtrE and MtrE E434K with a Kd of 2.2 μM and 0.13 μM respectively (Janganan et al., 2011a,b). Considering that MtrE binds the hairpin with relatively high affinity, we sought to determine if binding of the hairpin would be sufficient to induce opening of the MtrE channel. To test whether the hairpin could induce opening of the MtrE channel, we expressed it with a gIII signal sequence that targeted it to the periplasm (see Experimental procedures). Cells expressing the hairpin with MtrE or MtrE E434K were insensitive to vancomycin, indicating that it is incapable of inducing channel opening (Table 1). As a control, we tested and found that cells expressing NT-MtrC, targeted to the periplasm with a gIII signal sequence, were sensitive to vancomycin and failed to grow (Table 1). Thus, suggesting that the lipoyl, β-barrel and/or MP domains of MtrC, which interact with MtrD, relay conformational changes in MtrD to MtrE, inducing opening of its channel. Considering this behaviour we also tested the effect of expressing MtrD with MtrCE and MtrCE E434K, revealing that the cells were, insensitive and only moderately sensitive to (discs impregnated with 30 μg of) vancomycin respectively (Table 1). Furthermore, strains expressing pACYC-mtrCDE, and control strains with the pACYC vector, had an MIC of 512 μg ml−1, while cells expressing pACYC-mtrCDE-E434K and pACYC-mtrCE-E434K had MICs of 64 μg ml−1 and 16 μg ml−1 respectively (Janganan et al., 2011a). It is not surprising then that cells expressing mtrCDE grew without any apparent inhibition around a disc impregnated with 30 μg of vancomycin, while the cells expressing mtrCDE434K had a moderate zone of inhibition, and those expressing mtrCE434K failed to grow to any extent (Table 1). When compared with the susceptibility of cells expressing MtrCE and MtrCE E434K to vancomycin, these data indicate that MtrD retards vancomycin entry, presumably by either re-closing the MtrE channel and/or the occlusion of the periplasmic end of MtrE during complex formation.
Fig. 1.
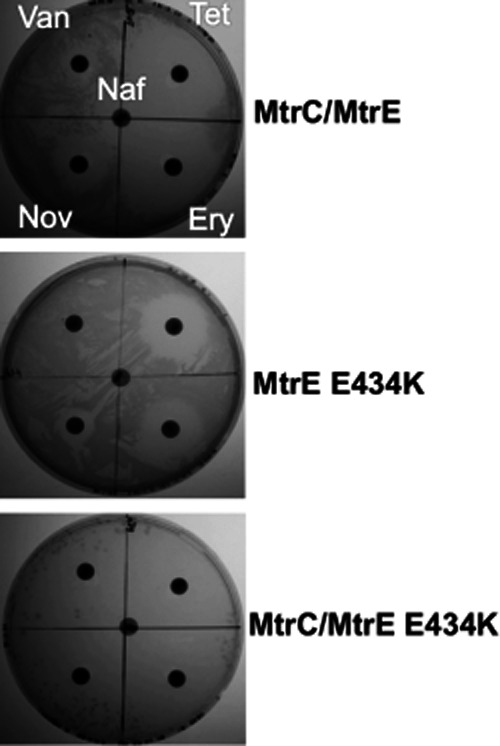
An E434K derivative of MtrE can be used to detect channel opening. Disc-diffusion assays were used to determine if KAM3(DE3) E. coli cells expressing mtrCDE genes and mutants were susceptible to vancomycin, which can be used as a marker of whether the OMP MtrE is open or closed, since it is too large to readily diffuse across the OM but can enter cells via an open OMP. A set of plates showing the disc-diffusion assays for cells expressing (i) MtrC and MtrE, (ii) MtrE E434K and (iii) MtrC and MtrE E434K. It is notable that cells expressing MtrE E434K alone were insensitive to vancomycin, MtrC/MtrE were moderately sensitive to vancomycin, and MtrC/MtrE E434K failed to grow in the presence of vancomycin. Disc-diffusion assays were performed in triplicate, generally assaying for growth around discs impregnated with the following antibiotics: vancomycin (Van; 30 μg), nafcillin (Naf; 1 μg), tetracycline (Tet; 30 μg), erythromycin (Ery; 10 μg) and novobiocin (Nov; 5 μg).
Table 1.
The effect of the MtrC hairpin on the vancomycin resistance of Escherichia coli cells expressing MtrE
| Strain | Vancomycin sensitivity (mm) |
|---|---|
| MtrE | NI |
| MtrE/MtrC | 9 ± 1 |
| MtrE E434K | NI |
| MtrE E434K/MtrC | NG |
| MtrE E434K/gIII-NT-MtrC | NG |
| MtrE/gIII-Hairpin | NI |
| MtrE E434K/gIII-Hairpin | NI |
| MtrC/MtrD/MtrE | NI |
| MtrC/MtrD/MtrE E434K | 10 ± 1 |
The indicated MtrE proteins were expressed from pACYC (and pET21a) and the MtrC and hairpin, with an N-terminal gIII signal sequence, proteins from pET24a in KAM3(DE3) E. coli cells in the presence of 30 μg of vancomycin. The inhibitory zone diameter is given in mm. All disc-diffusion assays were undertaken in triplicate and the average result given. Abbreviations: NI, no inhibition of growth; NG, no growth.
We sought to provide further evidence that vancomycin enters the cells through open MtrE. Large positively charged ions, such as hexamminecobalt (III) chloride (HC), have been shown to block the TolC channel via co-ordination with the ‘second selectivity’ gate residues Asp371 and Asp374, which form two concentric circles of negative charges in the inner cavity of TolC (Andersen et al., 2002b). Indeed TolC has been successfully crystallized in complex with HC (Higgings et al., 2004) and the binding has been shown to block the channel in electrophysiological studies (Andersen et al., 2002a,b). The affinity of the binding has been estimated to be in the range ∼ 20 nM from ITC measurements (Higgins et al., 2004). The TolC channel is effectively sealed for transport unless the Asp-ring is broken, which is suggested to be one of the roles of the MFP (Bavro et al., 2008). Hexammine cobalt stabilizes the Asp-ring, and hence renders the channel non-conductive to larger cargo. Using comparative sequence analysis and homology modelling studies of the MtrE channel (Janganan et al., 2011a), here we demonstrate that the Asp371 and Asp374 are conserved in MtrE, and the corresponding residues (Asp422 and Asp425 respectively) are exposed to the internal channel cavity, thus likely forming a similar arrangement to that in TolC (Fig. 2A). Hence, we capitalized on this to test whether blockage of the MtrE channel with HC would also result in a lowered sensitivity to vancomycin, as the latter is using the channel for cell entry. A growth curve analysis, using a series of increasing HC concentrations, revealed that concentrations above 50 μM were inhibitory for cell growth: in subsequent experiments 25 μM HC was used, which did not significantly inhibit cell growth (Fig. 2B). Consistent with HC blocking the MtrE channel, 25 μM HC caused a clear and significant decrease in the sensitivity of the cells, expressing both MtrC along with MtrE E434K, to 6.4 μg ml−1 vancomycin (Fig. 2B; left graph). Indeed, we found that the inclusion of 25 μM hexammine cobalt caused an increase in the MIC for vancomycin for cells expressing MtrCE E434K from 16 μg ml−1 to 64 μg ml−1. It was notable that HC appeared more efficient at blocking the entry of vancomycin into cells expressing MtrC and MtrE E434K with MtrD (Fig. 2B; right graph and bar chart). The vancomycin MIC for these cells increased from 64 μg ml−1 to 256 μg ml−1 when treated with 25 μM hexamine cobalt. Presumably, because both HC and MtrD can block the periplasmic end of the MtrE channel, they work together to more effectively block vancomycin entry into the cells.
Fig. 2.

Vancomycin entry into cells through MtrE is blocked by hexamminecobalt.
A. A homology model of the MtrE channel based upon a comparative sequence analysis of MtrE and TolC. This model demonstrates that Asp371 and Asp374 in TolC, which form a binding site for hexamminecobalt (III) chloride (HC), are conserved in MtrE and exposed to the internal channel cavity, thus likely forming a similar arrangement to that in TolC. The MtrE model is in blue; two subunits of the TolC–HC complex (1TQQ.pdb) are in magenta; HC is in spacefill; residues Asp371 and Asp374 from TolC, and the homologous Asp422 and Asp425 from MtrE, are shown as sticks.
B. A series of curves showing the relative growth, measured as the OD600, for E. coli cells C43(ΔacrB) transformed with pACYC-MtrC-MtrE(E434K) (left set of graphs) and pACYC-MtrC-MtrE(E434K) and pET21a-MtrD (right set of graphs). Cells were grown in the absence of drugs (×); or in the presence of HC (▪, 25 μM), vancomycin (♦, 6.4 μg ml−1), of HC and vancomycin (▴). Each bar is the average of at least three measurements, with error bars representing the standard deviation of these measurements from the average. Cells transformed with pACYC and pET grew slower than those transformed with pACYC only; but, as shown in the bar chart, attained a comparable OD after overnight growth.
Considering that the MtrC hairpin did not induce opening of the channel of MtrE suggested that it might have the potential to interfere with the assembly of the pump by preventing the binding of full-length MtrC to MtrE. However, we found that cells expressing MtrCDE with the hairpin consistently grew better than those expressing MtrCDE alone, in the presence of antibiotics; while cells expressing the hairpin either alone, or with MtrD, behaved as the control cells, which were sensitive to antibiotics and grew poorly (Fig. S1). Furthermore, we found that for the cells expressing the hairpin there was a significant decrease in their susceptibility to a range of antibiotics, and the cells were insensitive to vancomycin (Table 2). These data clearly indicate that the hairpin interacts with the pump, apparently activating it, rather than preventing its assembly. Providing a plausible explanation for this effect, chemical cross-linking, with EGS, revealed that the hairpin forms heterologous dimers with NT-MtrC (Fig. 3). Considering that there is mounting evidence that the MFP forms dimers (Janganan et al., 2011b; Tikhonova et al., 2011; Xu et al., 2011) that interact with three pairs of intra- and inter-protomer grooves on the surface of the surface of the trimeric OMP (Janganan et al., 2011b; Ferrandez et al., 2012), our data can be rationalized if the MFP binds to one of these grooves, while the hairpin bindings to the other, to activate the pump. Indeed it is expected that the two MFP-binding grooves on the surface of the OMP will represent two different, degenerate binding interfaces with different affinities, something that has also been previously suggested by ITC and SPR binding assays (Janganan et al., 2011a,b; Tikhonova et al., 2011). It is possible that while full-length MtrC is needed to initiate opening of the MtrE channel, the hairpin is capable of stabilizing the already open, activated state of the channel, but it is clearly not capable of conveying the opening on its own. Such data also agrees with the idea of the OMP having two different affinity sites that engage MFPs in a different fashion (Janganan et al., 2011b; Tikhonova et al., 2011).
Table 2.
The effect of the MtrC hairpin on the antibiotic susceptibility of Escherichia coli cells expressing components of the MtrCDE multidrug pump
| Constructs | Naf 1 μg | Tet 30 μg | Ery 10 μg | Nov 5 μg | Van 30 μg |
|---|---|---|---|---|---|
| pACYC-MtrCDE + pET24a | 13 ± 1 | 26 ± 1 | 21 ± 1 | 20 ± 1 | NI |
| pACYC-MtrCDE + pET24a-gIII MtrC hairpin | 9 ± 1 | 22 ± 1 | 18 ± 1 | 19 ± 2 | NI |
| pACYC + pET24a-gIII MtrC hairpin | 20 ± 1 | 36 ± 1 | 30 ± 0.1 | 20 ± 1 | NI |
| pACYC-MtrE | 15 ± 1 | 33 ± 3 | 27 ± 2 | 19 ± 1 | NI |
| pACYC-MtrE + pET24a-gIII MtrC hairpin | 15 ± 1 | 32 ± 1 | 26 ± 1 | 19 ± 1 | NI |
| pACYC MtrD | 16 ± 1 | 39 ± 3 | 30 ± 1 | 19 ± 2 | NI |
| pACYC MtrD + pET24a-gIII MtrC hairpin | 15 ± 1 | 40 ± 2 | 30 ± 1 | 20 + 1 | NI |
| pACYC Vector + pET24a | 18 ± 2 | 35 ± 2 | 32 ± 3 | 21 ± 1 | NI |
The indicated Mtr proteins were expressed from pACYC and the MtrC hairpin, with an N-terminal gIII signal sequence, from pET24a in KAM3(DE3) E. coli cells. The inhibitory zone diameter is given in mm. All disc-diffusion assays were undertaken in triplicate and the average result given. The amount of antibiotic impregnating each disc is given. Abbreviations: NI, no inhibition of growth; Naf, nafcillin; Tet, tetracycline; Ery, erythromycin; Nov, novobiocin; Van, vancomycin.
Fig. 3.
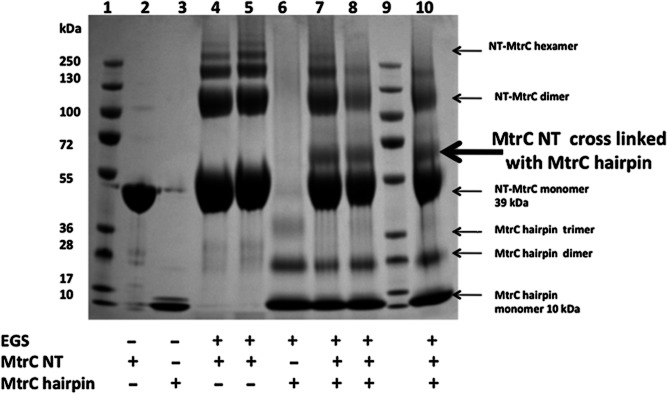
The hairpin of MtrC forms heterodimers with MtrC. To test for heterodimers of the MtrC hairpin with NT-MtrC, (1 mM) EGS was added to (8 μM) MtrC hairpin mixed with (8 μM) NT-MtrC (lanes 7, 8 and 10) and analysed by SDS-PAGE. The control lanes are as follows: Mr-standard (lanes 1 and 9), NT-MtrC (lane 2), hairpin (lane 3), NT-MtrC + EGS (lanes 4 and 5) and hairpin + EGS (lane 6). An arrow indicates the bands corresponding to the formation of heterodimers of hairpin/NT-MtrC.
The dissociation of MtrD from MtrE requires energy that is provided by proton translocation
Since our studies revealed that the C-terminal domain of MtrC, which interacts with MtrD, is required to facilitate opening of the MtrE channel, this suggested that it might play a role in conveying conformational changes from MtrD to MtrE. If this were the case, then presumably MtrD would need to be functional to facilitate the opening of MtrE. We sought to investigate this possibility by inactivating MtrD and determining if it could support the opening of MtrE. Previous studies of other RND pumps have revealed three residues (e.g. D407 and D408 in TMH 4, and K939 in TMH 10, of MexB; and the homologous D407, D408 and K939 residues of AcrB) that are essential and proposed to function as part of the proton-relay system (Guan and Nakae, 2001; Takatsuka and Nikaido, 2006; Seeger et al., 2009). Bioinformatic analysis indicates that the corresponding residues, D405, D406 and K948, are conserved in MtrD (Fig. S2), based on the close homology of MtrD with both AcrB from E. coli (48.3% identity) and MexB from Pseudomonas (48.6% identity) respectively. The secondary structural elements appear well preserved within the group as judged by ALIGN (Cohen, 1997).
Accordingly, we chose these residues for mutagenesis, with the aim of inactivating the pump. All of these residues were found to be essential for MtrCDE conferred resistance to antibiotics (Table 3). Furthermore, none of the double or triple mutants appeared more sensitive than any of the single mutants suggesting that they have a concerted function, presumably in relaying protons across the membrane (Eicher et al., 2009; Pos, 2009; Seeger et al., 2009). To further confirm the hypothesis that these residues could play a role of a proton-relay triad and are colocalized in the MtrD, and taking advantage of the number of high-resolution structures that are available for these proteins, we generated two homology models of MtrD, based on 2DHH.pdb (AcrB; Murakami et al., 2002) and 2V50.pdb (MexB; Sennhauser et al., 2009) structures respectively. When using the SSM superposition algorithm of COOT (Emsley and Cowtan, 2004), the two independent models align with a core RMSD of 2.3 Å, mainly due to a few flexible loops and hairpins, and are overall highly convergent, with the TM part in particular having an RMSD of less than 1 Å (Fig. 4A). As can be seen in Fig. 4A, in both models the residues are in close proximity and likely interacting with each other. This allows for an unambiguous location of the residues in the TM helices and, as can be seen in Fig. 4B, the D405, D406 and K948 appear within bonding distance in both models. In combination with our biochemical data, this provides very strong support to the idea of conservation of this charged network across different RND transporters and suggests that these residues perform in a similar fashion to those previously established in AcrB.
Table 3.
Identification of MtrD mutations that impair the function of the MtrCDE pump
| Constructs | Tet 30 μg | Ery 10 μg | Nov 5 μg | Van 30 μg |
|---|---|---|---|---|
| pACYC MtrCE + pET21a MtrD WT | 29 ± 2 | 20 ± 1 | 12 ± 2 | NI |
| pACYC MtrCE + pET21a MtrD D405K | 40 ± 1 | 30 ± 1 | 17 ± 1 | NI |
| pACYC MtrCE + pET21a MtrD D406K | 40 ± 1 | 27 ± 2 | 18 ± 1 | NI |
| pACYC MtrCE + pET21a MtrD K948E | 40 ± 1 | 27 ± 2 | 19 ± 1 | NI |
| pACYC MtrCE + pET21a MtrD D405K/D406K | 40 ± 1 | 30 ± 1 | 18 ± 1 | NI |
| pACYC MtrCE + pET21a MtrD D405K/D406K/K948E | 40 ± 2 | 30 ± 2 | 19 ± 1 | NI |
| pACYC + pET21a | 38 ± 1 | 29 ± 1 | 17 ± 1 | NI |
The MtrCE proteins were expressed from pACYC and the MtrD derivatives from pET21a in KAM3(DE3) E. coli cells. The inhibitory zone diameter is given in mm. All disc-diffusion assays were undertaken in triplicate and the average result given. The amount of antibiotic impregnating each disc is given. Abbreviations: NI, no inhibition of growth; WT, wild-type; Tet, tetracycline; Ery, erythromycin; Nov, novobiocin; Van, vancomycin.
Fig. 4.
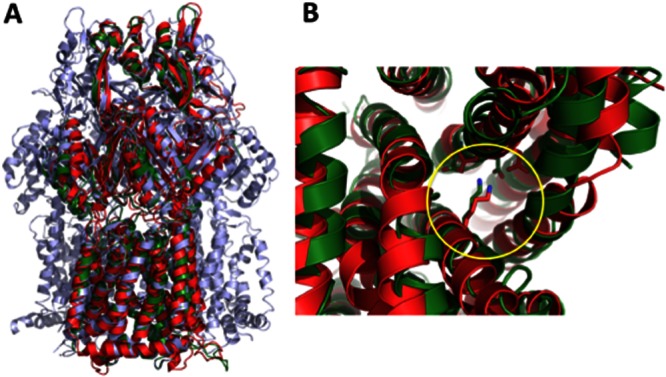
A homology model for MtrD.
A. An overlay of the MtrD models with AcrB trimer. The MexB-based model is in green; the AcrB-based model is in red; and the AcrB 2DHH structure is shown as a light blue trimer. This figure was generated using Pymol.
B. This homology modelling allows for an unambiguous location of the residues in the TM helices, and as can be seen, D405, D406 and K948 (highlighted by a yellow circle) appear within bonding distance in both models. In combination with our biochemical data, this provides very strong support to the idea of conservation of this charged network across different RND transporters and the clearly indicates that these residues perform in a similar fashion to these previously established in AcrB.
In common with cells expressing wild-type MtrCDE, all the mtrD mutants were insensitive to (discs impregnated with 30 μg of) vancomycin (Table 4), and had an MIC of 512 μg ml−1, indicating that the MtrE channel is closed or blocked. However, this behaviour could arise if, for the mtrD mutants, MtrD became uncoupled from MtrCE. To test for this possibility, we coexpressed the mtrD mutants with mtrC/mtrE E434K. As noted above, while cells expressing MtrC/MtrE E434K are sensitive to vancomycin and fail to grow, we found that cells expressing MtrC/MtrD/MtrE E434K are moderately sensitive (Table 4). If MtrD was decoupled from MtrE E434K, in the cells expressing the mtrD mutants with mtrC/mtrE E434K, then they should be highly sensitive to vancomycin and fail to grow. Importantly, the cells can only become resistant to vancomycin if they produce MtrD that can interact with MtrC/E E434K to prevent MtrC unlocking the MtrE E434K channel. Indeed, if MtrD failed to express at all, then the cells would only produce MtrC/MtrE E434K, and would fail to grow due to their sensitivity to vancomycin. Our analysis revealed that cells expressing MtrD D405K, D406K and/or K948E with MtrC/MtrE E434K were insensitive to (discs impregnated with 30 μg of) vancomycin (Table 4), and had an MIC of 512 μg ml−1. However, as expected, the mutants were sensitive to a range of antibiotics that are substrates of the pump (Table 4). Furthermore, pull-down assays revealed that MtrC interacts with both MtrE E434K and MtrD D405K/D406K/K948E, indicating that the mutations in these proteins did not impair their ability to interact (Fig. 5). These findings indicate that the MtrD derivatives interact with MtrC and MtrE E434K resulting in a seal of the MtrE channel. Consequently, access to the lumen of the MtrE channel requires functional MtrD. Considering that the D405K, D406K and K948E derivatives of MtrD are most likely compromised in their ability to translocate protons, suggests that proton translocation is coupled to exposure of the periplasmic end of the MtrE channel, which is brought about by the dissociation of MtrD from MtrE.
Table 4.
MtrD derivatives form a stable complex with MtrE, preventing vancomycin access into the cells
| Constructs | Naf 1 μg | Tet 30 μg | Ery 10 μg | Nov 5 μg | Van 30 μg |
|---|---|---|---|---|---|
| pACYC MtrCE + pET21a MtrD WT | NI* | 29 ± 2 | 20 ± 1 | 12 ± 2 | NI |
| pACYC MtrC/E (E434K) + pET21a MtrD WT | NI* | 45 ± 1 | 40 ± 1 | 21 ± 1 | 10 ± 1 |
| pACYC MtrC/E(E434K) + pET21a MtrD D405K | NI* | 40 ± 1 | 30 ± 2 | 20 ± 1 | NI |
| pACYC MtrC/E(E434K) + pET21a MtrD D406K | NI* | 39 ± 1 | 31 ± 1 | 17 ± 1 | NI |
| pACYC MtrC/E(E434K) + pET21a MtrD K948E | NI* | 38 ± 1 | 28 ± 1 | 17 ± 1 | NI |
| pACYC MtrC/E(E434K) + pET21a MtrD (D405K/D406K) | NI* | 39 ± 2 | 30 ± 1 | 20 ± 1 | NI |
| pACYC MtrC/E (E434K) + pET21a MtrD (D405K/D406K/K948E) | NI* | 41 ± 1 | 31 ± 1 | 20 ± 1 | NI |
| pACYC + pET21a | NI* | 38 ± 1 | 29 ± 2 | 17 ± 1 | NI |
| pACYC | 8 ± 1 | 36 ± 1 | 32 ± 1 | 17 ± 2 | NI |
| pACYC MtrC/MtrE WT | 24 ± 1 | 36 ± 1 | 37 ± 1 | 21 ± 1 | 9 ± 1 |
| pACYC MtrC/MtrE E434K | NG | 50 ± 2 | 40 ± 1 | NG | NG |
The MtrCE proteins were expressed from pACYC and the MtrD derivatives from pET21a in KAM3(DE3) E. coli cells. The inhibitory zone diameter is given in mm. All disc-diffusion assays were undertaken in triplicate and the average result given. The amount of antibiotic impregnating each disc is given. Abbreviations: NI, no inhibition of growth; NI*, pET21a plasmid encoded resistance; WT, wild-type; Naf, nafcillin; Tet, tetracycline; Ery, erythromycin; Nov, novobiocin; Van, vancomycin.
Fig. 5.
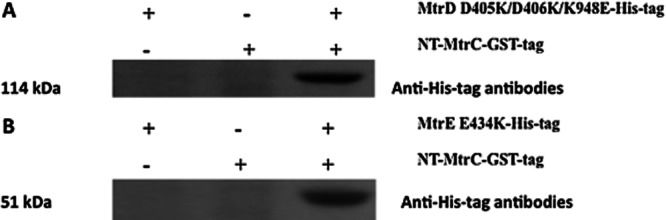
MtrC binds to both MtrE E434K and MtrD D405K/D406K/K948E. The purified NT-MtrC GST-tagged protein was immobilized on GST column beads, to which was added purified His-tagged MtrE E434K (B) or MtrD D405K/D406/K948E (A) prey protein. The beads were washed with Tris buffer, containing 0.1% w/v βDDM, then boiled at 90°C for 5 min to elute the complex. Western blotting, using antibodies to the His-tag, was used to detect the MtrE and MtrD prey proteins. As negative controls, either the bait protein or the prey protein was omitted from the assay (the presence and absence of proteins is indicated by ‘+’ and ‘−’). As shown in each assay when either of these proteins was omitted from the assay, the corresponding prey protein could not be detected. These assays indicate that MtrC interacts with MtrE E434K and MtrD D405A/D406A/K948K.
Discussion
Although our earlier studies provide strong evidence that MtrC plays a role in controlling the activity of the pump by binding to the intra- and inter-protomer grooves of MtrE, what was not clear from such studies was whether binding of the hairpin domain of MtrC is sufficient to induce opening of the MtrE channel. Since our previous in vitro studies revealed that the hairpin binds to both MtrE and its E434K derivative (Janganan et al., 2011a); herein, we utilized a gIII signal sequence to target the hairpin to the periplasm of cells express MtrE E434K, revealing that the hairpin is unable to unlock the MtrE channel on its own (Table 1). This contrasts with full-length MtrC, or NT-MtrC targeted to the periplasm with a gIII signal sequence, suggesting that the C-terminal domain constrains the conformation of the N-terminal hairpin domain, so that it can productively interact with MtrE E434K to open its channel. The hairpin domain is sandwiched between sequences that contribute residues to the lipoyl, the β-barrel and the membrane proximal (MP) domains, presumably restraining the flexibility of the hairpin (Symmons et al., 2009). In the case of AcrA, cross-linking studies have revealed that the α-hairpin domain interacts with TolC (Lobedanz et al., 2007), while the other three domains interact with AcrB (Symmons et al., 2009). The crystal structure of AcrA (Mikolosko et al., 2006), and molecular dynamic simulations (MDS) (Wang et al., 2012), revealed that it has large conformational flexibility that largely arises from the relative motions between the α-hairpin and the other domains. Interestingly, a G336C substitution in the MP domain of AcrA inhibited the AcrABTolC pump, indicating that interactions between this domain of AcrA and AcrB are essential for the operation of the pump (Ge et al., 2009; Tikhonova et al., 2011). Furthermore, while the AcrAB complex cannot interact with a TolC derivative in which the periplasmic end of the channel is open, a complex of AcrB with the G336C derivative of AcrA can interact with this open-state TolC derivative (Tikhonova et al., 2011), suggesting that AcrAB interacts with the closed-state TolC and that conformational changes in the MP domain of AcrA are associated with opening of the TolC channel. It seems highly plausible that the C-terminal end, and more precisely the MP domain, of the MFP is necessary for active coupling and energy transduction from the IMP to the OMP.
Active proton translocation by the RND pump would appear to result in the progressive peristaltic motion of the pump (Murakami et al., 2006; Seeger et al., 2006;2008), which is propagated to the surface of the extrusion protomer (Seeger et al., 2008; Nakashima et al., 2011; Eicher et al., 2012) and hence conceivably communicated to, and resulting in equally dramatic conformational changes, in the MFP. In turn, these conformational changes in the MFP could be used to alter its interaction with the OMP; to induce either opening of the OMP channel or the disengagement of the MFP from the OMP, allowing the IMP/MFP to dissociate from the OMP and hence decoupling of the pump post-drug extrusion. To determine if MtrD communicates with MtrE, we produced a set of functionally impaired mtrD mutants and tested the effect of these derivatives on the MtrE channels ability to conduct vancomycin. Cells expressing mtrC and mtrE E434K along with the non-functional mtrD mutants were rendered insensitive to vancomycin, as were cells expressing mtrE E434K alone, while cells expressing mtrC and mtrE E434K were highly sensitive to vancomycin (Table 4). One possibility for this behaviour is that, while MtrC can unlock the closed channel of MtrE E434K, the non-functional MtrD derivatives interact with MtrC, preventing it from unlocking the channel of MtrE E434K. However, since the MtrD derivatives would not be expected to undergo proton-driven conformational changes, it seems unlikely that they could induce the closure of the MtrE E434K channel, which would otherwise be open. An alternative, perhaps more plausible, possibility is that the MtrD derivatives fail to disengage from MtrE E434K, effectively sealing the channel to vancomycin entry. Such an intriguing interpretation would also suggest that channel unlocking by the hairpins of MtrC is energy-independent, while the proton-motive force is used to actively dissociate the complex and re-seal the periplasmic end of MtrE. Considering that cells expressing MtrCDE E434K are sensitive to vancomycin, which can be reversed by the binding of hexamminecobalt to the periplasmic end of MtrE, implies that MtrD can dissociate from MtrE E434K, which remains in the open state; conversely, cells expressing MtrCDE are insensitive to vancomycin, implying that the dissociation of MtrD is concomitant with closure of the MtrE channel. The insensitivity of the cells expressing the non-functional MtrD mutants to vancomycin would be consistent with them failing to dissociate from MtrE E434K. Indeed, in contrast to the substantive changes in the conformation of the periplasmic domain of the wild-type protein, there are only small changes in the conformation of the periplasmic domain of the AcrB D407A, D408A and K940A derivatives (Su et al., 2006; Takatsuka and Nikaido, 2006; Nikaido and Takatsuka, 2009); consequently, our MtrD derivatives would not be expected to induce the substantive conformational changes in MtrC that would presumably be necessary to induce its disengagement from MtrE.
In summary, our studies utilizing MtrE E434K have identified a number of additional conformational states (Fig. 6A), which can be incorporated into a model for the pump mechanism (Fig. 6B). The integral inner membrane protein MtrD, presumably with bound drug, and MtrC form a complex, in which a ring of six MtrC protomers assemble around the MtrD trimer (state 1). This complex can then dock with the MtrE trimer (state 2), which results in opening of the secondary selectivity gate (the aspartate ring) in the MtrE channel, but not the fully open state. The docking of MtrE and MtrD promoted by MtrC leads to final unlocking of the primary selectivity gate of MtrE by the β-hairpins of MtrD, which has already been suggested for TolC-AcrB (Andersen et al., 2002c; Tamura et al., 2005; Bavro et al., 2008). We can only speculate at this stage on the exact sequence of events during the engagement of MtrC with MtrE, but similar to the AcrA/TolC interaction (Bavro et al., 2008; Tikhonova et al., 2011), sequential engagement of the intra- and inter-protomer grooves is likely. Indeed it seems likely that the inter-protomer groove becomes available only upon the initial binding of the first MtrC monomer in the high-affinity intra-protomer groove, and that this is associated with the disruption of the aspartate gate, while the binding of the second hairpin, to the low-affinity inter-protomer groove, is just to stabilize the open assembly. Conceivably, the second hairpin only engages with MtrE after it has bound to MtrD, to stabilize the MtrE channel in the open conformation. The role of the hairpin in unlocking of the aspartate gate can also be implied by comparative studies of the Borellia efflux pump BesABC (Bunikis et al., 2008), where the MFP BesA uncharacteristically lacks a hairpin. It is notable however that the corresponding second selectivity gate in the OMP BesC is also not present. The binding of protons to the TM domain of MtrD, and their subsequent passage through the proton relay circuit, composed of the residues D405, D406 and K948, induces conformational changes in the periplasmic domain of MtrD that results in transfer of bound drugs towards the periplasmic end of the MtrE channel which has to be fully open by this stage (state 3). The deep interpenetration of the MFP hairpins with the helical coiled-coils of the TolC family members indicate that this is likely a very stable interaction, which is further confirmed by the fact that this extended binding interface tolerates single mutations and from binding studies that reveal a nanomolar affinity (Touzé et al., 2004; Bavro et al., 2008; Tikhonova et al., 2009; 2011; Janganan et al., 2011a,b). Furthermore, these interactions are about an order of magnitude stronger than the corresponding IMP/OMP interactions and are energy-independent (Touzé et al., 2004; Janganan et al., 2011a; Tikhonova et al., 2011). Dissociating such an extended binding interface is likely to require energy, and it is clear that only the IMP can provide this energy. In the absence of energy input, the complex will stay associated permanently, which is supported by our studies that suggest that vancomycin entry is blocked by our inactivated MtrD derivatives, which fail to dissociate from MtrE. Thus, this work provides a tantalizing suggestion that there is at least one more energy-dependent step in the drug expulsion process, and that this is the dissociation of MtrC from MtrE (state 4). Our in vivo experiments of the interaction of MtrC with MtrE E434K also clearly imply that MtrC is capable of binding to MtrE in the absence of an energized pump, which also results in channel opening. Thus the OMP–MFP interaction appears to be energy-independent and likely results in a low-energy state that is stabilized by the extensive helical coiled-coils interfaces. Although it is clear that MtrD must provide the energy for dissociation of the MtrCDE tripartite complex, it is not known whether this occurs concomitantly with drug extrusion. Once MtrC is removed from MtrE, MtrD will be able to dissociate from MtrE, and the outermost selectivity gate of MtrE will spontaneously collapse, while MtrC and MtrD may remain associated together for another drug cycle. It seems plausible that the proton-driven conformational changes in MtrD are coupled to conformational changes in MtrC that trigger its dissociation from MtrE.
Fig. 6.

A schematic model for the assembly and function of the MtrCDE tripartite pump.
A. Additional conformational states for assemblies of the components of the MtrCDE tripartite pump identified using the E434K, vancomycin leaky, derivative of MtrE. In the case of the MtrE E434K derivative, both the primary and the secondary gates of the channel are open, so that it can leak vancomycin. However, MtrD is capable of sealing the channel, to prevent vancomycin entry, until it dissociates from MtrE E434K.
B. A schematic model for the action of the MtrCDE pump. MtrC and MtrD form a complex (state 1) that in response to drug binding interacts with MtrE (state 2). The binding of MtrC to the intra and inter-protomers grooves, and then of MtrD, to the periplasmic tip, of MtrE opens the secondary and primary gates, respectively, of the MtrE channel. The binding of protons to the membrane domain of MtrD induces conformational changes in the periplasmic domain of MtrD that results in transfer of bound drugs to the open MtrE channel (state 3). A further energy-dependent step, possibly involving a second proton translocation event, is then needed to drive the dis-engagement of MtrCD from MtrE (state 4), allowing MtrE to collapse back to its original closed conformation (state 1). The protomers are labelled P1 and P2 (while P3 is hidden from view). The movement pathway of the drug (blue hexagon) and the proton (red circle) is shown as a red curved arrow; while that the sequential binding (marked 1 and 2) of the MtrC hairpin domains to the intra- and inter-protomers grooves of MtrE as yellow curved arrows.
Experimental procedures
Strains and plasmids
The E. coli strains and plasmids used are described in Table S1, and the primers used for construction of plasmid vectors in Table S2. For in vivo studies of the effects of MtrC and its hairpin upon MtrE and its E434 derivative, wild-type and mutant mtrE were expressed from pACYC Duet, while NT-mtrC and its hairpin, both bearing a gIII signal sequence to target the proteins to the periplasm, were expressed from pET24a. These experiments were also repeated using (i) pACYC Duet to express both mtrE and mtrC/gIII-hairpin, and (ii) pET21a and pET24a to express mtrE and mtrC/gIII-hairpin respectively. Such studies confirmed that the resulting effects were independent of the vector pair used for coexpression of these proteins (data not shown). For in vivo studies of the effect of introducing mutations into mtrD, the pET21a-mtrD plasmid was used as the template for site-directed mutagenesis using a QuikChange XL kit from Stratagene, with the primers given in Table S3.
Protein overexpression and purification
The MtrD, MtrE, NT-MtrC and MtrC hairpin proteins were purified as fusion proteins with a six-histidine tag, while GST-tagged NT-MtrC was used for pull-down assays. E. coli cells [strain BL21(DE3) transformed with the relevant pET21a vector for expression MtrD, MtrE, NT-MtrC and MtrC hairpin proteins] were grown at 37°C in 2× YT media, with shaking at 220 r.p.m., until the cells density reached an A600 of 0.6, when 0.5 mM IPTG was added, the temperature adjusted to 25°C and cells left to overexpress proteins for 6 h for membrane proteins and 3 h for soluble proteins. Cells were harvested by centrifugation at 4°C and resuspended in 20 mM Tris-HCl buffer, 300 mM NaCl, pH 7.5, 10% v/v glycerol. Complete protease inhibitor (Roche) and DNase I (Sigma) was added to the cell paste before lysing the cells, via three passes (e.g. 1 × 10 kpsi and 2 × 25 kpsi) using a (Z-plus 1.1 kW) Constant Systems cell disrupter. Cell debris was removed by centrifugation at 37K g (45 min at 4°C), and cell membranes isolated by ultracentrifugation at 125K g (90 min at 4°C) for membrane proteins, while the supernatant was collected for soluble proteins. Membranes pellets were then solubilized in 20 mM Tris-HCl buffer, pH 7.5, 100 mM NaCl, 10% (v/v) glycerol, 2% w/v β-d-dodecyl maltoside (βDDM) (Anatrace). For the purification of MtrD and MtrE proteins the membranes pellets were dissolved in 2% w/v βDDM and the protein were purified under 0.1% w/v βDDM by affinity chromatography using a Ni2+-charged HiTrap chelating column (GE Healthcare). After desalting, samples were subjected to ion exchange chromatography, using either a (GE Healthcare) HiTrap Q or a SP column, for the MtrD and MtrE proteins respectively, which were eluted using a NaCl gradient. For the purification of His-tagged Δ1–34-MtrC (NT-MtrC) and 100–183 α-helical hairpin of MtrC, the supernatant was purified by affinity chromatography using a HiTrap Ni2+-chelating column, followed by gel filtration chromatography, using a 120 ml Hiload 16/600 Superdex 200 prep grade column, in 20 mM Tris-HCl buffer, pH 7.5, 300 mM NaCl, 10% v/v glycerol. In addition, we found that the hairpin could be purified from cells that had been subjected to an osmotic shock to release periplasmic proteins: confirming the presence of the hairpin in the periplasm. For the purification of GST-tagged NT-MtrC, the protein was overexpressed by growing the E. coli cells [strain BL21(DE3) transformed with pGEX6p-3-NT-MtrC] at 25°C overnight with 0.2 mM IPTG. After cell lysis (in PBS) and centrifugation at 37K g (30 min at 4°C), the supernatant was mixed with GST resin and incubated at 4°C for 30 min. Then the resin was packed into a gravity flow column, washed with PBS and finally eluted with 10 mM glutathione (in PBS). The eluted protein was concentrated (to give a volume of about 3 ml) using a 50 kDa cut-off Vivaspin concentrator, and loaded onto a gel filtration column (Hiload 16/600 Superdex 200) for further purification.
Disc diffusion assays
Escherichia coli cells [strain KAM3(DE3) (Mima et al., 2007)] were grown to an OD600 of about 0.5, induced with 0.1 mM IPTG, the cells were diluted to an OD600 of 0.3 and then 350 μl of cell culture was spread on an MHA plate previously incorporated with 0.1–0.2 mM IPTG; antibiotic discs were placed on the plate medium and incubated at 37°C overnight. The zone of culture growth inhibition was determined. The discs used were impregnated with 1 μg of nafcillin, 30 μg of tetracycline, 10 μg of erythromycin, 5 μg of novobiocin and 30 μg of vancomycin. Western blotting with protein sequence-specific antibodies and/or antibodies to the His-tag was routinely used to test the cells used for antibiotic sensitivity assays for the expression of the MtrC, MtrD and MtrE proteins. None of the mutants used in our studies had protein levels substantively different than found in cells expressing the wild-type proteins; and consequently the differences in antibiotic sensitivity are not attributable to differences in the protein expression levels.
Growth curve assays
Escherichia coli cells (ΔacrB strains KAM3 or C43) were grown at 37°C in LB, supplemented with 30 μg ml−1 chloramphenicol, for selection of pACYC, and/or 100 μg ml−1 carbenicillin, for selection of pET, to an OD600 of about 0.4, and then induced with 1 mM IPTG for 2–2.5 h at 37°C. The cells were diluted to an OD600 of about 0.1, with LB, supplemented with appropriate antibiotics (e.g. 30 μg ml−1 chloramphenicol and/or 100 μg ml−1 carbenicillin) and for some cultures hexammine cobalt (III) chloride (final concentration 25 μM) and/or vancomycin (final concentration 6.4 μg ml−1), and the cells grown at 37°C, while recording their OD every 30 min, until they reached stationary phase. Similarly, after IPTG induction, the cells were diluted with 2× YT media containing antibiotic and monitored for growth after 24 h for MIC measurements.
Pull-down assays
For pull-down assays, we used the GST-tagged NT-MtrC as the bait protein, to pull-down the His-tagged MtrD and MtrE prey proteins. The purified NT-MtrC GST-tagged protein was immobilized on GST column beads (GE Healthcare), to which was added purified His-tagged MtrE E434K or MtrD D405K/D406/K948E prey protein (5 mg protein in 0.5 ml). The beads were washed with Tris buffer, containing 0.1% w/v βDDM, and then boiled at 90°C for 5 min to elute the complex. The proteins in samples were run on an SDS-PAGE gel and Western blotting was performed, with antibodies to the His-tag, to detect if the MtrD and MtrE prey proteins were present in the samples. Furthermore, the samples were subjected to MALDI-ToF PMF analyses, performed using a Voyager-DE™ STR BioSpectrometry™Workstation (Applied Biosystems, Warrington, UK). De-isotoped and calibrated spectra were then used to generate peak lists, which were searched using MASCOT (http://www.matrixscience.com) mass spectrometry database search software to identify the proteins. As a control, the GST protein was immobilized on GST column beads and tested for any interaction with the His-tagged prey proteins. No interaction of the His-tagged MtrD and MtrE prey proteins with GST was detected (data not shown).
Cross-linking of MtrC and hairpin
NT-MtrC and MtrC hairpin proteins were first buffer exchanged with Na2HP04, 100 mM NaCl, 10% v/v glycerol and 1 mM EDTA using mini chromatography column (Bio-RAD): 8 μM of each protein was incubated with 1 mM ethylene glycol bis-succinimidyl-succinate (EGS) at room temperature for 1 h. Then the cross-linking reaction was stopped with 100 mM Tris HCl, the sample was reduced with 50 mM DTT and mixed with 4× LDS sample buffer, boiled at 100°C for 10 min and separated on a 4–12% SDS-PAGE gradient gel with MOPS buffer.
Homology modelling of MtrE and MtrD
The homology model for MtrE has previously been reported (Janganan et al., 2011a,b). Two independent homology models of MtrD were created using ESyPred3D (Lambert et al., 2002) and utilizing the MODELLER (Sali and Blundell, 1993) homology-modelling engine based on the high-resolution experimental structures of AcrB (2DHH.pdb) and MexB (2V50.pdb) respectively. Models were superposed and analysed using Coot (Emsley and Cowtan, 2004) and structural alignments executed with Espript (Gouet et al., 1999).
Acknowledgments
This work was supported by grants from the Wellcome Trust. We would like to thank Prof. Per Bullough (University of Sheffield) for his assistance in completing the work for the manuscript.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
References
- Andersen C, Koronakis E, Hughes C, Koronakis V. An aspartate ring at the TolC tunnel entrance determines ion selectivity and presents a target for blocking by large cations. Mol Microbiol. 2002a;44:1131–1139. doi: 10.1046/j.1365-2958.2002.02898.x. [DOI] [PubMed] [Google Scholar]
- Andersen C, Hughes C, Koronakis V. Electrophysiological behavior of the TolC channel-tunnel in planar lipid bilayers. J Membr Biol. 2002b;185:83–92. doi: 10.1007/s00232-001-0113-2. [DOI] [PubMed] [Google Scholar]
- Andersen C, Koronakis E, Bokma E, Eswaran J, Humphreys D, Hughes C, Koronakis V. Transition to the open state of the TolC periplasmic tunnel entrance. Proc Natl Acad Sci USA. 2002c;99:11103–11108. doi: 10.1073/pnas.162039399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavro VN, Pietras Z, Furnham N, Pérez-Cano L, Fernández-Recio J, Pei XY, et al. Assembly and channel opening in a bacterial drug efflux machine. Mol Cell. 2008;30:114–121. doi: 10.1016/j.molcel.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair JM, Piddock LJ. Structure, function and inhibition of RND efflux pumps in Gram-negative bacteria: an update. Curr Opin Microbiol. 2009;12:512–519. doi: 10.1016/j.mib.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Bunikis I, Denker K, Ostberg Y, Andersen C, Benz R, Bergström S. An RND-type efflux system in Borrelia burgdorferi is involved in virulence and resistance to antimicrobial compounds. PLoS Pathog. 2008;4:e1000009. doi: 10.1371/journal.ppat.1000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen GH. ALIGN: a program to superimpose protein coordinates, accounting for insertions and deletions. J Appl Crystallogr. 1997;30:1160–1161. [Google Scholar]
- Eicher T, Brandstätter L, Pos KM. Structural and functional aspects of the multidrug efflux pump AcrB. Biol Chem. 2009;390:693–699. doi: 10.1515/BC.2009.090. [DOI] [PubMed] [Google Scholar]
- Eicher T, Cha HJ, Seeger MA, Brandstätter L, El-Delik J, Bohnert JA, et al. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc Natl Acad Sci USA. 2012;109:5687–5692. doi: 10.1073/pnas.1114944109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ferrandez Y, Monlezun L, Phan G, Benabdelhak H, Benas P, Ulryck N, et al. Stoichiometry of the MexA-OprM binding, as investigated by blue native gel electrophoresis. Electrophoresis. 2012;33:1282–1287. doi: 10.1002/elps.201100541. [DOI] [PubMed] [Google Scholar]
- Ge Q, Yamada Y, Zgurskaya H. The C-terminal domain of AcrA is essential for the assembly and function of the multidrug efflux pump AcrAB-TolC. J Bacteriol. 2009;191:4365–4371. doi: 10.1128/JB.00204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- Guan L, Nakae T. Identification of essential charged residues in transmembrane segments of the multidrug transporter MexB of Pseudomonas aeruginosa. J Bacteriol. 2001;183:1734–1739. doi: 10.1128/JB.183.5.1734-1739.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins MK, Bokma E, Koronakis E, Hughes C, Koronakis V. Structure of the periplasmic component of a bacterial drug efflux pump. Proc Natl Acad Sci USA. 2004;101:9994–9999. doi: 10.1073/pnas.0400375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland IB, Schmitt L, Young J. Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway. Mol Membr Biol. 2005;22:29–39. doi: 10.1080/09687860500042013. [DOI] [PubMed] [Google Scholar]
- Janganan TK, Zhang L, Bavro VN, Matak-Vinkovic D, Barrera NP, Burton MF, et al. Opening of the outer membrane protein channel in tripartite efflux pumps is induced by interaction with the membrane fusion partner. J Biol Chem. 2011a;286:5484–5493. doi: 10.1074/jbc.M110.187658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janganan TK, Bavro VN, Zhang L, Matak-Vinkovic D, Barrera NP, Venien-Bryan C, et al. Evidence for the assembly of a bacterial tripartite multidrug pump with a stoichiometry of 3:6:3. J Biol Chem. 2011b;286:26900–26912. doi: 10.1074/jbc.M111.246595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Nishino K, Yamaguchi A. Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J Bacteriol. 2001;183:5639–5644. doi: 10.1128/JB.183.19.5639-5644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- Kulathila R, Kulathila R, Indic M, van den Berg B. Crystal structure of Escherichia coli CusC, the outer membrane component of a heavy metal efflux pump. PLoS ONE. 2011;6:e15610. doi: 10.1371/journal.pone.0015610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert C, Leonard N, De Bolle X, Depiereux E. ESyPred3D: prediction of proteins 3D structures. Bioinformatics. 2002;18:1250–1256. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- Lobedanz S, Bokma E, Symmons MF, Koronakis E, Hughes C, Koronakis V. A periplasmic coiled-coil interface underlying TolC recruitment and the assembly of bacterial drug efflux pumps. Proc Natl Acad Sci USA. 2007;104:4612–4617. doi: 10.1073/pnas.0610160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomovskaya O, Lewis K. Emr, an Escherichia coli locus for multidrug resistance. Proc Natl Acad Sci USA. 1992;89:8938–8942. doi: 10.1073/pnas.89.19.8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F, Su CC, Zimmermann MT, Boyken SE, Rajashankar KR, Jernigan RL, Yu EW. Crystal structures of the CusA efflux pump suggest methionine-mediated metal transport. Nature. 2010;467:484–488. doi: 10.1038/nature09395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolosko J, Bobyk K, Zgurskaya HI, Ghosh P. Conformational flexibility in the multidrug efflux system protein AcrA. Structure. 2006;14:577–587. doi: 10.1016/j.str.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima T, Joshi S, Gomez-Escalada M, Schweizer HP. Identification and characterization of TriABC-OpmH, a triclosan efflux pump of Pseudomonas aeruginosa requiring two membrane fusion proteins. J Bacteriol. 2007;189:7600–7609. doi: 10.1128/JB.00850-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra R, Bavro VN. Assembly and transport mechanism of tripartite drug efflux systems. Biochim Biophys Acta. 2009;1794:817–825. doi: 10.1016/j.bbapap.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita Y, Kodama K, Shiota S, Mine T, Kataoka A, Mizushima T, Tsuchiya T. NorM, a putative multidrug efflux protein, of Vibrio parahaemolyticus and its homolog in Escherichia coli. Antimicrob Agents Chemother. 1998;42:1778–1782. doi: 10.1128/aac.42.7.1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–179. doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- Nagakubo S, Nishino K, Hirata T, Yamaguchi A. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J Bacteriol. 2002;184:4161–4167. doi: 10.1128/JB.184.15.4161-4167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature. 2011;480:565–569. doi: 10.1038/nature10641. [DOI] [PubMed] [Google Scholar]
- Nikaido H, Takatsuka Y. Mechanisms of RND multidrug efflux pumps. Biochim Biophys Acta. 2009;1794:769–781. doi: 10.1016/j.bbapap.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras Z, Bavro VN, Furnham N, Pellegrini-Calace M, Milner-White EJ, Luisi BF. Structure and mechanism of drug efflux machinery in Gram negative bacteria. Curr Drug Targets. 2008;9:719–728. doi: 10.2174/138945008785747743. [DOI] [PubMed] [Google Scholar]
- Pos KM. Drug transport mechanism of the AcrB efflux pump. Biochim Biophys Acta. 2009;1794:782–793. doi: 10.1016/j.bbapap.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- Seeger MA, von Ballmoos C, Eicher T, Brandstätter L, Verrey F, Diederichs K, Pos KM. Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nat Struct Mol Biol. 2008;15:199–205. doi: 10.1038/nsmb.1379. [DOI] [PubMed] [Google Scholar]
- Seeger MA, von Ballmoos C, Verrey F, Pos KM. Crucial role of Asp408 in the proton translocation pathway of multidrug transporter AcrB: evidence from site-directed mutagenesis and carbodiimide labeling. Biochemistry. 2009;48:5801–5812. doi: 10.1021/bi900446j. [DOI] [PubMed] [Google Scholar]
- Sennhauser G, Bukowska MA, Briand C, Grütter MG. Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa. J Mol Biol. 2009;389:134–145. doi: 10.1016/j.jmb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J Bacteriol. 2006;188:7290–7296. doi: 10.1128/JB.00684-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Yang F, Long F, Reyon D, Routh MD, Kuo DW, et al. Crystal structure of the membrane fusion protein CusB from Escherichia coli. J Mol Biol. 2009;393:342–355. doi: 10.1016/j.jmb.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Long F, Yu EW. The Cus efflux system removes toxic ions via a methionine shuttle. Protein Sci. 2011a;20:6–18. doi: 10.1002/pro.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su CC, Long F, Zimmermann MT, Rajashankar KR, Jernigan RL, Yu EW. Crystal structure of the CusBA heavy-metal efflux complex of Escherichia coli. Nature. 2011b;470:558–562. doi: 10.1038/nature09743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons MF, Bokma E, Koronakis E, Hughes C, Koronakis V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc Natl Acad Sci USA. 2009;106:7173–7178. doi: 10.1073/pnas.0900693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsuka Y, Nikaido H. Threonine-978 in the transmembrane segment of the multidrug efflux pump AcrB of Escherichia coli is crucial for drug transport as a probable component of the proton relay network. J Bacteriol. 2006;188:7284–7289. doi: 10.1128/JB.00683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura N, Murakami S, Oyama Y, Ishiguro M, Yamaguchi A. Direct interaction of multidrug efflux transporter AcrB and outer membrane channel TolC detected via site-directed disulfide cross-linking. Biochemistry. 2005;44:11115–11121. doi: 10.1021/bi050452u. [DOI] [PubMed] [Google Scholar]
- Tikhonova EB, Zgurskaya HI. AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J Biol Chem. 2004;279:32116–32124. doi: 10.1074/jbc.M402230200. [DOI] [PubMed] [Google Scholar]
- Tikhonova EB, Dastidar V, Rybenkov VV, Zgurskaya HI. Kinetic control of TolC recruitment by multidrug efflux complexes. Proc Natl Acad Sci USA. 2009;106:16416–16421. doi: 10.1073/pnas.0906601106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova EB, Yamada Y, Zgurskaya HI. Sequential mechanism of assembly of multidrug efflux pump AcrAB-TolC. Chem Biol. 2011;18:454–463. doi: 10.1016/j.chembiol.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzé T, Eswaran J, Bokma E, Koronakis E, Hughes C, Koronakis V. Interactions underlying assembly of the Escherichia coli AcrAB-TolC multidrug efflux system. Mol Microbiol. 2004;53:697–706. doi: 10.1111/j.1365-2958.2004.04158.x. [DOI] [PubMed] [Google Scholar]
- Wang B, Weng J, Fan K, Wang W. Interdomain flexibility and pH-induced conformational changes of AcrA revealed by molecular dynamics simulations. J Phys Chem B Mater Surf Interfaces Biophys. 2012;116:3411–3420. doi: 10.1021/jp212221v. [DOI] [PubMed] [Google Scholar]
- Xu Y, Song S, Moeller A, Kim N, Piao S, Sim SH, et al. Functional implications of an intermeshing cogwheel-like interaction between TolC and MacA in the action of macrolide-specific efflux pump MacAB-TolC. J Biol Chem. 2011;286:17910–17920. doi: 10.1074/jbc.M110.202598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE., Jr Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.