

Abstract
The ability of the innate immune system to quickly recognize and respond to an invading pathogen is essential for controlling the infection. For this purpose, cells of the immune system express receptors which recognize evolutionarily conserved structures expressed by various pathogens but absent from host cells. In this review we focus on the non-classical C-type lectin receptors including Dectin-1 whose role has been extensively characterized in the recognition and response to fungal pathogens. Dectin-1 is a type II transmembrane protein which binds β-1,3 and β-1,6 glucans. It is expressed on most cells of the innate immune system and has been implicated in phagocytosis as well as killing of fungi by macrophages, neutrophils and dendritic cells. The Dectin-1 cytoplasmic tail contains an immunoreceptor tyrosine based activation motif (ITAM) that signals in part through the spleen tyrosine kinase and in collaboration with Toll-like receptors. Although the main research focus has been on Dectin-1’s role as a fungal and yeast pathogen recognition receptor, more recent studies suggest that Dectin-1 may have a broader function in pathogen recognition including a role in directing a macrophage response to mycobacterial infections.
Keywords: Pattern recognition receptors, Dectin-1, signaling, mycobacteria, fungi, glucans, C-type lectin
INTRODUCTION
The immune system has the unenviable task of distinguishing friend from foe. It requires the ability to mount an effective immune response against antigens of an invading pathogen while remaining tolerant of self-antigens. To do so the acquired immune system has evolved mechanisms to eliminate or tolerize self-reactive T and B cells, while maintaining a large repertoire of lymphocytes which can recognize foreign antigens. The innate immune response, which primarily consists of effector functions of the professional phagocytic cells (monocytes/ macrophages, dendritic cells and neutrophils), can be distinguished from the acquired response in regards to mechanism of recognition and response to foreign antigens. The phagocytes express pattern recognition receptors (PRRs) either on the cell surface or within the cell which recognize evolutionarily conserved structures expressed by various pathogens but absent from host cells. Some examples of these pathogen associated molecular patterns (PAMPs) include: bacterial DNA, viral RNA, peptidoglycans, flagellins and β-glucans. The binding of these molecules to PRRs leads to a rapid response by cells of the innate immune system. This results in the activation of several signaling pathways leading to production of cytokines and chemokines which promote the adaptive immune response as well as increase the phagocytic cells’ ability to ingest and kill the invading microbe. Significant work over the past decade has lead to the discovery of the different PRRs, the PAMPs they recognize, and how the cell responds to their binding (reviewed in [1,2]).
Toll-like receptors (TLRs) as members of the PRRs are important in signaling responses to microbes and microbial products and work primarily, although not exclusively, through activation of the mitogen activated protein kinases (MAPK) and the NF-κB family of transcription factors [3]. Recent studies have also implicated TLRs in phagosome maturation (i.e. the “maturation of a phagosome into a phagolysosome) [4], although this is a subject of some debate [5]. Although a significant number of studies have addressed the role of TLRs in the recognition and response to pathogens much less work has been performed on non-TLR PRRs. Nevertheless, recent studies have implicated non-TLRs in responding to pathogens and initiating an innate immune response. The cytoplasmic NOD-like receptors, which number approximately 20 members, contain three domains: the C-terminal Leucine Rich Repeats (LRR), the central NACHT domain and the N-terminal effector domain. Binding of ligand to the LRRs is thought to release the auto-inhibition allowing the NACHT domain to oligomerize and subsequent binding of signaling molecules to the N-terminal effector domain. Although the ligands for many of the NOD-like receptors remain undefined, these receptors have been implicated in the recognition of cytoplasmic PAMPs including PGN, bacterial RNA and flagellin [6,7]. Another group of PRRs expressed at high levels in myeloid cells is the C-type lectin receptors which can be separated into two sub-families: the classical C-type lectin receptors which contain one or more carbohydrate recognition domains and require calcium for carbohydrate binding and the non-classical C-type lectin receptors which contain a modified carbohydrate recognition domain. The general role for C-type lectins in innate immunity has been addressed in an excellent review by Torrelles et al. in this issue of Current Drug Targets and will not be discussed further. In this review we will concentrate on the non-classical C-type lectin receptors with a focus on Dectin-1 including recent studies which indicate a role for this PRR in the recognition and response to mycobacteria.
NON-CLASSICAL C-TYPE LECTIN RECEPTORS
The non-classical C-type lectin receptors also known as the NK cell receptor-like C-type lectin receptors are primarily involved in regulating cellular cytotoxicity mostly through interaction with the MHC class I molecule and are expressed principally on NK and T cells. This family of type II transmembrane receptors contain a single extracellular carbohydrate recognition domain (CRD) usually extending from the plasma membrane via an amino acid “stalk” of variable length. Despite the presence of a CRD, the non-classical C-type lectin receptors in general do not bind carbohydrate since they lack conserved amino acids present in the classical C-type lectin receptors required for binding. Ligand binding also appears to be cation independent, in contrast to the classic C-type lectin receptors. The receptors often dimerize and either can initiate signaling directly or through association with adaptor molecules [8].
A subset of these receptors are of particular interest since they are expressed mainly on myeloid cells and have diverse functions and ligand specificity as compared to other more classic members of the NK-cell receptor-like receptor family. Members of this subset of C-type lectin receptors include MDL-1, LOX-1, DCAL-1, DCAL-2 (MICL), CLEC-1, CLEC-2 and Dectin-1. The information on most of these receptors is rather limited with the exception of LOX-1 and Dectin-1. LOX-1 which is an abbreviation for lectin-like oxidized low density lipoprotein receptor-1 is best known as a receptor for oxidized LDL and other host molecules such as fibronectin [9]. However, it has also been implicated in binding both gram-negative and gram-positive bacteria by endothelial cells and this binding appears to be mechanistically similar to binding by class A scavenger receptors [10]. Despite the limited ligand and functional information on the other members, sequence data suggests potential signaling motifs within their cytoplasmic tails.
DECTIN-1 STRUCTURE AND FUNCTION
Dectin-1 is a 28 KDa type II transmembrane protein with a single CRD domain. It was originally defined as a receptor on dendritic cells (DC) hence its name ‘Dendritic-cell-associated C-type lectin-1’ but later was found to be expressed on macrophages, monocytes, neutrophils and some T cells [11,12]. In humans, Dectin-1 is also expressed in eosinophils and B cells; the functional importance of this difference in expression patterns is not presently clear [13,14]. The expression pattern can differ between tissue and cell type but in general Dectin-1 is expressed at high levels in lung and intestine as well as areas of high T cell levels such as the spleen, thymus and lymph nodes [15]. This distribution suggests a role for Dectin-1 in pathogen recognition. Protein expression can also be regulated by various cytokines including IL-4 and GM-CSF as well as by microbial components.
Mice express two isoforms of Dectin-1 while in humans there are eight; however, only two of the human isoforms have a CRD. The difference between the two major isoforms in both human and mouse is the presence or absence of the stalk region that separates the CRD from the plasma membrane [16,17]. Nevertheless, both isoforms have been shown to be functional in vitro and in transfection studies [14,16]. Mouse Dectin-1 has 2 potential N-linked glycosylation sites within the CRD while the human β-glucan receptor lacks these sites but contains one potential site in the stalk region. Although glycosylation is not required for Dectin-1 ligand binding, a recent study by Kato et al. indicated that maximum cell surface expression and NF-κB activation is dependent on the presence of the glycosylation sites in the context of Dectin-1 expression studies in HEK293 cells [18]. Crystal structural data also indicates that Dectin-1 maintains a metal binding site characteristic of the snake venom coagulation factor IX binding protein. However, metal ion binding by Dectin-1 appears not to be required for proper protein folding or ligand association [19].
Within the cytoplasmic tail of both Dectin-1 and CLEC-2 is a motif characteristic of an immunoreceptor tyrosine based activation motif (ITAM). As discussed below, this ITAM functions to mediate a ligand induced activation of a signaling response leading to the production of various immune modulators. Another unique feature of Dectin-1 is its ability to bind carbohydrates. At present the only known ligand for Dectin-1 is β-1,3 and β-1,6 linked glucans which are expressed by a number of organisms including yeast, fungi, plants and a small number of bacteria [8]. Although 50% of the fungal cell wall consist of β-glucans, it was previously believed that the β-glucans were buried under a layer of mannosylated proteins. However, more recent studies suggest that in some regions of the cell wall β-glucans are exposed and accessible to bind Dectin-1 [20]. How Dectin-1 binds β-glucans is unclear since as a member of the non-classical C-type lectin receptor family they lack the conserved residues with the CRD required for carbohydrate binding. Nevertheless, mutagenesis studies indicate that Trp 221 and His 223 within the CRD are crucial for β-glucan binding [21,19]. Structural data indicates that these residues are present within a groove that constitutes a potential binding site for β-glucans [19]. Charge analysis of this groove suggests β-glucan binding is driven mainly by hydrophobic forces [22]. Although there is ample evidence that Dectin-1 binds β-glucans, less clear is what constitutes the minimal length of β-glucan required for binding. It appears that monomers or short oligosaccharides are not sufficient. Instead, affinity for Dectin-1 depends on whether the β-glucans adapt a helical or triple-helical confirmation, the degree of branching and the molecular mass [23]. In addition, β-glucans may not be the only ligands for Dectin-1. Studies by Ariizumi et al. demonstrated a Dectin-1 mediated interaction between DC and an unknown but apparent non-carbohydrate ligand on CD4+ and CD8+ T cells. They also showed that recombinant Dectin-1 can bind to T cells and promote anti-CD3 induced proliferation [11]. However, this Dectin-1 ligand expressed on T cells remains undefined although proteolysis data suggest it is protein in nature [11]. Recent studies from our laboratory indicate that mycobacteria express a Dectin-1 ligand [24]. However, there is no published report that the mycobacteria cell wall contains β-glucans.
DECTIN-1 SIGNALING
Dectin-1 maintains a motif typical of an ITAM. A classical ITAM consist of two intracellular YXXL/I sequences separated by 6 to 12 amino acids. During activation both Tyr resides are phosphorylated usually by Src family kinases leading to the association of Syk or Zap70 kinases via their Src-homology (SH2) domains [25,26]. These kinases initiate the signaling cascade that is coupled with the particular receptor-ligand interaction. The presence and phosphorylation of both Tyr residues within the ITAM motif is thought to be required for kinase activation. Interestingly, Dectin-1 requires only one YXXL sequence within its cytoplasmic tail and therefore it was initially unclear whether phosphorylation of this single Tyr residue could recruit and activate Syk. However, recent studies indicate that Dectin-1 can signal a transcriptional response and that Syk is required for at least some of these responses. Additional studies indicated that zymosan-induced Dectin-1 signaling, as defined through antibody blocking experiments, lead to the expression of cytokines IL-10 and IL-2 [27,28]. The diminished production of IL-10 and IL-2 in Syk deficient DC exposed to zymosan suggests a role for this kinase in Dectin-1 signaling [27]. Further, phosphorylated Syk has been localized to Dectin-1 and the removal of the Tyr residue within the YXXL resulted in loss of both colocalization and Dectin-1 mediated signaling [28]. Finally, a peptide containing this YXXL motif and surrounding amino acids of Dectin-1 binds Syk in vitro suggesting a direct interaction between Dectin-1 and Syk [28].
How is Dectin-1 binding Syk with only one YXXL motif? The interaction between Dectin-1 and Syk could be stabilized by specific residues within the Dectin-1 cytoplasmic tail. Another possibility is that Dectin-1 dimerizes or forms higher-order complexes once bound by ligand. In this context, it is interesting that small β-glucans polymers do not induce Dectin-1 signaling. This may reflect differences in affinity for Dectin-1 between large and small β-glucans or it may reflect the larger β-glucans ability to bridge/cluster Dectin-1. This could bring Dectin-1’s phosphorylated Tyr residues in alignment allowing the two SH2 domains of Syk to bind each Dectin-1 monomer. Recent studies with the non-classical C-type lectin receptor CLEC-2, which also has only one YXXL motif, have shown that signaling via CLEC-2 requires both SH2 domains of Syk to be functional. Mutating either SH2 domain of Syk resulted in a loss in CLEC-2-mediated signaling response suggesting that both Syk SH2 domains are engaged. The simplest explanation for these results is that each SH2 domain of Syk engages a phosphorylated Tyr residue on each of two CLEC-2 cytoplasmic tails. Additional studies are needed to determine whether both SH2 domains of Syk are required for Dectin-1 signaling. It is important to note, however, that Dectin-1 does not contain the Cys residue within its stalk region required for dimerization as defined for other C-type lectin receptors [11]. Therefore, if Dectin-1 dimerizes it must do so by another mechanism.
Although Syk is important in many Dectin-1-mediated signaling responses, DC production of IL-12 upon yeast infection [28] as well as phagocytosis of zymosan and fungi by macrophages are Syk independent [29]. A number of studies have also linked Dectin-1 signaling to TLR2/Myd88. Initial studies indicated that optimum production of TNF-α, ROS and IL-12 by zymosan-treated DC is dependent on both TLR2 and Dectin-1 [30]. Studies by Brown et al. indicated a link between TLR2 and Dectin-1 signaling in macrophage production of TNF-α, MIP-2 and IL-12 p40 following stimulation with zymosan or infection with C. albicans conidia [31]. The cooperation between Dectin-1 and TLR2 appears not to involve direct physical interaction although both have been localized to the phagocytic cup containing zymosan [30]. This suggests a linkage within the signaling pathways downstream of the two receptors. While TLR2 and Dectin-1 are linked in some signaling responses there are likely other receptors whose interaction with Dectin-1 also promotes an immune response. For example, the tetraspanin protein CD37 has been found to negatively regulate Dectin-1, as macrophages deficient in CD37 produce significantly more Dectin-1 dependent IL-6 and TNF-α in response to zymosan [32].
What constitutes the link between the Dectin-1 and TLR2 pathways and how are the signals transmitted from Dectin-1 to produce the different immune modulators? Until recently answers to these questions remained elusive. A major break-through came in studies by Gross et al. who demonstrated that DC production of TNF-α, IL-6 and IL-2 induced by zymosan was dependent on CARD9 [33]. CARD9 is an adapter molecule that has an N-terminal caspase-recruitment domain (CARD). Other family members including Carma1 (also known as CARD11) have been linked to NF-κB activation induced by T- and B-cell antigen receptors [34]. Like Carma1, NF-κB activation in DC upon Dectin-1/Syk activation was dependent on CARD9. Also like Carma1, the CARD9 mediated activation of NF-κB was dependent on the formation of a CARD9-Bcl-10-MALT1 complex [33,35]. Previous studies have shown MALT1 to activate the adaptors TRAF2 and TRAF6 leading to activation of the inhibitor of NF-κB kinase complex, degradation of the inhibitor of NF-κB and subsequent NF-κB nuclear translocation [36]. The link between Syk activation and CARD9 is presently unknown but it may again follow Carma1: i.e. Syk activation of phospholipase C (γ1 or γ2) leading to diacylglycerol-mediated activation of PKC isoforms and subsequent PKC activation of Carma1 [37] (Fig. 1). These results also suggest a mechanism by which TLR2/MyD88 and Dectin-1 signaling could work in concert to promote production of immune modulators since transcription for many of these genes are regulated by NF-κB. However, this hypothesis does not explain the results from several studies including the absolute requirement for TLR2 signaling for zymosan induced TNF-α production by macrophages. It is likely that other transcription factors required for TNF-α transcription are not activated by the Dectin-1/Syk pathway but require TLR engagement. TLR signaling leads to the activation of a number of transcription factors including NF-κB, AP-1 and IRF-3 in part through activation of the MAPK pathway. Nevertheless, recent studies have demonstrated Dectin-1 mediated activation of the transcription factor NFAT following Candida or zymosan ingestion by macrophages or DC as well as the importance of NFAT in the production of various immune modulators including COX-2, IL-2, IL-10 and IL-12p70 [38]. Further, Leiband Gut-Landmann et al. demonstrated that bone marrow-derived DC treated with curdlan (a pure β-glucan which does not stimulate through TLR/MyD88) activates the MAPKs p38 and ERK1/2 as well as NF-κB leading to IL-6, TNF-α and IL-12p40 production and DC maturation [39].
Fig. 1. Dectin-1 mediated signal transduction.

Upon ligand binding Dectin-1 is Tyr phosphorylated within the ITAM-like motif leading to Syk binding and activation. At present it is unclear whether two phosphorylated Tyr resides (i.e. dimerization of Dectin-1) are required for Syk activation since this kinase normally requires two phospho-Tyr for binding. Syk activation leads to a signaling cascade potentially involving PLC-gamma and PKC-mediated CARD9 activation. Phosphorylated CARD9 recruits and activates Bcl-10 and Malt1 leading to TRAF2 and TRAF6 activation and the subsequent IκB degradation and NF-κB nuclear translocation. The intersection of Dectin-1 and TLR signaling may be at the step of TRAF6 whose activation by TLR2 is mediated through IRAK1. Dashed arrows: hypothesized signaling pathways; solid arrows: demonstrated pathways.
DECTIN-1 AS A PHAGOCYTIC RECEPTOR
Dectin-1 functions in mediating phagocytosis of fungi, yeast and yeast products such as zymosan. This requires both a cluster of acidic residues in the cytoplasmic tail as well as a membrane-proximal Tyr residue [23]. In DC, actin polymerization and formation of the phagocytic cup requires Sykmediated activation of the Rho GTPases Cdc42 and Rac. In contrast, Dectin-1 mediated activation of Syk is not required for macrophage phagocytosis of zymosan; however, it does require the ITAM motif as well as Cdc42 and Rac1 [29]. Herre et al. suggest that Dectin-1 mediated phagocytosis in transfected NIH-3T3 cells most resembles FcγR mediated phagocytosis [29]. Dectin-1, internalized by phagocytosis, is targeted to the lysosome for degradation while the endocytosed receptor, for example by the addition of laminarin, is recycled to the plasma membrane. The different fates likely result from differences in uptake mechanisms: actin-mediated phagocytosis vs. clathrin-mediated endocytosis. The requirement of Dectin-1 for phagocytosis depends on the composition of the microbe or particle in question. For example, fungi have a cell wall consisting primarily of carbohydrates of which 50% are β-1,3 linked glucans. Therefore, the repertoire of receptors engaged by fungi is restricted and includes the Mannose receptor, Dectin-1, scavenger receptors, the lectin site of CR3, DC-SIGN, TLRs and lactosylceramide [8,40].
DECTIN-1 AND THE IMMUNE RESPONSE
The role of Dectin-1 in regulating an immune response has only been addressed in the context of anti-fungal activity due in large part to β-glucans being the only described ligand to date for Dectin-1. Anti-fungal immunity is known to involve a Th1 response as well as the generation of ROS by phagocytic cells such as macrophages and neutrophils. Recognition of fungi by macrophages, DC and other cells of the innate immune system involves various PRRs including the mannose receptor, TLRs and Dectin-1. TLR2 and Dectin-1 appear to play particularly prominent roles in inducing an immune response to various fungi including the opportunistic pathogens C. albicans and Pneumocystis carinii [41,42]. Dectin-1 mediated uptake of C. albicans and P. carinii leads to killing of the ingested fungus in part through the production of ROS as well as production of inflammatory mediators such as TNF-α and the chemokine CXCL2 [43]. A recent study, however, indicated that C. albicans does not use Dectin-1 to invade human monocyte-derived DC and does not induce NADPH oxidase activity [44]. As previously mentioned, TLR2 and Dectin-1 cooperate in the macrophage production of various immune modulators including TNF-α, MIP-2, IL-12p40 and ROS [31]. Dectin-1 has also been shown to promote alveolar macrophage production of TNF-α, IL-12, IL-1β, IL-6, MIP-1α, MIP-2, G-CSF and GM-CSF following infection with Aspergillus fumigatus [43]. Dectin-1 may also play a role in T cell activation. DCs stimulated with the β-glucan curdlan produced IL-23 and this stimulation was independent of MyD88. IL-23 is critical for the expansion of IL-17 producing T-cells (i.e. TH17 cells). The same study found that the IL-23 production and subsequent expansion of the IL-17 producing T-cells was dependent on Dectin-1, Syk and CARD9 [39]
Direct evidence for the importance of Dectin-1 in controlling fungal infections has recently been obtained using Dectin-1 deficient mice. In studies by Taylor et al. the Dectin-1−/− mice appeared to maintain a normal immune system and contained no gross developmental abnormalities. As expected, thioglycollate-elicited macrophages from the Dectin-1−/− mice showed impaired production of TNF-α and H2O2 upon incubation with zymosan. Interestingly, prior opsonization with complement lead to zymosan uptake in a Dectin-1 independent manner but production of TNF-α was still Dectin-1 dependent [42]. In this same study, Dectin-1−/− mice intravenous infected with C. albicans SC5314 showed decreased survival and increased fungal load throughout the intestine compared to infected WT 129/Sv mice. Macrophages isolated from the Dectin-1−/− mice also showed decreased recognition and impaired inflammatory response to C. albicans. Finally, the Taylor study showed that Dectin-1−/− mice intra-peritoneal infected with C. albicans demonstrated diminished recruitment of leukocytes and reduced production of chemokines CCL2 and CCL3 compared to infected WT mice. Together, the data suggest a defect in the early innate immune response to C. albican infection in Dectin-1−/− mice. An analogous study was performed by Saijo et al. using Dectin-1 deficient mice generated independently in their laboratory. Similar to studies described above, these Dectin-1−/− mice developed normally. Interestingly, although bone marrow-derived DC from Dectin-1−/− mice did not produce IL-12 or TNF-α upon exposure to a soluble β-1,6 branched β-1,3 glucan from Sparassis crispa, the production of TNF-α and IL-12 upon zymosan treatment was not reduced in Dectin-1−/− mice relative to WT mice. However, production of IL-10 was Dectin-1 dependent. Similar results were observed using thioglycollate-elicited macrophages, with IL-10 and ROS but not TNF-α production being dependent on Dectin-1 following zymosan treatment. Instead, TNF-α production was completely MyD88 dependent. The difference between these two studies using Dectin-1−/− macrophages and their response to zymosan is unclear, although one possibility is the strain differences; 129/Sv mice were used for the Taylor study compared to C57BL/6 for the Saijo study. Even more surprising, results from the C. albicans infection experiments also differed significantly between the two studies. In contrast to above, the Saijo study found no increased fungal burden or decreased survival between WT and Dectin-1−/− mice following various intravenous infection doses (2 × 104 – 1 × 106). Nor did this study find a difference between WT and Dectin-1−/− mice in TNF-α, IL-12 or ROS production following a C. albican infection. However, they did observe increased fungal burden as well as decreased ROS production in the Dectin-1−/− mice infected with P. carinii at early time points. Again the reasons for the differences between the two studies is unclear and may reflect C. albicans strain as well as mouse strain differences. In support of the Taylor study, CARD9 deficient mice were also more susceptible to C. albican infection [33]; however, recent studies indicate that CARD9 is also linked to activation of NF-κB through a variety of receptors including NOD2 as well as TLR-mediated activation of the MAPKs [45].
MYCOBACTERIA AND DECTIN-1
As outlined above, Dectin-1 functions in the recognition and response to fungi, yeast and some cell wall products. What is less clear is its role as a PRR for other microbes and potential pathogens. Until recently the only link between Dectin-1 and non-fungal pathogens involved the bacteria Haemophilus influenza. Studies by Ahren et al. demonstrated a Dectin-1 dependent binding of H. influenza to eosinophils as well as a Dectin-1 dependent production of IL-8 and respiratory burst [13]. More recently we have made the interesting observation that the production of inflammatory mediators by mycobacteria-infected murine macrophages was dependent, at least in part, on Dectin-1. Macrophages infected with the non-pathogenic mycobacteria M. smegmatis produce significantly greater TNF-α compared to cells infected with pathogenic mycobacteria and this was inhibited approximately 60% using either a blocking antibody to Dectin-1 or by blocking the receptor with the soluble Dectin-1 ligand laminarin [24]. The importance of Dectin-1 in the induction of TNF-α extended to macrophages infected with a number of mycobacterial species and strains including the non-pathogenic M. phlei, the attenuated M. bovis BCG, and the avirulent M. avium 2151 and M. tuberculosis H37Ra strains. Interestingly, the limited TNF-α produced by macrophages infected with the virulent M. tuberculosis strain H37Rv was not Dectin-1 dependent. The importance of Dectin-1 extended to other immune mediators as the macrophage production of IL-6, RANTES and G-CSF induced by M. smegmatis or M. bovis BCG infection was significantly inhibited by the Dectin-1 blocking antibody [24]. Similar to the results obtained with macrophages upon zymosan treatment, we observed that the Dectin-1 mediated TNF-κ production in M. smegmatis infected macrophages was dependent on TLR2, as the Dectin-1 blocking antibody had no effect on TNF-α production when TLR2−/− macrophages were used for the infection. A similar result was seen with IL-12p40 production (Fig. 2). It is important to note that in contrast to macrophage phagocytosis of fungi, Dectin-1 appears not to be required for phagocytosis of mycobacteria [24]. The likely explanation is that since mycobacteria engage a number of phagocytic receptors a loss of one receptor would be compensated for by others.
Fig. 2. Dectin-1-mediated IL-12p40 production by M. smegmatis infected macrophages requires TLR2 expression.
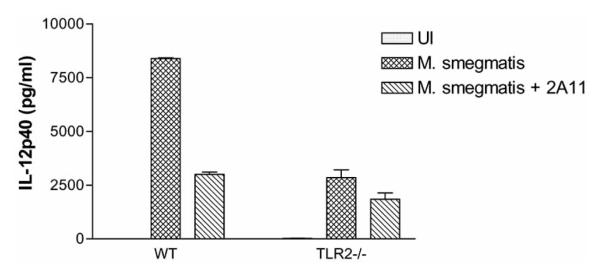
C57BL/6 bonemarrow derived macrophages were left untreated or pre-treated with 50 μg/ml anti-Dectin-1 blocking antibody 2A11 (Serotec, Oxford, England). 1 hour post-antibody treatment, the macrophages were infected with M. smegmatis. IL-12p40 released into the culture supernatant 9 hours post-infection was quantified by ELISA. Results represent mean ± SD of duplicate ELISA wells. UI: uninfected macrophages.
The results with the mycobacteria lead us to two fundamental questions: 1) what is the Dectin-1 ligand on mycobacteria and 2) what is the mechanism of Dectin-1 signaling and its relation to TLR2 signaling upon mycobacterial infection? In regard to the Dectin-1 ligand, there are no published reports of mycobacteria expressing β-1,3-glucans, although mycobacteria have been shown to express α-glucans [46,47]. However, biochemical studies do not support a role for α-glucans as Dectin-1 ligands [48]. Nevertheless, as shown with the β-glucans, the molecular weight, branching and tertiary structure of a specific α-glucan may dictate its binding affinity for Dectin-1. Determining whether the α-glucans or some other mycobacteria cell wall component serves as the Dectin-1 ligand awaits its purification. Preliminary proteolysis results suggest that the ligand is not proteinaceous (unpublished data). Once bound to Dectin-1, the mycobacteria induce a signaling response, and as predict from the zymosan experiments, our studies indicate that Src and Syk activation are required for optimum TNF-α production by macrophages upon M. smegmatis infection (Fig. 3). Whether the mycobacteria-induced activation of Syk leads to the formation and activation of the CARD9/Bcl-10/MALT1 complex awaits further investigation.
Fig. (3. Syk and Src are required for optimal TNF-α production in M. smegmatis infected macrophages.
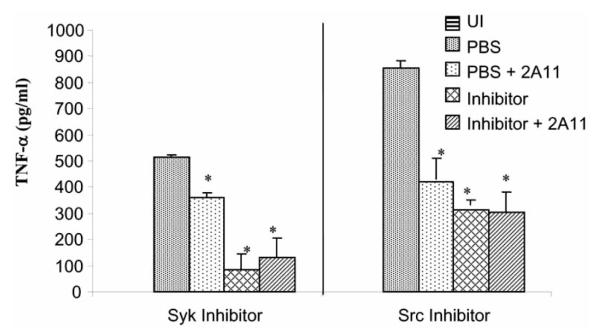
Balb/C bone-marrow derived macrophages were left untreated or pre-treated with 10 μM “Syk inhibitor” or PP2 (Src inhibitor) (CalBiochem, LaJolla, CA) and/or with 50 μg/ml anti-Dectin-1 blocking antibody 2A11. 1 hour post-antibody or 20 minutes post-inhibitor treatment, the macrophages were infected with M. smegmatis. TNF-α released into the culture supernatant 24 hours post-infection was quantified by ELISA. Results represent mean ± SD of duplicate ELISA wells. The Syk and Src inhibitor experiments were performed independently. *P<0.05. UI: uninfected macrophages.
CONCLUSION
Our understanding of Dectin-1 has increased significantly since the seminal observation that Dectin-1 is the β-glucan receptor [49]. We know a great deal more from recent studies about the signaling responses down-stream of Dectin-1 and how this converges with TLR signaling. The generation of knockout mice has also expanded our understanding of Dectin-1’s role in controlling fungal infections although there are differences between studies that need to be addressed. However, where information is lacking is how Dectin-1 may function as a PRR beyond its role in fungal and yeast recognition. Moreover, recent studies also suggest that Dectin-1 may recognize ligands in addition to β-glucans. The ability of PRRs to bind multiple ligands expressed on different pathogens is the norm and therefore it is likely that Dectin-1 will function in a similar manner. Unfortunately, until we define these different ligands it will be difficult to address Dectin-1’s importance in the innate immune response to diverse pathogens. Nevertheless, based on the recent results with mycobacteria it is clear that we need to expand our umbrella of potential pathogens recognized by Dectin-1 beyond the current focus on fungi and yeast. A thorough understanding of the relative contributions of Dectin-1 and other PRR in modulating immunity should provide insight into the development of effective adjuvants and therapeutics for a variety of infectious diseases.
ACKNOWLEDGEMENTS
This work was supported through grants AI056979 and AI052439 from the National Institute of Allergy and Infectious Diseases.
REFERENCES
- [1].Trinchieri G, Sher A. Nat. Rev. Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- [2].Kabelitz D, Medzhitov R. Curr. Opin. Immunol. 2007;19:1–3. doi: 10.1016/j.coi.2006.11.018. [DOI] [PubMed] [Google Scholar]
- [3].Akira S, Takeda K, Kaisho T. Nat. Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- [4].Blander JM, Medzhitov R. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- [5].Yates RM, Russell DG. Immunity. 2005;23:409–417. doi: 10.1016/j.immuni.2005.09.007. [DOI] [PubMed] [Google Scholar]
- [6].Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nat. Immunol. 2006;7:1250–1257. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- [7].Mitchell JA, Paul-Clark MJ, Clarke GW, McMaster SK, Cartwright N. J. Endocrinol. 2007;193:323–330. doi: 10.1677/JOE-07-0067. [DOI] [PubMed] [Google Scholar]
- [8].Brown GD. Nat. Rev. 2006;6:33–43. doi: 10.1038/nri1745. [DOI] [PubMed] [Google Scholar]
- [9].Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, Tanaka T, Miwa S, Katsura Y, Kita T, Masaki T. Nature. 1997;386:73–77. doi: 10.1038/386073a0. [DOI] [PubMed] [Google Scholar]
- [10].Shimaoka T, Kume N, Minami M, Hayashida K, Sawamura T, Kita T, Yonehara S. J. Immunol. 2001;166:5108–5114. doi: 10.4049/jimmunol.166.8.5108. [DOI] [PubMed] [Google Scholar]
- [11].Ariizumi K, Shen GL, Shikano S, Xu S, Ritter R, 3rd, Kumamoto T, Edelbaum D, Morita A, Bergstresser PR, Takashima A. J. Biol. Chem. 2000;275:20157–20167. doi: 10.1074/jbc.M909512199. [DOI] [PubMed] [Google Scholar]
- [12].Taylor PR, Brown GD, Reid DM, Willment JA, Martinez-Pomares L, Gordon S, Wong SY. J. Immunol. 2002;169:3876–3882. doi: 10.4049/jimmunol.169.7.3876. [DOI] [PubMed] [Google Scholar]
- [13].Ahren IL, Eriksson E, Egesten A, Riesbeck K. Am. J. Respir. Cell Mol. Biol. 2003;29:598–605. doi: 10.1165/rcmb.2002-0138OC. [DOI] [PubMed] [Google Scholar]
- [14].Willment JA, Marshall AS, Reid DM, Williams DL, Wong SY, Gordon S, Brown GD. Eur. J. Immunol. 2005;35:1539–1547. doi: 10.1002/eji.200425725. [DOI] [PubMed] [Google Scholar]
- [15].Reid DM, Montoya M, Taylor PR, Borrow P, Gordon S, Brown GD, Wong SY. J. Leukoc. Biol. 2004;76:86–94. doi: 10.1189/jlb.0104031. [DOI] [PubMed] [Google Scholar]
- [16].Willment JA, Gordon S, Brown GD. J. Biol. Chem. 2001;276:43818–43823. doi: 10.1074/jbc.M107715200. [DOI] [PubMed] [Google Scholar]
- [17].Yokota K, Takashima A, Bergstresser PR, Ariizumi K. Gene. 2001;272:51–60. doi: 10.1016/s0378-1119(01)00528-5. [DOI] [PubMed] [Google Scholar]
- [18].Kato Y, Adachi Y, Ohno N. Biol. Pharm. Bull. 2006;29:1580–1586. doi: 10.1248/bpb.29.1580. [DOI] [PubMed] [Google Scholar]
- [19].Brown J, O’Callaghan CA, Marshall AS, Gilbert RJ, Siebold C, Gordon S, Brown GD, Jones EY. Protein Sci. 2007;16:1042–1052. doi: 10.1110/ps.072791207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gantner BN, Simmons RM, Underhill DM. Embo. J. 2005;24:1277–1286. doi: 10.1038/sj.emboj.7600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Adachi Y, Ishii T, Ikeda Y, Hoshino A, Tamura H, Aketagawa J, Tanaka S, Ohno N. Infect. Immun. 2004;72:4159–4171. doi: 10.1128/IAI.72.7.4159-4171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Weis WI, Drickamer K. Annu. Rev. Biochem. 1996;65:441–473. doi: 10.1146/annurev.bi.65.070196.002301. [DOI] [PubMed] [Google Scholar]
- [23].Herre J, Gordon S, Brown GD. Mol. Immunol. 2004;40:869–876. doi: 10.1016/j.molimm.2003.10.007. [DOI] [PubMed] [Google Scholar]
- [24].Yadav M, Schorey JS. Blood. 2006;108:3168–3175. doi: 10.1182/blood-2006-05-024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Koyasu S, Tse AG, Moingeon P, Hussey RE, Mildonian A, Hannisian J, Clayton LK, Reinherz EL. Proc. Natl. Acad. Sci. U. S. A. 1994;91:6693–6697. doi: 10.1073/pnas.91.14.6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Abram CL, Lowell CA. Sci. STKE. 2007;2007:re2. doi: 10.1126/stke.3772007re2. [DOI] [PubMed] [Google Scholar]
- [27].Slack EC, Robinson MJ, Hernanz-Falcon P, Brown GD, Williams DL, Schweighoffer E, Tybulewicz VL, Reis e Sousa C. Eur. J. Immunol. 2007;37:1600–1612. doi: 10.1002/eji.200636830. [DOI] [PubMed] [Google Scholar]
- [28].Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, Schweighoffer E, Williams DL, Gordon S, Tybulewicz VL, Brown GD, Reis ESC. Immunity. 2005;22:507–517. doi: 10.1016/j.immuni.2005.03.004. [DOI] [PubMed] [Google Scholar]
- [29].Herre J, Marshall AS, Caron E, Edwards AD, Williams DL, Schweighoffer E, Tybulewicz V, Reis e Sousa C, Gordon S, Brown GD. Blood. 2004;104:4038–4045. doi: 10.1182/blood-2004-03-1140. [DOI] [PubMed] [Google Scholar]
- [30].Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. J. Exp. Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. J. Exp. Med. 2003;197:1119–1124. doi: 10.1084/jem.20021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Meyer-Wentrup F, Figdor CG, Ansems M, Brossart P, Wright MD, Adema GJ, van Spriel AB. J. Immunol. 2007;178:154–162. doi: 10.4049/jimmunol.178.1.154. [DOI] [PubMed] [Google Scholar]
- [33].Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, Forster I, Ruland J. Nature. 2006;442:651–656. doi: 10.1038/nature04926. [DOI] [PubMed] [Google Scholar]
- [34].Matsumoto R, Wang D, Blonska M, Li H, Kobayashi M, Pappu B, Chen Y, Wang D, Lin X. Immunity. 2005;23:575–585. doi: 10.1016/j.immuni.2005.10.007. [DOI] [PubMed] [Google Scholar]
- [35].Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, Iwakura Y, Ohno N, Koseki H, Yoshida H, Penninger JM, Saito T. Nat. Immunol. 2007;8:619–629. doi: 10.1038/ni1466. [DOI] [PubMed] [Google Scholar]
- [36].Bidere N, Snow AL, Sakai K, Zheng L, Lenardo MJ. Curr. Biol. 2006;16:1666–1671. doi: 10.1016/j.cub.2006.06.062. [DOI] [PubMed] [Google Scholar]
- [37].Schmitz ML, Bacher S, Dienz O. FASEB J. 2003;17:2187–2193. doi: 10.1096/fj.02-1100rev. [DOI] [PubMed] [Google Scholar]
- [38].Astarie-Dequeker C, N’Diaye EN, Le Cabec V, Rittig MG, Prandi J, Maridonneau-Parini I. Infect. Immun. 1999;67:469–477. doi: 10.1128/iai.67.2.469-477.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Leibundgut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, Reis ESC. Nat. Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- [40].Netea MG, Ferwerda G, van der Graaf CA, Van der Meer JW, Kullberg BJ. Curr. Pharm. Des. 2006;12:4195–4201. doi: 10.2174/138161206778743538. [DOI] [PubMed] [Google Scholar]
- [41].Saijo S, Fujikado N, Furuta T, Chung SH, Kotaki H, Seki K, Sudo K, Akira S, Adachi Y, Ohno N, Kinjo T, Nakamura K, Kawakami K, Iwakura Y. Nat. Immunol. 2007;8:39–46. doi: 10.1038/ni1425. [DOI] [PubMed] [Google Scholar]
- [42].Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, Brown GD. Nat. Immunol. 2007;8:31–38. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Steele C, Rapaka RR, Metz A, Pop SM, Williams DL, Gordon S, Kolls JK, Brown GD. PLoS Pathog. 2005;1:e42. doi: 10.1371/journal.ppat.0010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Donini M, Zenaro E, Tamassia N, Dusi S. Eur. J. Immunol. 2007;37:1194–1203. doi: 10.1002/eji.200636532. [DOI] [PubMed] [Google Scholar]
- [45].Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin XF, Dong C, Lin X. Nat. Immunol. 2007;8:198–205. doi: 10.1038/ni1426. [DOI] [PubMed] [Google Scholar]
- [46].Lemassu A, Daffe M. Biochem. J. 1994;297(Pt 2):351–357. doi: 10.1042/bj2970351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dinadayala P, Lemassu A, Granovski P, Cerantola S, Winter N, Daffe M. J. Biol. Chem. 2004;279:12369–12378. doi: 10.1074/jbc.M308908200. [DOI] [PubMed] [Google Scholar]
- [48].Palma AS, Feizi T, Zhang Y, Stoll MS, Lawson AM, Diaz-Rodriguez E, Campanero-Rhodes MA, Costa J, Gordon S, Brown GD, Chai W. J. Biol. Chem. 2006;281:5771–5779. doi: 10.1074/jbc.M511461200. [DOI] [PubMed] [Google Scholar]
- [49].Brown GD, Gordon S. Nature. 2001;413:36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]