

Abstract
Cell encapsulation in hydrogels is widely used in tissue engineering applications, including encapsulation of islets or other insulin-secreting cells in pancreatic substitutes. Use of adhesive, bio-functionalized hydrogels is receiving increasing attention, as cell-matrix interactions in 3-D can be important for various cell processes. With pancreatic substitutes, studies have indicated benefits of 3-D adhesion on the viability and/or function of insulin-secreting cells. As long-term storage of microencapsulated cells is critical for their clinical translation, cryopreservation of cells in hydrogels is actively being investigated. Previous studies have examined the cryopreservation response of cells encapsulated in non-adhesive hydrogels using conventional freezing and/or vitrification (ice-free cryopreservation), however, none have systematically compared the two cryopreservation methods with cells encapsulated within an adhesive 3-D environment. The latter would be significant, as evidence suggests adhesion influences cellular response to cryopreservation. Thus, the objective of this study was to determine the response to conventional freezing and vitrification of insulin-secreting cells encapsulated in an adhesive biomimetic hydrogel. Recombinant insulin-secreting C2C12 myoblasts were encapsulated in oxidized RGD-alginate and cultured 1 or 4 days post-encapsulation, cryopreserved, and assessed up to 3 days post-warming for metabolic activity and insulin secretion, and one day post-warming for cell morphology. Besides certain transient differences of the vitrified group relative to the Fresh control, both conventional freezing and vitrification maintained metabolism, secretion and morphology of the recombinant C2C12 cells. Thus, due to a simpler procedure and slightly superior results, conventional freezing is recommended over vitrification for the cryopreservation of C2C12 cells in oxidized RGD-modified alginate.
Keywords: cell encapsulation, cryopreservation, vitrification, alginate, RGD peptide, pancreatic substitute
1. Introduction
Cell microencapsulation in hydrogels has been widely used for various tissue engineered constructs, with alginate commonly being used as the encapsulation material [1–3]. To better control cell fate in 3-D, bio-functionalized hydrogels are increasingly being studied, with additions of bioactive motifs to help control cellular processes such as adhesion [4, 5]. Cell-matrix interactions in 3-D have been shown to be important for cell survival, proliferation, and differentiation among other cell processes [6]. Specifically, hydrogels containing the RGD adhesive peptide motif have been used for encapsulation of a variety of cell types, including myoblasts [7, 8], bone marrow stromal cells [9], pre-osteoblasts [8], and human embryonic stem cells [10].
For long-term storage and clinical translation of microencapsulated cell systems, cryopreservation is critical. The two main methods of cryopreservation are conventional freezing and ice-free cryopreservation, or vitrification. Although extracellular ice formation is generally not detrimental to single cells in suspension, ice formation during freezing may cause significant damage to multicellular systems and tissues [11, 12]. Thus, vitrification has been investigated for preservation of various natural tissues [11, 13, 14] as well as tissue engineered constructs [15–21]. However, vitrification may potentially lead to excessive cell osmotic excursions as well as cytotoxicity due to the high concentration of cryoprotectants used in the procedure [11, 22]. Therefore, as both methods have their potential drawbacks, it is important to investigate both vitrification and freezing in order to determine the best method of preservation for a given system.
With respect to cryopreservation of encapsulated cells, many studies have examined cellular response in non-adhesive hydrogel systems [18–20, 23–32]. However, with pancreatic substitutes, cell-matrix interactions in 3-D have been shown to be beneficial for cell viability and/or function of encapsulated β cells or islets [33–35] and may become critical as anchorage-dependent, non-β cell types are being explored for use in an encapsulated cell therapy for diabetes. Studying the cryopreservation response of cells encapsulated in an adhesive environment is, therefore, necessary, especially as previous studies have indicated that adhesion to a substrate affects cryopreservation in different and reportedly conflicting ways [36–39]. Some studies have indicated an increased likelihood of cryoinjury due to adhesion [36, 39], while others have indicated benefits to cryopreservation of attached cells compared to cells cryopreserved in suspension [38] or in non-adhesive matrices [37]. Previous studies with cells encapsulated in adhesive hydrogels derived from natural extracellular matrices showed that freezing leads to changes in cell morphology [40, 41] and decreases in cell viability [40] or function [41], relative to non-preserved controls. Vitrification of similar constructs, on the other hand, resulted in viability similar to non-preserved controls [21]. However, no studies have systematically compared freezing and vitrification with cells encapsulated in the same adhesive hydrogel system.
In this work, we studied and compared conventional freezing and vitrification of a model pancreatic substitute consisting of murine C2C12 myoblasts, stably transfected to secrete insulin, encapsulated in partially oxidized, RGD-modified alginate hydrogels. Parental C2C12 cells have been well characterized in RGD-alginate, displaying the ability to survive, proliferate, and differentiate on 2-D [42] as well as in 3-D hydrogel systems [7]. Also, as previous studies have indicated benefits of longer-term culture of encapsulated cells pre-preservation [38, 41], we evaluated the effects of cryopreservation 1 and 4 days post-encapsulation. Bead integrity, cell metabolic activity and morphology, and insulin secretion after cryopreservation were quantified and compared to those of Fresh controls. The implications of our findings in identifying appropriate cryopreservation procedures for cells in adhesive 3-D hydrogels are discussed.
2. Materials and Methods
2.1 Alginate Modification
All chemicals were obtained from Sigma (St. Louis, MO), unless otherwise indicated. Alginate was partially oxidized with sodium periodate based on previously published protocols [7, 43, 44]. Briefly, Pronova ultrapure low viscosity high mannuronic acid (LVM) alginate (FMC Biopolymer, Philadelphia, PA) with 43% guluronic acid content and viscosity of 24 mPa•s was dissolved at a concentration of 1% (w/v) in ultrapure water. Subsequently, sodium periodate (Acros Organics, Geel, Belgium) was added to the alginate solution at 21.6 mg/g alginate [7] and stirred in the dark at room temperature for 19 hours. Subsequently, to ensure quenching of the oxidation reaction, a two-fold molar excess, compared to the sodium periodate used, of ethylene glycol was added to the alginate solution and stirred for an additional 2 hours [44]. Alginate was then placed in 2000 MWCO dialysis cassettes (Thermo Fisher Scientific, Waltham, MA) and dialyzed against ultrapure water for 3 days prior to lyophilization.
Partially oxidized alginate was subsequently reconstituted and conjugated with GGGGRGDSP [7] or GGGGRGESP (Biomatik Corporation, Wilmington, DE) at 10mg/g alginate using aqueous carbodiimide chemistry [42, 45]. Briefly, 1% (w/v) partially oxidized alginate was dissolved in 0.1 M MES buffer with 0.3 M NaCl (pH 6.5) for four hours at room temperature. Subsequently, 1-Ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) (Thermo Fisher Scientific) and N-hydroxysulfosuccinimide (sulfo-NHS) (Thermo Fisher Scientific) were dissolved in MES buffer immediately prior to the reaction and added to the alginate to obtain a 2:1 sulfo-NHS:alginate molar ratio. The alginate solution was allowed to zreact for 5 minutes prior to addition of peptide [9]. The resulting solution was continuously mixed at room temperature for 20 hours prior to being transferred to 3500 MWCO dialysis cassettes (Thermo Fisher Scientific) for three-day dialysis against ultrapure water prior to lyophilization.
2.2 Cell Culture and Encapsulation
Stable C2C12 cells were previously prepared by transfection with a furin-cleavable, B10-modified human insulin gene expressed downstream of a CMV promoter and by selection with puromycin [46]. Stable C2C12 cells were cultured in 25 mM glucose Dulbecco’s Modified Eagle’s Medium (DMEM, Mediatech, Manassas, VA) supplemented with 10% Fetal Bovine Serum (FBS, Gemini Bioproducts, West Sacramento, CA), 1% penicillin/streptomycin (P/S, Mediatech) and 1 μg/ml puromycin (Sigma) in order to maintain constant selective pressure on the cells. Cells were cultured under subconfluent conditions in order to prevent cell differentiation [42] in a 37°C humidified incubator with 95% air/5% CO2. All salt solutions used for encapsulation/coating were adjusted to 300 mOsm by changing the amount of the major salt component in the solution. For encapsulation, cells were detached from flasks using 0.25% trypsin (Mediatech) and cell number determined using trypan blue dye exclusion. The cell suspension was then centrifuged, and 3.5 % oxidized RGD-modified LVM, reconstituted in 0.85% NaCl (w/v), was added to the cell pellet to obtain an encapsulation density of 3x106 cells/ml alginate. This low cell density was used to promote cell-matrix interactions over cell-cell interactions, thus allowing us to study the effect of the former and minimize the effect of the latter on the cryopreservation outcome. Beads of 300–600 μm diameter were formed using an electrostatic bead generator (Nisco Engineering AG, Zurich, Switzerland) and crosslinked in a 1.1% (w/v) CaCl2 bath. Beads were immediately coated according to the procedure of Sun [47], with modifications. Beads were placed in 0.1% (w/v) CHES in 1.1% (w/v) CaCl2 for 3 minutes, washed with 1.1% (w/v) CaCl2, and then incubated in 0.1% poly-L-lysine (PLL) (MW 15,000–30,000, Sigma) in 0.85% (w/v) NaCl, with mixing, for 5 minutes. Beads were then exposed to successive washes in 0.1% (w/v) CHES in 1.1% (w/v) CaCl2, 1.1% (w/v) CaCl2, and 0.85% (w/v) NaCl (two washes). Beads were then incubated in 0.2% w/v Pronova UP LVM (unmodified) alginate in 0.85% (w/v) NaCl for 4 minutes. After one additional wash in 0.85% (w/v) NaCl, beads were washed with culture medium and placed in T-25 flasks. Beads were not exposed to sodium citrate. Flasks were placed on a platform rocker (Stovall, Greensboro, NC) in an incubator at 37°C. Encapsulated cells were cultured in the same type of medium as monolayers.
2.3 Cryopreservation
2.3.1 Vitrification
Vitrification was carried out with the cryoprotectant (CPA) cocktail solution DPS as in [48]. DPS consisted of 3 M dimethylsulfoxide (DMSO), 3 M 1,2 propanediol, and 0.5 M sucrose in a modified version of the EuroCollins carrier solution, the latter consisting of 34.95 g/l glucose, 0.84 g/l NaHCO3 (Fisher Chemical, Fisher Scientific, Pittsburgh, PA), 1.12 g/l KCl and 1.68 g/l NaCl [48]. For CPA addition, approximately 0.7 ml beads were placed in 40 μm cell strainers (BD Biosciences, Bedford, MA), and sequentially transferred through CPA solutions of increasing concentration at 4°C in a six-well plate (BD Biosciences) (Table 1). After incubation in the last CPA solution, beads and solution were transferred to pre-siliconized 20 ml borosilicate glass vials (Fisherbrand/Fisher Scientific) and a layer of isopentane (EMD Chemicals, Gibbstown, NJ) was placed on top of the CPA and beads. Vials were then placed on ice until transferred to a pre-chilled rack. The rack was then placed in an isopentane bath in a −135°C freezer for fast cooling to approximately −100°C (~64°C/min) prior to transfer to a rack in the freezer for slow cooling (~2°C/min) to −130°C. Temperature was tracked during cooling using a thermocouple placed in a sample vial containing only CPA solution and isopentane. Vitrification was verified by visual observation after cooling and immediately prior to warming of vials, and there was no crystallization/devitrification visible upon rewarming [48, 49]. After overnight storage, vials were warmed rapidly in a 30% (v/v) DMSO in water bath at room temperature. Subsequently, vials were placed on ice and beads transferred to cell strainers for subsequent CPA removal in a stepwise fashion of decreasing CPA concentration at room temperature. Beads were then placed in T-12.5 flasks in fully supplemented DMEM at 37°C or used for immediate assay.
Table 1.
Cryoprotectant addition/removal protocol for DPS vitrification.
| Step | DMSO(M) | PD(M) | Sucrose (M) | Time (min) | Temperature (°C) |
|---|---|---|---|---|---|
| A1 | 1 | 1 | 0.15 | 2 | 4 |
| A2 | 2 | 2 | 0.3 | 2 | 4 |
| A3 | 3 | 3 | 0.5 | 2 | 4 |
| R1 | 2.25 | 2.25 | 0.3 | 2 | RT |
| R2 | 1.5 | 1.5 | 0.2 | 2 | RT |
| R3 | 0.75 | 0.75 | 0.1 | 2 | RT |
| R4 | 0 | 0 | 0 | 4 | RT |
A: addition; R: removal; RT: room temperature; PD; 1,2 propanediol
2.3.2 Conventional Freezing
For conventional freezing, 10% DMSO in fully supplemented DMEM at 4°C was added to beads, and beads were incubated for 10 minutes, as in [48]. Beads and solution were subsequently transferred to 2.0 ml cryogenic vials (Corning Inc., Corning, NY) which were placed in a Mr. Frosty isopropyl alcohol bath (Nalgene/Thermo Fisher Scientific, Rochester, NY) in a −80°C freezer (VWR International, Radnor, PA) for 1.5 hours. Subsequently, vials were plunged into liquid nitrogen. After overnight storage, beads were warmed rapidly in a 37°C water bath until no ice was visible. Beads were then placed in fully supplemented DMEM at 37°C for 10 minutes, followed by one wash in fully supplemented DMEM. Beads were subsequently transferred to T-12.5 flasks with fully supplemented DMEM and placed in the incubator at 37°C or used for immediate assay.
2.4 Metabolic Activity and Insulin Secretion
Metabolic activity of encapsulated cells was measured using alamarBlue® (Life Technologies, Carlsbad, CA). For the study comparing metabolic activity for the Fresh RGD/RGE/No peptide alginate encapsulated cells, 0.1 ml beads were added to 1 ml DMEM and 100 μl alamarBlue® stock solution in a 12-well plate. After incubation for four hours at 37°C, samples of supernatant from each well were placed in a black 96-well plate (Nalgene/Nunc) and fluorescence was measured using a Synergy H4 Multimode microplate reader (Biotek Instruments, Winooski, VT) using excitation and emission wavelengths of 544 nm and 590 nm, respectively. For cryopreservation studies, a lower volume of 50 μl of beads (to help prevent saturation of the fluorescence signal over the course of the experiment) was placed in 1 ml DMEM and 100 μl alamarBlue® stock solution and incubated at 37°C for 4 hours. Medium was sampled at the beginning and end of the alamarBlue® assay period for insulin as well as for measuring fluorescence, as mentioned above. Insulin concentrations in samples were measured using an ultrasensitive human insulin radioimmunoassay kit (Millipore, St. Louis, MO), according to manufacturer’s instructions.
2.5 LIVE/DEAD® Staining of Beads and Image Analysis
At one day post-warming, beads were stained with the LIVE/DEAD® viability kit (Life Technologies). Briefly, beads were washed 3 times in 0 mM glucose, phenol-free DMEM and then incubated in the same type of DMEM for 30 minutes with 2 μM calcein AM and 4 μM ethidium homodimer. Subsequently, beads were imaged using an LSM 510 NLO confocal microscope with the following excitation/emission settings: calcein AM (excitation: 488 nm/emission: 500–530 nm) and ethidium homodimer (excitation: 543 nm/emission: 560 nm) (Carl Zeiss, Thornwood, NY). A number of 10–20 beads were imaged per group, with Z-stacks taken every 10–20 μm. No movement in beads was detected during the imaging of each bead, and the beads were kept stationary using chamber slides.
For image analysis, Image J (NIH) was used to determine circularity of the cells in the beads, based on a procedure adapted from Yang et al.[50]. Briefly, one Z-stack was taken per bead; for each image in the Z-stack, the green channel was isolated and smoothed, a channel intensity threshold value of 30 was chosen, and particles with a size greater than 30 pixels squared were analyzed. The average circularity from each slice was calculated, and circularity values from all slices within a bead were averaged to obtain the cell circularity per bead.
2.6 Bead Integrity Immediately Post-Warming
Immediately post-warming, bead integrity was assessed using phase contrast light microscopy with an Olympus IX71 inverted light microscope (Olympus, Inc., Center Valley, PA). The percentage of broken beads was determined by counting beads in 6 independent fields of view for each group, with an average of 40 ± 3 beads per field of view. Averages were taken from 2–3 independent experiments.
2.7 Statistical Analysis
Data are represented as mean ± standard deviation. If necessary, data were normalized using the Box-Cox transformation [51] prior to statistical analysis. Data were analyzed using a two-tailed t-test or one-way ANOVA and Tukey’s post-hoc analysis in the General Linear Model in Minitab (Minitab Inc., State College, PA). When comparing cell circularity per bead among different groups, individual beads were nested within encapsulations. Values of p<0.05 were considered statistically significant.
3. Results
3.1 Metabolic Activity of Stable C2C12 Cells in Alginate Hydrogels
To determine the appropriate RGD-alginate matrix to allow for cell spreading in 3-D while maintaining overall bead integrity during culture and post-preservation, various bead preparations were tested (data not shown). Parameters varied included cross-linking ion type and concentration, alginate concentration, and the presence and concentration of poly-l-lysine (PLL) used during coating. Oxidized, RGD-modified LVM at 3.5% concentration, cross-linked with 100 mM CaCl2 and coated with 0.1% PLL exhibited the required properties; this encapsulation matrix was used in all experiments in this study. Metabolic activity in encapsulated stable C2C12 cells was then evaluated in adhesive (RGD) vs. non-adhesive matrices (RGE/no peptide) over 4 days (Figure 1). Metabolic activity was maintained in cells encapsulated in RGD-modified alginate hydrogels from Day 1 to Day 4 (p>0.05). On the other hand, metabolic activity of cells encapsulated in non-adhesive (RGE-modified and no peptide) alginates declined over time, e.g., from Day 1 to Day 4 (p<0.05). Cells in RGD-alginate had a higher metabolic activity compared to cells in non-adhesive alginates from Day 2 onward (p<0.05). However, there was no difference between RGE and no peptide alginate groups at any given time point (p>0.05). Based on these findings, RGE-modified alginate was chosen as the non-adhesive alginate control.
Figure 1.

Metabolic activity over time for cells encapsulated in 3.5% RGD (white bars), RGE (light gray bars), and no peptide (dark gray bars) LVM alginate, and normalized to respective groups on Day 1. #p<0.05 compared to RGD group at same time point, *p<0.05 compared to Day 1 within same group, ^p<0.05 compared to same group on Day 2. n=3–4.
3.2 Bead Integrity Post-Preservation
In what follows, cells in RGD-alginate beads cultured for 1 day prior to cryopreservation are referred to as “RGD C1” and those cryopreserved after 4 days in culture are referred to as “RGD C4”. Cells in RGE alginate beads were always cultured for 1 day prior to cryopreservation. Bead integrity was observed by phase contrast microscopy. Fewer than 10% of beads in each population were broken for RGD C1, RGD C4, and RGE groups for Fresh and both Cryopreserved treatments immediately post-warming (Figure 2). Furthermore, there was also no apparent difference in bead morphology between the cryopreservation groups. No significant debris from broken beads was observed post-cryopreservation, and the occasional beads that appeared damaged were broken in half or had visible cracks/tears in them.
Figure 2.

Representative phase contrast light micrographs (4x) of Cryopreserved beads immediately post-warming and of Fresh controls. Arrows point to representative damaged beads. The scale bars represent 500 μm.
3.3 Cell Morphology in Fresh and Cryopreserved Beads
Figures 3A, 3B and 3C show representative LIVE/DEAD images from the RGD C1, RGD C4, and RGE groups, respectively, one day post-warming. Qualitative examination of these images indicated that there was no extensive cell death in any of the Cryopreserved groups when compared to their respective Fresh controls. Cell morphology data are shown in Figure 4. For the Fresh group, cells encapsulated in the non-adhesive matrix, RGE, exhibited a higher cell circularity per bead (more rounded shape) compared to the cells in both RGD C1 and RGD C4 groups (p<0.05, Figure 4A). In addition, cells in the RGD C4 beads exhibited a lower cell circularity per bead (more elongated shape) compared to both the RGE and the RGD C1 beads. Figures 4B and 4C show that for the RGD C1 and RGD C4 groups, respectively, cryopreservation by either freezing or vitrification did not alter the cell circularity per bead relative to the Fresh controls. In the non-adhesive matrix, RGE, there was no difference between the cell circularity per bead in the Fresh control and either Frozen or Vitrified groups (p>0.05, Figure 4D). However, the DPS-vitrified group exhibited a higher cell circularity per bead compared to the Frozen group (p<0.05). In addition, when directly comparing cell circularity per bead in the Cryopreserved RGD C1 and RGD C4 groups, cells in the RGD C4 group had a lower circularity per bead (p<0.05) or were more elongated than cells in the RGD C1 group one day post-warming (data not shown).
Figure 3.

Representative LIVE/DEAD® images (10x) from 3-D projections of beads one day post-warming for Cryopreserved groups and respective Fresh controls for A) RGD C1, B) RGD C4, and C) RGE groups. Scale bar represents 100 μm. Beads from RGD C1 and C4 groups were cryopreserved one and four days post-encapsulation, respectively.
Figure 4.

Box plots displaying circularity per bead for A) Fresh RGE, RGD C1 and RGD C4, B) RGD C1 Fresh and Cryopreserved, C) RGD C4 Fresh and Cryopreserved, and D) RGE Fresh and Cryopreserved groups. ● average bead circularity for each group, and whiskers on each plot extend from minimum to maximum values of bead circularity within a given group. The middle line in each box represents the median and the bottom and top of each box represent the 25th and 75th percentile, respectively. n=3 for 3 independent encapsulations/cryopreservations. Ten to twenty beads per treatment were analyzed. Beads from RGD C1 and C4 groups were cryopreserved one and four days post-encapsulation, respectively.
3.4 Metabolic Activity in Fresh and Cryopreserved RGD-Alginate Hydrogels
Studies with stable C2C12 cells encapsulated in RGD and RGE-alginate hydrogels indicated that their metabolic activities normalized to the respective Fresh controls were similar to each other immediately and one day post-warming (results not shown). However, as the function of stable C2C12 cells in RGE-alginate was compromised over time (Figure 1), longer-term studies were carried out with RGD-alginate only, which is also the relevant and appropriate system for encapsulation of these cells in 3-D.
Figure 5 shows the metabolic activity of Fresh and Cryopreserved groups normalized to the respective Fresh t0 groups for RGD C1 (Figure 5A) and RGD C4 (Figure 5B). For the RGD C1 group there was no difference in metabolic activity between Fresh and Frozen beads at any time point assayed (p>0.05) (Figure 5A). However, the DPS-vitrified beads exhibited lower metabolic activity than the Fresh control immediately post-warming (p<0.05) but not at 1 and 3 days post-warming. With respect to RGD C4 (Figure 5B), the DPS-vitrified group had lower metabolic activity compared to the Fresh and Frozen groups immediately post-warming (p<0.05). There was no difference in metabolic activity between the Frozen and Fresh groups at any time point. All groups exhibited an increase in metabolic activity from 0 to 3 days post-warming (p<0.05). To more directly assess the effect of pre-preservation culture time on cryopreservation response, the same data used in Figure 5 were re-analyzed and plotted in Figure 6 with Cryopreserved groups normalized to the same day Fresh control, and responses from RGD C1 and RGD C4 groups at each time point were directly compared for Vitrified (Figure 6A) and Frozen (Figure 6B) groups. There were no differences in metabolic activity post-preservation between the RGD C1 and RGD C4 groups.
Figure 5.
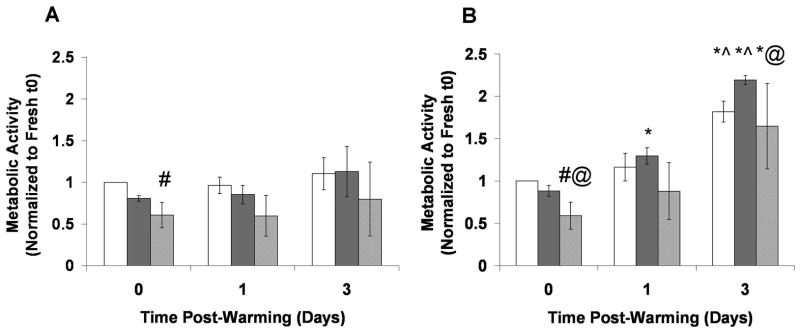
Metabolic activity over time in Fresh and Cryopreserved stable C2C12 cells encapsulated in hydrogels and cultured A) 1 (RGD C1) or B) 4 (RGD C4) days post-encapsulation. Cryopreserved groups and Fresh controls were assessed up to 3 days post-warming. All groups were normalized to the Fresh group at t0. The Fresh group is represented by the white bars, the Frozen group by the dark gray bars, and the DPS-vitrified group by the light gray bars. *p<0.05 compared to same group at t0, ^p<0.05 compared to same group at one day post-warming, #p<0.05 compared to Fresh group at same time point, @p<0.05 compared to Frozen group at same time point. n=3.
Figure 6.

Effect of culture time prior to cryopreservation on metabolic activity of A). Vitrified or B). Frozen RGD-alginate encapsulated stable C2C12 cells. *p<0.05 compared to the other group at same time point. n=3.
3.5 Insulin Secretory Function in Fresh and Cryopreserved RGD-Alginate Hydrogels
As with metabolic activity, insulin secretion normalized to the respective Fresh control was similar for stable C2C12 cells encapsulated in RGD and RGE-alginate hydrogels immediately and one day post-warming. For the reasons explained in the previous section, longer-term studies were carried out with RGD-alginate encapsulated cells only.
Figure 7 shows insulin secretion rates (ISR) of Fresh and Cryopreserved encapsulated cells from RGD C1 and RGD C4 groups, normalized to the respective Fresh t0 groups, up to 3 days post-warming. The average ISR values for the Fresh groups at t0 were similar, with values of 5.82 ± 0.80 and 4.26 ± 0.85 μU/(0.05 ml beads*hr) for RGD C1 and RGD C4, respectively. For the RGD C1 group, there were no differences between the Fresh and Frozen groups and Fresh and DPS-vitrified group at any time point (p>0.05, Figure 7A). For the RGD C4 group, both Fresh and Cryopreserved beads exhibited an increase in normalized ISR from immediately post-warming to 3 days post-warming and from one day post-warming to 3 days post-warming (p<0.05). The DPS group transiently had higher insulin secretion than the Fresh group immediately post-warming (p<0.05). As with the RGD C1 group, there were no differences in insulin secretion between the Fresh and Frozen groups at any time point analyzed. To more directly assess the effect of pre-preservation culture period on ISR, the data used in Figure 7 were reanalyzed and plotted in Figure 8 to directly compare responses from RGD C1 and RGD C4 groups. There was no difference in normalized ISR post-warming for DPS-vitrified beads between RGD C1 and RGD C4 groups (Figure 8A). With Frozen beads at one day post-warming, the RGD C1 group exhibited a higher normalized ISR relative to the RGD C4 group (p<0.05), but there were no differences immediately and 3 days post-warming (Figure 8B).
Figure 7.

Cryopreservation effects on insulin secretory function of stable C2C12 cells encapsulated in RGD-alginate hydrogels cultured A) One or B) Four days post-encapsulation, prior to cryopreservation. Cryopreserved groups and Fresh controls were assessed up to 3 days post-warming. All groups were normalized to the Fresh group at t0. The Fresh group is represented by the white bars, the Frozen group by the dark gray bars, and the DPS-vitrified group by the light gray bars. *p<0.05 compared to same group at t0, ^p<0.05 compared to same group at one day post-warming, #p<0.05 compared to Fresh group at same time point, @p<0.05 compared to Frozen group at same time point. n=3.
Figure 8.

Effect of culture time prior to cryopreservation on insulin secretory function of A). Vitrified or B). Frozen RGD-alginate encapsulated stable C2C12 cells. *p<0.05 compared to the other group at same time point. n=3.
4. Discussion
Cryopreservation is critical for clinical realization of tissue engineered constructs [11, 52]. Studying cellular responses after exposure to different cryopreservation methods in an adhesive hydrogel environment is particularly important, as adhesion has been shown to affect cellular response to cryopreservation [36–39]. The added complexity of adhesion in 3-D may affect cellular response, but it has not been extensively investigated in biomimetic hydrogels. In this study, we characterized cellular responses to cryopreservation of insulin-secreting, stable C2C12 myoblasts encapsulated in a 3-D adhesive RGD-alginate system. Overall, metabolic activity, insulin secretory function, and cell morphology were maintained in stable C2C12 cells encapsulated in partially oxidized, RGD-modified alginate hydrogels after conventional freezing. Although DPS-vitrification led to differences in metabolic activity and insulin secretory function immediately post-warming compared to Fresh controls, these differences were transient in nature and were abolished 1 and 3 days post-warming.
Cell-matrix interactions were important for cell survival in our system, as indicated by measurements of metabolic activity over time. Specifically, the decline in metabolic activity for stable C2C12 cells encapsulated in non-adhesive (RGE-modified and no peptide) alginate from Day 1 to Day 4, the higher metabolic activity in the RGD vs. non-adhesive hydrogels from Day 2 onward, and the maintenance of metabolic activity in the RGD-alginate beads from Day 1 to Day 4, all indicate that the encapsulated stable C2C12 cells performed better in the adhesive environment at the cell density used. Additionally, as there was no difference in metabolic activity between stable C2C12 cells encapsulated in RGE and no peptide alginate hydrogels at any time point assayed, there was likely no negative effect of incorporation of RGE into the alginate matrix.
Developing a hydrogel system that allowed for cell spreading in 3-D, while maintaining bead integrity in culture and after cryopreservation, was challenging. To achieve cell spreading, lower hydrogel stiffness appeared necessary, whereas beads still had to be robust enough to survive both methods of cryopreservation. Bead breakage immediately post-warming was indeed low in the current experiments (less than 10%). The cause of this breakage is unclear; however, one possibility is that it was due, at least in part, to the agitation of the beads in the cell strainer during the CPA addition and removal procedures.
Cell morphology was quantitatively assessed, via circularity measurements, after cryopreservation, as cell spreading of myoblasts encapsulated in partially oxidized, RGD-modified hydrogels has been associated with differentiation and fusion of myoblasts into multinucleated myotubes [7]. For a model pancreatic substitute containing recombinant myoblasts, terminal differentiation of myoblasts and fusion into myotubes could be beneficial prior to use in vivo, as this would prevent excessive cell growth after implantation. As expected, the circularity per bead for the Fresh RGE group was higher than that for both the Fresh RGD C1 and RGD C4 groups. This is likely due to the inability of the cells to attach and subsequently spread in the RGE-functionalized matrix, as opposed to their ability to bind to the RGD-alginate. Soluble RGD peptide competition studies have indicated that C2C12 cells can bind in an RGD-specific manner to RGD-conjugated alginate [42, 45].
Additionally, the cell circularity per bead was similar between the Cryopreserved groups and the Fresh control for the RGD and the RGE groups one day post-warming. Although a quantitative measure of cell shape after cryopreservation in 3-D hydrogels is lacking in the literature, some studies have qualitatively assessed cell morphology after freezing and warming in collagen gels [40, 41]. Koebe et al. [41] reported that only approximately 30% of hepatocytes cultured in a collagen sandwich configuration and conventionally frozen had a morphology similar to CPA-treated, non-frozen controls 2 and 11 days post-warming. Teo et al. [40], showed that immediately after freezing-thawing, fibroblasts encapsulated in collagen matrices experienced damage to cellular extensions as well as cell detachment, indicating damage to cell-matrix adhesions in addition to changes in the microstructure of the matrix. In our system, it is possible that changes in cell morphology present immediately post-warming were corrected, hence not detected, by day one when cell morphology was measured. However, it is also possible that cell morphology was better maintained in the RGD-alginate relative to cells encapsulated and cryopreserved in collagen gels, as stresses during freezing may have affected the collagen fibrils more than the RGD peptides in the flexible alginate hydrogel. Freezing has been shown to affect the matrix structure of collagen gels, specifically by increasing the mean void area, or porosity, of the matrix [40]. Similarly, the structure of collagen fibers from heart valve tissue has been shown to be damaged by freezing and to a smaller extent by vitrification [53, 54]. Since we did not use a gel consisting of a structural protein, but rather a polysaccharide conjugated with a small peptide, the damage to the peptide itself after freezing or vitrification may have been less than that to a gel containing a dense extracellular matrix protein structure. Another plausible reason for the maintenance of cell morphology in the current study, especially after freezing, is the possible low level of ice formation. Indeed, polysaccharide gels such as dextran [55] and alginate [56] have been shown to reduce ice crystal formation during cryopreservation. As such, encapsulation in alginate better maintained morphology of cell clusters including islets [23] and neurospheres [28] after conventional freezing compared to non-encapsulated controls.
Importantly, we examined cryopreservation response in RGD-alginate hydrogels up to 3 days post-warming, to account for recovery over time and because results immediately post-warming may give overestimates of cell viability due to delayed-onset cell death after cryopreservation [57]. As the metabolic activity for the DPS-vitrified group was lower than Fresh immediately post-warming (p<0.05), and there were no differences in metabolic activity between Fresh and Frozen constructs, freezing was generally better than vitrification at maintaining metabolic activity. Overall, insulin secretion rate was also maintained in Frozen constructs. This is in contrast to Murua et al. [30], who saw a 42% decrease in erythropoietin secretion, compared to the Fresh control, after freezing stably transfected C2C12 cells encapsulated in alginate-poly-L-lysine-alginate beads. Although one difference between their study and the current study is the presence of adhesive ligands in the alginate, we observed no differences in ISR for Cryopreserved RGE-alginate encapsulated cells relative to Fresh controls (data not shown). However, the alginate type and freezing protocol used by Murua et al. [30] were different than those used in the current study, and this may have contributed to the reduced secretion.
Interestingly, when directly comparing cryopreservation response for RGD-modified beads cultured one day (RGD C1) vs. four days (RGD C4) prior to cryopreservation (Figures 6 and 8), no differences in metabolic activity or insulin secretion were detected, except for a small difference in insulin secretion between RGD C1 and RGD C4 groups for the Frozen beads one day post-warming. In contrast, Koebe et al. [41] found overall more prolonged albumin secretion post-warming for encapsulated rat hepatocytes cultured 7 or 11 days prior to freezing compared to constructs cultured only 3 days prior to cryopreservation. Additionally, Ji et al. [38] found that cryopreserving adherent human embryonic stem cell colonies 24 hours after placement of a top matrigel layer led to a higher viability compared to colonies that were cryopreserved only one hour after placement of the matrigel layer. However, there were no differences in viability between groups cryopreserved 24 and 48 hours after matrigel layer placement on top of cells [38]. In the current study, it is possible that differences may have been detected with beads cryopreserved earlier than 1 day post-encapsulation; however, cryopreserving cells shortly after encapsulation would likely place them under undue stress from the combined encapsulation and cryopreservation processes. Additionally, differences in cryopreservation response may have occurred if RGD alginate beads had been cultured longer than 4 days before preservation. Longer culture periods were not investigated, however, due to the development of large cell clusters at later time points (data not shown); these would have led to increased cell-cell interactions, which were undesirable, as the focus of the present study was on the effect of cell-matrix interactions rather than cell-cell interactions on cryopreservation response.
5. Conclusions
In summary, partially oxidized RGD–modified alginate hydrogels better maintained metabolic activity for encapsulated stable C2C12 myoblasts compared to non-adhesive matrices. Except for small transient differences in metabolic activity and insulin secretion rate of the DPS-vitrified group immediately post-warming relative to Fresh controls, overall, conventional freezing and DPS-vitrification maintained metabolic activity, insulin secretion rate, and cell morphology for stable C2C12 cells encapsulated in RGD-alginate hydrogels. In addition, when directly comparing the effect of pre-preservation culture period on RGD-alginate-encapsulated cell response post-warming, metabolic activity and insulin secretory response were similar irrespective of whether the pre-preservation culture period was 1 or 4 days long. Due to simplicity of procedure and slightly superior results relative to Fresh controls, freezing appears appropriate for cryopreservation of stable C2C12 cells encapsulated in a partially oxidized, RGD-alginate matrix.
Acknowledgments
This work was funded by NIH R01DK73991. The authors are grateful for this support. The authors also wish to thank Dr. Peter Yang for his assistance with image analysis and Dr. Brani Vidakovich for his assistance with the statistical analysis of the imaging results.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Drury JL, Mooney DJ. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 2003;24:4337–51. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 2.Hernandez RM, Orive G, Murua A, Pedraz JL. Microcapsules and microcarriers for in situ cell delivery. Adv Drug Deliv Rev. 2010;62:711–30. doi: 10.1016/j.addr.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Orive G, Tam SK, Pedraz JL, Halle JP. Biocompatibility of alginate-poly-L-lysine microcapsules for cell therapy. Biomaterials. 2006;27:3691–700. doi: 10.1016/j.biomaterials.2006.02.048. [DOI] [PubMed] [Google Scholar]
- 4.Jabbari E. Bioconjugation of hydrogels for tissue engineering. Curr Opin Biotechnol. 2011;22:655–60. doi: 10.1016/j.copbio.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 6.Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol. 2007;213:565–73. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- 7.Boontheekul T, Hill EE, Kong HJ, Mooney DJ. Regulating myoblast phenotype through controlled gel stiffness and degradation. Tissue Eng. 2007;13:1431–42. doi: 10.1089/ten.2006.0356. [DOI] [PubMed] [Google Scholar]
- 8.Kong HJ, Boontheekul T, Mooney DJ. Quantifying the relation between adhesion ligand-receptor bond formation and cell phenotype. Proc Natl Acad Sci U S A. 2006;103:18534–9. doi: 10.1073/pnas.0605960103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connelly JT, Garcia AJ, Levenston ME. Inhibition of in vitro chondrogenesis in RGD-modified three-dimensional alginate gels. Biomaterials. 2007;28:1071–83. doi: 10.1016/j.biomaterials.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Hwang NS, Varghese S, Zhang Z, Elisseeff J. Chondrogenic differentiation of human embryonic stem cell-derived cells in arginine-glycine-aspartate modified hydrogels. Tissue Eng. 2006;12:2695–706. doi: 10.1089/ten.2006.12.2695. [DOI] [PubMed] [Google Scholar]
- 11.Brockbank KGM, Walsh JR, Song YC, Taylor MJ. Vitrification: Preservation of Celluar Implants. In: Ashammakhi N, Ferretti P, editors. Topics in Tissue Engineering. 2003. pp. 2–26. [Google Scholar]
- 12.Fahy GM, Wowk B, Wu J. Cryopreservation of complex systems: The missing link in the regenerative medicine supply chain (vol 9, pg 279, 2006) Rejuv Res. 2006;9:509. doi: 10.1089/rej.2006.9.279. [DOI] [PubMed] [Google Scholar]
- 13.Song YC, Khirabadi BS, Lightfoot F, Brockbank KGM, Taylor MJ. Vitreous cryopreservation maintains the function of vascular grafts. Nat Biotechnol. 2000;18:296–9. doi: 10.1038/73737. [DOI] [PubMed] [Google Scholar]
- 14.Brockbank KGM, Song PC, Greene ED, Taylor MJ. Quantitative analyses of vitrified autologous venous arterial bypass graft explants. Cell Preserv Technol. 2007;5:68–76. [Google Scholar]
- 15.Kuleshova LL, Gouk SS, Hutmacher DW. Vitrification as a prospect for cryopreservation of tissue-engineered constructs. Biomaterials. 2007;28:1585–96. doi: 10.1016/j.biomaterials.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 16.Dahl SLM, Chen ZZ, Solan AK, Brockbank KGM, Niklason LE, Song YC. Feasibility of vitrification as a storage method for tissue-engineered blood vessels. Tissue Eng. 2006;12:291–300. doi: 10.1089/ten.2006.12.291. [DOI] [PubMed] [Google Scholar]
- 17.Bhakta G, Lee KH, Magalhaes R, Wen F, Gouk SS, Hutmacher DW, et al. Cryoreservation of alginate-fibrin beads involving bone marrow derived mesenchymal stromal cells by vitrification. Biomaterials. 2009;30:336–43. doi: 10.1016/j.biomaterials.2008.09.030. [DOI] [PubMed] [Google Scholar]
- 18.Agudelo CA, Iwata H. The development of alternative vitrification solutions for microencapsulated islets. Biomaterials. 2008;29:1167–76. doi: 10.1016/j.biomaterials.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 19.Agudelo CA, Teramura Y, Iwata H. Cryopreserved Agarose-Encapsulated Islets As Bioartificial Pancreas: A Feasibility Study. Transplantation. 2009;87:29–34. doi: 10.1097/TP.0b013e318191b24b. [DOI] [PubMed] [Google Scholar]
- 20.Mukherjee N, Chen ZZ, Sambanis A, Song Y. Effects of cryopreservation on cell viability and insulin secretion in a model tissue-engineered pancreatic substitute (TEPS) Cell Transplant. 2005;14:449–56. doi: 10.3727/000000005783982882. [DOI] [PubMed] [Google Scholar]
- 21.Wu YN, Yu HR, Chang S, Magalhaes R, Kuleshova LL. Vitreous cryopreservation of cell-biomaterial constructs involving encapsulated hepatocytes. Tissue Eng. 2007;13:649–58. doi: 10.1089/ten.2006.0075. [DOI] [PubMed] [Google Scholar]
- 22.Fowler A, Toner M. Cryo-injury and biopreservation. Ann Ny Acad Sci. 2005;1066:119–35. doi: 10.1196/annals.1363.010. [DOI] [PubMed] [Google Scholar]
- 23.Inaba K, Zhou D, Yang B, Vacek I, Sun AM. Normalization of diabetes by xenotransplantation of cryopreserved microencapsulated pancreatic islets - Application of a new strategy in islet banking. Transplantation. 1996;61:175–9. doi: 10.1097/00007890-199601270-00001. [DOI] [PubMed] [Google Scholar]
- 24.Schneider S, Klein HH. Preserved insulin secretion capacity and graft function of cryostored encapsulated rat islets. Regul Peptides. 2011;166:135–8. doi: 10.1016/j.regpep.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Stiegler PB, Stadlbauer V, Schaffellner S, Halwachs G, Lackner C, Hauser O, et al. Cryopreservation of insulin-producing cells microencapsulated in sodium cellulose sulfate. Transplant P. 2006;38:3026–30. doi: 10.1016/j.transproceed.2006.08.188. [DOI] [PubMed] [Google Scholar]
- 26.Zhou DB, Vacek I, Sun AM. Cryopreservation of microencapsulated porcine pancreatic islets - In vitro and in vivo studies. Transplantation. 1997;64:1112–6. doi: 10.1097/00007890-199710270-00005. [DOI] [PubMed] [Google Scholar]
- 27.Stensvaag V, Furmanek T, Lonning K, Terzis AJA, Bjerkvig R, Visted T. Cryopreservation of alginate-encapsulated recombinant cells for antiangiogenic therapy. Cell Transplant. 2004;13:35–44. doi: 10.3727/000000004772664879. [DOI] [PubMed] [Google Scholar]
- 28.Malpique R, Osorio LM, Ferreira DS, Ehrhart F, Brito C, Zimmermann H, et al. Alginate encapsulation as a novel strategy for the cryopreservation of neurospheres. Tissue Eng Part C Methods. 2010;16:965–77. doi: 10.1089/ten.TEC.2009.0660. [DOI] [PubMed] [Google Scholar]
- 29.Umemura E, Yamada Y, Nakamura S, Ito K, Hara K, Ueda M. Viable cryopreserving tissue-engineered cell-biomaterial for cell banking therapy in an effective cryoprotectant. Tissue Eng Part C Methods. 2011;17:799–807. doi: 10.1089/ten.tec.2011.0003. [DOI] [PubMed] [Google Scholar]
- 30.Murua A, Orive G, Hernandez RM, Pedraz JL. Cryopreservation based on freezing protocols for the long-term storage of microencapsulated myoblasts. Biomaterials. 2009;30:3495–501. doi: 10.1016/j.biomaterials.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 31.Hang H, Shi X, Gu G, Wu Y, Gu J, Ding Y. In vitro analysis of cryopreserved alginate-poly-L-lysine-alginate-microencapsulated human hepatocytes. Liver Int. 2010;30:611–22. doi: 10.1111/j.1478-3231.2009.02197.x. [DOI] [PubMed] [Google Scholar]
- 32.Schneider S, Klein HH. Long -term Graft Function of Cryostored Alginate Encapsulated Rat Islets. Eur J Med Res. 2011;16:396–400. doi: 10.1186/2047-783X-16-9-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber LM, Anseth KS. Hydrogel encapsulation environments functionalized with extracellular matrix interactions increase islet insulin secretion. Matrix Biology. 2008;27:667–73. doi: 10.1016/j.matbio.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weber LM, Hayda KN, Anseth KS. Cell-matrix interactions improve beta-cell survival and insulin secretion in three-dimensional culture. Tissue Eng Part A. 2008;14:1959–68. doi: 10.1089/ten.tea.2007.0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber LM, Hayda KN, Haskins K, Anseth KS. The effects of cell-matrix interactions on encapsulated beta-cell function within hydrogels functionalized with matrix-derived adhesive peptides. Biomaterials. 2007;28:3004–11. doi: 10.1016/j.biomaterials.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Acker JP, Larese A, Yang H, Petrenko A, McGann LE. Intracellular ice formation is affected by cell interactions. Cryobiology. 1999;38:363–71. doi: 10.1006/cryo.1999.2179. [DOI] [PubMed] [Google Scholar]
- 37.Sambu S, Xu X, Schiffter HA, Cui ZF, Ye H. Rgds-Fuctionalized Alginates Improve the Survival Rate of Encapsulated Embryonic Stem Cells during Cryopreservation. Cryoletters. 2011;32:389–401. [PubMed] [Google Scholar]
- 38.Ji L, de Pablo JJ, Palecek SP. Cryopreservation of adherent human embryonic stem cells. Biotechnol Bioeng. 2004;88:299–312. doi: 10.1002/bit.20243. [DOI] [PubMed] [Google Scholar]
- 39.Choi J, Bischof JC. Cooling rate dependent biophysical and viability response shift with attachment state in human dermal fibroblast cells. Cryobiology. 2011;63:285–91. doi: 10.1016/j.cryobiol.2011.09.142. [DOI] [PubMed] [Google Scholar]
- 40.Teo KY, DeHoyos TO, Dutton JC, Grinnell F, Han B. Effects of freezing-induced cell-fluid-matrix interactions on the cells and extracellular matrix of engineered tissues. Biomaterials. 2011;32:5380–90. doi: 10.1016/j.biomaterials.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koebe HG, Dunn JC, Toner M, Sterling LM, Hubel A, Cravalho EG, et al. A new approach to the cryopreservation of hepatocytes in a sandwich culture configuration. Cryobiology. 1990;27:576–84. doi: 10.1016/0011-2240(90)90045-6. [DOI] [PubMed] [Google Scholar]
- 42.Rowley JA, Mooney DJ. Alginate type and RGD density control myoblast phenotype. J Biomed Mater Res. 2002;60:217–23. doi: 10.1002/jbm.1287. [DOI] [PubMed] [Google Scholar]
- 43.Boontheekul T, Kong HJ, Mooney DJ. Controlling alginate gel degradation utilizing partial oxidation and bimodal molecular weight distribution. Biomaterials. 2005;26:2455–65. doi: 10.1016/j.biomaterials.2004.06.044. [DOI] [PubMed] [Google Scholar]
- 44.Bouhadir KH, Hausman DS, Mooney DJ. Synthesis of cross-linked poly(aldehyde guluronate) hydrogels. Polymer. 1999;40:3575–84. [Google Scholar]
- 45.Rowley JA, Madlambayan G, Mooney DJ. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials. 1999;20:45–53. doi: 10.1016/s0142-9612(98)00107-0. [DOI] [PubMed] [Google Scholar]
- 46.Cheng SY, Constantinidis I, Sambanis A. Use of glucose-responsive material to regulate insulin release from constitutively secreting cells. Biotechnol Bioeng. 2006;93:1079–88. doi: 10.1002/bit.20817. [DOI] [PubMed] [Google Scholar]
- 47.Sun AM. Microencapsulation of Pancreatic-Islet Cells - a Bioartificial Endocrine Pancreas. Method Enzymol. 1988;137:575–80. doi: 10.1016/0076-6879(88)37053-9. [DOI] [PubMed] [Google Scholar]
- 48.Ahmad HF, Simpson NE, Lawson AN, Sambanis A. Cryopreservation Effects on Intermediary Metabolism in a Pancreatic Substitute: A (13)C Nuclear Magnetic Resonance Study. Tissue Eng Part A. 2012;18:2323–31. doi: 10.1089/ten.tea.2011.0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weiss AD, Forbes JF, Scheuerman A, Law GK, Elliott JA, McGann LE, et al. Statistical prediction of the vitrifiability and glass stability of multi-component cryoprotective agent solutions. Cryobiology. 2010;61:123–7. doi: 10.1016/j.cryobiol.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 50.Yang PJ, Levenston ME, Temenoff JS. Modulation of Mesenchymal Stem Cell Shape in Enzyme-Sensitive Hydrogels Is Decoupled from Upregulation of Fibroblast Markers Under Cyclic Tension. Tissue Eng Part A. 2012;18:2365–75. doi: 10.1089/ten.tea.2011.0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Box G, Cox DRT. An analysis of transformations. Journal of the Royal Statistical Society Series B. 1964:211–52. [Google Scholar]
- 52.Karlsson JOM, Toner M. Long-term storage of tissues by cryopreservation: Critical issues. Biomaterials. 1996;17:243–56. doi: 10.1016/0142-9612(96)85562-1. [DOI] [PubMed] [Google Scholar]
- 53.Schenke-Layland K, Xie J, Heydarkhan-Hagvall S, Hamm-Alvarez SF, Stock UA, Brockbank KG, et al. Optimized preservation of extracellular matrix in cardiac tissues: implications for long-term graft durability. Ann Thorac Surg. 2007;83:1641–50. doi: 10.1016/j.athoracsur.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 54.Votteler M, Berrio DAC, Pudlas M, Walles H, Stock UA, Schenke-Layland K. Raman spectroscopy for the non-contact and non-destructive monitoring of collagen damage within tissues. J Biophotonics. 2012;5:47–56. doi: 10.1002/jbio.201100068. [DOI] [PubMed] [Google Scholar]
- 55.Murase N, Abe S, Takahashi H, Katagiri C, Kikegawa T. Two-dimensional diffraction study of ice crystallisation in polymer gels. Cryo Letters. 2004;25:227–34. [PubMed] [Google Scholar]
- 56.Zhang W, Yang G, Zhang A, Xu LX, He X. Preferential vitrification of water in small alginate microcapsules significantly augments cell cryopreservation by vitrification. Biomed Microdevices. 2010;12:89–96. doi: 10.1007/s10544-009-9363-z. [DOI] [PubMed] [Google Scholar]
- 57.Baust JM, Vogel MJ, Van Buskirk R, Baust JG. A molecular basis of cryopreservation failure and its modulation to improve cell survival. Cell Transplant. 2001;10:561–71. [PubMed] [Google Scholar]