

Abstract
Although radiotherapy improves survival in patients, GBMs tend to relapse with augmented tumor migration and invasion even after irradiation (IR). Aberrant NF-κB and Stat3 activation and interaction has been suggested in several human tumors. However, possible NF-κB/Stat3 interaction and the role of Stat3 in maintenance of NF-κB nuclear retention in glioblastoma (GBM) still remain unknown. Stat3 and NF-κB (p65) physically interact with one another in the nucleus in glioma tumors. Most importantly, GST pull-down assays identified that Stat3 binds to the p65 transactivation domain (TAD) and is present in the NF-κB DNA-binding complex. Irradiation significantly elevated nuclear phospho-p65/phospho-Stat3 interaction in correlation with increased ICAM-1 and sICAM-1 levels, migration and invasion in human glioma xenograft cell lines 4910 and 5310. ChIP and promoter luciferase activity assays confirmed the critical role of adjacent NF-κB (+399) and Stat3 (+479) binding motifs in the proximal intron-1 in elevating IR-induced ICAM-1 expression. Specific inhibition of Stat3 and NF-κB with Stat3.siRNA or JSH-23 severely inhibited IR-induced p65 recruitment onto ICAM-1 intron-1 and suppressed migratory properties in both cell lines. On the other hand, Stat3C- or IR-induced Stat3 promoter recruitment was significantly decreased in p65-knockdown cells, thereby suggesting the reciprocal regulation between p65 and Stat3. We also observed a significant increase in NF-κB enrichment on ICAM-1 intron-1 and ICAM-1 transactivation in Stat3C overexpressing cells. In in vivo orthotopic experiments, suppression of tumor growth in Stat3.si+IR-treated mice was associated with the inhibition of IR-induced p-p65/p-Stat3 nuclear-colocalization and ICAM-1 levels. To our knowledge, this is the first study showing the crucial role of NF-κB/Stat3 nuclear association in IR-induced ICAM-1 regulation and implies that targeting NF-κB/Stat3 interaction may have future therapeutic significance in glioma treatment.
Keywords: Chromatin immunoprecipitation (ChIP), Glioma, GST pull-down, NF-κB, Stat3, Transactivation domain (TAD)
Introduction
Glioblastoma multiforme (GBM) is a lethal human brain tumor with a mean survival rate of less than one year despite the use of aggressive postsurgical multimodal treatments including ionizing-radiation (IR) and chemotherapy (1–3). Although locoregional radiotherapy improves survival in patients, GBMs tend to relapse in close proximity to the resection cavity after IR treatment (4). Tumor recurrence due to escape of IR-induced growth arrest is, in part, attributed to the excessive secretion of proteases, cytokines, and growth factors from the irradiated tumor cells into the extracellular milieu accompanied by inflammation (5, 6). Nuclear factor-κB (NF-κB) is a central signaling hub in inflammation-induced carcinogenesis and maintenance of established cancers (7–9). NF-κB is a family of five dimeric transcription factors (TFs) which contain Rel-homology domain (RHD). Among these, RelA (p65), RelB and c-Rel possess an additional c-terminal transactivation domain (TAD). Various stimuli including UV or X-ray irradiation cause genotoxic stress, IKK-mediated ubiquitination and degradation of IκBs and lead to nuclear translocation of NF-κB, which undergoes further post-translational modifications and binds to the κB consensus elements in the target gene promoters (10–12). Acetyltransferase p300/CBP cofactors mediate acetylation of p65 which, in turn, is deacetylated by histone deacetylases (13, 14). This reversible acetylation is crucial for high nuclear import/export ratios and constant in/out shuttling of NF-κB (15).
Signal transducer and activator 3 (Stat3) is another pro-oncogenic TF playing a role in glioma initiation and progression. Stat3 translocates to the nucleus in a signal-coupled manner and binds to gamma-interferon activated sequences (GAS) in the promoters of target genes which encode multiple anti-apoptotic or oncogenic proteins (16, 17). Recent evidences suggest the possible Stat3 and NF-κB collaboration in different human malignancies (18). However, the Stat3 binding region within the NF-κB (p65) protein is still not identified. In addition, knowledge about the possible functional significance of nuclear Stat3/NF-κB interaction in GBM and post-irradiated glioma malignancy is still lacking.
Intercellular adhesion molecule-1 (ICAM-1/CD54) is an inducible cell-surface glycoprotein that mediates adhesion-dependent cellular interactions, and its high expression correlates with increased tumor malignancy and poor prognosis (19). GBMs are characterized by significant ICAM-1 expression levels and IR elevates serum soluble-ICAM-1 levels, suggesting a role of ICAM-1 in post-irradiation tumor malignancy (20–22). ICAM-1 promoter possesses consensus elements for SP-1, AP-1, NFATc1 and NF-κB transcription factors upstream to the transcriptional start site (23, 24). The significance of p65/NFATc1 cooperativity by binding to ICAM-1 intron in the regulation of thrombin-induced ICAM-1 was reported in endothelial cells (25). Our analysis of the ICAM-1 5′-regulatory region revealed the presence of putative Stat-and NF-κB-binding sites in the intron-1 of ICAM-1. In this study, we show that the p65-TAD domain is essential for Stat3 interaction. Promoter reporter studies suggested that the intronic region possessing putative adjacent NF-κB (+399) and Stat3 (+479) consensus elements is central to IR-induced ICAM-1 expression. Our in vitro and in vivo studies indicated that the IR activated Stat3 and NF-κB in glioma xenograft cells and suggested the crucial role of NF-κB/Stat3 physical association in the regulation of ICAM-1-mediated migration and invasion.
Results
Stat3 directly interacts with NF-κB (p65)
Co-immunoprecipitation studies indicated the endogenous Stat3 and p65 complex formation in 4910 glioma cells (Figure 1A). IP with anti-Flag in Stat3C-overexpressing 4910 cells also indicated that p65 interacts with Stat3 (Figure 1B). In vitro translated biotin-labeled Stat3 and p65 proteins showed direct interaction with GST-p65 (left panel) and GST-Stat3 (right panel), respectively (Figure 1C). To further map the specific interacting regions on Stat3 and p65, we tested the ability of different biotin-labeled p65 and Stat3 truncated mutants to bind to GST-Stat3 and GST-p65, respectively. GST-Stat3 specifically interacted with the c-terminal region of the p65 protein comprising a transactivation (TAD, 291–551 aa) domain. Conversely, Stat3 binding to the RHD domain (1–290 aa) was not detected (Right panel, Figure 1D). On the other hand, GST-p65 bound to the 1–460 aa region of Stat3. Further, the region 1–120 aa did not show binding with p65, indicating the importance of coiled-coil and DNA-binding-domains of Stat3 in p65 coupling (Left panel, Figure 1D). Collectively, these results confirm that Stat3 directly binds to the TAD domain within p65 (Figure 1E). To check whether the NF-κB DNA-binding activity requires its interaction with Stat3 or p300, EMSA was performed using 4910 and 5310 nuclear extracts. Pre-incubation with anti-Stat3 and anti-p300 antibodies significantly abrogated the NF-κB binding activity and resulted in a gel super-shift, thereby confirming the presence of Stat3 and p300 in the NF-κB-DNA complex (Figure 1F). These results suggest that Stat3 interaction with p65-TAD is required for the maintenance of constitutive NF-κB DNA-binding activity in glioma xenograft cells.
Figure 1.

Direct interaction of Stat3 and NF-κB (p65) in glioma. (A) Complex formation of p65/Stat3 in 4910 cells was analyzed by co-IP in NEs (500 μg) using specific antibodies against Stat3 and non-specific IgG. IP samples were subjected to WB and probed with anti-p65 ab. Reciprocal IP with p65 and subsequent immunoblotting with Stat3 also confirmed the Stat3/p65 association. Input samples indicate 10% of pre-immunoprecipitated samples (50 μg). (B) The Stat3-p65 complex formation in Stat3C overexpressing 4910 cells was analyzed by IP with anti-Flag antibody, followed by immunoblotting with anti-p65 and anti-Stat3 antibodies. Pre-immnuoprecipitated inputs were subjected to WB to determine Stat3 protein levels. (C) Recombinant GST-p65, GST-Stat3 fusion proteins were separated on SDS-PAGE, transferred to nitrocellulose membrane and visualized by Ponceau staining. In vitro synthesized biotin-labeled Stat3 and p65 truncated proteins were incubated with GST-p65 and GST-Stat3, respectively. Elutes were then subjected to SDS-PAGE, analyzed and representative blots from three independent experiments were presented. Ten percent of the biotin-labeled proteins were loaded as input controls in the binding reaction. (D) Mapping of the minimal interacting regions on p65 and Stat3. Bacterially expressed GST, GST-p65 and GST-Stat3 were purified using MagneGST™ Pull-Down System following manufacturer’s instructions. Biotin-labeled truncated mutants of Stat3 and p65 proteins were synthesized using a TNT® Quick Coupled Transcription/Translation Systems as described in Materials and Methods. Labeled-Stat3 mutants were incubated with GST-p65 (Top left panel). The mixtures were washed and elutes were SDS-PAGE separated, transferred to nitrocellulose membrane and interacting partners were then detected. Input samples (10% input) were analyzed by SDS-PAGE (Bottom left panel). Biotin-labeled p65 truncated proteins were incubated with GST-Stat3 (Top right panel). Input samples were analyzed by SDS-PAGE (Bottom right panel). (E) Top panel-schematic representations of p65 and its truncation mutants used in GST pull-downs to localize the Stat3 interaction domain within p65. RHD-Rel Homology Domain, TAD-Transactivation Domain. Bottom panel-schematic representations of Stat3 and its truncation mutants used in GST pull-down experiments to localize the p65 interaction domain within Stat3. The following domains were represented with corresponding amino acid positions: ID-Interaction Domain; CCD-Coilied Coil Domain; DBD-DNA Binding Domain; SH2 dimerization domain and TAD-Transactivation Domain. (F) EMSA was performed to detect the NF-κB DNA-binding activity. Antibodies specific for p65, Stat3 and p300 were incubated with the control sample before adding labeled probe to observe supershift (SS). Representative blots from three independent experiments were shown.
Enhanced nuclear Stat3/p65 binding in IR-treated glioma xenograft cells
IR-treated cells displayed enhanced nuclear translocation of both Stat3 and NF-κB (p65) after 24 h (Figure 2A). We observed a dose-dependent increase in p-Stat3, p-p65 and ace-p65 levels with an optimum elevation observed at 8 Gy in both cell lines (Figure S1A). Based on previous reports on the role of p65/Stat3 interaction in maintaining consistent p65 activation and nuclear retention, we attempted to check the effect of IR on p65/Stat3 interaction in possible correlation with post-IR enhanced malignancy (18, 26). Immunofluorescence analysis showed elevated nuclear p65/Stat3 colocalization in the 6 h and 12 h IR-treated cells (Figure S1B). Nuclear colocalization of p65/Stat3 was also evident after 24 h in both cell lines, suggesting that IR leads to constitutive nuclear interaction of these TFs (Figure 2B). Further, co-IP using NEs showed elevated p-Stat3/p-p65 and p-Stat3/ace-p65 binding in IR-treated cells compared to controls (Figure 2C). There was also an increased co-precipitation of p-p65 with p-Stat3 and p300 acetyl transferase in IR-treated cells (Figure 2D). These results were also verified by co-IP with acetyl-p65 antibodies (Figure 2E). Nuclear p-Stat3/p-p65 complex formation was also confirmed by co-IP in glioma cell lines U87MG, U251 and SNB19 (Figure S2A). Co-immunoprecipitation with anti-Stat3 and anti-p65 antibodies in normal brain tissues (NB) and glioma patient biopsies (GBM) also confirmed high p65/Stat3 nuclear interaction in GBM samples (Figure S2B). Additionally, studies with human glioma tissue array showed significant nuclear colocalization of p-Stat3/p-p65 proteins, suggesting a possible functional role of NF-κB/Stat3 association in GBM tumors (Figure S2C). Immunopositivity of p-Stat3/p-p65 colocalization was increased in correlation with increasing GBM pathological grades (Figure S2D).
Figure 2.

Stat3 and NF-κB (p65) nuclear interaction is enhanced in irradiated human glioma xenograft cell lines. (A) The cytoplasmic and nuclear extracts from IR-treated (0 to 8 Gy) 4910 and 5310 cells were subjected to WB. Blots were reprobed with antibodies against GAPDH and HDAC-1 to monitor equal loading of cytoplasmic and nuclear fractions, respectively. (B) The control and IR-treated cells were immunostained to check the expression and sub-cellular distribution of p65 (red) and Stat3 (green). DAPI was used for nuclear counterstaining and representative pictures from three individual experiments were presented. (C) Co-IP experiments were carried out by precipitating the NEs (500 μg) with non-specific IgG and p-Stat3 antibodies using μMACS™ protein G microbeads and MACS separation columns following the manufacturer’s instructions. WB was carried out using immunoprecipitates. Blots were stripped and reprobed with antibody used for IP to confirm the antibody specificity. Input control samples (50 μg of pre-immunoprecipitated samples) were subjected to IB and input blots were also probed with HDAC-1 to monitor equal loading. (D) IP was performed using p-p65 antibody and immunopecipitates were subjected WB as described above. (E) IP experiments were carried out with ace-p65 antibody. Representative blots from three independent experiments were presented.
Radiation enhanced adhesion, invasion and migratory properties of cells in correlation with increased cellular-ICAM-1 and soluble-ICAM-1 levels
We observed noticeable increases in the invasive potential of IR-treated 4910 and 5310 cells to ~39.0% and ~37.5%, respectively (Figure 3A). The relative migration distances in IR-treated cells were increased by ~38% (4910) and ~37% (5310) (Figure 3B). Additionally, the percentage of cell adhesion in IR-treated 4910 (~29%, ~48%, ~46% and ~43%) and 5310 cells (~30%, 44%, 43% and 39%) was significantly increased on CN, VN, FN and MG, respectively (Figure S3). IR remarkably enhanced the VN-adhesion followed by adhesion on FN among the different ECM components tested. Increased invasiveness in IR-treated cells was in correlation with significant elevation in ICAM-1 transcription (Figure 3C) and protein levels (Figure 3D). Western blotting using CM indicated a dose-dependent upregulation of sICAM-1 in IR-treated cells after 24 hours. As shown in Figure 3E, IR elevated ICAM-1 expression and prominent membrane localization at the migratory edges compared to basal expression levels in untreated controls. Significant increase in ICAM-1 membrane localization in IR-treated cells was also confirmed by WB using membrane proteins (Figure 3G). Radiation-induced ICAM-1 upregulation directly indicated its promoter transactivation by enhanced TF-binding in IR-treated cells.
Figure 3.

IR upregulated ICAM-1 expression and enhanced migration and invasion in human glioma xenograft cell lines. (A) The 4910 and 5310 cells were subjected to IR (0 and 8 Gy) and after 24 hours, Matrigel invasion assay was performed as described in Materials and Methods. Representative micrographs of invaded cells after 12 h in three independent experiments were shown (Upper panel). The mean±SE values of percentage of cell invasion obtained from three experiments were depicted in the bar diagram and significance was denoted by * at p<0.01 (Lower panel). (B) Representative pictures from wound healing migration assay were shown (Upper panel). The relative migration distances were determined in three independent experiments, plotted as mean±SE and the significant difference was depicted by * at p<0.01 (Lower panel). (C) Quantitative RT-PCR was performed and relative ICAM-1 mRNA levels normalized to internal GAPDH were shown as mean±SE obtained from three experiments. (D) The whole cell lysates (WCL) and tumor conditioned media (CM) were subjected to Western blotting. (E) The mock and IR (24 h) treated 4910 and 5310 cells were subjected to immunostaining to visualize ICAM-1 expression. Arrows denote the ICAM-1 membrane localization in IR-treated cells. (F) Cells were irradiated with 0, 4 and 8 Gy for 24 hours. The membrane proteins were extracted using MEM-PER Eukaryotic Membrane Protein Extraction Kit following the manufacturer’s instructions and subjected to WB to analyze ICAM-1 expression levels.
Strong recruitment of NF-κB and Stat3 to the ICAM-1 proximal intron-1 in IR-treated cells
Based on IR-induced ICAM-1 upregulation, we attempted to elucidate the underlying molecular mechanism involved in the potentiating effect of IR on ICAM-1 transactivation by promoter analysis. We identified putative Stat- and NF-κB-binding sites in a span of ~1.0 kb (http://www.cbrc.jp-/research/db/TFSEARCH). Figure 4A schematically represents the putative NF-κB (κB) and Stat (GAS) binding motifs on the human ICAM-1 gene (Table S1). Based on the presence of three potential Stat- and four NF-κB-binding sites, we divided the ICAM-1 promoter (−1165 to +994) into 6 different fragments (A to F) and prepared the respective luciferase reporter constructs. We also performed the ChIP assays for four different regions (R-1 [promoter], R-2 [exon], R-3 [proximal intron-1] and R-4 [distal intron-1]). Putative Stat3-binding motifs occurred adjacent to NF-κB-binding sites in R-2 and R-3 whereas the overlapping binding sites occurred in the R-4 region. Based on this promoter study, we reasoned that the Stat3/NF-κB complex is likely to be involved in the modulation of ICAM-1 expression in gliomas. ChIP assays indicated a basal level of p65 binding to different regions with the highest binding affinity to R-1 followed by R-3, R-2 and R-4, with no obvious binding recorded to the–ve fragment in the control cells (Figure 4B). After IR treatment, the p65-occupancy to all the regions was increased with a maximum increase in R-3 (~16.1 fold; proximal intron-1) followed by R-4 (~9.5 fold; distal intron-1) over basal levels. Stat3 showed more binding affinity towards R-3 followed by R-4 and R-2, whereas no obvious binding occurred to R-1. On the other hand, Stat3 promoter recruitment was significantly increased on R-3 (~15.5-fold), R-4 (~8.5-fold) and R-2 (~3.1-fold) regions in IR-treated cells (Figure 4C). Luciferase reporter assays showed the highest promoter activity with Fragment F (D+E), followed by D (proximal intron with adjacent NF-κB- and Stat3-binding sites) and E (distal intron with overlapping NF-κB-, Stat3-elements) of ICAM-1 (Figure 4D). However, Fragment C (exon with adjacent NF-κB-, Stat3- binding elements) showed no increase in promoter activity compared to intronic Fragments D, E and F. Conversely, both fragments A and B showed moderate increases in activity in both cell lines (~1.5–2.0 fold). Disruption of putative NF-κB-binding sites in D and E fragments remarkably reduced the promoter activity (Figure 4E, 4F). Similarly, transfections with pGL3 plasmids carrying mutated Stat3 consensus sequences in Fragments D and E significantly inhibited IR-induced promoter activity in both 4910 and 5310 cell lines (Figure 4G). These results highlight the potential role of preferential p65/Stat3 recruitment to the intron-1 in IR-induced ICAM-1 activation in glioma.
Figure 4.

Enhanced NF-κB (p65) and Stat3 recruitment on the ICAM-1 intronic consensus elements in IR-treated glioma xenograft cells. (A) Schematic representation of potential DNA-binding elements present in the promoter and intron-1 of the ICAM-1 gene. ICAM-1 promoter contains multiple putative NF-κB binding sites (denoted by gray color filled rectangles) and the intron-1 comprised two potential binding sites. The putative Stat-binding elements (black filled circles) were identified in the exon-1 and intron-1 of ICAM-1. The fragments A, B, C, D, E and F (dotted arrows) were cloned in pGL3-basic to construct promoter-reporter vectors. ChIP primers specific for R-1, R-2, R-3 and R-4 regions were used to determine p65 and Stat3 occupancy on ICAM-1 promoter. (B) IR-treatment elevated recruitment of p65 to the proximal intron-1 of the ICAM-1 in glioma xenograft cells. ChIP assay was performed using anti-p65 and anti-IgG antibodies and relative amplification levels of DNA fragments were represented as percent of inputs in 4910 (upper panel) and 5310 (lower panel) cells. The mean±SE values obtained from three independent experiments were presented and statistical significance was indicated by *p<0.05, **p<0.01 and ***p<0.001. (C) Enhanced Stat3 occupancy to the intron-1 of ICAM-1 gene after IR-treatment in 4910 and 5310 cells. ChIP assay was performed using anti-Stat3 and anti-IgG antibodies. IP samples were analyzed with specific primers for R-1, R-2, R-3, R-4 and –ve regions. Relative amplification levels of DNA fragments were represented as percent of input controls in 4910 (upper panel) and 5310 (lower panel) cells. The mean±SE values of obtained from three experimental replicates were presented, *p<0.05, **p<0.01 and ***p<0.001. (D) Transient transfection studies were performed with firefly basic luciferase pGL3 vectors containing different fragments (A–F) as described in Materials and methods. At 18 h post-transfection, cells were irradiated and incubated for another 6 h. Luciferase assay was performed with cell lysates using Dual-Luciferase Reporter Assay System following the manufacturer’s instructions. Relative activity values obtained from three experiments were represented as mean±SE (*p<0.05 and **p<0.01). (E) Specific nucleotide mutations (underlined) were incorporated into the putative NF-κB and Stat3 consensus sequences in pGL3-D and pGL3- E vectors using QuikChange® II XL Site-Directed Mutagenesis Kit following manufacturer’s instructions. (F) Promoter reporter activity assays were performed in IR-treated cells using respective pGL3 vectors as described above. Relative luciferase activities obtained from three experimental replicates were presented as mean±SE and significance was indicated by *p<0.05 and **p<0.01. (G) The relative luciferase activities obtained from three independent experiments were shown as mean ± SE (*p<0.05 and **p<0.01).
Coordinated regulation of IR-induced ICAM-1 transactivation by p65 and Stat3
To further assess the biological role of Stat3/NF-κB interaction on ICAM-1 expression and to activate Stat3 and NF-κB, we treated the cells with Stat3C and TNF-α respectively. In parallel, specific knockdown experiments were also performed using Stat3.si and JSH-23 or p65.siRNA treatments in combination with IR or TNF-α.
Individual treatments with IR and TNF-α enhanced nuclear expression levels of p-Stat3, p-p65 and ace-p65 in both 4910 and 5310 cells. Despite the IR and TNF-α treatments given in Stat3.si+IR and Stat3.si+TNF-α, Stat3 downregulation significantly decreased nuclear p-p65 and ace-p65 levels, when compared to pSV+IR or pSV+TNF-α treatments (Figure 5A). Additionally, Stat3.si significantly inhibited the IR- and TNF-α-induced ICAM-1 transcription (Figure S4A). Western blotting indicated that the Stat3.si treatment significantly reversed IR- or TNF-α-induced ICAM-1 expression in both cell lines (Figure 5B). The sICAM-1 levels were also decreased by Stat3.si, suggesting the requirement of coordinated regulation of Stat3/NF-κB for IR-induced ICAM-1 upregulation (Figure 5C). To further evaluate the role of Stat3 in the regulation of p65 recruitment onto the intron-1 of ICAM-1, we performed ChIP assay using R-3 primers. Stat3.si remarkably inhibited the IR- and TNF-α-induced p65 recruitment to the R-3 region (Figure 5D). This reversal of p65 binding was in correlation with IR- or TNF-α induced nuclear p-Stat3 expression shown in Figure 5A. Conversely, elevated Stat3 recruitment to the R-3 region was significantly decreased by p65.si treatment in these cells (Figure 5E). These data suggest the reciprocal regulation between p65 and Stat3 for recruitment on the intron-1 of ICAM-1 in glioma.
Figure 5.

Abrogation of Stat3 inhibited IR- and TNF-α-induced NF-κB (p65) nuclear translocation and DNA-binding activity in glioma xenograft cells. (A) The 4910 and 5310 cells were transfected with mock, pSV and Stat3.si, and at 18 h post-transfection, cells were treated with either IR (8 Gy) or TNF-α (10 ng/mL) and incubated for another 6 h. Nuclear extracts were subjected to WB and blots were subsequently reprobed with HDAC-1 to confirm equal loading. (B) WB was performed using whole cell lysates and representative blots from three independent repetitions were shown. GAPDH served as loading control. (C) Conditioned media were subjected to WB to detect sICAM-1 levels. (D) The ChIP quantitative PCR values of p65 recruitment on R-3 region were quantified against input controls in different treatments. The mean±SE values obtained from three experimental replicates were presented (*p<0.01). (E) The ChIP PCR values of Stat3 recruitment on R-3 were quantified against input values. The mean±SE values from three independent repetitions were presented in the bar diagram and significance was indicated by *p<0.01.
Disruption of p65/Stat3 binding inhibited IR-induced ICAM-1-mediated migration and invasion in glioma
We next examined the effect of NF-κB suppression on IR-induced Stat3 activation. Stat3C-overexpressing and IR-treated 4910 and 5310 cells showed elevated nuclear p-Stat3, p-p65 and ace-p65 levels (Figure 6A). Alternatively, the JSH-23 overturned the Stat3C- and IR-induced p-Stat3 levels, thereby implying that NF-κB suppression led to the depletion of Stat3 in the nucleus. This might be due to a reciprocal nuclear regulation of NF-κB on Stat3 or an indirect effect of NF-κB target protein suppression including IL-6 or IL-10, which are required to activate Stat3 signaling. In addition, JSH-23 drastically inhibited Stat3C-induced ICAM-1 levels in both cell lines. Mirroring these results, EMSA results also showed hindrance of NF-κB activity in Stat3-knockdown cells. In addition, Stat3.si counteracted IR- or TNF-α-induced NF-κB activity (Figure S4B). ChIP DNA binding assays confirmed the significant elevation in p65 recruitment on ICAM-1 R-3 region in Stat3C overexpressing cells. Conversely, Stat3C-induced p65-DNA-binding is reversed and inhibited in Stat3C+JSH-23 treatments (Figure 6B). The percentage of cell invasion was considerably hindered in Stat3.si or JSH-23 treatments and Stat3.si+IR or JSH-23+IR treatments counteracted the IR-induced invasion in both cell lines (Figure 6C). Stat3.si and JSH-23 also inhibited IR-induced migration in these cells (Figure 6D). These results strongly suggest the cooperative role of p65/Stat3 interaction in the regulation of migration and invasion in glioma.
Figure 6.

Inhibition of NF-κB activation downregulated Stat3 and reduced ICAM-1-mediated migration and invasion in IR-treated cells. (A) Effect of JSH-23 on ICAM-1 expression levels in Stat3C-overexpressing and IR-treated cells. At the end of different treatments, the 4910 and 5310 NEs and whole cell lysates were subjected to WB. HDAC-1 and GAPDH served as loading controls for NEs and whole cell lysates, respectively. (B) Quantitative-ChIP assay was performed using anti-p65 antibody to analyze the recruitment of p65 on R-3 region in 4910 and 5310 cells and values were quantified against input controls. The mean±SE values were obtained from three independent experiments (*p<0.05 and **p<0.01). (C) Cells were pre-treated with mock, pSV, Stat3.si, JSH-23, pSV+IR, Stat3.si+IR and JSH-23+IR and Matrigel invasion assay was performed as described in Materials and Methods. The percentage of cell invasion obtained in different treatments was represented as mean±SE obtained from three repetitions (* at p<0.05). (D) Wound healing migration assay was performed and the relative migration distances were measured after 12 h and plotted as mean±SE obtained from at least three independent experiments. Significance was depicted by * at p<0.01. (E) The 4910 and 5310 cells were initially treated with pSV or ICAM.si for 18 h. After 18 h, medium was aspirated and cells were individually treated with IR (8 Gy) or rhICAM-1 (2 μg) and cultured for another 6 hours. Whole cell lysates were subjected to WB and blots were subsequently stripped and reprobed with GAPDH.
To further determine the specific role of ICAM-1 in IR-induced glioma invasion and migration, we treated the cells with ICAM.si or rhICAM-1. ICAM.si significantly reduced IR-induced ICAM-1 levels in both cell lines (Figure 6E). Conversely, rhICAM-1 elevated IR-induced ICAM-1 in these cells. The loss of ICAM-1 severely hindered cell invasion and the ICAM.si reversed IR- or rhICAM-1-induced invasiveness (Figure S5A). ICAM-1 downregulation also inhibited IR- and rhICAM-1-induced migration in both 4910 and 5310 cells (Figure S5B). These data suggest the crucial role of ICAM-1 in the regulation of IR-induced migration and invasion of glioma xenograft cells.
Stat3.si inhibits in vivo tumor growth by abrogating nuclear p65/Stat3 interactions and ICAM-1 expression
We next evaluated the effect of Stat3.si on in vivo orthotropic tumor growth in nude mice. Stat3.si significantly reduced the tumor size (~53%) compared to pSV-controls, which showed tumors with prominent and dense regions of proliferation (Figure 7A). On the other hand, IR (8 Gy) treatment alone inhibited tumor development (~29%) and the presence of less dense cells was evident compared to pSV-controls. Additionally, Stat3.si treatment prior to IR significantly decreased tumor development (~69%) in comparison to pSV-control treatments (Figure 7B). Immunofluorescence revealed the inhibition in p-Stat3/p-p65 nuclear expression and colocalization in Stat3.si-treated tumors. Further, IR-induced in vivo nuclear colocalization of p-Stat3/p-p65 was reduced by Stat3.si in Stat3.si+IR-treated tumors (Figure 7C). Stat3.si also showed an inhibitory effect on IR-induced ICAM-1 expression in tumor sections (Figure 7D). Further, IR-induced mRNA levels of ICAM-1 were significantly decreased in Stat3.si+IR-treated subcutaneous tumors (Figure S6A). IR-induced nuclear expression levels of p-p65, p-Stat3 and subsequent elevation in ICAM-1 levels were noticeably decreased in Stat3.si+IR-treated tumors (Figure S6B). Inhibition of p65/Stat3-mediated ICAM-1 levels in Stat3.si+IR-treated tumors was in correlation with suppressed tumor growth in vivo (Figure S6C). Figure 8 schematically summarizes the potential mechanism of IR-induced nuclear p65/Stat3 interaction and their eventual binding to ICAM-1 intron-1, with functional significance in enhancing post-IR glioma malignancy. These in vivo studies validate our in vitro findings, and established the prominent role of Stat3/NF-κB direct nuclear binding in the ICAM-1-mediated invasion and migration, and also suggest the essential role of Stat3 in maintaining NF-κB nuclear retention in glioma.
Figure 7.

Stat3.si inhibited p65/Stat3 interactions, ICAM-1 expression and in vivo orthotopic tumor growth in nude mice. (A) H&E staining in tumor sections (Top panel-10×). Yellow insets portion of the tumor is enlarged to show 60 × magnifications (Bottom panel). (B) The relative tumor volumes were determined as described in Materials and Methods and represented graphically as mean±SE; *p<0.05 and **p<0.01 (n=10). (C) Confocal microscopy showing the expression of p-p65 (green) and p-Stat3 (red) in 4910 and 5310 tumor sections (40×). Representative florescence images from different microscopic fields were presented (n=10). Nuclei were counterstained with DAPI. Insets indicate immunostaining with non-specific IgG for negative control. The p-p65+p-Stat3 co-localization was shown in the enlarged pictures with 60× magnifications (Insets with white-dotted line, Top panel). (D) DAB staining shows the ICAM-1 expression levels in tumor sections. Nuclei were counterstained by hematoxylin.
Figure 8.
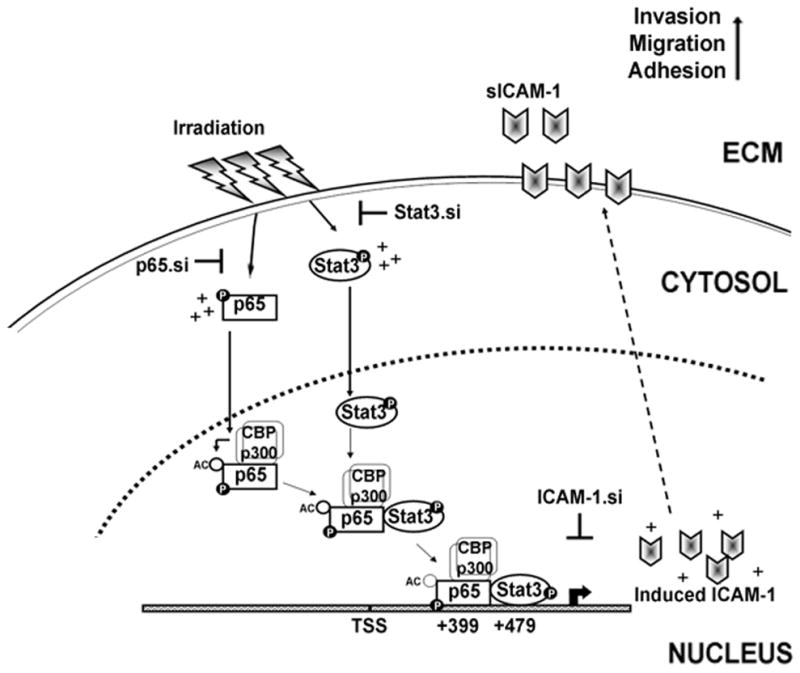
Schematic representation of direct nuclear interaction and reciprocal regulation of NF-κB (p65) and Stat3 in glioma. IR treatment increased phosphorylation of both p65 and Stat3 which eventually translocate into the nucleus. In the nucleus, CBP/p300 acetyl transferases bind to the p-p65, transfer acetyl group and acetylate p65. The p-Stat3 directly interacts with p-p65 at transactivation domain and forms p-p65/p-Stat3 complex in the nucleus. IR elevates the p-p65/p-Stat3 complex formation and subsequent binding to the adjacent NF-κB (+399) and Stat3 (+479) consensus sequences in the ICAM-1 intron-1. As a result, there is a significant increase in ICAM-1 transcription, membrane localization and an increase in sICAM-1 levels in the extracellular milieu of IR-treated cells. This mechanism leads to enhanced adhesion, invasion and migratory properties in post-irradiated cells. On the other hand, the p65/Stat3 interactions were abrogated by specific siRNA-mediated knockdown of p65 and Stat3. IR-induced ICAM-1 upregulation, cell invasion and migration were abrogated by using ICAM-1.siRNA. In summary, our results demonstrated that the elevated functional association of p65 and Stat3 as a function of IR in enhancing glioma malignancy through ICAM-1 upregulation. Hence, blockade of p65 and Stat3 binding provides a potential therapeutic approach in the future treatment of glioma.
Discussion
Individually, both NF-κB and Stat3 regulate the expression of a large number of target genes involved in tumor cell proliferation, migration and invasion. The increased number of overlapping target genes regulated by positive or negative crosstalk between NF-κB and Stat3 suggest the requirement of combined targeting therapies of these TFs (27–30). Conversely, the potential role of Stat3/NF-κB interaction in GBM pathogenesis still remains to be investigated. NF-κB is also reported to be involved in a complex formation with several other TFs. In addition to the classical regulation pathway of NF-κB through IκB proteins, recent studies evidenced that an increasing number of proteins, including negative regulators PIAS3 (31) and SOCS1 (32) as well as positive regulators BRCA1 (33) CAPERα (34) and SP1 (35), interact with and regulate NF-κB. Though earlier studies suggested that p65 binds to the Stat3-DBD, the Stat binding region within the p65 protein is unidentified. Our studies indicated that Stat3 binds to the p65-TAD and augments NF-κB activity. Previous studies demonstrated the functional significance of SP-1/NF-κB coupling and binding onto the DNMT1 and KIT oncogene promoter elements. Moreover, treatments with Bortezomib or SP-1 shRNA massively decreased DNMT and/or KIT expression levels in acute myeloid leukemia (35, 36). The possible functional collaboration between NF-κB/Stat3 in the regulation of several genes, including iNOS and Th-1 has been reported (26, 30, 37–40). However, the NF-κB/Stat3 crosstalk has been reported to repress the IL-6 and IL-12p40 genes (41). Conversely, our results indicated the cooperative role of NF-κB and Stat3 in ICAM-1 activation in glioma. Earlier studies demonstrated the NF-κB/Stat3/PI3K crosstalk in the regulation of the Myc gene, and inhibition of any of these pathways abrogated Myc gene activation (37). Xue, et al. (25) showed the presence of potential NF-κB binding elements in intron-1 (+70 and +611) of the ICAM-1 gene and demonstrated that the physical interaction and preferential recruitment of p65/NFATc1 (nuclear factor of activated T cells) was essential for the thrombin-induced ICAM-1 gene expression in endothelial cells.
As GBM tumor recurrence primarily lies in proximity to the field of radiation, any method inhibiting the recurrence of the primary tumor after radiotherapy has potential significance in GBM treatment (42). Radiation-induced cytotoxic effect is conferred by the induction of DNA double strand breaks. On the other hand, IR-induced ROS-mediated activation of NF-κB facilitates cells to escape apoptotic elimination, thereby resulting in enhanced malignancy (11). Based on the previous reports indicating the elevated serum-ICAM-1 levels in GBM patients, it is interesting to study the exact molecular mechanism of ICAM-1 transactivation in irradiated tumor cells (20, 22). Earlier reports characterized the role of ICAM-1 in tumor malignancy and addressed the importance of inducible TF-binding sites in the ICAM-1 promoter (25, 43). The presence of consensus binding sites for different transcription factors, including Ap1, Ets, NF-κB, c/EBP, TRE, AP2 and AP3, Stat1 in the ICAM-1 promoter were identified previously (44, 45). The TNF-α- and LPS-induced (−187 and −178) and thrombin-induced (−231 to −211) binding of the p65/RelA homodimer has been shown to be essential for the activation of ICAM-1 in endothelial cells (24, 46).
We analyzed the ICAM-1 5′ regulatory region for presence of TF consensus elements and studied the significance of occurrence both NF-κB and Stat3 binding sites in the intron-1. Further, disruption of either NF-κB or Stat3 binding sequences in D (proximal intron-1) or E (distal intron-1) fragments significantly abrogated the promoter activity and inhibited Stat3 and p65 promoter recruitment. The distal intronic region containing overlapped NF-κB/Stat3 binding elements showed less IR-induced activity, indicating the significance of NF-κB (+399) and Stat3 (+479) consensus elements in NF-κB and Stat3 enrichment on ICAM-1 intron-1 in post-IR cells compared to controls. However, further studies are required to find the exact mechanism of how p65 and Stat3 bind to and regulate the ICAM-1 gene expression. Independent treatments with Stat3.si, Stat3C or p65.siRNA further indicated the reciprocal regulation of Stat3 and p65 on ICAM-1-intron-1 occupancy and gene activation. Our studies identified that Stat3 binds to the p65 transactivation domain and enhanced NF-κB DNA-binding activity. To our knowledge, this is the first attempt to study the role of IR-induced functional interplay between Stat3 and NF-κB in the regulation of ICAM-1-mediated migration and invasion in glioma. In conclusion, our study provides evidence for the possible therapeutic efficacy of targeting NF-κB/Stat3 liaison to prevent malignancy in post-irradiated glioma tumors.
Materials and Methods
Cell culture, reagents, irradiation and transfection studies
Human glioma xenograft cell lines 4910 and 5310 (generously provided by Dr. David James, University of California, San Francisco), which are highly invasive in the mouse brain were developed and maintained in mice (47). Cells were cultured in RPMI 1640 (Mediatech Inc., Herndon, VA) supplemented with 10% FBS (Invitrogen, Carlsbad, CA), 50 units/mL penicillin, and 50 μg/mL streptomycin. Human astrocytes were cultured in astrocyte medium augmented with 2% FBS, 1% penicillin/streptomycin and 1% astrocyte growth supplements (Sciencell Research Laboratories, Carlsbad, CA). Human glioma cell lines U87MG, U251 and SNB19 were maintained in DMEM supplemented with 10% FBS. Transient transfections (24 h) were performed after 6 h serum starvation using mock (1×PBS), pcDNA3.0-Stat3.siRNA (Stat3.si, 5′-GCAGTTTCTTCAGAGCAGGTAGAGTCGTCGTACCTGCTCTGAAGAAACTGC-3′), ICAM-1.siRNA (Santa Cruz Biotechnology, Santa Cruz, CA), SignalSilence® NF-κB p65 siRNA (#6261, Cell Signaling) or non-target scrambled-vectors (pSV) using FuGene HD transfection reagent following the manufacturer’s instructions (Roche Applied Science, Indianapolis, IN). We developed constitutively active Stat3C (Addgene: plasmid 8722; Bromberg et al. 1999) stable 4910 and 5310 cells following the procedure described earlier (48). Cells were irradiated with different doses (Gy) using a RS2000 Biological Irradiator (Rad Source Technologies Inc., Boca Raton, FL). For combination treatments, cells were either treated with TNF-α (10 ng/mL) or 10 mM JSH-23 (NF-κB Activation Inhibitor II) or recombinant human ICAM-1 protein (rhICAM-1, 2 μg/mL) at 18 h post-transfection incubated for another 6 h and harvested. We used TNF-α, JSH-23 (Calbiochem, La Jolla, CA) and recombinant human ICAM-1 protein (rhICAM-1) (Novus Biologicals, Littleton, CO). We used p-p65 (Ser 536), p-Stat3 (Tyr 705), ICAM-1, GAPDH, phospho-Tyrosine, HDAC-1, and HRP/Alexa Fluor-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). We also used acetyl-p65-K310 (Abcam, Cambridge, MA), NF-κB (p65)α, Stat3α, p300 and anti-FLAG (DYKDDDDK Tag) antibodies (Cell Signal Technology, Boston, MA) in our study.
Quantitative-PCR and chromatin immunoprecipitation (ChIP) assay
Quantitative RT-PCR was performed as described earlier (32) with following parameters: 1 cycle of 95 °C-10 min and 40 cycles of 95 °C-15 s, 60 °C-30 s, 72 °C-30 s, followed by 1 cycle of 72 °C-10 min. Data were analyzed using iCycler IQ version-3.1 software (Bio-Rad) and Ct values were converted into fold change of expression using 2-ΔΔCt method (ΔΔCT=ΔCT of treatment–ΔCT of control) (49). ChIP assay was done with anti-Stat3, anti-p65 or IgG antibodies using ChIP-IT™ Express Magnetic Chromatin-Immunoprecipitation kit (Active Motif, Carlsbad, CA). The following sequence-specific ChIP quantitative PCR primers were used to amplify different ICAM-1 promoter regions (R-1, R-2, R-3 and R-4): Region-1 (~158 bp) sense-5′-CAGGATTTTCCCAGGCCTT-3′, antisense-5′-CTCGGTCATTCCCAAGGAA-3′; Region-2 (~164 bp) sense-5′-CCCGATTGCTTTAGCTTGGA-3 ′, a n t i s e n s e-5′-AAGGGGCGGTGCTGCTT-3′; Region-3 (~121 bp) sense-5′-TAGCCCCTCCTTCCCATAA-3′, antisense-5′-TGAGTCGGGGTGGGGATT-3′; Region-4 (~160 bp) sense-5′-CAGGACTCGATCATGATGGTT-3′ and antisense-5′-TGGAATTCCCCCAAGTAGAA-3′. The negative control region which does not possess any putative Stat3 or NF-κB binding motifs (Fragment ve, ~214 bp) was amplified with sense-5′-AGCAAACACCCGCTCATAT-3′ and antisense-5′-ACTGAGGCAGCTAGCTTGGA primers. Pre-immunoprecipitated input DNA (5% of sample) was used as a control in each reaction. Specific GAPDH primers sense-5′-CGGTGCGTGCCCAGTTG-3′ and antisense-5′-GCGACGCAAAAGAAGATG-3′ were used for input PCR (49). The relative-ChIP amplification levels of each fragment were presented as a percent of total inputs in three experiments.
Preparation of ICAM-1 luciferase reporter constructs, transient-transfections and luciferase assay
The human ICAM-1 5′ regulatory region including the intron-1 (~2160 bp) was PCR amplified from the genomic DNA of human mammary epithelial cells using sense-5′-CAGGATTTTCCCAGGCCTTC-3′ and antisense-5′-TAAGTCAGCTCTGGAATTCCC-3′ primers and cloned in pGL3-basic vector (Fragment-A, −1165 to +994, cloned at KpnI and NheI). Sequence of Fragment-A is divided in to 4 different regions (R-1, R-2, R-3 and R-4), which are enriched with the presence of NF-κB binding sites. The following primer sets were used to amplify different fragments containing NF-κB binding motifs: Fragment-B (R-1; −1165 to −226, KpnI a n d XhoI) sense-5′-CAGGATTTTCCCAGGCCTTC-3′, antisense-5′-CTCCCTCCACTGAGGGATG-3′; Fragment-C (R-2; +75 to +230, KpnI and XhoI) sense-5′-CCCGATTGCTTTAGCTTGGA-3 ′, a n t i s e n s e-5′-TGCTGCTTTCCCGGAAACCT-3′; Fragment-D (R-3; +384 to +492, KpnI and XhoI) sense-5′-TGAGTCGGGGTGGGGATT-3′, antisense-5′-TAGCCCCTCCTTCCCATAAA-3′; Fragment-E (R-4; +675 to +994, KpnI and NheI) sense-5′-TTACCTCCTGCCTCAGCCT-3 ′, a n t i s e n s e-5′-AAGTCAGCTCTGGAATTCCC-3′ and Fragment-F (R-3 and R-4; +384 to +994, KpnI and SalI) s e n s e-5′-TGAGTCGGGGTGGGGATT-3 ′, a n t i s e n s e-5′-AAGTCAGCTCTGGAATTCCC-3′. The negative control fragment (Fragment–ve; -212 to -67), containing no putative NF-κB- or Stat-binding elements, was also cloned and used in the study. The putative NF-κB consensus sites in D (D-p65) and E (E-p65) fragments were modified from 5′-GGGGATTGCC-3′ and 5′-GGGGAATTCC-3′ to D-Mut-p65-5′-AAGGATTGCC-3′ and E-Mut-p65-5′-TTGGAATTCC-3′, respectively, using QuikChange® II XL Site-Directed Mutagenesis Kit (La Jolla, CA) as described earlier (49). Putative Stat3 binding sites in D (D-Stat3) and E (E-Stat3) fragments were changed from 5′-TTATGGGAAG-3′ and 5′-TTGGGGGAA-3′ to D-Mut-Stat3-5′-GGTGGGAAG-3′ and E-Mut-Stat3-5′-CCGGGGGAA- 3′, respectively. Independent transient-transfections were carried out in 4910 and 5310 cells with pGL3 plasmid constructs (harboring ICAM-1 promoter Fragments A–F) as described above. At 24 h post-transfection, cells were further treated with IR (8 Gy), TNF-α, or JSH-23 and incubated for another 6 h. To normalize transfection efficiency, the cells were co-transfected with pRL-TK construct (Promega) and luciferase activities were determined using the Dual-Luciferase Reporter Assay System (Promega) following the manufacturer’s protocol.
In vitro transcription and translation and GST pull-down assays
Various truncated mutants of p65 and Stat3 were in vitro synthesized from specific PCR-amplified fragments using Transcend™ Biotin-Lysyl-tRNA and TNT® Quick Coupled Transcription/Translation System (Promega) following the manufacturer’s instructions. The full-length PCR-amplified fragments of Stat3 (EcoRI and SalI) and p65 (BamHI and XhoI) were sub-cloned in pGEX-5X-1 (GE Healthcare, Piscataway, NJ), sequencing-verified, following the induction of log-phase cultures of E. coli (DH5α) in LB media with 1.0 mg/ml IPTG for 6–12 h at 30°C. Recombinant GST-fusion proteins GST-p65, GST-Stat3 and GST were purified using MagneGST™ Pull-Down System (Promega) following manufacturer’s protocol and verified by SDS-PAGE. The aliquotes of diluted protein-packed GST particles (~2 μg) were incubated with biotin-labeled p65 and -Stat3 truncated proteins overnight at 4°C. Beads were washed thoroughly, eluted in 20 μl of pre-heated sample buffer, SDS-PAGE-separated, transferred to nitrocellulose membrane and detected using Transcend™ Non Radioactive Translation Detection Systems following manufacturer’s instructions (Promega).
Nuclear/Cytoplasmic protein fractionation, Western blotting (WB) and co- immunoprecipitation (co-IP)
Whole cell lysates and conditioned media (CM) were prepared as described previously (49). The nuclear (NE) and cytoplasmic extracts in cells and tissue samples were fractionated using NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific, Rockford, IL) following the manufacturer’s instructions. Membrane proteins were extracted using the MEM-PER Eukaryotic Membrane Protein Extraction Kit (Pierce Biotechnology, Inc., Rockford, IL) following manufacturer’s instructions. Co-IP was performed with NEs (500 μg) incubated with p-p65, p-Stat3, ace-p65-K310 or non-specific-IgG (Nsp-IgG) antibodies using μMACS™ protein-G microbeads and MACS Separation Columns following the manufacturer’s instructions (Miltenyi Biotec, Germany) (49).
Electrophoretic mobility shift assay
EMSA assays for determining p65 DNA binding were performed using Panomics EMSA Kit (Panomics, Inc., Fremont, CA) following the manufacturer’s instructions (48). For supershift, each antibody (2 μg) was pre-incubated with nuclear protein for 30 min prior to the addition of the labeled probe.
Wound healing migration and Matrigel invasion assays
For wound healing migration assay, cells were treated as described above for 24 h. By considering this point as 0 h, a straight scratch was made in individual wells using a 200 μL pipette-tip and after 12 h, the plates were microscopically observed for wound healing and the average cell migration distances were measured using an ocular micrometer. Matrigel invasion assay was performed as described earlier (50).
Immunofluorescence (IF) and immunohistochemical (IHC) analyses
The immunocytochemical and immunohistochemical analyses were performed exactly as described earlier (51).
In vivo orthotropic animal experiments
Intracranial stereotactic injections of 4910 and 5310 cells (1×106 cells/mouse) into nu/nu mice brains were performed as described previously (51). After 10 days, animals (n=10) were treated with mock, pSV, Stat3.si (dose: 6–8 mg/kg body weight), IR and Stat3.si+IR using Alzet osmotic pumps (Model 2004, ALZET Osmotic Pumps, Cupertino, CA) (50). After 48 h of plasmid delivery, the animals were given radiation treatments (8 Gy) exclusively to the tumor area by covering the rest of the body with a lead sheet. Animals were monitored for 60 days, after which the brains tumor sections were H&E-stained and total tumor volumes were calculated using Image Pro Discovery Program software (Media Cybernatics, Inc., Silver Spring, MD) as explained earlier (49).
Statistical analysis
Data are analyzed with one-way ANOVA using the Neumann-Keuls method of Sigmastat 3.1 software and expressed as mean±SE values obtained from three repetitive experiments. Significant difference was indicated at p<0.05, p<0.01 or p<0.001.
Supplementary Material
Acknowledgments
Funding: This research was supported by award NS64535-01A2 (to J.S.R.) from the National Institute of Neurological Disorders and Stroke. Contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
We thank Noorjehan Ali for technical assistance, Shelle Abraham for manuscript preparation, Diana Meister and Sushma Jasti for manuscript review.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
Reference List
- 1.Fine HA, Dear KB, Loeffler JS, Black PM, Canellos GP. Meta-analysis of radiation therapy with and without adjuvant chemotherapy for malignant gliomas in adults. Cancer. 1993;71:2585–2597. doi: 10.1002/1097-0142(19930415)71:8<2585::aid-cncr2820710825>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 3.Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer. 2003;3:489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- 4.Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res. 2001;61:2744–2750. [PubMed] [Google Scholar]
- 5.Kargiotis O, Geka A, Rao JS, Kyritsis AP. Effects of irradiation on tumor cell survival, invasion and angiogenesis. J Neurooncol. 2010;100:323–338. doi: 10.1007/s11060-010-0199-4. [DOI] [PubMed] [Google Scholar]
- 6.Li T, Zeng ZC, Wang L, Qiu SJ, Zhou JW, Zhi XT, et al. Radiation enhances long-term metastasis potential of residual hepatocellular carcinoma in nude mice through TMPRSS4-induced epithelial-mesenchymal transition. Cancer Gene Ther. 2011 doi: 10.1038/cgt.2011.29. [DOI] [PubMed] [Google Scholar]
- 7.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 8.Lowe JM, Cha H, Yang Q, Fornace AJ., Jr Nuclear factor-kappaB (NF-kappaB) is a novel positive transcriptional regulator of the oncogenic Wip1 phosphatase. J Biol Chem. 2010;285:5249–5257. doi: 10.1074/jbc.M109.034579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 11.Magne N, Toillon RA, Bottero V, Didelot C, Houtte PV, Gerard JP, et al. NF-kappaB modulation and ionizing radiation: mechanisms and future directions for cancer treatment. Cancer Lett. 2006;231:158–168. doi: 10.1016/j.canlet.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 12.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 13.Chen LF, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 14.Ghizzoni M, Haisma HJ, Maarsingh H, Dekker FJ. Histone acetyltransferases are crucial regulators in NF-kappaB mediated inflammation. Drug Discov Today. 2011;16:504–511. doi: 10.1016/j.drudis.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 16.Brantley EC, Benveniste EN. Signal transducer and activator of transcription-3: a molecular hub for signaling pathways in gliomas. Mol Cancer Res. 2008;6:675–684. doi: 10.1158/1541-7786.MCR-07-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 18.Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–293. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roland CL, Harken AH, Sarr MG, Barnett CC., Jr ICAM-1 expression determines malignant potential of cancer. Surgery. 2007;141:705–707. doi: 10.1016/j.surg.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 20.Maenpaa A, Kovanen PE, Paetau A, Jaaskelainen J, Timonen T. Lymphocyte adhesion molecule ligands and extracellular matrix proteins in gliomas and normal brain: expression of VCAM-1 in gliomas. Acta Neuropathol. 1997;94:216–225. doi: 10.1007/s004010050696. [DOI] [PubMed] [Google Scholar]
- 21.Burim RV, Teixeira SA, Colli BO, Peria FM, Tirapelli LF, Marie SK, et al. ICAM-1 (Lys469Glu) and PECAM-1 (Leu125Val) polymorphisms in diffuse astrocytomas. Clin Exp Med. 2009;9:157–163. doi: 10.1007/s10238-009-0040-6. [DOI] [PubMed] [Google Scholar]
- 22.Berens ME, Giese A. “...those left behind”” Biology and oncology of invasive glioma cells. Neoplasia. 1999;1:208–219. doi: 10.1038/sj.neo.7900034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voraberger G, Schafer R, Stratowa C. Cloning of the human gene for intercellular adhesion molecule 1 and analysis of its 5′-regulatory region. Induction by cytokines and phorbol ester. J Immunol. 1991;147:2777–2786. [PubMed] [Google Scholar]
- 24.Ledebur HC, Parks TP. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-kappa B site and p65 homodimers. J Biol Chem. 1995;270:933–943. doi: 10.1074/jbc.270.2.933. [DOI] [PubMed] [Google Scholar]
- 25.Xue J, Thippegowda PB, Hu G, Bachmaier K, Christman JW, Malik AB, et al. NF-kappaB regulates thrombin-induced ICAM-1 gene expression in cooperation with NFAT by binding to the intronic NF-kappaB site in the ICAM-1 gene. Physiol Genomics. 2009;38:42–53. doi: 10.1152/physiolgenomics.00012.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L, et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood. 2008;111:3701–3713. doi: 10.1182/blood-2007-09-111948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atkinson GP, Nozell SE, Benveniste ET. NF-kappaB and STAT3 signaling in glioma: targets for future therapies. Expert Rev Neurother. 2010;10:575–586. doi: 10.1586/ern.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10:1314–1319. doi: 10.1038/embor.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–19. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H, Deng J, Xin H, Liu Y, Pardoll D, Yu H. A requirement of STAT3 DNA binding precludes Th-1 immunostimulatory gene expression by NF-kappaB in tumors. Cancer Res. 2011;71:3772–3780. doi: 10.1158/0008-5472.CAN-10-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jang HD, Yoon K, Shin YJ, Kim J, Lee SY. PIAS3 suppresses NF-kappaB-mediated transcription by interacting with the p65/RelA subunit. J Biol Chem. 2004;279:24873–24880. doi: 10.1074/jbc.M313018200. [DOI] [PubMed] [Google Scholar]
- 32.Strebovsky J, Walker P, Lang R, Dalpke AH. Suppressor of cytokine signaling 1 (SOCS1) limits NFkappaB signaling by decreasing p65 stability within the cell nucleus. FASEB J. 2011;25:863–874. doi: 10.1096/fj.10-170597. [DOI] [PubMed] [Google Scholar]
- 33.Benezra M, Chevallier N, Morrison DJ, MacLachlan TK, el-Deiry WS, Licht JD. BRCA1 augments transcription by the NF-kappaB transcription factor by binding to the Rel domain of the p65/RelA subunit. J Biol Chem. 2003;278:26333–26341. doi: 10.1074/jbc.M303076200. [DOI] [PubMed] [Google Scholar]
- 34.Dutta J, Fan G, Gelinas C. CAPERalpha is a novel Rel-TAD-interacting factor that inhibits lymphocyte transformation by the potent Rel/NF-kappaB oncoprotein v-Rel. J Virol. 2008;82:10792–10802. doi: 10.1128/JVI.00903-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu S, Wu LC, Pang J, Santhanam R, Schwind S, Wu YZ, et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell. 2010;17:333–347. doi: 10.1016/j.ccr.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S, Liu Z, Xie Z, Pang J, Yu J, Lehmann E, et al. Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF-kappaB-dependent DNA methyltransferase activity in acute myeloid leukemia. Blood. 2008;111:2364–2373. doi: 10.1182/blood-2007-08-110171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han SS, Yun H, Son DJ, Tompkins VS, Peng L, Chung ST, et al. NF-kappaB/STAT3/PI3K signaling crosstalk in iMyc E mu B lymphoma. Mol Cancer. 2010;9:97. doi: 10.1186/1476-4598-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Surh YJ, Bode AM, Dong Z. Breaking the NF-kappaB and STAT3 alliance inhibits inflammation and pancreatic tumorigenesis. Cancer Prev Res (Phila) 2010;3:1379–1381. doi: 10.1158/1940-6207.CAPR-10-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17:286–297. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He G, Karin M. NF-kappaB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Squarize CH, Castilho RM, Sriuranpong V, Pinto DS, Jr, Gutkind JS. Molecular cross-talk between the NFkappaB and STAT3 signaling pathways in head and neck squamous cell carcinoma. Neoplasia. 2006;8:733–746. doi: 10.1593/neo.06274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McDonald MW, Shu HK, Curran WJ, Jr, Crocker IR. Pattern of failure after limited margin radiotherapy and temozolomide for glioblastoma. Int J Radiat Oncol Biol Phys. 2011;79:130–136. doi: 10.1016/j.ijrobp.2009.10.048. [DOI] [PubMed] [Google Scholar]
- 43.Anwar KN, Fazal F, Malik AB, Rahman A. RhoA/Rho-associated kinase pathway selectively regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via activation of I kappa B kinase beta and phosphorylation of RelA/p65. J Immunol. 2004;173:6965–6972. doi: 10.4049/jimmunol.173.11.6965. [DOI] [PubMed] [Google Scholar]
- 44.Roebuck KA. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: differential activation and binding of the transcription factors AP-1 and NF-kappaB (Review) Int J Mol Med. 1999;4:223–230. doi: 10.3892/ijmm.4.3.223. [DOI] [PubMed] [Google Scholar]
- 45.Roy J, Audette M, Tremblay MJ. Intercellular adhesion molecule-1 (ICAM-1) gene expression in human T cells is regulated by phosphotyrosyl phosphatase activity. Involvement of NF-kappaB, Ets, and palindromic interferon-gamma-responsive element-binding sites. J Biol Chem. 2001;276:14553–14561. doi: 10.1074/jbc.M005067200. [DOI] [PubMed] [Google Scholar]
- 46.Rahman A, True AL, Anwar KN, Ye RD, Voyno-Yasenetskaya TA, Malik AB. Galpha(q) and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circ Res. 2002;91:398–405. doi: 10.1161/01.res.0000033520.95242.a2. [DOI] [PubMed] [Google Scholar]
- 47.Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro-oncol. 2005;7:164–176. doi: 10.1215/S1152851704000821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kesanakurti D, Sareddy GR, Babu PP, Kirti PB. Mustard NPR1, a mammalian IkappaB homologue inhibits NF-kappaB activation in human GBM cell lines. Biochem Biophys Res Commun. 2009;390:427–433. doi: 10.1016/j.bbrc.2009.09.046. [DOI] [PubMed] [Google Scholar]
- 49.Kesanakurti D, Chetty C, Dinh DH, Gujrati M, Rao JS. Role of MMP-2 in the regulation of IL-6/Stat3 survival signaling via interaction with a5B1 integrin in glioma. Oncogene. 2012 doi: 10.1038/onc.2012.52. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Badiga AV, Chetty C, Kesanakurti D, Are D, Gujrati M, Klopfenstein JD, et al. MMP-2 siRNA Inhibits Radiation-Enhanced Invasiveness in Glioma Cells. PLoS One. 2011;6:e20614. doi: 10.1371/journal.pone.0020614. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Kesanakurti D, Chetty C, Bhoopathi P, Lakka SS, Gorantla B, Tsung AJ, et al. Suppression of MMP-2 Attenuates TNF-alpha Induced NF-kappaB Activation and Leads to JNK Mediated Cell Death in Glioma. PLoS One. 2011;6:e19341. doi: 10.1371/journal.pone.0019341. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.