

Abstract
Although low exercise capacity is a risk factor for stroke, the exact mechanisms that underlie this connection are not known. As a model system for exploring the association between aerobic capacity and disease risks we applied two-way artificial selection over numerous generations in rats to produce low capacity runners (LCR) and high capacity runners (HCR). Here we compared intracerebral hemorrhage (ICH)-induced brain injury in both genders of these rat lines. HCR and LCR rats had 100 μl blood injected into the right caudate and were killed at days 1, 3, 7 and 28 for brain water content determination, immunohistochemistry, histology, Western blot, and behavioral tests. Compared to male HCRs, male LCRs had more severe ICH-induced brain injury including worse brain edema, necroptosis, brain atrophy, and neurological deficits, but not increased numbers of Fluoro-Jade C positive cells or elevated cleaved caspase-3 levels. This was associated with greater microglial activation, and heme oxygenase-1 and protease activated receptor (PAR)-1 upregulation. In females, edema was also greater in LCRs than in HCRs, although it was less severe in females than in males for both LCRs and HCRs. Thus, ICH-induced brain injury was more severe in LCRs, a model of low exercise capacity, than in HCRs. Increased activation of microglia and PAR-1 may participate mechanistically in increased ICH-susceptibility. Females were protected against ICH-induced brain edema formation in both HCRs and LCRs.
Keywords: Metabolic syndrome, Intracerebral hemorrhage, Brain injury, PAR-1
Introduction
Large-scale clinical studies show that low exercise capacity is a stronger predictor of morbidity and mortality relative to other commonly reported risk factors including hypertension, type II diabetes, obesity, or smoking (Blair et al., 1996; Kavanagh et al., 2003; Kokkinos et al., 2008; Myers et al., 2002). These clinical association studies led to us formulate the idea that variation in capacity for oxygen metabolism is a central mechanistic determinant of the divide between complex disease and health that we termed the Aerobic Hypothesis (Koch and Britton, 2008). Starting in 1996, we prospectively tested this hypothesis by applying divergent (two-way) artificial selection for low and high aerobic treadmill running capacity using the genetically heterogeneous N/NIH rats as the founder population (Koch and Britton, 2001). As expected, selection produced low capacity runners (LCRs) and high capacity runners (HCRs) that differ markedly for running performance. Consistent with the aerobic hypothesis numerous disease risks segregated with selection in the LCR and resistance to disease risks segregated with selection for HCR. The LCR scored higher than the HCR for risk factors including diminished aging and longevity (Koch et al., 2011), the metabolic syndrome (Wisloff et al., 2005), hepatic steatosis (Thyfault et al., 2009), and disordered capacity to oxidize lipids (Lessard et al., 2009; Rivas et al., 2011).
While low exercise capacity is a risk factor for stroke (Kurl et al., 2009) as well as other forms of cardiovascular disease (Kavanagh et al., 2003; Kokkinos et al., 2008; Myers et al., 2002), the mechanistic connection has not been defined. Here we examined differences in brain injury in an experimental model of intracerebral hemorrhage (ICH) using LCR and HCR rats. Spontaneous ICH is a common and often fatal stroke subtype accounting for 10–15% of all strokes. Mortality rates for ICH are more than 40% and many survivors have significant neurological deficits (Mendelow et al., 2005).
We compared ICH-induced brain injury in LCRs and HCRs by measuring brain edema, brain atrophy, and behavioral deficits. We also examined a potential novel marker of ICH-induced injury, receptor-interacting protein 1 (RIP1), because this protein plays a critical role in necroptosis and contributes to renal ischemia/reperfusion injury (Linkermann et al., 2012). The mechanisms of brain injury after ICH also include coagulation cascade activation with thrombin production (Lee et al., 1997; Xi et al., 1998), inflammation (Aronowski and Zhao, 2011), and hemoglobin- and iron-induced toxicity (Huang et al., 2002; Nakamura et al., 2004). Therefore, we compared ICH-induced changes in LCRs and HCRs for: 1) protease activated receptor (PAR)-1, a thrombin receptor that is involved in thrombin-induced brain injury after hemorrhagic and ischemic stroke (Junge et al., 2003; Xue et al., 2009), 2) microglial activation that can exacerbate ICH-induced brain injury (Wu et al., 2008), and 3) heme oxygenase (HO)-1, an enzyme involved in the breakdown of heme and the release of iron (Gong et al., 2006).
There is evidence that low exercise capacity is a risk factor for cardiovascular disease in men and women (Kavanagh et al., 2003; Kokkinos et al., 2008; Myers et al., 2002) but gender affects ischemia- and hemorrhage-induced brain injury (Nakamura et al., 2005; Pelligrino et al., 1998) and there is evidence that the relationship between insulin resistance and age-associated coronary and cerebrovascular diseases could be mediated by estrogen receptors (Cardona-Gomez et al., 2002). Thus, the current study also compared ICH-induced brain injury in both male and female LCRs and HCRs. Our results demonstrate more severe ICH-induced brain injury in LCRs and that females were provided relative protection against ICH-induced brain edema formation for both LCRs and HCRs.
Materials and methods
Animals
A previous report (Wisloff et al., 2005) provides a detailed description on the development of the rat models for aerobic exercise capacity. In brief, divergent selected lines were generated from a founder population of 80 male and 88 female N-NIH stock rats based on intrinsic aerobic treadmill running capacity. Thirteen families for each line were set up for a within-family rotational breeding paradigm. This schedule permits <1% inbreeding per generation to maintain a heterogeneous genetic substrate within each selected line. At each generation young adult rats (11 weeks of age) were tested for their intrinsic (untrained) ability to perform forced speed-ramped treadmill running until exhausted. This test was performed daily over five consecutive days. The greatest distance in meters (m) achieved out of the five trials was considered the best estimate of an individual’s exercise capacity. The highest scored female and male from each of the thirteen families were selected as breeders for the next generation of HCRs. The same process was used with lowest scored females and males to generate LCRs. Rats were phenotyped for running capacity in the Koch/Britton laboratory and then transferred to the Hua/Keep laboratory for subsequent study. Rats were studied for response to ICH at least 20 weeks after their last run test. Comparison data between LCR and HCR groups are presented for male rats from generation (G) 25 (n=6 per group), G26 (n=14 per group), and G27 (n=12 per group) and for females from G 28 (n=18 per group). Rats were studied at 5–6 months of age.
Animal preparation and intracerebral infusion
All animal procedures were approved by the University Committee on Use and Care of Animals, University of Michigan. Rats were anesthetized with pentobarbital (50 mg/kg, i.p.) and the right femoral artery catheterized to monitor arterial blood pressure and to obtain blood for analysis of blood gases, pH, glucose, and hematocrit. Body temperature was maintained at 37.5 °C using a feedback-controlled heating pad. Rats were positioned in a stereotactic frame (Kopf Instrument, Tujunga, CA) and a cranial burr hole drilled near the right coronal suture 4.0 mm lateral to the midline. A 26-gauge needle was inserted stereotaxically into the right basal ganglia (coordinates for rats: 0.2 mm anterior, 5.5 mm ventral, and 4.0 mm lateral to the bregma) and blood infused into right basal ganglia at a rate of 10 μl/min using a microinfusion pump. After injection, the needle was removed and the skin incisions closed.
Experimental groups
This study was divided into two parts. First, male HCR and LCR rats had 100 μl blood injected into the right basal ganglia. Rats were euthanized at (1) day 3 (n=6) for brain water content determination, (2) at day 1 and day 3 (n=4) for immunohistochemistry, (3) at days 3 and 7 (n=4) for Western blot and (4) at day 28 (n=6) for histology. Rats had behavioral testing at days 3, 7, 14 and 28 after ICH. In the second part of the study, female HCR and LCR rats had 100 μl blood injected into the right basal ganglia and were killed at day 3. The brains were used for water content measurement (n=6) and Western blot (n=4).
Brain water content
Animals were reanesthetized with pentobarbital (60 mg/ kg, i.p.) and decapitated three days after ICH (Xi et al., 1999). Brains were removed and a 3-mm thick coronal brain slice cut 4-mm from the frontal pole, and dissected into ipsi- and contralateral cortex, and ipsi- and contralateral basal ganglia. The cerebellum was taken as a control. Samples were immediately weighed to obtain the wet weight, and then dried at 100 °C for 24 h to obtain the dry weight. Percent water content was determined as: ((wet weight—dry weight)/wet weight)*100%. In measurements of the basal ganglia contralateral to the ICH, the LCRs had significantly lower water content than HCRs (76.2±0.5 vs. 77.5±0.6%; p<0.01), a phenomenon that we have found previously in comparing aged and young rats (Gong et al., 2004). We also found that the water content of the cerebellum (a site distant from the ICH) was slightly lower in LCRs (76.9±0.1%) than in HCRs (77.3±0.4%), p<0.05. Therefore, edema was assessed as the difference in water content between ipsi- and contralateral hemispheres.
Immunohistochemistry
Immunohistochemistry was performed as described previously (Xi et al., 1999). Briefly, rats were anesthetized and perfused with 4% paraformaldehyde in 0.1 M phosphate-buffered saline (pH7.4). Brains were removed, kept in 4% paraformaldehyde for 6 h and then immersed in 25% sucrose for 3–4 days at 4 °C. After embedding in a mixture of 25% sucrose and OCT (SAKURA Finetek, USA), 18-μm sections were taken on a cryostat. The avidin–biotin complex technique was used for staining with hematoxylin as counter stain. Primary antibodies were mouse anti-rat OX6 (1:400 dilution, AbD Serotec) and rabbit anti-HO-1 (1:400 dilution, Stress Gen). Normal rabbit or mouse serum and the absence of primary antibody were negative controls.
Eighteen-micrometer thick coronal sections from both 1 mm anterior and 1 mm posterior to the blood injection site were used for cell counting. Three high-power images (×40 magnification) were taken adjacent to the hematoma and positive cells counted manually.
Western blot analysis
Western blot analysis was performed as previously described (Xi et al., 1999). Briefly, brain tissue was immersed in Western sample buffer and sonicated. Protein concentration was determined by the Bio-Rad protein assay kit and 50-μg protein from each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a Hybond-C pure nitrocellulose membrane (Amersham). Membranes were probed with the following primary antibodies: rabbit anti-RIP1 (1:1000, Abcam), rabbit anti-PAR-1 (1:1000), rabbit anti-HO-1 (1:2000), rabbit anti-LC3 (1:1000, Abgent) and rabbit anti-cleaved caspase-3 (1:1000, Cell Signaling). Antigen–antibody complexes were visualized with the ECL chemi-luminescence system (Amersham) and exposed to a Kodak X-OMAT film. The relative densities of bands were analyzed with NIH ImageJ.
Fluoro-Jade C staining
To assess neuronal degeneration, Fluoro-Jade C (FJC) staining was performed on brain coronal sections (Okauchi et al., 2009).
Cell counts
For cell counting, we used 18 μm thick coronal sections from either 1 mm anterior or 1 mm posterior to the injection site. Three high-power images (×40 magnification) were taken of the ipsilateral basal ganglia using a digital camera at pre-specified locations relative to the hematoma site (Wang et al., 2012). Fluoro-Jade C, HO-1, and OX-6 positive cells were counted on 3 perihematoma areas from each rat brain section by a blinded observer and expressed as cells/mm2 (Gong et al., 2004).
Behavioral tests
Three behavioral tests were used in this study: forelimb placing, forelimb use asymmetry, and corner turn tests, as described previously (Hua et al., 2002). All animals were scored by a behavioral tester who was blind to the neurological condition.
Statistical analysis
Student t test was used. Values are mean±SD. Statistical significance was set at p<0.05.
Results
As calculated from time (min) and speed (m/min), the mean distance (m) run at exhaustion was significantly higher in HCRs than in LCRs in both male and female rats (p<0.01). The HCRs ran about 7-fold further than the LCRs for both females and males (Table 1). The selective breeding for running capacity produced heavier body weight in LCRs than in HCRs (p<0.01), and the males were heavier than females (p<0.01). Because the rats ran uphill at a fixed slope of 15°, the vertical work performed that takes into account body weight could be calculated for each run. In joules (kg m2/s2) the HCR performed about 6-fold more work than the LCR for both males and females. Blood gases, pressure, glucose, and hematocrit were in the normal range and there were no differences between LCR and HCR rats during surgery (Table 2).
Table 1.
Measures of the aerobic capacity and body weight for all the male and female HCR and LCR rats in this study. Values are means ± SD.
| Body weight (g) | Best time (min) | Best distance (m) | Best speed (m/min) | Vertical work (J) | ||
|---|---|---|---|---|---|---|
| Male | HCR (n = 32) | 246±29 | 70.4±5.9 | 1920±260 | 44.8±2.9 | 1189±151 |
| LCR (n = 32) | 336±41 | 16.2±2.7 | 221±48 | 17.5±1.3 | 187±38 | |
| p Value | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 | |
| Female | HCR (n = 18) | 168±15 | 71.5±6.1 | 1966±279 | 45.3±3.2 | 835±105 |
| LCR (n = 18) | 204±14 | 19.7±2.2 | 283±42 | 19.3±1.1 | 146±23 | |
| p Value | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
Table 2.
Physiological parameters for the male and female HCR and LCR rats in this study. Values are means ± SD; HCT = hematocrit.
| pH | pCO2 (mm Hg) | pO2 (mm Hg) | Blood glucose (mg/dL) | Blood pressure (mm Hg) | HCT (%) | ||
|---|---|---|---|---|---|---|---|
| Male | HCR | 7.38±0.04 | 49±5.0 | 82±7.4 | 120±21.8 | 98±10.6 | 44±3 |
| LCR | 7.40±0.04 | 50±3.3 | 83±6.4 | 127±14.5 | 110± 11.9 | 44±2 | |
| p | >0.05 | >0.05 | >0.05 | >0.05 | >0.05 | >0.05 | |
| Female | HCR | 7.38±0.04 | 48±4.7 | 81± 10.4 | 127±16.2 | 99.3±17.4 | 44±2 |
| LCR | 7.39±0.03 | 48±5.2 | 83±13 | 122±13.6 | 101±11.2 | 44±3 | |
| P | >0.05 | >0.05 | >0.05 | >0.05 | >0.05 | >0.05 |
Brain water content was measured three days after blood injection into right caudate. The change in brain water content (ipsilateral–contralateral) showed greater brain edema formation in the ipsilateral basal ganglia of LCRs than in HCRs (6.6±0.7 vs. 4.8±0.8%; p<0.01,Fig. 1A). The number of dying neurons, FJC positive cells around the hematoma showed no difference between LCRs and HCRs at both day 1 (250.6±30.4 vs. 246.4±53.2, p>0.05) and day 3 (81.2±84.8 vs. 126±58.4, p>0.05). The protein level of RIP1, a mediator of necroptosis, was upregulated by ICH at day 3 in HCRs (the ratio of RIP1/β-actin was 0.6±0.1 in ipsilateral vs. 0.05±0.05 in contralateral, p<0.01) and at day 7 (p<0.01, Fig. 1B), and was significantly higher in LCRs than HCRs at day 7 (0.66±0.19 vs. 0.25±0.13; p<0.05, Fig. 1B), but did not reach significant levels at day 3 (0.67±0.15 vs. 0.6±0.1, p>0.05). Ipsilateral lateral ventricle enlargement occurred in both HCRs and LCRs at day 28 after ICH. Both the contra- and ipsilateral ventricles were larger in LCRs than in HCRs (p<0.05, Fig. 1C). LCRs also had significantly worse neurological deficits (forelimb placing, p<0.01 and corner turn, p<0.05, but not asymmetry, Figs. 2A–C) compared to HCRs.
Fig. 1.

Graphs showing: (A) Brain edema (% water content, ipsiminus contralateral) in cortex and basal ganglia of HCR and LCR male rats at day 3 after ICH. Values are means±SD, n=6, #p<0.01 vs. HCR by student T test. (B) RIP1 protein levels in the ipsiand contralateral basal ganglia of male HCRs and the ipsilateral basal ganglia of male LCRs at day 7 after ICH. Values are means±SD, n=4, #p<0.01 and *p<0.05 vs. HCRs. (C) Lateral ventricular areas in HCR and LCR male rats at day 28 after ICH. Values are means±SD, *p<0.05 vs. HCR.
Fig. 2.

Bar graphs showing the results of forelimb placing (A, normal=100%), forelimb use asymmetry (B, normal=0%) and corner turn (C, normal=50%) tests in male HCR and LCR rats after ICH. Values are means±SD. *p<0.05, #p<0.01 vs. HCRs.
Perihematomal activated microglia (OX-6 positive) and HO-1 positive cells were found in both LCRs and HCRs at day 3 after ICH. However, the number of OX-6 positive cells (activated microglia) was significantly higher in LCRs (298±73 vs. 171±25 cells/mm2 in HCRs; p<0.05, Fig. 3A) as were the number of HO-1 positive cells (486±83 vs. 380±41 cells/mm2 in HCRs; p<0.05, Fig. 3B). A difference in HO-1 upregulation in the ipsilateral basal ganglia in LCRs was also found by Western blot at day 7 after ICH (ratio to β-actin: 0.87±0.09 vs. 0.69±0.4 in HCRs, p<0.05). In addition, the PAR-1 levels in the ipsilateral basal ganglia were higher in LCRs than in HCRs (PAR-1: 1.2±0.1 vs. 0.7±0.1, p<0.01, Fig. 3C).
Fig. 3.

OX-6 (A) and HO-1 (B) positive cells with quantification, and the ratio of PAR-1/β-actin (C) in the ipsilateral basal ganglia of male HCR and LCR rats 3 days after ICH. Scale bar=20 μm. Values are means±SD, *p<0.05, #p<0.01 vs. HCR.
In contrast to the differences in RIP-1, HO-1, PAR-1 and microglia activation, there were no differences in perihematomal cleaved caspase-3, a marker of apoptosis, between LCRs and HCRs (caspase-3/β-actin ratio at day 3: caspase-3/β-actin: 0.25±0.03 vs. 0.20±0.08; day 7: 0.34±0.22 vs. 0.23±0.1, p>0.05) nor were there differences in the conversion of LC3-I to LC3-II, a marker of autophagy, between LCRs and HCRs (LC3-II/LC3-I ratio at day 3: 2.58±0.32 vs. 1.8±0.46; at day 7: 1.16±0.18 vs. 0.98±0.005, p>0.05).
In female rats, the brain water content in HCRs and LCRs showed a similar pattern to males at day 3 after ICH. There was more severe brain edema formation in the ipsilateral basal ganglia of LCRs than HCRs (5.6±0.8 vs. 3.8±0.9%; p<0.01, Fig. 4A). Interestingly, there was less brain edema formation in female LCRs and HCRs compared to males (p<0.05; Figs. 4B and C). Also, the protein levels of PAR-1 in the ipsilateral basal ganglia were higher in the LCRs than in HCRs of females (p<0.05, Fig. 5A), and in males than in females of both LCRs (p<0.01, Fig. 5B) and HCRs (p<0.01, Fig. 5C).
Fig. 4.
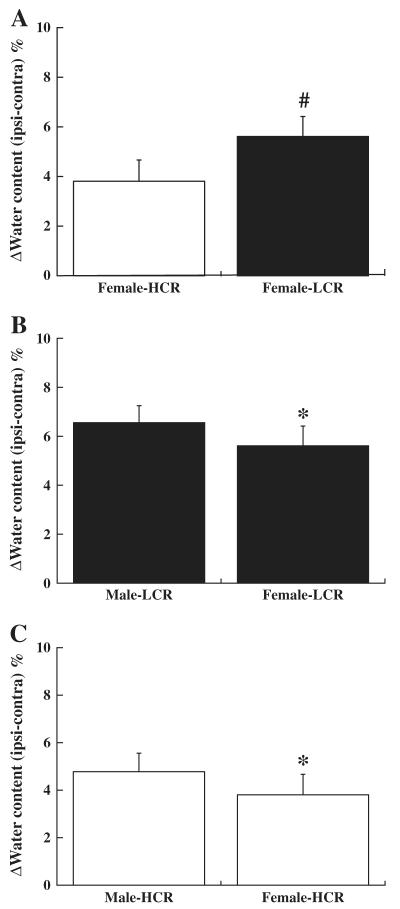
Brain edema (% water content, ipsilateral–contralateral) in (A) HCR or LCR female rats, (B) male or female LCR rats, and (C) male or female HCR rats three days after ICH. Values are mean±SD, #p<0.01 vs. HCRs and *p<0.05 vs. males.
Fig. 5.

The ratio of PAR-1/β-actin in (A) HCR or LCR female rats, (B) male or female LCR rats, and (C) male or female HCR rats three days after ICH. Values are mean±SD, *p<0.05 vs. HCRs and #p<0.01 vs. males.
Discussion
The present study demonstrates that ICH causes more severe brain edema, necroptosis, brain atrophy, and neurological deficits in rats bred for low intrinsic aerobic capacity compared to rats with high intrinsic capacity. This was associated with greater microglia activation, and HO-1 and PAR-1 upregulation. In addition, we discovered low capacity female rats had reduced brain edema formation and PAR-1 upregulation compared to males but worse brain injury compared to high capacity female rats.
Development of the LCR-HCR model system based upon the aerobic hypothesis provides polygenic-based experimental substrate for exploring both diagnostic and prescriptive translational clues that underlie disease risk factors. The major genetical hypothesis is that functional alleles at multiple interacting loci that affect intrinsic aerobic capacity have been enriched or fixed differentially between the LCR and HCR (Wisloff et al., 2005). Such contrasting model systems allow exploitation of the power of “Mendelian randomization” for the identification of allelic variants that are causative of phenotypic differences between the strains. For this, LCR and HCR strains can be crossed to develop an F2 population and then tested for segregation of allelic variants with phenotypic differences to define causation. The differential susceptibility between LCR and HCR for ICH makes this trait a candidate for such an experiment.
While there is much evidence that low aerobic capacity has a major impact on the incidence of all forms of cardiovascular disease, including stroke (Kavanagh et al., 2003; Kokkinos et al., 2008; Li and Siegrist, 2012; Myers et al., 2002), the exact molecular mechanisms have not been resolved. The current results demonstrate that low intrinsic exercise capacity has greater expression of receptor-interacting protein (RIP) 1 compared to HCR. RIP1 is a mediator of necroptosis and contributes to renal ischemia/reperfusion injury (Linkermann et al., 2012). Necroptosis plays a significant role in the pathogenesis of cell death and functional outcome after TBI, and necrostatin-1, a necroptosis inhibitor, may have therapeutic potential for TBI (You et al., 2008). Distinguishing cause and effect is very difficult, i.e. is the increase in RIP-1 the cause or the result of greater ICH-induced injury in LCRs. However, it should be noted that no significant differences were found between LCRs and HCRs in caspase-3 levels, a marker of apoptosis, or conversion of LC3-I and LC3-II, a marker of autophagy, suggesting that the effects on RIP-1 are specific. The role of necroptosis in ICH-induced brain injury merits further examination.
The coagulation cascade and the formation of thrombin have an important role in early edema formation after ICH (Xi et al., 2006). The current study showed significant increases in PAR-1, a thrombin receptor, after ICH in LCR rats compared to HCRs. After ICH, there is production of thrombin, a PAR-1 agonist, in the brain (Gong et al., 2008). Activation of PAR-1 has been linked to many intracellular signaling pathways and is related to brain injury after hemorrhagic and ischemic stroke (Junge et al., 2003; Keep et al., 2012; Xue et al., 2009). Thus, inhibiting thrombin or PAR-1 knockout can reduce ICH-induced brain injury (Kitaoka et al., 2002; Xue et al., 2009). Thrombin, via PAR-1, can activate Src kinase which is thought to contribute to mitogenic stress, excitoxicity (NMDA activation), vascular hyperpermeability and inflammation (Gingrich et al., 2000; Liu and Sharp, 2012).
There is evidence of increased inflammation in the LCR rats compared to HCR rats (Bowman et al., 2010; Koch et al., 2011) and in humans with low physical activity (Pedersen, 2009). Many studies have shown that inflammation plays a role in ICH-induced injury (Wu et al., 2008; Xi et al., 2006). ICH causes perihematomal activation of microglia as well as an influx of leukocytes from the bloodstream and microglial inhibition is known to reduce ICH-induced brain injury (Wu et al., 2009). In the current study, LCR rats had greater perihematomal microglial activation after ICH than HCR rats.
Much evidence indicates that iron overload plays a pivotal role in ICH-induced brain injury (Wu et al., 2003). The current study showed an increase in perihematomal HO-1 levels in LCR compared to HCR rats. HO-1 is involved in heme degradation after ICH and the release of iron which may contribute to oxidative damage (Gong et al., 2006).
Female animals usually sustain less severe brain damage after stroke or traumatic brain injury than males (Alkayed et al., 1998; Nakamura et al., 2002). We have previously shown in Sprague Dawley rats that females have less severe ICH-induced brain injury than males through an estrogen receptor-dependent mechanism (Nakamura et al., 2005). In the current study, female LCR and HCR rats had less brain edema than their male counterparts, but female LCR rats still had less edema than female HCRs, i.e. the detrimental effect found in LCRs was gender independent. This is in agreement with human studies where low exercise capacity has a detrimental effect on cardiovascular disease in both men and women (Kavanagh et al., 2003; Kokkinos et al., 2008; Myers et al., 2002). Similarly, the upregulation of PAR-1 in the brains of LCR rats compared to HCRs was also found in both sexes although, again, females had lower levels of PAR-1 than males for both LCRs and HCRs.
The LCR–HCR model system was developed based upon the aerobic hypothesis and represents a bottom-up approach for testing mechanistically the susceptibility (or resistance) to clinically relevant insults, such as ICH. This approach to model development is in line with the current shift from focus on manipulating individual genes and pathways to the consideration that biological function resides within highly interconnected modules of molecular networks (Jeong et al., 2000; Zhu et al., 2008). Such model systems may, given their genetic complexity, prove useful in determining the potential effect of different treatments (such as statins) given before or after a stroke. As noted above, the LCR rat demonstrates the hallmarks of metabolic syndrome with obesity, hyperinsulinemia, elevated triglycerides and small increases in conscious blood pressure (Wisloff et al., 2005). This model may be useful for examining treatments for stroke and other cardiovascular diseases in patients with that syndrome.
In conclusion, rats with a low aerobic capacity for running have more severe ICH-induced brain damage than those with a high aerobic capacity. This is associated with changes related to coagulation, inflammation, and iron-handling in the brain. These results suggest exercise capacity affects stroke outcome as well as stroke incidence (Kurl et al., 2009). The LCR/HCR rats provide a unique model for examining the effect of low aerobic capacity on the impact of different neurological conditions.
Acknowledgments
This work was supported by the National Institutes of Health grants NS057539 (YH), NS034709 (RFK) and NS039866 (GX) and a grant from the American Heart Association, 0840016N. The LCR–HCR rat model system was specifically funded by the National Center for Research Resources (NCRR) grant R24 RR017718 and is currently supported by the Office of Research Infrastructure Programs/OD grant ROD012098A (to L.G.K. and S.L.B.) from the National Institutes of Health. S.L.B. was also supported by the National Institutes of Health grant RO1 DK077200. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the AHA.
We acknowledge the expert care of the rat colony provided by Molly Kalahar and Lori Gilligan. The LCR and HCR model can be made available for collaborative study (contact: brittons@umich.edu or lgkoch@umich.edu).
Footnotes
Disclosure None.
References
- Alkayed NJ, et al. Gender-linked brain injury in experimental stroke. Stroke. 1998;29:159–165. doi: 10.1161/01.str.29.1.159. (discussion 166) [DOI] [PubMed] [Google Scholar]
- Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42:1781–1786. doi: 10.1161/STROKEAHA.110.596718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair SN, et al. Influences of cardiorespiratory fitness and other precursors on cardiovascular disease and all-cause mortality in men and women. JAMA. 1996;276:205–210. [PubMed] [Google Scholar]
- Bowman TA, et al. Caloric restriction reverses hepatic insulin resistance and steatosis in rats with low aerobic capacity. Endocrinology. 2010;151:5157–5164. doi: 10.1210/en.2010-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona-Gomez GP, et al. Interactions of estrogen and insulin-like growth factor-I in the brain: molecular mechanisms and functional implications. J. Steroid Biochem. Mol. Biol. 2002;83:211–217. doi: 10.1016/s0960-0760(02)00261-3. [DOI] [PubMed] [Google Scholar]
- Gingrich MB, et al. Potentiation of NMDA receptor function by the serine protease thrombin. J. Neurosci. 2000;20:4582–4595. doi: 10.1523/JNEUROSCI.20-12-04582.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, et al. Intracerebral hemorrhage: effects of aging on brain edema and neurological deficits. Stroke. 2004;35:2571–2575. doi: 10.1161/01.STR.0000145485.67827.d0. [DOI] [PubMed] [Google Scholar]
- Gong Y, et al. Systemic zinc protoporphyrin administration reduces intracerebral hemorrhage-induced brain injury. Acta Neurochir. Suppl. 2006;96:232–236. doi: 10.1007/3-211-30714-1_50. [DOI] [PubMed] [Google Scholar]
- Gong Y, et al. Increase in brain thrombin activity after experimental intracerebral hemorrhage. Acta Neurochir. Suppl. 2008;105:47–50. doi: 10.1007/978-3-211-09469-3_10. [DOI] [PubMed] [Google Scholar]
- Hua Y, et al. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33:2478–2484. doi: 10.1161/01.str.0000032302.91894.0f. [DOI] [PubMed] [Google Scholar]
- Huang F, et al. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J. Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- Jeong H, et al. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- Junge CE, et al. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13019–13024. doi: 10.1073/pnas.2235594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh T, et al. Peak oxygen intake and cardiac mortality in women referred for cardiac rehabilitation. J. Am. Coll. Cardiol. 2003;42:2139–2143. doi: 10.1016/j.jacc.2003.07.028. [DOI] [PubMed] [Google Scholar]
- Keep RF, et al. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11:720–731. doi: 10.1016/S1474-4422(12)70104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaoka T, et al. Delayed argatroban treatment reduces edema in a rat model of intracerebral hemorrhage. Stroke. 2002;33:3012–3018. doi: 10.1161/01.str.0000037673.17260.1b. [DOI] [PubMed] [Google Scholar]
- Koch LG, Britton SL. Artificial selection for intrinsic aerobic endurance running capacity in rats. Physiol. Genomics. 2001;5:45–52. doi: 10.1152/physiolgenomics.2001.5.1.45. [DOI] [PubMed] [Google Scholar]
- Koch LG, Britton SL. Aerobic metabolism underlies complexity and capacity. J. Physiol. 2008;586:83–95. doi: 10.1113/jphysiol.2007.144709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch LG, et al. Intrinsic aerobic capacity sets a divide for aging and longevity. Circ. Res. 2011;109:1162–1172. doi: 10.1161/CIRCRESAHA.111.253807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkinos P, et al. Exercise capacity and mortality in black and white men. Circulation. 2008;117:614–622. doi: 10.1161/CIRCULATIONAHA.107.734764. [DOI] [PubMed] [Google Scholar]
- Kurl S, et al. Exercise workload, cardiovascular risk factor evaluation and the risk of stroke in middle-aged men. J. Intern. Med. 2009;265:229–237. doi: 10.1111/j.1365-2796.2008.02006.x. [DOI] [PubMed] [Google Scholar]
- Lee KR, et al. Mechanisms of edema formation after intracerebral hemorrhage: effects of thrombin on cerebral blood flow, blood–brain barrier permeability, and cell survival in a rat model. J. Neurosurg. 1997;86:272–278. doi: 10.3171/jns.1997.86.2.0272. [DOI] [PubMed] [Google Scholar]
- Lessard SJ, et al. Impaired skeletal muscle beta-adrenergic activation and lipolysis are associated with whole-body insulin resistance in rats bred for low intrinsic exercise capacity. Endocrinology. 2009;150:4883–4891. doi: 10.1210/en.2009-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Siegrist J. Physical activity and risk of cardiovascular disease—a meta-analysis of prospective cohort studies. Int. J. Environ. Res. Public Health. 2012;9:391–407. doi: 10.3390/ijerph9020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, et al. Rip1 (Receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney International. 2012. [DOI] [PubMed]
- Liu D, Sharp F. Excitatory and mitogenic signaling in cell death, blood–brain barrier breakdown, and BBB repair after intracerebral hemorrhage. Transl. Stroke Res. 2012;3:S62–S69. doi: 10.1007/s12975-012-0147-z. [DOI] [PubMed] [Google Scholar]
- Mendelow AD, et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet. 2005;365:387–397. doi: 10.1016/S0140-6736(05)17826-X. [see comment] [DOI] [PubMed] [Google Scholar]
- Myers J, et al. Exercise capacity and mortality among men referred for exercise testing. N. Engl. J. Med. 2002;346:793–801. doi: 10.1056/NEJMoa011858. [DOI] [PubMed] [Google Scholar]
- Nakamura M, et al. Rapid tolerance to focal cerebral ischemia in rats is attenuated by adenosine A1 receptor antagonist. J. Cereb. Blood Flow Metab. 2002;22:161–170. doi: 10.1097/00004647-200202000-00004. [DOI] [PubMed] [Google Scholar]
- Nakamura T, et al. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J. Neurosurg. 2004;100:672–678. doi: 10.3171/jns.2004.100.4.0672. [DOI] [PubMed] [Google Scholar]
- Nakamura T, et al. Estrogen therapy for experimental intracerebral hemorrhage in rats. J. Neurosurg. 2005;103:97–103. doi: 10.3171/jns.2005.103.1.0097. [DOI] [PubMed] [Google Scholar]
- Okauchi M, et al. Tissue-type transglutaminase and the effects of cystamine on intracerebral hemorrhage-induced brain edema and neurological deficits. Brain Res. 2009;1249:229–236. doi: 10.1016/j.brainres.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK. The diseasome of physical inactivity—and the role of myokines in muscle–fat cross talk. J. Physiol. 2009;587:5559–5568. doi: 10.1113/jphysiol.2009.179515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelligrino DA, et al. Cerebral vasodilating capacity during forebrain ischemia: effects of chronic estrogen depletion and repletion and the role of neuronal nitric oxide synthase. Neuroreport. 1998;9:3285–3291. doi: 10.1097/00001756-199810050-00026. [DOI] [PubMed] [Google Scholar]
- Rivas DA, et al. Low intrinsic running capacity is associated with reduced skeletal muscle substrate oxidation and lower mitochondrial content in white skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;300:R835–R843. doi: 10.1152/ajpregu.00659.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyfault JP, et al. Rats selectively bred for low aerobic capacity have reduced hepatic mitochondrial oxidative capacity and susceptibility to hepatic steatosis and injury. J. Physiol. 2009;587:1805–1816. doi: 10.1113/jphysiol.2009.169060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, et al. Iron enhances the neurotoxicity of amyloid β. Transl. Stroke Res. 2012;3:107–113. doi: 10.1007/s12975-011-0099-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisloff U, et al. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science. 2005;307:418–420. doi: 10.1126/science.1108177. [DOI] [PubMed] [Google Scholar]
- Wu J, et al. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–2969. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]
- Wu J, et al. Microglial activation and brain injury after intracerebral hemorrhage. Acta Neurochir. Suppl. 2008;105:59–65. doi: 10.1007/978-3-211-09469-3_13. [DOI] [PubMed] [Google Scholar]
- Wu J, et al. Minocycline reduces intracerebral hemorrhage-induced brain injury. Neurol. Res. 2009;31:183–188. doi: 10.1179/174313209X385680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi G, et al. The role of blood clot formation on early edema development following experimental intracerebral hemorrhage. Stroke. 1998;29:2580–2586. doi: 10.1161/01.str.29.12.2580. [DOI] [PubMed] [Google Scholar]
- Xi G, et al. Attenuation of thrombin-induced brain edema by cerebral thrombin preconditioning. Stroke. 1999;30:1247–1255. doi: 10.1161/01.str.30.6.1247. [DOI] [PubMed] [Google Scholar]
- Xi G, et al. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- Xue M, et al. Relative importance of proteinase-activated receptor-1 versus matrix metalloproteinases in intracerebral hemorrhage-mediated neurotoxicity in mice. Stroke. 2009;40:2199–2204. doi: 10.1161/STROKEAHA.108.540393. [DOI] [PubMed] [Google Scholar]
- You Z, et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 2008;28:1564–1573. doi: 10.1038/jcbfm.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008;40:854–861. doi: 10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]