

Abstract
Prokaryotic cell wall biosynthesis is coordinated with cell growth and division, but the mechanisms regulating this dynamic process remain obscure. Here, we describe a phosphorylation-dependent regulatory complex that controls peptidoglycan (PG) biosynthesis in Mycobacterium tuberculosis. We found that PknB, a PG-responsive Ser-Thr protein kinase (STPK), initiates complex assembly by phosphorylating a kinase-like domain in the essential PG biosynthetic protein, MviN. This domain was structurally diverged from active kinases and did not mediate phosphotransfer. Threonine phosphorylation of the pseudokinase domain recruited the FhaA protein through its forkhead-associated (FHA) domain. The crystal structure of this phosphorylated pseudokinase–FHA domain complex revealed the basis of FHA domain recognition, which included unexpected contacts distal to the phosphorylated threonine. Conditional degradation of these proteins in mycobacteria demonstrated that MviN was essential for growth and PG biosynthesis and that FhaA regulated these processes at the cell poles and septum. Controlling this spatially localized PG regulatory complex is only one of several cellular roles ascribed to PknB, suggesting that the capacity to coordinate signaling across multiple processes is an important feature conserved between eukaryotic and prokaryotic STPK networks.
INTRODUCTION
The integrity, permeability, surface properties, and osmotic stability of bacterial cells are determined by the cell wall that surrounds the inner membrane. Inhibition of cell wall synthesis by penicillin or other antibiotics leads to rapid cell lysis, making cell wall homeostasis a proven target for antimicrobials. Peptidoglycan (PG), an essential component of nearly all prokaryotic cells, is composed of carbohydrate polymer chains cross-linked by short peptides. The stable covalent cross-linking of this structure belies the dynamic remodeling that occurs during cell growth and division. Regulating the synthesis and reorganization of this structure represents a fundamental biological problem because the extracellular synthesis of PG must be coordinated with cues from both the external environment and the cytosol, where growth and division are controlled.
Emphasizing the challenge of coupling intracellular and extracellular reactions, PG precursors are synthesized in the cytosol but are used by extracellular enzymes to build the PG polymer (1). Precursor export is a multistep process involving the conjugation of muropeptide precursors to a membrane-embedded isoprenoid carrier, modification of the lipid-linked precursor to form “lipid II,” physical inversion (flipping) of lipid II in the membrane to expose the disaccharide-pentapeptide monomer to the extracellular space, and recycling of the isoprenoid carrier after addition of the monomer to the growing PG chain. Several integral membrane proteins are required for this process, and these proteins likely form complexes specific to each subcellular site of PG synthesis (2). MraYand MurG synthesize the mature precursor. SEDS (shape, elongation, division, and sporulation) proteins such as FtsW, which has been proposed to have lipid II flippase activity (3), may catalyze inversion. The integral membrane protein MviN has also been proposed to invert lipid II (4, 5). Although biochemical evidence of this activity is lacking, MviN inhibition blocks PG polymerization and causes an accumulation of mature lipid-linked PG precursors in Escherichia coli, indicating an essential role in either the export of precursors or their addition to the growing PG meshwork.
It remains unclear which of these diverse PG biosynthetic activities are controlled to govern cell wall metabolism, and few regulatory proteins have been described. To understand this process in mycobacteria, we searched for possible regulatory domains in PG biosynthetic components. Unlike most MviN proteins, the mycobacterial MviN ortholog is fused to a module homologous to receptor Ser-Thr protein kinases (STPKs), suggesting a possible mechanism of control. Here, we show that this regulatory module, instead of mediating phosphorylation, affords an initial example of a bacterial pseudokinase. Despite lacking enzymatic activity, we determined that this domain plays a central role in regulating cell wall synthesis. Phosphorylation of the pseudokinase by the PG-responsive STPK, PknB, recruited a forkhead-associated (FHA) domain protein, FhaA. The crystal structures of the pseudokinase domain alone and in complex with the FHA domain revealed a divergent STPK fold in the pseudokinase domain of MviN that binds the FHA domain through energetically important, unanticipated three-dimensional contacts. FhaA binding modulates local PG synthesis at the cell poles and septum, providing a mechanism to explain the spatial regulation of PG biosynthesis during growth and division.
RESULTS
The mycobacterial MviN ortholog is essential for growth
All PG-producing organisms express at least one gene encoding a protein in the MviN family (4). In Mycobacterium tuberculosis (Mtb), this homology occurs in the N-terminal half of Rv3910, which consists of an MviN domain comprising 14 predicted transmembrane helices. However, amino acid sequence analysis showed that Rv3910 contains additional domains not found in previously characterized MviN homologs. C-terminal to helix 14, a predicted intracellular domain (ICD) with homology to STPKs, is linked through a single predicted transmembrane helix to an extracellular domain (ECD) that resembles carbohydrate-binding proteins. This arrangement is reminiscent of transmembrane receptor kinases, suggesting that the C-terminal domains might regulate the MviN homolog of Mtb. To determine whether Rv3910, which we refer to as Mt-MviN, represents the mycobacterial ortholog of the E. coli MviN protein, we determined whether Mt-mviN was essential for mycobacterial growth and PG biosynthesis.
As an initial test of essentiality, we determined whether transposon insertion mutations could be tolerated in different regions of the Mt-mviN gene. Using a data set consisting of more than 40 million transposon:chromosome junction sequences derived from 105 independent random insertion events in M. tuberculosis, we identified transposons in 44,350 of the 76,925 TA dinucleotide insertion sites in the genome (57% coverage). This distribution is consistent with the previously described random integration of this element (6). In the Mt-mviN locus, transposon insertions were not observed in any of the 28 potential TA insertion sites in the MviN domain (Fig. 1A). The probability of this distribution occurring by chance was <10−6, indicating that disruption of this domain produced nonviable mutants. However, 3' to the MviN domain coding region, insertions were detected in 65% of the TA insertion sites. This distribution suggested that the MviN domain was essential and the C-terminal domains were less important for in vitro growth.
Fig. 1.

Mt-MviN is essential for growth and PG biosynthesis. (A) Transposon insertion mapping in Mtb indicates that the MviN domain of Mt-MviN is essential for growth. Vertical bars represent the number of sequence reads corresponding to each TA insertion site. “TN∷576” indicates the position of the first transposon insertion. (B) Homologous recombination confirms the differential essentiality of Mt-MviN domains. The indicated targeted deletions from the 3′ end of Mt-mviN were attempted. Quotients indicate the number of deletion mutants identified over the total number of hygr colonies obtained. (C) Conditional depletion of Ms-MviN protein in M. smegmatis arrests growth. The indicated strains were cultured with or without ATc, and growth was assessed by optical density. Data represent three experiments. Error bars are smaller than the data points. Inset: Western analysis demonstrates that the tagged Ms-MviN protein runs as a doublet and is degraded within 6 hours of ATc addition. The same amount of protein (as measured with the BCA protein assay) was loaded in each lane. (D) Ms-MviN depletion alters cell morphology. Scanning electron micrographs of wild-type M. smegmatis and Ms-MviN-depleted cells. HIV-2 protease was expressed for 12 hours in both strains. (E) Ms-MviN depletion in M. smegmatis increases the intra-cellular pool of DAP-containing PG precursors. Histogram shows the quantity of solvent-extractable, DAP-containing precursors in wild-type or Ms-MviN-ID M. smegmatis induced for 6 hours with ATc (+ATc) or untreated (−ATc). Data represent the average of triplicate samples and the error bars indicate SDs.
To confirm these predictions, we determined whether the regions of the gene encoding each Mt-MviN domain could be deleted by homologous recombination (Fig. 1B). We obtained viable mutants of Mtb lacking the C-terminal ECD, the ECD and the previous transmembrane segment, and the ECD and ICD. However, no deletion mutants were obtained lacking any segment of the N-terminal MviN domain, consistent with a function critical for growth.
Mycobacterial MviN is required for cell wall integrity and PG biosynthesis
Because Mt-mviN is essential for growth, we analyzed the phenotypic effects of depleting the protein by conditionally degrading its single homolog (fig. S1) in the saprophytic mycobacterium, Mycobacterium smegmatis. We targeted Ms-MviN, using a protein knockdown system based on a masked degradation signal—the SsrA sequence linked to an HIV-2 protease site and a FLAG epitope—engineered at the C terminus of the protein (7). Using homologous recombination, we added this inducible degradation (ID) tag in-frame to the 3' end of the ICD sequence in the chromosomal Ms-mviN gene. The HIV-2 protease–encoding gene under the control of a tetracycline-inducible promoter was introduced separately into the chromosome. Addition of anhydrotetracycline (ATc) to this strain induced the production of HIV-2 protease, which cleaved the ID tag and exposed the C-terminal SsrA sequence that targeted the Ms-MviN protein for degradation by the ClpXP system.
Within 6 hours of ATc addition, the Ms-MviN protein was efficiently degraded (Fig. 1C, inset). Ms-MviN depletion caused a rapid inhibition of bacterial growth (Fig. 1C) and altered cell morphology (Fig. 1D). Irregularly shaped cells were observed with bulges at the ends and along the cell body. These phenotypes were not observed in the absence of ATc or in bacteria that did not express both ID-tagged Ms-MviN and the HIV-2 protease. Furthermore, the integration of a wild-type Mt-mviN allele at a distal chromosomal location restored normal growth upon depletion of the ID-tagged Ms-MviN protein (Fig. 1C). These results indicated an essential role for Ms-MviN in cell growth and morphogenesis.
To directly assess the role of Ms-MviN in PG synthesis, we quantified the accumulation of solvent-extractable PG precursors containing diaminopimelate (DAP), an amino acid exclusively used in PG synthesis. These precursors represent both nucleotide-linked muropeptides and more mature lipid-linked compounds, all of which are extractable only before their polymerization into the mature cell wall. Consistent with a role for this protein in precursor export or polymerization, depletion of Ms-MviN increased the accumulation of solvent-extractable precursors >10-fold compared to the levels observed in cells expressing this Ms-MviN (Fig. 1E). This dramatic accumulation verified that Ms-MviN functions as the MviN ortholog in mycobacteria, and it is required for the addition of PG precursors into the growing cell wall.
Mt-MviN ICD structure reveals a bacterial pseudokinase
Although the Mt-MviN ICD contains a predicted STPK domain, the absence of characteristic substrate binding and catalytic sequence motifs (fig. S2A) suggested that this domain might lack phosphotransfer activity. To investigate the functions of the Mt-MviN ICD, we determined the three-dimensional structure of the kinase homology domain (KHD, residues 681 to 963) in two distinct crystal forms (table S1).
The Mt-MviN KHD structure adopts a classic kinase-like fold (Fig. 2A) that is most closely related to the Mtb PknB kinase domain (KD) (Fig. 2B). However, the Mt-MviN KHD structure is highly diverged from that of an active kinase. Key kinase active-site motifs are absent in the Mt-MviN KHD structure. Strikingly, the canonical adenosine 5′-triphosphate (ATP) binding site is filled with aliphatic and aromatic residues (Fig. 2C and fig. S2B), indicating that nucleotides are unlikely to bind. The kinase P-loop motif (GXGXXG in PknB), which binds and orients the ATP phosphates in active STPKs, is replaced by the diverged sequence GGVPPL, which loops into the space that in an active kinase would form the nucleotide-binding site (Fig. 2C). In the Mt-MviN KHD, Thr is present in place of the Lys on the β3 strand that coordinates the α and β phosphates of ATP in most active kinases. The structural alignment of the Mt-MviN KHD and the PknB KD also reveals that Leu-Ser-Ile (LSI) and Tyr-Pro-Ala (YPA) tripeptides in Mt-MviN replace, respectively, the STPK His-Arg-Asp (HRD) (catalytic loop) and Asp-Phe-Gly (DFG) (Mg2+ATP binding) motifs (fig. S2A). A 10-residue connector from sheet β8 to helix αF in Mt-MviN (Fig. 2D) occurs in place of the 25-residue activation loop of PknB, a partially disordered segment that includes two Thr residues that are phosphorylated to activate the kinase (8). Moreover, a long loop unique to Mt-MviN emerges from the C-terminal domain and covers the canonical ATP binding cleft and presumed protein-substrate binding surface (Fig. 2E). This extended loop replaces the STPK G helix, which has been implicated in kinase substrate recognition (9). These critical differences from active kinases suggest that the Mt-MviN KHD is a pseudokinase that neither binds ATP nor catalyzes phosphotransfer.
Fig. 2.

The Mt-MviN KHD is a pseudokinase that fails to bind ATP. (A) Ribbon diagram of the Mt-MviN KHD reveals a diverged eukaryotic-like kinase fold. The N lobe contains largely β sheet and the C lobe is largely α-helical. (B) Superposition of the Mt-MviN KHD (blue) and the Mtb PknB KD (orange) shows similar folds, with 222 residues aligning with a Cα rmsd of 3.5 Å. The N and C lobes are more closed in the Mt-MviN pseudokinase compared to these lobes in PknB bound to ATP. (C) Aromatic and aliphatic side chains (including Phe724, Trp789, and Tyr849) in the Mt-MviN pseudokinase domain occlude the canonical STPK ATP binding site and complete the C spine. The Mt-MviN P loop also occludes the ATP binding site. ATP, with the solvent-accessible surface shown in gray, is superimposed from the nucleotide-bound complex of the PknB KD. (D) A 10-residue linker in the Mt-MviN KHD (blue) replaces the activation loop. The typical 27-residue activation loop in the superimposed human protein kinase C [red; Protein Data Bank (PDB) file 1XJD] is shown. (E) The Mt-MviN KHD lacks the G helix involved in substrate recognition and instead contains a unique inserted loop that meanders over the location of the substrate and nucleotide-binding sites in active kinases. The G helix from PknB (orange) is shown for comparison. (F) Fluorescence polarization shows BODIPY-ATP, which commonly binds to STPKs, binding to the PknB KD but not the Mt-MviN pseudokinase domain.
To verify that the Mt-MviN ICD is not a cryptic active kinase, we tested for nucleotide binding and catalytic activity of the largest folded KHD fragment (612 to 963) identified by limited proteolysis of the full-length ICD. Fluorescence polarization measurements provided no evidence of binding to the fluorescent ATP analog, boron dipyrromethene (BODIPY)-ATP, which commonly binds STPKs (Fig. 2F). Consistent with the apparent absence of detectable nucleotide-analog binding, we observed no phosphoryl transfer activity when the Mt-MviN ICD was incubated with [γ-32P]ATP and the generic kinase substrate, myelin basic protein, in the presence of Mg2+, Mn2+, or EDTA (fig. S2C). This lack of auto- or transphosphorylation activity is consistent with the absence of conserved sequence motifs that mediate ATP binding, substrate binding, and catalysis in authentic STPKs. We conclude that this domain encodes a catalytically inactive pseudokinase.
Mt-MviN pseudokinase forms a canonical bacterial receptor STPK dimer
The absence of phosphotransfer activity begged the question of what kinase-associated function was preserved during the divergence of the Mt-MviN pseudokinase domain. Despite sharing only 18% identity with the PknB KD, the Mt-MviN pseudokinase formed a dimer that closely resembled the active dimers of transmembrane receptor kinases, such as PknB and PknE (Fig. 3A). The key residues involved in this interface—including Arg-Leu-Asp triads that make intersubunit contacts—are conserved in both the primary sequence (figs. S2A and S3A) and the three-dimensional structure (Fig. 3B and fig. S3, B and C). These residues in PknB (Arg10, Leu33, and Asp76) are essential for kinase activation in vitro and in vivo (10).
Fig. 3.

The Mt-MviN pseudokinase domain dimerizes like active Mtb receptor kinases. (A) Comparison of the dimer structures of the Mt-MviN pseudokinase domain (blue) and the KD of PknB (orange). The pseudokinase forms a back-to-back dimer analogous to sensor kinases. (B) A conserved cluster of residues mediates intersubunit interactions in the dimer interfaces of the Mt-MviN pseudokinase domain (left) and the PknB KD (right). (C) Gel exclusion chromatogram showing that the 612 to 963 Mt-MviN pseudokinase construct (37 kD) eluted as a dimer, whereas the shorter 681 to 963 construct (30 kD) eluted as a monomer. Full-length ICD (residues 541 to 975) also eluted as a dimer (fig. S3D). Numbers and down arrows indicate the elution volumes of molecular mass standards (kD). AU, absorbance units at 280 nm.
In contrast to the PknB KD, which forms an unstable dimer with a dissociation constant (Kd) in the millimolar range (10), gel exclusion chromatography showed that the full-length Mt-MviN ICD formed a stable dimer in solution (fig. S3D). An ICD construct containing residues 612 to 963 also eluted as a dimer, whereas a construct comprising residues 681 to 963 eluted as a monomer (Fig. 3C). Thus, the 612 to 680 segment N-terminal to the pseudokinase domain stabilizes the Mt-MviN dimer. This segment has no analog in PknB. The R709A and D775A mutations in the Mt-MviN KHD dimer interface (equivalent to Arg10 and Asp76 in PknB) dramatically reduced production of the protein in E. coli, suggesting that dimerization also promotes structural stability. Despite the sequence similarity of the dimer interfaces, the structures differ in detail (fig. S3C), and we detected no glutaraldehyde-cross-linked heterodimers in a mixture of the Mt-MviN ICD and the PknB KD.
PknB phosphorylates the Mt-MviN pseudokinase domain
About 10% of human kinase homologs are predicted to be pseudokinases (11). Although these proteins lack enzymatic activity, they are nevertheless highly conserved and serve important roles as kinase targets and scaffolds for phosphorylation-dependent protein complexes. To determine whether this bacterial pseudokinase serves a similar function, we investigated whether the Mt-MviN ICD could be phosphorylated in vitro by any of the 11 mycobacterial STPKs (Fig. 4A). The purified PknB KD efficiently phosphorylated the Mt-MviN ICD. Weaker or negligible phosphorylation was observed with other KDs. Tandem mass spectrometry (MS/MS) analysis of the ICD phosphorylated to completion by the PknB KD revealed a single site of phosphorylation at Thr947 (Fig. 4B). Substitution of Thr947 with Ala abolished phosphorylation by the PknB KD (Fig. 4B, inset), indicating that this STPK phosphorylates this single site on the Mt-MviN pseudokinase domain in vitro. The substitution also blocked in vitro phosphorylation by the PknA, PknE, and PknH KDs (fig. S4).
Fig. 4.

Mt-MviN pseudokinase forms a phosphorylation-dependent heterodimer with the FhaA FHA domain. (A) Phosphorylation of the full-length Mt-MviN ICD (residues 541 to 975) by active constructs of all 11 Mtb STPKs. Autoradiogram shows rapid, quantitative phosphorylation by the PknB KD. PknA, PknE, PknH, PknJ, and PknL phosphorylate the ICD more slowly. Autophosphorylation of inactive PknB is shown for comparison. (B) MS/MS of a tryptic digest of the PknB-phosphorylated ICD shows modification of Thr947 in the peptide 929-SASTLLNLMQQATAVADRpTEVLGPIDEAPVSAAPR-963. The T947A substitution in the Mt-MviN ICD abolishes in vitro phosphorylation by PknB (inset). KDs indicates kinase domain of PknB. Abbreviations for the amino acids are as follows: A, Ala; D, Asp; E, Glu; G, Gly; I, Ile; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; and V, Val. (C) SDS-PAGE shows that a 52-kD protein from M. smegmatis ly-sate binds to the PknB-phosphorylated (left) but not unphosphorylated (right) GST–Mt-MviN ICD fusion. The 52-kD protein was identified by MS as the M. smegmatis FhaA homolog (fig. S5A). (D) Phosphorylation-dependent binding of the Mt-MviN ICD to the purified FhaA FHA domain. SDS-PAGE shows binding of the FHA domain of FhaA, but not Rv1747 or GarA, to the phosphorylated GST–Mt-MviN ICD fusion.
Phosphorylated Mt-MviN pseudokinase recruits a regulatory FHA domain protein
Because the Mt-MviN pseudokinase lacks enzymatic activity that could be regulated by phosphorylation, we tested the hypothesis that phosphorylation creates a binding site for another mycobacterial protein. Using affinity chromatography, we identified a single ~52-kD protein in M. smegmatis cell lysate that bound efficiently to the Mt-MviN ICD in a phosphorylation-dependent manner (Fig. 4C). MS/MS analysis revealed that this Mt-MviN-binding protein is the ortholog of M. tuberculosis FhaA (Rv0020c) (fig. S5, A and B), an FHA domain-containing protein encoded in the same operon as PknB.
FHA domains in eukaryotes bind specific phospho-Thr (pThr)-containing peptides, forming phosphorylation-dependent protein complexes. To verify that the FHA domain of FhaA binds phosphorylated Mt-MviN and to investigate the specificity of the interaction, we tested binding of the Mt-MviN pseudokinase to three of the seven FHA domains encoded in the Mtb genome. The FHA domain (residues 390 to 525) of FhaA was sufficient to bind phosphorylated, but not unphosphorylated, Mt-MviN ICD (Fig. 4D). In contrast, neither Mtb GarA, which contains an FHA domain, nor the first FHA domain of Rv1747 associated with the Mt-MviN ICD.
Quantitative measurements of the Mt-MviN-FhaA interaction showed that the FhaA FHA domain binds unusually tightly to the phosphorylated Mt-MviN ICD. Analysis using isothermal titration calorimetry (ITC) (fig. S5C) revealed an apparent dissociation constant of 58 ± 2 nM (table S2). This binding is 4 to 172 times tighter than the typical Kd values of 0.23 to 10 μM reported for phosphopeptide recognition by other FHA domains (12–14).
The pseudokinase and FHA domain complex forms a three-dimensional interface
To define the mechanism of specific phosphoprotein recognition by FhaA, we determined the crystal structure of the phosphorylated Mt-MviN pseudo-kinase domain in complex with the FhaA FHA domain (Fig. 5A, table S1, and fig. S5D). The C-terminal residues of the Mt-MviN pseudokinase containing pThr947 bound in an extended conformation (Fig. 5B), with the conserved “SR motif” (Ser473 and Arg474) of the FHA domain engaging the pThr947 residue. Mt-MviN Leu950 at the pThr +3 position in the ICD fits into a pocket on the FHA domain surface, similar to the arrangements observed previously in peptide complexes with other FHA domains (13, 15).
Fig. 5.
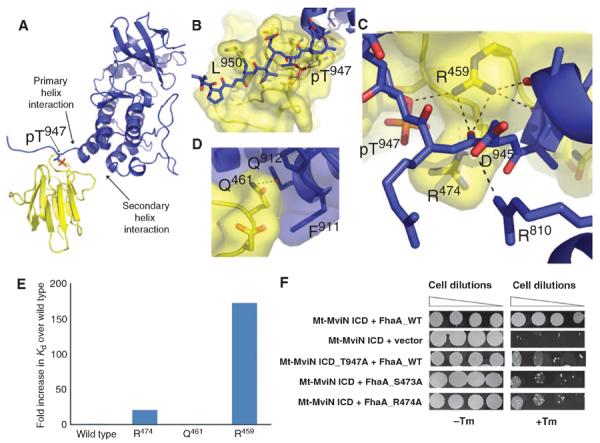
Three-dimensional contacts augment phosphopeptide recognition in the structure of the phosphorylated Mt-MviN pseudokinase domain complex with the FhaA FHA domain. (A) Ribbon diagram of the pseudokinase–FHA domain heterodimer structure showing the C terminus of the Mt-MviN ICD (blue) bound in an extended conformation to the FhaA FHA domain (yellow). The pThr is shown in stick form. (B) Recognition of pThr947 and the neighboring residues of the pseudokinase domain (blue) by the FhaA FHA domain (yellow). The pThr and Leu950 at the +3 position bind in pockets on the FHA domain. Asp945 at the −2 position forms an intramolecular ion pair with the Arg810 in the complex. (C) FhaA Arg459 (yellow) caps the C-terminal helix of the Mt-MviN pseudokinase domain (blue) and forms a network of hydrogen bonds (dotted lines) to the pseudokinase helix (right), the main-chain carbonyl of the first residue after the helix (center), and the pThr947 bridging oxygen (left). (D) Contacts of FhaA Gln461 away from the pThr site with Pro910, Phe911, and Gln912 of the Mt-MviN ICD set the relative orientation of the domains. (E) Relative affinities of FhaA variants for the phosphorylated Mt-MviN ICD determined by ITC at 25°C. Affinity is expressed as the fold increase in Kd from the wild-type interaction. (F) Phosphorylation-dependent recognition of Mt-MviN pThr947 by FhaA FHA domain in vivo. Interaction between the Mt-MviN ICD and wild-type FhaA confers Tm-resistant growth as a result of the association of the appended murine DHFR fragments in M. smegmatis. No interaction was observed when the Mt-MviN ICD was co-expressed with the control vector expressing only the mDHFR fragment. The T947A mutation at the Mt-MviN phosphorylation site or substitutions of the FhaA SR motif in the pThr-binding site reduced Tm resistance.
In addition to these characteristic contacts neighboring the pThr site, the FhaA FHA domain makes unanticipated contacts with the core pseudokinase domain. The conserved Arg459 in the FHA domain, for example, caps the C-terminal pseudokinase helix at Ala944, effectively measuring the distance between this helix and the pThr (Fig. 5C). In contrast, the end of this helix in the isolated Mt-MviN pseudokinase domain shows evidence of flexibility, terminating at different residues (942 to 950) in the four independent molecules in the asymmetric unit. A second unexpected contact surface is formed by the loop containing Gln461 in the FHA domain interacting with Pro910, Phe911, and Gln912 at the N terminus of the penultimate helix of the Mt-MviN pseudokinase (Fig. 5D). Mt-MviN residues Arg810 and Asp945 also form an intramolecular ion pair in the FhaA-containing complex that is not seen in the free pseudokinase structures (Fig. 5C).
The “three-dimensional” contacts between the Mt-MviN ICD and the FhaA FHA domain were not predicted by the common view that FHA domain specificity is determined solely by the sequence surrounding the pThr site (16). To determine the importance of the structural contacts, we used ITC to measure the effects of alanine substitutions in the Mt-MviN–FHA domain interface (table S2). The FhaA Q461A substitution, which disrupts contacts with the penultimate helix of the Mt-MviN pseudokinase, had minimal effects on the affinity of the complex, suggesting that this side chain is not contributing appreciably to the binding energy. In contrast, the R459A substitution, which eliminates the contact to the pseudokinase C-terminal helix, reduced the binding affinity more than 170-fold, compared to a reduction of 41-fold by R474A substitution in the canonical FHA SR motif in the pThr binding site (Fig. 5E).
FhaA interacts with the phosphorylated Mt-MviN ICD in vivo
To determine whether these specific features of the FhaA–Mt-MviN interactions are recapitulated in vivo, we used a mycobacterial protein fragment complementation assay (17). Interactions were detected by fusing the FhaA protein and Mt-MviN ICD to separate segments of the murine dihydrofolate reductase (mDHFR) enzyme. Dimerization of the appended domains mediates assembly of active mDHFR and confers resistance to the bacterial DHFR inhibitor trimethoprim (Tm). Using this assay, we found that the wild-type Mt-MviN ICD interacts with FhaA in M. smegmatis (Fig. 5F). This interaction was nearly abrogated by the T947A replacement of the Mt-MviN phosphorylation site. Similar decreases in Tm resistance were observed upon mutation of the FhaA “SR” motif that interacts directly with the pThr residue in the crystal structure (Fig. 5F). These results showed that the FhaA FHA domain can bind the Mt-MviN pseudokinase domain in vivo and that this interaction depended on the phosphorylation of Mt-MviN Thr947, presumably by the endogenous M. smegmatis PknB kinase.
PknB and FhaA regulate cell wall synthesis
The specific, phosphorylation-dependent, high-affinity binding of FhaA to Mt-MviN may be a mechanism by which PknB-mediated phosphorylation of Mt-MviN regulates PG metabolism. To test this hypothesis, we investigated the effect of modulating PknB abundance on cell growth and PG synthesis. We found that the inducible PknB overexpression in M. smegmatis caused rapid and complete cessation of cell growth, which was accompanied by the accumulation of extractable DAP-containing PG precursors (Fig. 6A). Both phenotypes resembled the effects of Ms-MviN depletion (Fig. 1), suggesting a link between PknB signaling and inhibition of PG synthesis.
Fig. 6.

FhaA localizes to the sites of cell growth and division and regulates PG synthesis. (A) PknB over-expression in M. smegmatis increases the cellular pool of DAP-containing PG precursors. M. smegmatis carrying an ATc-inducible pknB overexpression plasmid cultured without or with ATc. Bars indicate the amount of solvent-extractable DAP-containing precursors measured with GC-MS after 6 hours of ATc treatment. (B) FhaA-mVenus is found at cell poles and septa. Superimposed fluorescence and differential interference contrast images of M. smegmatis expressing an FhaA-mVenus fusion. (C) FhaA depletion causes a marginal growth defect. The indicated strains of M. smegmatis were cultured with or without ATc, and growth was quantified by optical density. Inset: Western analysis shows that FhaA-ID fusion protein doublet was degraded within 6 hours after induction of HIV-2 protease. The same amount of protein (as measured with the BCA protein assay) was loaded in each lane. (D) FhaA depletion causes morphological changes. Scanning electron micrographs of fhaA wild type or fhaA-ID M. smegmatis after 12 hours of ATc treatment. Both strains express HIV-2 protease. (E) FhaA depletion increases BODIPY-vancomycin binding. The kinetics of BODIPY-vancomycin binding was measured in wild-type or FhaA-depleted bacteria. Inhibition of metabolism with sodium azide had no effect on initial staining but substantially reduced the rate of BODIPY-vancomycin accumulation. (F) Nascent PG accumulates at the poles after FhaA depletion in M. smegmatis. Wild-type bacteria (cytosol labeled with red fluorescent protein) or FhaA-ID cells were treated with ATc for 6 hours. Both cell populations were stained with BODIPY-vancomycin (green) and imaged. In representative cells, vancomycin-stained poles and septa are indicated with arrowheads and arrows, respectively. Data points in (A), (C), and (E) depict the means of three independent measurements. Error bars indicating SD are included for all points but are often too small to be visible.
To investigate the specific role of FhaA in this regulation, we determined whether FhaA is located at sites of cell wall synthesis. The chromosomal fhaA open reading frame in M. smegmatis was fused to the fluorescent protein mVenus. The FhaA-mVenus fusion protein was functional because it supported normal growth rate and cell morphology. Microscopic observation of live cells revealed that the FhaA fusion protein was enriched at the cell poles and septa (Fig. 6B and fig. S6), which are the major sites of cell wall biosynthesis in mycobacteria (18, 19).
To reduce the amount of FhaA in vivo, we degraded the FhaA fusion with the ID tag incorporated into the fluorescent protein. Depletion of FhaA, as judged by Western blotting and loss of polar and septal fluorescence, had only a minimal effect on bacterial growth (Fig. 6C) and did not reproducibly alter the abundance of extractable DAP-containing PG precursors. These phenotypes were consistent with the nonessential nature of the Mt-MviN regulatory domain to which FhaA binds. However, FhaA depletion dramatically altered cell morphology, producing irregularly shaped cells that were markedly shorter and wider than wild-type M. smegmatis (Fig. 6D). Thus, both the localization of FhaA and its role in maintaining normal cell shape indicated a function in regulating cell wall biosynthesis.
Because Mt-MviN was required for PG biosynthesis, we used a fluorescent derivative of vancomycin to determine whether FhaA depletion altered the production or structure of nascent PG at the cell surface. This membrane-impermeable antibiotic binds to the d-Ala–d-Ala moiety of nascent PG and lipid II, inhibiting subunit incorporation into growing PG and subsequent peptide cross-linking. Association of BODIPY-vancomycin with M. smegmatis increased over hours and required continual cellular metabolism (Fig. 6E), indicating that the accumulation of fluorescence could be used to estimate the rate at which the d-Ala–d-Ala moiety appeared at the cell surface. With this assay, FhaA depletion significantly increased the rate of BODIPY-vancomycin staining at the cell poles and septa (Fig. 6, E and F, and fig. S7). Polar staining may reflect binding of vancomycin to stem peptides in uncross-linked PG, sequestration of lipid II at the pole, or both. This localized increase in nascent PG abundance indicated that morphological defects observed upon FhaA depletion resulted from misregulated PG biosynthesis.
DISCUSSION
This work describes the discovery of a regulatory complex consisting of an essential PG biosynthetic protein, Mt-MviN, and an FHA domain–containing regulatory protein, FhaA. Our data suggest that the STPK PknB, which is essential for the growth and proper cell morphology of mycobacteria (20), initiates the formation of this complex. The PknB extracellular sensor domain contains four tandem PASTA repeats that bind synthetic PG fragments (21, 22). PASTA domains in the orthologous STPK of Bacillus subtilis, PrkC, bind weakly to sites in mature PG and more strongly to soluble PG fragments (23, 24). The competition between these two interactions may provide a conserved mechanism that regulates PknB by altering dimerization or localization of the intracellular KDs (10, 22, 25). We found that PknB quantitatively phosphorylated Thr947 at the C terminus of the pseudokinase domain of Mt-MviN, which is among the sites phosphorylated in Mtb in vivo (26).
This phosphorylation site occurs in the juxtamembrane linker C-terminal to the Mt-MviN pseudokinase domain. The crystal structure of the Mt-MviN domain provided an initial viewof a bacterial pseudokinase and revealed the most deeply diverged pseudokinase described to date (fig. S8). Aromatic residues in Mt-MviN fill the canonical ATP binding site and play a structural role by completing the C spine, a stabilizing nonpolar network that extends between the lobes (27). The absence of an ATP binding site suggests that Mt-MviN will be refractory to ATP-competitive kinase inhibitors.
Our analysis showed that the Mt-MviN pseudokinase retains at least two functional characteristics of homologous STPKs—the formation of a back-to-back dimer and phosphorylation in the juxtamembrane linker following the KHD (28). A segment N-terminal to the KHD that is absent from catalytically active bacterial receptor STPKs augmented dimer formation. Because Mtb receptor STPKs are also phosphorylated at juxtamembrane linker sites (29), the phosphorylation-dependent binding of FhaA to the Mt-MviN linker may reflect a regulatory mechanism that was retained (30) or enhanced in the divergence of Mt-MviN.
Phosphorylation of Mt-MviN recruits FhaA through an interaction with the FHA domain. Both in vitro and in vivo, this association was tight and specific and depended on Thr947 phosphorylation. Unlike previous structures of FHA domains bound to cognate peptides, the Mt-MviN–FhaA interface contains protein-protein contacts that are remote from the pThr binding site. The most important of these interactions appears to be FhaA Arg459, which caps the C-terminal helix in the pseudokinase three amino acids before pThr947 and bridges to the Mt-MviN Thr947 oxygen. In complexes of FHA domains with target phosphopeptides, a homologous Arg makes a distinct interaction with the main chain in the −2 position of the bound peptide (13, 31). Considering the FhaA–Mt-MviN structure, it will be of interest to discover whether this conserved Arg serves a general function as a “ruler” that measures three residues from the end of a helix to an exposed pThr site.
The Mt-MviN pseudokinase–FhaA FHA complex was up to 170-fold more stable than previously characterized interactions of diverse FHA domains with cognate phosphopeptides and had a twofold greater affinity than a short peptide optimized for binding to FhaA (15). The nonbiological sequence used in that work, TAPpTEKI (compared to ADRpTEVL in Mt-MviN), forms a structurally similar complex with the FhaA FHA domain [root mean square deviation (rmsd) for all atoms = 0.34 Å]. Nonetheless, the optimized peptide makes distinct interactions—including −1 and −2 main-chain contacts with the SR motif Arg, reduced chain flexibility of Pro at −1 and Ile at +3, and augmented hydrophobic and polar contacts of Lys at +2—that may mediate tight binding. In contrast, our affinity measurements indicated that the additional contacts, which are remote from the pThr peptide, contributed to the Mt-MviN:FhaA association. These three-dimensional protein contacts constitute a previously unrecognized feature of phosphoprotein recognition by an FHA domain.
Because Mt-MviN is required for Mtb growth, the cellular role of these interactions in vivo was difficult to assess with standard loss-of-function genetics. Instead, we used a conditional degradation system (7) to determine the effects of specific protein depletion over time. Degradation of Ms-MviN and FhaA had distinct effects, suggesting that these proteins serve opposing roles. Ms-MviN depletion inhibited cell growth and PG synthesis. In contrast, FhaA depletion only mildly affected growth but dramatically increased the accumulation of nascent PG stem peptides at the cell poles and septum. Moreover, FhaA, like PknB (21), localized to the cell poles and septum. This spatial pattern places the kinase and the regulator at the sites of PG biosynthesis, rather than randomly distributed throughout the cell. Although more complicated models can be imagined, our results support the interpretation that FhaA binds Thr947-phosphorylated Mt-MviN and inhibits the late stages of PG biosynthesis. This PknB-initiated complex between Mt-MviN and FhaA embodies functional relationships characteristic of a regulatory feedback loop that controls PG biosynthesis in response to extracellular PG substructures (Fig. 7).
Fig. 7.
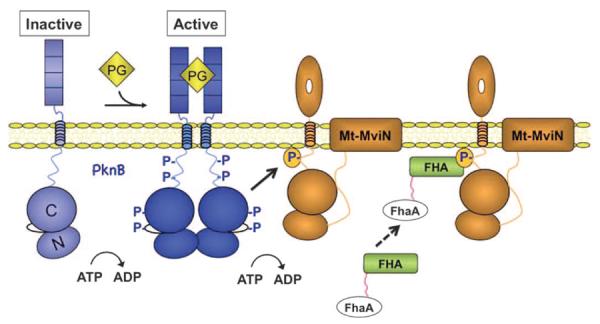
Model for the PknB-dependent formation of the Mt-MviN-FhaA complex. The extracellular PASTA domains of PknB bind to PG fragments or discrete cell wall structures, activating the intracellular KDs. The N and C lobes of the KDs are indicated. The active kinase phosphorylates Thr947 of Mt-MviN. Specific binding of the FHA domain recruits FhaA to phospho–Mt-MviN, thereby inhibiting PG synthesis. This complex creates a feedback loop in which extracellular PG binding coordinates cell wall biosynthesis with other processes regulated by PknB. Additional layers of regulation could be imparted by FhaA phosphorylation and binding to PknB (30, 32), which are not depicted in the figure.
The regulatory circuits initiated by PknB extend beyond the control of cell wall biosynthesis through phosphorylation of MviN. For example, PknB phosphorylates FhaA, which also binds PknB (30, 32), raising the possibility of further kinase regulation of the circuit. Moreover, PknB has been reported to regulate additional cell wall synthetic steps and other processes in Mtb (33). Reported PknB substrates include GlmU (34), which is involved in biosynthesis of the carbohydrate component of PG, and enzymes that participate in synthesis of the mycolic acid cell wall layer (35, 36). PknB phosphorylates the PknA STPK, which has been implicated in cytokinesis (37, 38), as well as regulators of transcription, translation, and cell division (20, 26, 33, 39, 40). Thus, the signaling pathway described in this work is part of a wider network controlled by PknB, which coordinates a number of fundamental cellular processes.
Although the first prokaryotic STPK was discovered 2 decades ago (41), the essential functions of these regulators distinct from the roles of bacterial histidine kinases have not been entirely appreciated (42). In various bacteria, Thr phosphorylation is required to mediate FHA domain binding (43). However, many STPK phosphorylations apparently function independently of FHA domain proteins (33). Thus, in addition to engaging FHA domain regulators, the stable and coordinated phosphorylation and dephosphorylation of diverse proteins can have distinct functional effects. The linear pathways composed of the differentially labile modifications downstream of a histidine kinase do not mediate such broad effects in concert. In this light, our work begins to reveal that the ability to initiate complex, synchronized, stable signaling networks may be an essential characteristic that is conserved among eukaryotic and prokaryotic STPKs.
MATERIALS AND METHODS
Protein purification
Mt-MviN ICD constructs (full-length, 541 to 975, and truncations 612 to 955, 612 to 963, 681 to 955, and 681 to 963) were cloned into the Gateway vectors pHGWA, pHMGWA (44), and pDEST15 (Invitrogen) with an N-terminal tobacco etch virus (TEV)–cleavable His tag, His-MBP (maltose binding protein) tag, or glutathione S-transferase (GST) tag, respectively. The FhaA FHA domain (residues 390 to 525) was cloned into pET28 with a thrombin-cleavable, N-terminal His tag, and the FHA domains of Rv1747 and Rv1837 were tagged with GST (32). Mutations were introduced by the QuikChange method (Stratagene).
Proteins were expressed using autoinduction (45) at 30°C or 18°C. Cells from 1-liter cultures containing GST fusions were resuspended in ~50 ml of buffer A [20 mM tris-HCl (pH 7.5), 150 mM NaCl, and 10% glycerol] containing protease inhibitors 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), leupeptin, and E64 and were lysed by sonication. Lysates cleared by centrifugation were mixed with glutathione Sepharose beads (GE Healthcare) for 1 hour at 4°C. The beads were washed with buffer A and eluted with 20 mM glutathione. Fusions were purified by ion exchange chromatography on MonoQ (GE Healthcare) with a 50 to 500 mM NaCl gradient elution in 20 mM tris-HCl (pH 8.0) and 10% glycerol. The Mt-MviN was applied to a Sephadex S75 column (GE Healthcare) in buffer A. Untagged proteins were released from the glutathione beads by TEV protease cleavage overnight at 4°C and purified as described for the tagged proteins. The selenomethionine (SeMet)–containing L784M Mt-MviN612–955 was expressed and purified with the addition of 1 mM reduced dithiothreitol to the buffers.
Cells containing His or His-MBP fusions were resuspended in 20 mM tris-HCl (pH 7.5), 300 mM NaCl, 20% glycerol, and 20 mM imidazole. Cleared supernatant was applied to a 5-ml Ni-NTA column (GE Healthcare), and the column was washed and eluted with a gradient of 50 to 300 mM imidazole. Pseudokinase fusion constructs were purified with MonoQ and Sephadex S75. Untagged proteins were produced by cleavage with TEV protease overnight at 4°C, dialyzed to remove imidazole, chromatographed on a Ni-NTA column to remove the tag, and purified by ion exchange and gel exclusion as described above.
The FhaA FHA domain (32), the PknB KD (28), and active forms of all 11 Mtb kinases (46, 47) were expressed and purified as described.
Phosphorylation and nucleotide binding assays
Assays were performed with [γ-32P]ATP as described (47). The Mt-MviN ICD was phosphorylated for binding and structural studies for 24 to 48 hours at 4°C with the ICD at 0.1 to 1 mg/ml in tris buffer (pH 7.0), 1 mM ATP, 1 mM MnCl2, 0.1 mM EDTA, and protease inhibitors AEBSF, leupeptin, and E64. PknB KD was added at 1:10 to 1:20 kinase-substrate ratio. The kinase was removed with a 1-ml HisTrap column (GE Healthcare), and the ATP and MnCl2 were removed by dialysis. Phosphorylation was confirmed with Pro-Q Diamond stain (Molecular Probes) and by altered SDS–polyacrylamide gel electrophoresis (SDS-PAGE) migration for constructs <30 kD.
Purified PknB or the Mt-MviN ICD at 0, 0.1, 0.5, 1, 2, 5, or 10 μM in 60 mM Mops, 0.1 mM EDTA, 1 mM MgCl2, 1 mM MnCl2, and 4% glycerol was mixed with 25 nM BODIPY–ATP-γ-S [adenosine 5′-O-(3-thiotriphosphate)] analog. The fluorescence polarization was measured using an excitation energy of 485 nm and a 535-nm emission filter with a Victor 3V 1420 multilabel counter (PerkinElmer).
Crystallization and x-ray structure determination
Native and SeMet-labeled KHD (7 to 20 mg/ml) were crystallized by vapor diffusion at 18°C in 1:1 protein-precipitant solution. In some cases, 1:1000 trypsin was added to facilitate crystallization. The P21 crystal form grew in 0.7 to 0.8 M succinate (pH 7.0), and the P3121 native crystals formed in 60% tacsimate with 4% 1,1,1,3,3,3 hexafluoro-2-propanol or 3 M NaCl, and 0.1 M tris-HCl (pH 7.5 to 8.5). The L784M SeMet crystals grew in 3 M NaCl and 0.1 M tris-HCl (pH 7.5 to 8.5).
The phospho–Mt-MviN681–955 KHD–FhaA FHA domain complex was formed using KHD phosphorylated overnight at 4°C with PknB KD. Needle crystals grew from a 1:1 protein mixture (20 mg/ml) in 0.2 M CaCl2, 0.1 M MES (pH 6), 1 mM MnCl2, and 20% PEG 6000 (polyethylene glycol, molecular weight 6000).
X-ray data were collected at Beamline 8.3.1 at the Advanced Light Source from crystals flash-frozen in 20% xylitol or glycerol. Diffraction data were integrated and scaled with HKL 2000 (48). The Mt-MviN pseudokinase structure was solved by SAD phasing of the L784M SeMet derivative in the P3121 crystal form at 3.5 Å resolution with SOLVE/RESOLVE (49). The phases were extended in a 3.4 Å native data set and solvent-flattened with DM (50). The polyalanine/glycine main-chain fragments output from RESOLVE and a PHYRE homology model (51) were used to guide manual model building with Coot (52). Models were refined with the maximum-likelihood method in PHENIX (53) using the 3.4 Å resolution native data set.
This preliminary pseudokinase-domain structure was used as a search model in Phaser to calculate phases for a 3.1 Å resolution data set and a 2.25 Å resolution data set from a crystal that formed after 1 year. Both of these monoclinic crystal forms had four molecules in the asymmetric unit. The 2.25 Å resolution form had slightly different crystal contacts, and the protein was truncated at the C terminus compared to the 3.1 Å form. Refinement using PHENIX, first with noncrystallographic symmetry restraints and then without, and Coot resulted in largely complete models. Chains in the 2.25 Å resolution model were moved as rigid bodies into the electron density map of the 3.1 Å resolution P21 crystal form and molecule B was placed in the 3.4 Å electron density map of the P3121 crystal form. These models were refined with PHENIX.
The structure of the pseudokinase-FHA complex was solved at 2.7 Å resolution by molecular replacement with Phaser (54). Molecule B from the P21 form of the pseudokinase domain was the search model for the Mt-MviN KHD. The FHA domain search model was derived from Mtb EmbR (2FEZ), with sequence differences truncated at the Cγ position with CHAINSAW (55). The model was adjusted with Coot and refined with PHENIX. Model validation was carried out with MolProbity (56).
Phosphorylation site mapping
The full-length Mt-MviN ICD phosphorylated with PknB was purified by high-performance liquid chromatography (HPLC). The intact mass was determined by electrospray ionization–ion trap MS (Bruker-Agilent). The phosphorylation site was determined by MS/MS sequencing of a tryptic digest (9).
Identification of a phospho–Mt-MviN binding partner
M. smegmatis culture (50 ml) grown to an optical density at 600 nm (OD600) of ~1 in 7H9 medium was centrifuged, resuspended in 0.75 ml of extraction buffer (20 mM tris-HCl, 150 mM NaCl, 10% glycerol, and 1 mM AEBSF), and lysed by bead beating for 2 min on ice. The supernatant was retained, and bead beating was repeated in 200 μl of extraction buffer. Pooled supernatant (100 μl) was mixed for 30 to 60 min with glutathione resin saturated with unphosphorylated or PknB-phosphorylated GST–Mt-MviN ICD. After four washes with 500 μl of extraction buffer, resin-bound proteins were separated by SDS-PAGE. The candidate band stained with Colloidal Blue was sliced into ~1-mm3 pieces and washed four times in 25 mM ammonium bicarbonate/50% acetonitrile. The gel pieces were dried (~30 min in a vacuum centrifuge), rehydrated in 1 volume of trypsin (0.04 mg/ml) in 25 mM ammonium bicarbonate, and incubated 16 to 20 hours at 37°C. Peptides were extracted with two volumes of water and twice more with two volumes of 5% trifluoroacetic acid (TFA)/50% acetonitrile. After the volume was reduced to 0.5 ml in a vacuum centrifuge, the peptides were desalted on C18 ZipTips (Millipore) for MS analysis.
Isothermal titration calorimetry
Proteins (FhaA and phosphorylated KHD681–963) were dialyzed into 20 mM tris-HCl, 150 mM NaCl, and 10% glycerol. Measurements were obtained in a VP-ITC instrument (MicroCal) at 25°C by injecting the FHA domain (40 to 100 μM) into the monomeric phospho-KHD681–963 (4 to 12 μM). Injection into buffer controlled for dilution effects. The data were fitted to a single binding site model with the Origin software package (MicroCal).
Mycobacterial protein fragment complementation assay
The GCN4 leucine zipper domains in the mycobacterial two-hybrid protein complementing plasmids, pUAB100 and pUAB200 (17), were replaced, respectively, with Mt-MviN ICD residues 548 to 978 and full-length FhaA. Dilutions of co-transformants of M. smegmatis carrying both plasmids were spotted on 7H11 + Hyg + Kan agar plates containing Tm (0 or 15 μg/ml).
Inducible protein expression and degradation in M. smegmatis
Using phage Che9c-mediated recombineering (57), we introduced ID tags in-frame in the chromosome at the end of the ICD coding sequence of Ms-MviN or the 3′ end of fhaA. The ID tag encoded a codon-optimized mCherry, an SsrA tag (GLAA), the HIV-2 protease cleavage site PQFS, FLAG, and 6×His epitopes, and a hygr marker (7). A kanr plasmid expressing HIV-2 protease from a tet-inducible promoter (7) was integrated in the L5 phage attachment site. Complementing plasmids encoding either fhaA with its native promoter or the MSMEG6928–Ms-MviN operon were based on a gentamycinr derivative of pTTP1A, which integrates at the mycobacteriophage Tweety site (58). These strains were grown in 7H9 broth at 37°C to OD600 ~0.25, and protein depletion was initiated by addition of ATc (50 ng/ml) to induce HIV2 protease. Mycobacterial cell lysates for Western blots were prepared as described (59). To extract membrane proteins, we incubated the lysates with 1.0% SDS for 15 min at 50°C before centrifugation. Equal amounts of total protein [determined with the BCA (bicinchoninic acid) kit (Pierce)] from each lysate were subjected to SDS-PAGE. Coomassie staining confirmed equal protein loading. Immunoblots were developed with horseradish peroxidase–conjugated anti–red fluorescent protein (RFP) (mCherry) antibodies (Abcam) with the ECL system (GE Healthcare).
For pknB overexpression, M. smegmatis harboring the Mt-pknB gene on a tet-inducible mycobacterial vector pUV15-tetOtetR (60) was grown in 7H9 + hygromycin (50 μg/ml) to OD600 of 0.25, and ATc (50 ng/ml) was added to induce Mt-pknB.
Mtb mutagenesis
Genome-wide transposon libraries were generated and characterized as described, and the complete data set is available in (61). To calculate the statistical probability of MviN essentiality, we compared the observed gap in transposon coverage to the longest gap expected by chance, using the cumulative value of the extreme value distribution (61). Recombineering in Mtb used an approach similar to that for M. smegmatis, with the following differences. Che9c expression was induced (18 hours; 1 μM isovaleronitrile), and the cultures were treated with 0.2 M glycine for 12 hours before making electrocompetent cells (62). After electroporation, the cells were incubated for 24 hours in 7H9 broth and plated on 7H10 containing hygromycin (50 μg/ml). Each hygr colony was screened with three polymerase chain reaction (PCR) assays to detect the novel 5′ and 3′ recombination junctions and the deletion of an internal chromosomal fragment. The PCR products containing the 5′ and 3′ junctions were sequenced to verify the junctions.
Diaminopimelic acid analysis
Bacteria were centrifuged in a preweighed glass tube and washed with phosphate-buffered saline (PBS). Norvaline was added as internal standard, samples were dried in a vacuum centrifuge (Savant) for 6 hours, and the dry weight of the sample was determined. Samples were extracted sequentially with n-butanol/6 M pyridinium acetate [pH 4.2, 4:1 (v/v)] and ethanol/water [1:1 (v/v)]. Extracts were pooled and filtered with polytetrafluoroethylene syringe filters (Millipore). The filtrates were dried under vacuum and hydrolyzed in sealed tubes in 0.2 ml of 6 M HCl at 110°C for 18 hours. The hydrolysates were derivatized with the EZ Fast GC (gas chromatography)–MS kit (Phenomenex). Samples were analyzed on a Varian CP-3800 gas chromatograph fitted with a ZB-5 column (30 m × 0.25 mm × 0.25 μm) connected to a Varian 320 MS-TQ mass spectrometer. DAP quantitation was normalized to the dry weight of the sample and norvaline recovery.
Fluorescent vancomycin staining and FhaA localization
Nascent PG was labeled on the cell surface by adding a mixture of 1 μg/ml each of BODIPY-vancomycin (Invitrogen) and unlabeled vancomycin to wild-type or FhaA-depleted (6 hours with ATc) cells for 90 min at 37°C (19). Bacteria were concentrated, placed on a 1.2% LB agarose pad, and visualized on a Leica SP5 laser scanning microscope. Images were captured and fluorescence was quantified in regions of interest (ROIs) around the poles with LASAF software. An equal area proximal to the ROIs provided corrections for background intensity. FhaA-mVenus was localized in an M. smegmatis strain with the codon-optimized fluorescent protein tag integrated at the end of the chromosomal fhaA gene by means of the Che9c-medated recombineering approach (57).
The kinetics of cell surface stem-peptide accumulation was determined after the addition of vancomycin–BODIPY-vancomycin (1 μg/ml each). Cells were centrifuged, washed twice with PBS, and resuspended in PBS in the original culture volume. The role of active metabolism was determined by addition of sodium azide (10 mM) for 20 min before the addition of vancomycin. Fluorescence was measured (excitation 485/20 and emission 528/20) with a Synergy Hybrid Multimode Microplate Reader (BioTeK). The fluorescence values were normalized to the OD of the same samples.
Supplementary Material
Acknowledgments
We thank G. Hendricks for electron microscopy assistance and K. Murphy for the development of recombineering methods. We are grateful to J. Holton, G. Meigs, and J. Tanamachi for help with x-ray data collection. S. Smerdon provided results before publication. We thank A. Craney, R. Beckwith, I. Hood, and M. Seeliger for help analyzing Mt-MviN: FhaA interactions.
Funding: This work was supported by NIH grants to D.C.C. (AI049151), E.J.R. (AI071881), C.M.S. (AI073509 and AI064282), and T.A. (GM70962 and AI68135). The Bill and Melinda Gates Foundation supported E.J.R. and C.M.S. The Howard Hughes Med ical Institute supported A.M.F., D.S.K., and C.M.S.
Footnotes
SUPPLEMENTARY MATERIALS www.sciencesignaling.org/cgi/content/full/5/208/ra7/DC1
Author contributions: All authors planned the experiments. C.L.G., K.G.P., S.R.B., C.E.B., A.M.F., D.S.K., J.E.G., H.V., A.Z., J.-R.W., and R.K.D. performed the experiments. All authors analyzed the data and wrote the paper.
Competing interests: The authors declare that they have no competing interests.
Accession numbers: The coordinates and structure factors were deposited in the PDB [accession numbers 3OTV, 3OUK, 3UQC (pseudokinase), and 3OUN (phospho-pseudokinase–FHA)].
REFERENCES AND NOTES
- 1.Bouhss A, Trunkfield AE, Bugg TD, Mengin-Lecreulx D. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol. Rev. 2008;32:208–233. doi: 10.1111/j.1574-6976.2007.00089.x. [DOI] [PubMed] [Google Scholar]
- 2.White CL, Kitich A, Gober JW. Positioning cell wall synthetic complexes by the bacterial morphogenetic proteins MreB and MreD. Mol. Microbiol. 2010;76:616–633. doi: 10.1111/j.1365-2958.2010.07108.x. [DOI] [PubMed] [Google Scholar]
- 3.Mohammadi T, van Dam V, Sijbrandi R, Vernet T, Zapun A, Bouhss A, Diepeveende Bruin M, Nguyen-Distèche M, de Kruijff B, Breukink E. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 2011;30:1425–1432. doi: 10.1038/emboj.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruiz N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2008;105:15553–15557. doi: 10.1073/pnas.0808352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inoue A, Murata Y, Takahashi H, Tsuji N, Fujisaki S, Kato J. Involvement of an essential gene, mviN, in murein synthesis in Escherichia coli. J. Bacteriol. 2008;190:7298–7301. doi: 10.1128/JB.00551-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12712–12717. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei JR, Krishnamoorthy V, Murphy K, Kim JH, Schnappinger D, Alber T, Sassetti CM, Rhee KY, Rubin EJ. Depletion of antibiotic targets has widely varying effects on growth. Proc. Natl. Acad. Sci. U.S.A. 2011;108:4176–4181. doi: 10.1073/pnas.1018301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boitel B, Ortiz-Lombardia M, Durán R, Pompeo F, Cole ST, Cerveñansky C, Alzari PM. PknB kinase activity is regulated by phosphorylation in two Thr residues and dephosphorylation by PstP, the cognate phospho-Ser/Thr phosphatase, in Mycobacterium tuberculosis. Mol. Microbiol. 2003;49:1493–1508. doi: 10.1046/j.1365-2958.2003.03657.x. [DOI] [PubMed] [Google Scholar]
- 9.Mieczkowski C, Iavarone AT, Alber T. Auto-activation mechanism of the Mycobacterium tuberculosis PknB receptor Ser/Thr kinase. EMBO J. 2008;27:3186–3197. doi: 10.1038/emboj.2008.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lombana TN, Echols N, Good MC, Thomsen ND, Ng HL, Greenstein AE, Falick AM, King DS, Alber T. Allosteric activation mechanism of the Mycobacterium tuberculosis receptor Ser/Thr protein kinase, PknB. Structure. 2010;18:1667–1677. doi: 10.1016/j.str.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. Trends Cell Biol. 2006;16:443–452. doi: 10.1016/j.tcb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 12.England P, Wehenkel A, Martins S, Hoos S, André-Leroux G, Villarino A, Alzari PM. The FHA-containing protein GarA acts as a phosphorylation-dependent molecular switch in mycobacterial signaling. FEBS Lett. 2009;583:301–307. doi: 10.1016/j.febslet.2008.12.036. [DOI] [PubMed] [Google Scholar]
- 13.Durocher D, Taylor IA, Sarbassova D, Haire LF, Westcott SL, Jackson SP, Smerdon SJ, Yaffe MB. The molecular basis of FHA domain: Phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol. Cell. 2000;6:1169–1182. doi: 10.1016/s1097-2765(00)00114-3. [DOI] [PubMed] [Google Scholar]
- 14.Ding Z, Wang H, Liang X, Morris ER, Gallazzi F, Pandit S, Skolnick J, Walker JC, Van Doren SR. Phosphoprotein and phosphopeptide interactions with the FHA domain from Arabidopsis kinase-associated protein phosphatase. Biochemistry. 2007;46:2684–2696. doi: 10.1021/bi061763n. [DOI] [PubMed] [Google Scholar]
- 15.Pennell S, Westcott S, Ortiz-Lombardia M, Patel D, Li J, Nott TJ, Mohammed D, Buxton RS, Yaffe MB, Verma C, Smerdon SJ. Structural and functional analysis of phosphothreonine-dependent FHA domain interactions. Structure. 2010;18:1587–1595. doi: 10.1016/j.str.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 16.Durocher D, Jackson SP. The FHA domain. FEBS Lett. 2002;513:58–66. doi: 10.1016/s0014-5793(01)03294-x. [DOI] [PubMed] [Google Scholar]
- 17.Singh A, Mai D, Kumar A, Steyn AJ. Dissecting virulence pathways of Mycobacterium tuberculosis through protein–protein association. Proc. Natl. Acad. Sci. U.S.A. 2006;103:11346–11351. doi: 10.1073/pnas.0602817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hett EC, Chao MC, Rubin EJ. Interaction and modulation of two antagonistic cell wall enzymes of mycobacteria. PLoS Pathog. 2010;6:e1001020. doi: 10.1371/journal.ppat.1001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thanky NR, Young DB, Robertson BD. Unusual features of the cell cycle in mycobacteria: Polar-restricted growth and the snapping-model of cell division. Tuberculosis. 2007;87:231–236. doi: 10.1016/j.tube.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Kang CM, Abbott DW, Park ST, Dascher CC, Cantley LC, Husson RN. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: Substrate identification and regulation of cell shape. Genes Dev. 2005;19:1692–1704. doi: 10.1101/gad.1311105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mir M, Asong J, Li X, Cardot J, Boons GJ, Husson RN. The extracytoplasmic domain of the Mycobacterium tuberculosis Ser/Thr kinase PknB binds specific muropeptides and is required for PknB localization. PLoS Pathog. 2011;7:e1002182. doi: 10.1371/journal.ppat.1002182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barthe P, Mukamolova GV, Roumestand C, Cohen-Gonsaud M. The structure of PknB extracellular PASTA domain from mycobacterium tuberculosis suggests a ligand-dependent kinase activation. Structure. 2010;18:606–615. doi: 10.1016/j.str.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Lee M, Hesek D, Shah IM, Oliver AG, Dworkin J, Mobashery S. Synthetic peptidoglycan motifs for germination of bacterial spores. Chembiochem. 2010;11:2525–2529. doi: 10.1002/cbic.201000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah IM, Laaberki MH, Popham DL, Dworkin J. A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell. 2008;135:486–496. doi: 10.1016/j.cell.2008.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alber T. Signaling mechanisms of the Mycobacterium tuberculosis receptor Ser/Thr protein kinases. Curr. Opin. Struct. Biol. 2009;19:650–657. doi: 10.1016/j.sbi.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prisic S, Dankwa S, Schwartz D, Chou MF, Locasale JW, Kang CM, Bemis G, Church GM, Steen H, Husson RN. Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U.S.A. 2010;107:7521–7526. doi: 10.1073/pnas.0913482107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ten Eyck LF, Taylor SS, Kornev AP. Conserved spatial patterns across the protein kinase family. Biochim. Biophys. Acta. 2008;1784:238–243. doi: 10.1016/j.bbapap.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Young TA, Delagoutte B, Endrizzi JA, Falick AM, Alber T. Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat. Struct. Biol. 2003;10:168–174. doi: 10.1038/nsb897. [DOI] [PubMed] [Google Scholar]
- 29.Duran R, Villarino A, Bellinzoni M, Wehenkel A, Fernandez P, Boitel B, Cole ST, Alzari PM, Cerveñansky C. Conserved autophosphorylation pattern in activation loops and juxtamembrane regions of Mycobacterium tuberculosis Ser/Thr protein kinases. Biochem. Biophys. Res. Commun. 2005;333:858–867. doi: 10.1016/j.bbrc.2005.05.173. [DOI] [PubMed] [Google Scholar]
- 30.Roumestand C, Leiba J, Galophe N, Margeat E, Padilla A, Bessin Y, Barthe P, Molle V, Cohen-Gonsaud M. Structural insight into the Mycobacterium tuberculosis Rv0020c protein and its interaction with the PknB kinase. Structure. 2011;19:1525–1534. doi: 10.1016/j.str.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 31.Ali AA, Jukes RM, Pearl LH, Oliver AW. Specific recognition of a multiply phosphorylated motif in the DNA repair scaffold XRCC1 by the FHA domain of human PNK. Nucleic Acids Res. 2009;37:1701–1712. doi: 10.1093/nar/gkn1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grundner C, Gay LM, Alber T. Mycobacterium tuberculosis serine/threonine kinases PknB, PknD, PknE, and PknF phosphorylate multiple FHA domains. Protein Sci. 2005;14:1918–1921. doi: 10.1110/ps.051413405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chao J, Wong D, Zheng X, Poirier V, Bach H, Hmama Z, Av-Gay Y. Protein kinase and phosphatase signaling in Mycobacterium tuberculosis physiology and pathogenesis. Biochim. Biophys. Acta. 2010;1804:620–627. doi: 10.1016/j.bbapap.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 34.Parikh A, Verma SK, Khan S, Prakash B, Nandicoori VK. PknB-mediated phosphorylation of a novel substrate, N-acetylglucosamine-1-phosphate uridyltransferase, modulates its acetyltransferase activity. J. Mol. Biol. 2009;386:451–464. doi: 10.1016/j.jmb.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 35.Molle V, Brown AK, Besra GS, Cozzone AJ, Kremer L. The condensing activities of the Mycobacterium tuberculosis type II fatty acid synthase are differentially regulated by phosphorylation. J. Biol. Chem. 2006;281:30094–30103. doi: 10.1074/jbc.M601691200. [DOI] [PubMed] [Google Scholar]
- 36.Molle V, Gulten G, Vilcheze C, Veyron-Churlet R, Zanella-Cléon I, Sacchettini JC, Jacobs WR, Jr., Kremer L. Phosphorylation of InhA inhibits mycolic acid bio-synthesis and growth of Mycobacterium tuberculosis. Mol. Microbiol. 2010;78:1591–1605. doi: 10.1111/j.1365-2958.2010.07446.x. [DOI] [PubMed] [Google Scholar]
- 37.Sureka K, Hossain T, Mukherjee P, Chatterjee P, Datta P, Kundu M, Basu J. Novel role of phosphorylation-dependent interaction between FtsZ and FipA in mycobacterial cell division. PLoS One. 2010;5:e8590. doi: 10.1371/journal.pone.0008590. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Thakur M, Chakraborti PK. GTPase activity of mycobacterial FtsZ is impaired due to its transphosphorylation by the eukaryotic-type Ser/Thr kinase, PknA. J. Biol. Chem. 2006;281:40107–40113. doi: 10.1074/jbc.M607216200. [DOI] [PubMed] [Google Scholar]
- 39.Park ST, Kang CM, Husson RN. Regulation of the SigH stress response regulon by an essential protein kinase in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 2008;105:13105–13110. doi: 10.1073/pnas.0801143105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jani C, Eoh H, Lee JJ, Hamasha K, Sahana MB, Han JS, Nyayapathy S, Lee JY, Suh JW, Lee SH, Rehse SJ, Crick DC, Kang CM. Regulation of polar peptidoglycan biosynthesis by Wag31 phosphorylation in mycobacteria. BMC Microbiol. 2010;10:327. doi: 10.1186/1471-2180-10-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muñoz-Dorado J, Inouye S, Inouye M. A gene encoding a protein serine/threonine kinase is required for normal development of M. xanthus, a gram-negative bacterium. Cell. 1991;67:995–1006. doi: 10.1016/0092-8674(91)90372-6. [DOI] [PubMed] [Google Scholar]
- 42.Greenstein AE, Grundner C, Echols N, Gay LM, Lombana TN, Miecskowski CA, Pullen KE, Sung PY, Alber T. Structure/function studies of Ser/Thr and Tyr protein phosphorylation in Mycobacterium tuberculosis. J. Mol. Microbiol. Biotechnol. 2005;9:167–181. doi: 10.1159/000089645. [DOI] [PubMed] [Google Scholar]
- 43.Pallen M, Chaudhuri R, Khan A. Bacterial FHA domains: Neglected players in the phospho-threonine signalling game? Trends Microbiol. 2002;10:556–563. doi: 10.1016/s0966-842x(02)02476-9. [DOI] [PubMed] [Google Scholar]
- 44.Busso D, Delagoutte-Busso B, Moras D. Construction of a set Gateway-based destination vectors for high-throughput cloning and expression screening in Escherichia coli. Anal. Biochem. 2005;343:313–321. doi: 10.1016/j.ab.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 45.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 46.Baer CE. thesis. University of California; Berkeley, CA: 2010. Mechanisms of Mycobacterium tuberculosis Serine/Threonine Protein Kinase Activation. [Google Scholar]
- 47.Sharma AK, Ye L, Baer CE, Shanmugasudaram K, Alber T, Alper SL, Rigby AC. Solution structure of the guanine nucleotide-binding STAS domain of SLC26-related SulP protein Rv1739c from Mycobacterium tuberculosis. J. Biol. Chem. 2011;286:8534–8544. doi: 10.1074/jbc.M110.165449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr., editor. Methods in Enzymology. vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 49.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang B-C. Resolution of phase ambiguity in macromolecular crystallography. In: Wyckoff HW, Hirs CHW, Timasheff SN, editors. Methods in Enzymology. vol. 115. Academic Press; New York: 1985. pp. 90–112. [DOI] [PubMed] [Google Scholar]
- 51.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 52.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 53.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stein N. CHAINSAW: A program for mutating pdb files used as templates in molecular replacement. J. Appl. Cryst. 2008;41:641–643. [Google Scholar]
- 56.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Kessel JC, Hatfull GF. Recombineering in Mycobacterium tuberculosis. Nat. Methods. 2007;4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- 58.Pham TT, Jacobs-Sera D, Pedulla ML, Hendrix RW, Hatfull GF. Comparative genomic analysis of mycobacteriophage Tweety: Evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology. 2007;153:2711–2723. doi: 10.1099/mic.0.2007/008904-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papavinasasundaram KG, Anderson C, Brooks PC, Thomas NA, Movahedzadeh F, Jenner PJ, Colston MJ, Davis EO. Slow induction of RecA by DNA damage in Mycobacterium tuberculosis. Microbiology. 2001;147:3271–3279. doi: 10.1099/00221287-147-12-3271. [DOI] [PubMed] [Google Scholar]
- 60.Ehrt S, Guo XV, Hickey CM, Ryou M, Monteleone M, Riley LW, Schnappinger D. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 2005;33:e21. doi: 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parish T, Stoker NG. Electroporation of mycobacteria. Methods Mol. Biol. 1998;101:129–144. doi: 10.1385/0-89603-471-2:129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.