

Abstract
A secreted glycoprotein YKL-40 also named chitinase-3-like-1 is normally expressed by multiple cell types such as macrophages, chondrocytes, and vascular smooth muscle cells. However, a prominently high level of YKL-40 was found in a wide spectrum of human diseases including cancers and chronic inflammatory diseases where it was strongly expressed by cancerous cells and infiltrating macrophages. Here, we summarized recent important findings of YKL-40 derived from cancerous cells and smooth muscle cells during tumor angiogenesis and development. YKL-40 is a potent angiogenic factor capable of stimulating tumor vascularization mediated by endothelial cells and maintaining vascular integrity supported by smooth muscle cells. In addition, YKL-40 induces FAK-MAPK signaling and up-regulates VEGF receptor 2 in endothelial cells; but a neutralizing antibody (mAY) against YKL-40 inhibits its angiogenic activity. While YKL-40 is essential for angiogenesis, little is known about its functional role in tumor-associated macrophage (TAM)-mediated tumor development. Therefore, significant efforts are urgently needed to identify pathophysiological function of YKL-40 in the dynamic interaction between tumor cells and TAMs in the tumor microenvironment, which may offer substantial mechanistic insights into tumor angiogenesis and metastasis, and also point to a therapeutic target for treatment of cancers and other diseases.
Keywords: YKL-40, angiogenesis, VEGF, tumor cells, vascular endothelial cells, tumor-associated macrophages, tumor microenvironment, neutralizing anti-YKL-40 antibody
Introduction
YKL-40 is a 40-kDa secreted glycoprotein discovered as a heparin-binding protein and belongs to the chitinase gene family that binds to chitin-like oligosaccharides (Shackelton et al., 1995; Hu et al., 1996; Fusetti et al., 2003). However, it does not have chitinase/hydrolase activity because of the substitution of an essential glutamic acid with leucine in the chitinase-3-like catalytic domain (Renkema et al., 1998; Fusetti et al., 2003). YKL-40 is normally expressed by a number of different cell types including chondrocytes (Hu et al., 1996), synoviocytes (Nyirkos and Golds, 1990), vascular smooth muscle cells (Shackelton et al., 1995), macrophages (Rehli et al., 1997), and neutrophils (Kzhyshkowska et al., 2007), and it has been recognized as a growth factor capable of stimulating connective tissue cell growth and endothelial cell migration, and inhibiting mammary epithelial cell differentiation (Malinda et al., 1999; De Ceuninck et al., 2001; Recklies et al., 2002; Scully et al., 2011). However, the pathophysiological function of YKL-40 is still not fully understood.
Growing evidence has indicated that expression levels of YKL-40 are elevated in multiple human diseases including type 2 diabetes (Persson et al., 2012), obesity and insulin resistance in children (Kyrgios et al., 2012), Alzheimers' diseases (Perrin et al., 2011), heart failure (Harutyunyan et al., 2012), and other cardiovascular disorders (Kjaergaard et al., 2010). In addition, elevated YKL-40 was found in a vast array of inflammatory diseases that contain bacterial infections (Kronborg et al., 2002), rheumatoid arthritis (Nielsen et al., 2011), osteoarthritis (Volck et al., 2001), hepatic fibrosis (Pizano-Martinez et al., 2011), and hepatitis (Johansen et al., 2000; Fontana et al., 2010), asthma and chronic obstructive pulmonary diseases (Park et al., 2010), neuroinflammation (Bonneh-Barkay et al., 2010), and bowel lesion (Vind et al., 2003). In the chronic inflammatory diseases, YKL-40 is appreciated to mediate infiltration, differentiation, and maturation of macrophages, the primary leukocytes in response to inflammation (Boot et al., 1995; Rehli et al., 1997; Renkema et al., 1998; Rehli et al., 2003). The cytokines colony-stimulating factor-1 and granulocyte macrophage colony-stimulating factor, essential for macrophage recruitment, displayed the ability to induce 180–200 fold higher levels of YKL-40 mRNA transcripts in macrophages, thus rendering infiltrating macrophages mature (Hashimoto et al., 1999; Suzuki et al., 2000). Studies with YKL-40 deficient mice offered strong evidence supporting the role of YKL-40 in macrophage activity, as these mice exhibited markedly diminished antigen-induced Th2 inflammation and impaired macrophage activation and differentiation (Lee et al., 2009).
Over the past decades, multiple independent studies have demonstrated that high serum levels of YKL-40 are correlated with metastasis and poor survival in a variety of human carcinomas such as breast cancer (Jensen et al., 2003), colorectal cancer (Cintin et al., 1999), ovarian cancer (Hogdall et al., 2003), leukemia (Bergmann et al., 2005), lymphoma (Hottinger et al., 2011), and glioblastoma (GBM) (Pelloski et al., 2005), suggesting that serum levels of YKL-40 serve as a diagnostic and prognostic cancer biomarker. YKL-40 is expressed by both tumor cells and their surrounding tumor infiltrating macrophages also named tumor-associated macrophages (TAM) that produce various tumor-promoting factors including angiogenic factors [vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), basic fibroblastic growth factor (bFGF), platelet-derived growth factor (PDGF)] (Chong et al., 1999; Ganapathy et al., 2010), cytokines (IL-1, IL-6) (Wang et al., 2009; Pini et al., 2012), and chemokines (CCL-2, CCL-18, CXCL-12) (Dubinett et al., 2010; Chen et al., 2011a,b,c; Fridlender et al., 2011; Boimel et al., 2012). Although the overall pathological role and molecular mechanisms of YKL-40 in tumorigenesis remain to be established, an angiogenic feature has been reported to regulate tumor development in breast cancer, colon cancer, and GBM (Shao et al., 2009; Francescone et al., 2011; Kawada et al., 2012). Here, this review primarily focused on the angiogenic signature of YKL-40 derived from tumor cells and smooth muscle cells, as a model is illustrated in Figure 1, while a potential distinct role of YKL-40 in TAM-mediated tumor development warrants further investigation.
Figure 1.

A scheme for YKL-40-induced tumor angiogenesis. YKL-40 secreted from tumor cells stimulates vascular endothelial cell activation to induce tumor angiogenesis through membrane receptor coupling of syndecan-1 with integrin. YKL-40 regulates VEGF in tumor cells and both may synergistically promote endothelial cell angiogenesis. YKL-40 derived from smooth muscle cells also controls vessel stability and permeability via inducing association of N- and VE-cad with β-catenin (β-cate) expressed by smooth muscle cells and endothelial cells, respectively. TAMs participate in the vascular development probably through YKL-40, which warrants further investigation. Green balls indicate secreted YKL-40 and brown triangles represent VEGF.
An angiogenic signature of YKL-40
Due to lack of its chitinase activity, the pathological role of YKL-40 in cancer development has not been substantially explored yet. Gp38k, a YKL-40 homolog, was found to induce endothelial cell migration, indicative of angiogenic activity (Nishikawa and Millis, 2003). To evaluate if YKL-40 possesses the same angiogenic activity in cancer, a breast cancer line MDA-MB-231 and colon cancer lines HCT-116 and SW480 were engineered to express ectopic YKL-40 (Shao et al., 2009; Kawada et al., 2012). Xenotransplantation of YKL-40-expressing tumor cells gave rise to 4–8 fold larger tumors than ones formed from their corresponding control cells, while acquired expression of YKL-40 did not predispose these cells toward increased proliferation in the cultured condition. Immunohistochemical studies indicated that levels of blood vasculature formed in YKL-40-expressing MDA-MB-231, HCT-116, and SW480 tumors were 1.8–2.0 fold greater than those in control tumors, suggesting that YKL-40 acts as an angiogenic factor to promote vessel formation and tumor growth. Such an angiogenic capability of YKL-40 was also validated in GBM (Francescone et al., 2011), the most lethal primary brain tumor characterized by vigorous vascularization (Wen and Kesari, 2008). For example, YKL-40-directed gene knockdown in GBM-derived U87 cells notably suppressed tumor angiogenesis, as the vessel density of YKL-40 shRNA tumors was decreased to 44% of vasculature relative to control tumors and tumor volume was accordingly reduced to approximately 30% of control counterparts (Shao et al., 2009). All of these multiple in vivo approaches demonstrate the angiogenic signature of YKL-40 in the tumor development, based on these xenografts carrying different levels of YKL-40. However, this angiogenic phenotype may also involve tumor-promoting function of host-derived cells in the tumor microenvironment, as increased infiltrating macrophages were observed in the YKL-40-expressing tumors, but not in the control tumors (Kawada et al., 2012). It will be interesting to know if these macrophages also increase to produce YKL-40 that enhances the angiogenesis induced by tumor-derived YKL-40.
To monitor its direct effects on vascular endothelial cells, conditioned media derived from both MDA-MB-231 and HCT-116 cells ectopically expressing YKL-40 or vector were introduced to human microvascular endothelial cells (HMVEC) and tested for endothelial cell angiogenic activity in vitro. Analogous to the findings in animals, both YKL-40-producing tumor cells induced endothelial cell migration and tube formation (Shao et al., 2009). Likewise, SW480 over-expressing YKL-40 also enhanced migration and tube formation of human umbilical vein endothelial cells by 1.4–2 fold greater than the control cells expressing vector (Kawada et al., 2012). YKL-40 gene knockdown abrogated these angiogenic activities. In addition, conditioned medium of U87 cells expressing YKL-40 shRNA inhibited the angiogenic activities of HMVEC vs. control cell medium (Francescone et al., 2011). To further support these in vitro data and firmly establish the angiogenic signature for YKL-40, recombinant YKL-40 was created and characterized for the angiogenic activity. YKL-40 stimulated endothelial cell migration and tube formation approximately 3–4 fold greater than control cells, the angiogenic capability identical to VEGF, one of the most potent angiogenic factors (Shao et al., 2009). It was noted that most of these cultured concentrations of YKL-40 between 100 and 200 ng/ml were based on serum levels of YKL-40 observed in cancer patients (Jensen et al., 2003; Johansen et al., 2003). However, it is unclear if these concentrations indeed reflect YKL-40 levels in the local tumor, because the serum levels are probably derived from multiple organs and also involve the dilution effect. Therefore, a cautious interpretation from these cultured systems should be considered in stimulating YKL-40's action in vivo. Nevertheless, all these animal and cultured data suggest that YKL-40 acts as an angiogenic factor to trigger tumor vascular development.
Relationship between YKL-40 and VEGF
In the tumor microenvironment, a significant amount of angiogenic factors are secreted from tumor cells and activate adjacent vascular endothelial cells to induce angiogenic responses by means of a paracrine loop (Hanahan and Weinberg, 2010). YKL-40 and VEGF are believed to be mainly derived from tumor cells and both display strong angiogenic activities in tumor development, but their regulatory relationship has not been revealed until recently. YKL-40-induced endothelial cell angiogenic responses in culture were VEGF-independent, as an anti-VEGF neutralizing antibody failed to impede YKL-40-induced migration and tube formation of HMVECs (Shao et al., 2009). This data suggests that YKL-40 and VEGF individually promote endothelial cell angiogenesis. U87 brain tumor cells were found to express high levels of YKL-40 and VEGF (Francescone et al., 2011). When YKL-40 expression was inhibited via small hairpin RNA (shRNA), a reduction of VEGF was subsequently obtained in these tumor cells, indicative of a regulatory role of YKL-40 in VEGF production. In light of a potential similar role of VEGF in YKL-40 expression, transient neutralization of VEGF using a neutralizing anti-VEGF antibody for 24 h did not have impacts in YKL-40 production. Interestingly, inhibition of VEGF for 1 week noticeably induced expression of YKL-40, the unexpected event identical to the documented evidence using VEGF shRNA in U87 cells (Saidi et al., 2008). These results imply that VEGF does not regulate YKL-40, but a long-term blockade of VEGF may result in angiogenic compensative activities of tumor cells by inducing YKL-40. It is most likely that a long course of the stress caused by blockade of one growth factor and/or angiogenic factor commits the cells to induce expression of other potent angiogenic factors in order for cell survival and function. It was noted that these tumor cells such as brain tumor cells express a high level of angiogenic factors able to promote vascular development (Junker et al., 2005a,b; Francescone et al., 2011). This phenomenon was identically observed in a number of tumor models treated chronically with a single anti-angiogenic drug, the event known as angiogenic rebound (see below). However, it needs to determine if this angiogenic switch is unique for highly angiogenic tumors, but not for other non-angiogenic tumors.
Apart from their relationship defined earlier in cultured cancer cell lines, studies on human cancers also suggest the similar association of YKL-40 with VEGF in tumor angiogenesis. Tumor specimens from 12 cases of patients with GBM were used to test the relationship between YKL-40 and VEGF (Francescone et al., 2011). Expression of YKL-40 and VEGF in tumor samples displayed a trend toward positive correlation (p = 0.062), but a larger sample pool sufficient to establish their relationship is required. In context with the findings in vitro, all the evidence suggests that YKL-40 regulates VEGF in tumor cells and both may exert a synergistic impact in tumor vascularization (Figure 1).
A chronic course of angiogenic blockade in either YKL-40 or VEGF may not receive a full elimination of tumor angiogenesis; instead, an unexpected compensation by the other factor may lead to an opposite outcome including resistance to the single-factor treatment and angiogenic rebound. Indeed, the theme of this anti-angiogenic bypass upon a chronic single treatment has been supported by a number of strong evidence documented in pre-clinical and clinical trials. For instance, individual anti-angiogenic treatment with bevacizumab (anti-VEGF antibody, Avastin), DC101 (anti-VEGF receptor antibody), or sunitinib (anti-VEGF receptor kinase inhibitor) can elicit vascular rebound and tumor cell invasiveness and metastasis in several animal models (Casanovas et al., 2005; Ebos et al., 2009; Paez-Ribes et al., 2009). In clinical trials, the benefit of anti-angiogenic agents (e.g., sunitinib, bevacizumab) appears to be transitory in the treatment of several types of advanced cancers, as drug resistance, tumor regrowth, and extensive vascular recovery rapidly develop, once the therapy is terminated (Bergers and Hanahan, 2008; Burstein et al., 2008; Verhoeff et al., 2009; Wick et al., 2010). In addition, it is noteworthy that bevacizumab has been removed by the Food and Drug Administration from monotreatment of metastatic breast cancers, based on insufficient amelioration of patient overall survival. While it is emerging that a monotherapy against a single factor could unexpectedly result in conflicting outcomes, it is still enigmatic if YKL-40 acts as a major factor to contribute to the angiogenic rebound in these patients that are treated with one drug such as bevacizumab. Nevertheless, to prevent either anti-VEGF or possible anti-YKL-40 resistance, it should be taken into account for a combined regimen with anti-VEGF and anti-YKL-40 therapies in cancer patients.
Molecular mechanisms of YKL-40 in endothelial cells and tumor cells
Although membrane receptors specific for YKL-40 binding remain to be identified, heparin-binding affinity of YKL-40 seems to be essential for its activity, resembling the heparin-binding property of other secreted proteins such as extracellular matrix protein vitronectin and angiogenic factors bFGF and VEGF (Bernfield et al., 1999; Beauvais et al., 2004). The heparin binding affinity is at least approximately 100-fold lower (disassociation constant Kd ~10−8 – 10−9 M) than their specific receptor binding (Kd ~10−11 – 10−12 M), but this binding can facilitate their adjacent specific receptor binding (Baird et al., 1988; Park et al., 2000; Prince et al., 2010). Syndecan-1, a transmembrane receptor, is the major source of cell surface heparan sulfate (HS). There is compelling evidence demonstrating that endowed with the HS chain on its ectodomains, syndecan-1 acts as a matrix co-receptor with adjacent membrane-bound receptors such as integrins to mediate cell adhesion and/or spreading (McQuade et al., 2006). This co-membrane receptor model of syndecan-1 with integrin was found to play an indispensable role in mediating YKL-40-induced angiogenic responses (Shao et al., 2009). YKL-40 can induce coupling of syndecan-1 with integrin αvβ3 through binding to HS and then activate intracellular signaling effectors focal adhesion kinase (FAK861) and mitogen-activated protein kinase (MAPK) that regulate endothelial cell adhesion and motility (Figure 2). In addition, treatment of HMVEC with recombinant YKL-40 increases protein expression and active form of both VEGF receptor 2 (Flk-1) and intracellular extracellular signal-regulated kinase (Erk 1 and 2) that in turn enhance angiogenic signaling pathways (Faibish et al., 2011; Lee et al., 2011). Furthermore, an additional phosphoinositide 3-kinase-protein kinase B (PI3K-AKT) pathway responsible for YKL-40's action in vascular endothelial cells is proposed, but no date has confirmed it yet.
Figure 2.
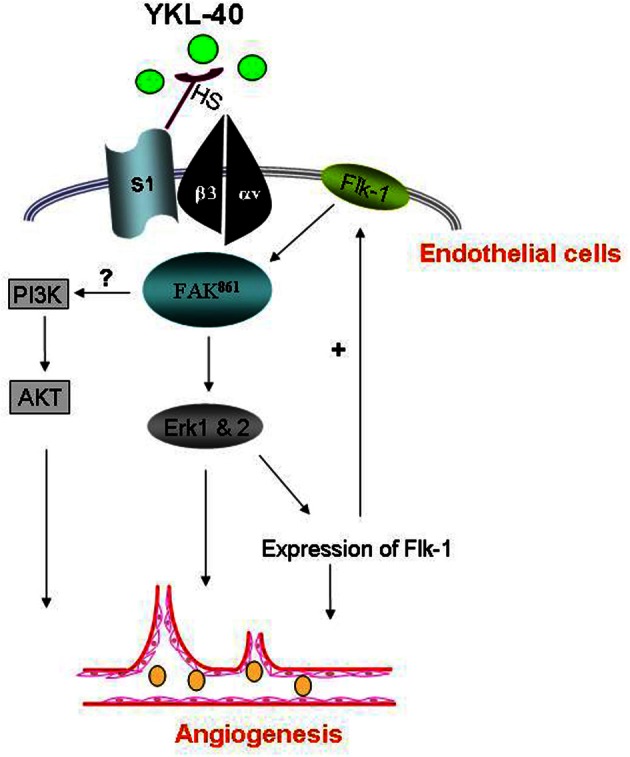
YKL-40 induces angiogenic signaling in endothelial cells. YKL-40 induces the coordination of syndecan-1 (S1) and integrin αvβ3 through binding heparan sulfate chains (HS) of S1 on cell surface. The intracellular signaling pathway includes pFAK861 and downstream MAP kinase Erk 1 and 2, leading to angiogenic responses and angiogenic gene expression as well (e.g., Flk-1). Flk-1 up-regulation in turns activates the signal transduction cascade, constituting a positive feedback loop to enhance angiogenic responses. Elevated Flk-1 may also sensitize angiogenic responses to VEGF. An additional PI3K-AKT pathway participating in YKL-40-induced angiogenesis in endothelial cells warrants further investigation.
The signaling activation by YKL-40 in endothelial cells was similarly identified in the tumor line U87 cells, in which YKL-40 induces strong association of syndecan-1 with different integrin αvβ5 and downstream activation of FAK397 and Erk 1 and 2, thus targeting VEGF expression that evokes endothelial cell angiogenesis (Francescone et al., 2011). In apoptotic responses, YKL-40 was found to prevent U87 cell death from γ-irradiation through activation of PI3K-AKT pathways, the signal transduction identical to the cascade that mediates YKL-40-induced mitogenesis in connective tissue cells (Recklies et al., 2002). In SW480 cells, YKL-40 also regulated MAPK including Erk 1, 2, and JNK that induce expression of IL-8 and monocyte chemoattractant protein-1, facilitating angiogenesis (Kawada et al., 2012). Therefore, YKL-40 acts as an angiogenic factor and a growth factor to induce distinct signaling cascades in endothelial cell angiogenesis and tumor cell survival, respectively.
Activity of YKL-40 in vascular smooth muscle cells
As discussed earlier, neo-vascular development is mainly ascribed to the activation of vascular endothelial cells, the primary component of blood vessels. However, a functional role of YKL-40 in smooth muscle cells or vascular pericytes, another subset of vascular cell populations that support vessel integrity and stability, is poorly understood, even those cells express YKL-40. Using a smooth muscle cell model named glioblastoma stem-differentiated cells (GSDC), we found that YKL-40 enhances both GSDC and HMVEC contacts, restricts vascular leakage, and stabilizes vascular networks (Francescone et al., 2013). Furthermore, the vascular sprouting and stability mediated by smooth muscle-like cells are dependent on signaling activation induced by YKL-40, which includes interaction of membrane adhesion molecules neural cadherin (N-cad) with β-catenin and downstream intracellular cytoskeleton smooth muscle alpha actin (Figure 1). Likewise, adhesion and permeability of HMVECs regulated by YKL-40 rely on the interaction of vascular endothelial cadherin (VE-cad) with β-catenin and downstream effector actin. YKL-40 gene knockdown in GSDCs leads to disruption of association of VE-cad with β-catenin and increases endothelial cell permeability via a paracrine manner. In GSDCs, YKL-40 shRNA also inhibits interaction of N-cad with β-catenin and reduces GSDC-mediated vessel stability, suggesting that both vascular cell populations regulated by YKL-40 coordinately contribute to the angiogenesis. Furthermore, xenotransplantation of GSDCs expressing YKL-40 shRNA in mice gives rise to impaired blood vessel integrity with collapsed vessel lumens and diminished smooth muscle-like cell coverage; whereas control GSDCs develop extensive and stable blood vessels covered with more smooth muscle-like cells, highlighting a unique role of YKL-40 derived from smooth muscle-like cells in the maintenance of vascular permeability, stability, and angiogenesis. Although the interaction between cadherins and catenin is vital for YKL-40's function, it is still unknown whether or not this interaction is dependent on pre-activation of syndecan-1 that is for YKL-40 binding on the membrane.
Functional blockade of YKL-40—a potential tool for anti-angiogenic therapy
A neutralizing anti-YKL-40 antibody (named mAY) from mice immunized against recombinant YKL-40 was recently established (Faibish et al., 2011). HMVEC migration and tube formation induced by YKL-40 in a dose-dependent fashion were markedly suppressed by mAY. mAY was also found to abolish YKL-40-induced activation of Flk-1 and intracellular signaling MAP kinase Erk 1 and Erk 2 in HMVEC. In addition, mAY facilitated death responses of the U87 glioblastoma cell line to γ-irradiation through decreased expression of pAKT and AKT (Faibish et al., 2011). Consistent with these data from cultured cells, tumor angiogenesis developed from xenografted U87 cells expressing YKL-40 was abrogated in mice treated with mAY, whereas vigorous angiogenesis was observed in mIgG-treated control tumors. Similar studies focusing on YKL-40 neutralization in the angiogenesis of colon cancer unveiled the identical importance for the anti-YKL-40 antibody (Kawada et al., 2012). Therefore, the evidence from such pre-clinical trials has hold therapeutic promise for formulating a humanized anti-YKL-40 antibody in the treatment of cancer patients as well as other possible diseases.
Chitin can bind to both chitinases that have hydrolase activity and chitinase-like proteins that lack the enzymatic activity such as YKL-40 (Lee et al., 2008). Size difference of chitin exhibits distinct capabilities of inducing host immune responses, as small particles (<10 μm) can induce TH1 type immune responses whereas large ones (>50 μm) activate TH2 type responses (Shibata et al., 1997; Lee et al., 2008). Recently, Iragavarapu-Charyulu's group has utilized small chitin to test a hypothesis that saturation of YKL-40's binding can alleviate its direct tumor-promoting effects on tumors (Libreros et al., 2012). Chitin (1–10 μm) has strong binding affinity with YKL-40 and is associated with activation of M1 type macrophages. This binding between chitin and YKL-40 may induce immune response shift from pro-tumorigenic TH2 type (M2 macrophage activation) to anti-tumorigenic TH1 type (M1 macrophage activation). They found that the treatment of mammary tumor-bearing mice with chitin significantly decreased serum levels and splenic macrophages of YKL-40, CXCL2, and MMP-9, thereof impeding lung metastasis. It remains to be determined if the reduction of YKL-40 expression and subsequent inhibition of tumor progression are different from treatment with large chitin. Other alternative possible approaches that block YKL-40 signaling pathways may also suffice to prevent YKL-40 activity or be synergistic in conjunction therapies with YKL-40-directed inhibitors. Nonetheless, the recent multiple animal approaches to blocking YKL-40 function offer therapeutic value potential for modalities of clinical patients.
YKL-40 in human tumor angiogenesis
A multitude of clinical studies have revealed that serum levels of YKL-40 were elevated in patients with a series of carcinomas including breast (Jensen et al., 2003), colorectum (Cintin et al., 1999), ovary (Hogdall et al., 2003), prostate (Kucur et al., 2008), brain (Pelloski et al., 2005), and blood (Bergmann et al., 2005). These increased levels were correlated with poorer survival of cancer patients (Cintin et al., 1999, 2002; Hogdall et al., 2003; Jensen et al., 2003; Johansen et al., 2003; Bergmann et al., 2005; Pelloski et al., 2005), suggesting that serum levels of YKL-40 serve as a prognostic cancer biomarker (Johansen et al., 2009).
While amounting evidence was documented in the study of serum levels of YKL-40, there is relatively limited evidence focusing on YKL-40 expression in cancers, particularly for its association with angiogenesis. Thirty-eight cases of breast infiltrating ductal carcinomas were surveyed for relationship of YKL-40 with vessel formation using immunohistochemistry of CD34, a vascular endothelial cell marker (Shao et al., 2009). Of those 38 cancers, 23.7% (9 cases) contained high expression levels of YKL-40 and 23.7% (9 cases) displayed medium levels of YKL-40; whereas 52.6% (20 cases) were negative or low. These three groups with different expression levels of YKL-40 were found to be significantly correlated with different degrees of vascularization with CD34-positive vessels in tumor sections (p = 0.006), in which the blood vessel density of the two groups that demonstrated high and medium levels of YKL-40 were 2.1 and 1.6-fold greater than the group expressing low YKL-40, respectively. Consistent with this finding, of 61 colorectal cancer samples, 37 and 24 cases expressing strong YKL-40 and weak YKL-40 exhibited 2.0 and 1.6-fold higher microvessel density than did 12 normal subjects, respectively (Kawada et al., 2012). In addition, studying 11 cases of patients with GBM revealed that the higher the YKL-40 expression, the more extensive the vessels appeared to be (Francescone et al., 2011). All of the evidence demonstrates that YKL-40 expression in cancer is associated with vascular network development, underscoring the angiogenic property of YKL-40 identified in pre-clinical (cultured cells and xenografted animal models) and clinical studies.
In the study of YKL-40 expression and clinical outcomes, several independent studies with large breast cancer cohorts from different laboratories including ours demonstrate that YKL-40 expressed by breast cancer is associated with estrogen receptor (ER−), progesterone receptor (PR−), and human epidermal growth factor receptor 2 (Her2/neu) (Kim et al., 2007; Roslind et al., 2007b; Shao et al., 2011). Unexpectedly, cancer tissue expression, contrary to its levels in the blood, was not correlated with patient overall survival or disease-free survival in 8-year follow-up studies (Shao et al., 2011). This finding was reinforced by the others surveying 630 breast cancer patients (Roslind et al., 2007b). Interestingly, strong expression levels of YKL-40 were identified in TAMs in both breast cancer and lung cancer, as these TAMs surrounding tumor cells co-expressed YKL-40 and CD68, a marker of macrophages (Junker et al., 2005a,b; Roslind et al., 2007a; Stearman et al., 2008). It is well established that infiltrating macrophages play an essential role for angiogenesis in both inflammatory diseases and tumor development, because increased infiltration of macrophages leads to accumulation of multiple growth factors (TGF-β, EGF, bFGF, VEGF, and PDGF) that modulate tissue repair and angiogenesis (Chong et al., 1999; Ganapathy et al., 2010). Furthermore, increased macrophage density in cancers correlates with tumor angiogenesis and poorer patient survival (Leek et al., 1996, 2000; Bingle et al., 2002; Tsutsui et al., 2005). However, it remains to be clarified if the expression of YKL-40 by TAMs is associated with cancer metastasis and patient survival. Validating their relationship may provide a key role of TAMs in the contribution to cancer malignancy.
Unanswered questions
It has been established that chronic inflammation is a key component of cancer development and metastasis (Coussens and Werb, 2002). TAMs, the primary infiltrating leukocytes, act as a core mediator to regulate inflammatory responses that exacerbate the pathogenesis of cancers (Coussens and Werb, 2002; Pollard, 2004; Lewis and Pollard, 2006). Although tumor-derived YKL-40 was reported to be associated with macrophage recruitment and angiogenesis in colorectal cancer (Kawada et al., 2012), we still lack sufficient knowledge regarding the functional role and molecular mechanisms of YKL-40 in TAM-mediated tumorigenesis. TAMs have the ability to render tumor cells invasive through up-regulation of multiple inflammatory factors such as cytokines, growth factors, chemokines, and metalloproteinases (MMPs). It is noted that YKL-40 is essential for macrophage differentiation and maturation (Rehli et al., 1997, 2003). Thus, it is intriguing to interrogate if the inflammatory responses mediated by these factors in the tumor microenvironment are dependent on TAM-derived YKL-40. For example, little is known if YKL-40 up-regulates inflammatory cytokines in TAMs, even though YKL-40 is recognized as an inflammatory factor and can induce IL-8 from tumor cells (Rathcke and Vestergaard, 2006; Qin et al., 2007; Kawada et al., 2012). YKL-40 can induce VEGF in tumor cells as discussed earlier, but the similar relationship in TAMs remains to be established. In addition, it is still unclear regarding the molecular mechanisms by which YKL-40 regulates macrophage recruitment, differentiation, and maturation. Identification of potential signaling mediators in TAMs may provide alternative approaches to block activities of TAMs, thus impeding tumor progression. Besides TAMs, other YKL-40-producing cells surrounding tumor cells and TAMs (e.g., neutrophils) should be not neglected in the tumor microenvironment, as these cell populations likely coordinate with tumor cells, TAMs, and vessel cells to facilitate tumor cell ectopic dissemination. The function of cell-associated YKL-40 in tumor may be different from free form of YKL-40 in the blood, because we currently do not know receptors or ligands for YKL-40 binding. This may also explain the difference between serum levels and cancer cell levels of YKL-40 in association with tumor malignancy. Thus, characterization of their relationship will aid in establishing a new therapeutic target for treatment.
YKL-40 harbors chitinase-3-like catalytic domains, but does not possess chitinase activities. Therefore, its functional domain(s) are still unclear. Chen at al., recently reported that a chitin-binding motif located between 325 and 339 amino acid residues at the C terminus of YKL-40 is critical for YKL-40 inflammatory activities including AKT-mediated cytokine production (IL-8 and TNF-α) in colonic epithelial cells (Chen et al., 2011a,b). This motif may be also vital for other activities of YKL-40 such as angiogenic function, tumor cell survival, and inflammatory responses of TAMs, all of which need to be proven in individual cell types. Moreover, if a single amino acid residue within this motif is found to mainly contribute to YKL-40's function, this could help screen new therapeutic agents aiming at this specific element. Finally, one of the most challenging research approaches is to identify the membrane receptor(s) specific for YKL-40 binding, which would not only provide new mechanistic insights into YKL-40's action, but also establish proof-of-principle for offering a novel mechanistically-directed target in treatment of a wide spectrum of cancers as well as other types of diseases. Therefore, gaining such important knowledge about pathological activities and molecular mechanisms of YKL-40 will unequivocally pave a fundamental way toward an advanced platform able to notably improve the current diagnosis, prognosis, and therapy of multiple human diseases that are associated with increased levels of YKL-40.
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NCI R01 CA120659, DOD W81XWH-06-1-0563, and Collaborative Biomedical Research Program, Baystate Medical Center/University of Massachusetts at Amherst.
Glossary
Abbreviations
- VEGF
vascular endothelial growth factor
- Flk-1
VEGF receptor 2
- PDGF
platelet-derived growth factor
- EGF
epidermal growth factor
- bFGF
basic fibroblastic growth factor
- mAY
neutralizing anti-YKL-40 antibody
- FAK
focal adhesion kinase
- MAPK
mitogen-activated protein kinase
- Erk
Extracellular signal-regulated kinase
- PI3K
phosphoinositide 3-kinase
- AKT
protein kinase B
- JNK
c-Jun N-terminal kinase
- TAM
tumor-associated microphage
- GBM
glioblastoma
- GSDC
glioblastoma stem-differentiated cells
- HMVEC
human microvascular endothelial cells
- VE-cad
vascular endothelial cadherin
- N-cad
neural cadherin
- ER
estrogen receptor
- PR
progesterone receptor
- Her2/neu
human epidermal growth factor receptor 2
- MMP
metalloproteinase
- shRNA
mall hairpin
- HS
heparan sulfate.
References
- Baird A., Schubert D., Ling N., Guillemin R. (1988). Receptor- and heparin-binding domains of basic fibroblast growth factor. Proc. Natl. Acad. Sci. U.S.A. 85, 2324–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvais D. M., Burbach B. J., Rapraeger A. C. (2004). The syndecan-1 ectodomain regulates alphavbeta3 integrin activity in human mammary carcinoma cells. J. Cell Biol. 167, 171–181 10.1083/jcb.200404171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G., Hanahan D. (2008). Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603 10.1038/nrc2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann O. J., Johansen J. S., Klausen T. W., Mylin A. K., Kristensen J. S., Kjeldsen E., et al. (2005). High serum concentration of YKL-40 is associated with short survival in patients with acute myeloid leukemia. Clin. Cancer Res. 11, 8644–8652 10.1158/1078-0432.CCR-05-1317 [DOI] [PubMed] [Google Scholar]
- Bernfield M., Gotte M., Park P. W., Reizes O., Fitzgerald M. L., Lincecum J., et al. (1999). Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 68, 729–777 10.1146/annurev.biochem.68.1.729 [DOI] [PubMed] [Google Scholar]
- Bingle L., Brown N. J., Lewis C. E. (2002). The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J. Pathol. 196, 254–265 10.1002/path.1027 [DOI] [PubMed] [Google Scholar]
- Boimel P. J., Smirnova T., Zhou Z. N., Wyckoff J., Park H. I., Coniglio S. J., et al. (2012). Contribution of CXCL12 secretion to invasion of breast cancer cells. Breast Cancer Res. 14, R23 10.1186/bcr3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneh-Barkay D., Wang G., Starkey A., Hamilton R. L., Wiley C. A. (2010). In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J. Neuroinflamm. 7:34 10.1186/1742-2094-7-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boot R. G., Renkema G. H., Strijland A., van Zonneveld A. J., Aerts J. M. (1995). Cloning of a cDNA encoding chitotriosidase, a human chitinase produced by macrophages. J. Biol. Chem. 270, 26252–26256 10.1074/jbc.270.44.26252 [DOI] [PubMed] [Google Scholar]
- Burstein H. J., Elias A. D., Rugo H. S., Cobleigh M. A., Wolff A. C., Eisenberg P. D., et al. (2008). Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J. Clin. Oncol. 26, 1810–1816 10.1200/JCO.2007.14.5375 [DOI] [PubMed] [Google Scholar]
- Casanovas O., Hicklin D. J., Bergers G., Hanahan D. (2005). Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. In Cancer Cell, pp. 299–309 10.1016/j.ccr.2005.09.005 [DOI] [PubMed] [Google Scholar]
- Chen C. C., Llado V., Eurich K., Tran H. T., Mizoguchi E. (2011a). Carbohydrate-binding motif in chitinase 3-like 1 (CHI3L1/YKL-40) specifically activates Akt signaling pathway in colonic epithelial cells. Clin. Immunol. 140, 268–275 10.1016/j.clim.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. C., Pekow J., Llado V., Kanneganti M., Lau C. W., Mizoguchi A., et al. (2011b). Chitinase 3-like-1 expression in colonic epithelial cells as a potentially novel marker for colitis-associated neoplasia. Am. J. Pathol. 179, 1494–1503 10.1016/j.ajpath.2011.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Yao Y., Gong C., Yu F., Su S., Chen J., et al. (2011c). CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 19, 541–555 10.1016/j.ccr.2011.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H., Vodovotz Y., Cox G. W., Barcellos-Hoff M. H. (1999). Immunocytochemical localization of latent transforming growth factor-beta1 activation by stimulated macrophages. J. Cell. Physiol. 178, 275–283 [DOI] [PubMed] [Google Scholar]
- Cintin C., Johansen J. S., Christensen I. J., Price P. A., Sorensen S., Nielsen H. J. (1999). Serum YKL-40 and colorectal cancer. Br. J. Cancer 79, 1494–1499 10.1038/sj.bjc.6690238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cintin C., Johansen J. S., Christensen I. J., Price P. A., Sorensen S., Nielsen H. J. (2002). High serum YKL-40 level after surgery for colorectal carcinoma is related to short survival. Cancer 95, 267–274 10.1002/cncr.10644 [DOI] [PubMed] [Google Scholar]
- Coussens L. M., Werb Z. (2002). Inflammation and cancer.[see comment]. Nature 420, 860–867 10.1038/nature01322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ceuninck F., Gaufillier S., Bonnaud A., Sabatini M., Lesur C., Pastoureau P. (2001). YKL-40 (cartilage gp-39) induces proliferative events in cultured chondrocytes and synoviocytes and increases glycosaminoglycan synthesis in chondrocytes. Biochem. Biophys. Res. Commun. 285, 926–931 10.1006/bbrc.2001.5253 [DOI] [PubMed] [Google Scholar]
- Dubinett S. M., Lee J. M., Sharma S., Mule J. J. (2010). Chemokines: can effector cells be redirected to the site of the tumor? Cancer J. 16, 325–335 10.1097/PPO.0b013e3181eb33bc [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebos J. M., Lee C. R., Cruz-Munoz W., Bjarnason G. A., Christensen J. G., Kerbel R. S. (2009). Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis.[see comment]. Cancer Cell 15, 232–239 10.1016/j.ccr.2009.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faibish M., Francescone R., Bentley B., Yan W., Shao R. (2011). A YKL-40–neutralizing antibody blocks tumor angiogenesis and progression: a potential therapeutic agent in cancers. Mol. Cancer Therap. 10, 742–751 10.1158/1535-7163.MCT-10-0868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana R. J., Dienstag J. L., Bonkovsky H. L., Sterling R. K., Naishadham D., Goodman Z. D., et al. (2010). Serum fibrosis markers are associated with liver disease progression in non-responder patients with chronic hepatitis C. Gut 59, 1401–1409 10.1136/gut.2010.207423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francescone R. A., Ngernyuang N., Yan W., Bentley B., Shao R. (2013). Tumor-derived mural-like cells coordinate with endothelial cells: role of YKL-40 in mural cell-mediated angiogenesis. Oncogene. [Epub ahead of print]. 10.1038/onc.2013.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francescone R. A., Scully S., Faibish M., Taylor S. L., Oh D., Moral L., et al. (2011). Role of YKL-40 in the angiogenesis, radioresistance, and progression of glioblastoma. J. Biol. Chem. 286, 15332–15343 10.1074/jbc.M110.212514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlender Z. G., Kapoor V., Buchlis G., Cheng G., Sun J., Wang L. C., et al. (2011). Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am. J. Res. Cell Mol. Biol. 44, 230–237 10.1165/rcmb.2010-0080OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusetti F., Pijning T., Kalk K. H., Bos E., Dijkstra B. W. (2003). Crystal structure and carbohydrate-binding properties of the human cartilage glycoprotein-39. J. Biol. Chem. 278, 37753–37760 pp.37753-60 10.1074/jbc.M303137200 [DOI] [PubMed] [Google Scholar]
- Ganapathy V., Ge R., Grazioli A., Xie W., Banach-Petrosky W., Kang Y., et al. (2010). Targeting the Transforming Growth Factor-beta pathway inhibits human basal-like breast cancer metastasis. Mol. Cancer 9:122 10.1186/1476-4598-9-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2010). Hallmarks of cancer: the next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Harutyunyan M., Christiansen M., Johansen J. S., Kober L., Torp-Petersen C., Kastrup J. (2012). The inflammatory biomarker YKL-40 as a new prognostic marker for all-cause mortality in patients with heart failure. Immunobiology 217, 652–656 10.1016/j.imbio.2011.11.003 [DOI] [PubMed] [Google Scholar]
- Hashimoto S., Suzuki T., Dong H. Y., Yamazaki N., Matsushima K. (1999). Serial analysis of gene expression in human monocytes and macrophages. Blood 94, 837–844 [PubMed] [Google Scholar]
- Hogdall E. V., Johansen J. S., Kjaer S. K., Price P. A., Christensen L., Blaakaer J., et al. (2003). High plasma YKL-40 level in patients with ovarian cancer stage III is related to shorter survival. Oncol. Rep. 10, 1535–1538 [PubMed] [Google Scholar]
- Hottinger A. F., Iwamoto F. M., Karimi S., Riedel E., Dantis J., Park J., et al. (2011). YKL-40 and MMP-9 as serum markers for patients with primary central nervous system lymphoma. Ann. Neurol. 70, 163–169 10.1002/ana.22360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B., Trinh K., Figueira W. F., Price P. A. (1996). Isolation and sequence of a novel human chondrocyte protein related to mammalian members of the chitinase protein family. J. Biol. Chem. 271, 19415–19420 10.1074/jbc.271.32.19415 [DOI] [PubMed] [Google Scholar]
- Jensen B. V., Johansen J. S., Price P. A. (2003). High levels of serum HER-2/neu and YKL-40 independently reflect aggressiveness of metastatic breast cancer. Clin. Cancer Res. 9, 4423–4434 [PubMed] [Google Scholar]
- Johansen J. S., Christensen I. J., Riisbro R., Greenall M., Han C., Price P. A., et al. (2003). High serum YKL-40 levels in patients with primary breast cancer is related to short recurrence free survival. Breast Cancer Res. Treat. 80, 15–21 10.1023/A:1024431000710 [DOI] [PubMed] [Google Scholar]
- Johansen J. S., Christoffersen P., Moller S., Price P. A., Henriksen J. H., Garbarsch C., et al. (2000). Serum YKL-40 is increased in patients with hepatic fibrosis. J. Hepatol. 32, 911–920 [DOI] [PubMed] [Google Scholar]
- Johansen J. S., Schultz N. A., Jensen B. V. (2009). Plasma YKL-40: a potential new cancer biomarker? Future Oncol. 5, 1065–1082 10.2217/fon.09.66 [DOI] [PubMed] [Google Scholar]
- Junker N., Johansen J. S., Andersen C. B., Kristjansen P. E. (2005a). Expression of YKL-40 by peritumoral macrophages in human small cell lung cancer. Lung Cancer 48, 223–231 10.1016/j.lungcan.2004.11.011 [DOI] [PubMed] [Google Scholar]
- Junker N., Johansen J. S., Hansen L. T., Lund E. L., Kristjansen P. E. (2005b). Regulation of YKL-40 expression during genotoxic or microenvironmental stress in human glioblastoma cells. Cancer Sci. 96, 183–190 10.1111/j.1349-7006.2005.00026.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada M., Seno H., Kanda K., Nakanishi Y., Akitake R., Komekado H., et al. (2012). Chitinase 3-like 1 promotes macrophage recruitment and angiogenesis in colorectal cancer. Oncogene 31, 3111–3123 10.1038/onc.2011.498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H., Das K., Noreen S., Coffman F., Hameed M. (2007). Prognostic implications of immunohistochemically detected YKL-40 expression in breast cancer. World J. Surg. Oncol. 5:17 10.1186/1477-7819-5-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjaergaard A. D., Bojesen S. E., Johansen J. S., Nordestgaard B. G. (2010). Elevated plasma YKL-40 levels and ischemic stroke in the general population. Ann. Neurol. 68, 672–680 10.1002/ana.22220 [DOI] [PubMed] [Google Scholar]
- Kronborg G., Ostergaard C., Weis N., Nielsen H., Obel N., Pedersen S. S., et al. (2002). Serum level of YKL-40 is elevated in patients with Streptococcus pneumoniae bacteremia and is associated with the outcome of the disease. Scand. J. Infect. Dis. 34, 323–326 10.1080/00365540110080233 [DOI] [PubMed] [Google Scholar]
- Kucur M., Isman F. K., Balci C., Onal B., Hacibekiroglu M., Ozkan F., et al. (2008). Serum YKL-40 levels and chitotriosidase activity as potential biomarkers in primary prostate cancer and benign prostatic hyperplasia. Urol. Oncol. 26, 47–52 10.1016/j.urolonc.2007.07.020 [DOI] [PubMed] [Google Scholar]
- Kyrgios I., Galli-Tsinopoulou A., Stylianou C., Papakonstantinou E., Arvanitidou M., Haidich A. B. (2012). Elevated circulating levels of the serum acute-phase protein YKL-40 (chitinase 3-like protein 1) are a marker of obesity and insulin resistance in prepubertal children. Metabolism. 61, 562–568 10.1016/j.metabol.2011.09.004 [DOI] [PubMed] [Google Scholar]
- Kzhyshkowska J., Gratchev A., Goerdt S. (2007). Human chitinases and chitinase-like proteins as indicators for inflammation and cancer. Biomark. Insights 2, 128–146 [PMC free article] [PubMed] [Google Scholar]
- Lee C. G., Da Silva C. A., Dela Cruz C. S., Ahangari F., Ma B., Kang M. J., et al. (2011). Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol. 73, 479–501 10.1146/annurev-physiol-012110-142250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. G., Da Silva C. A., Lee J. Y., Hartl D., Elias J. A. (2008). Chitin regulation of immune responses: an old molecule with new roles. Curr. Opin. Immunol. 20, 684–689 10.1016/j.coi.2008.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. G., Hartl D., Lee G. R., Koller B., Matsuura H., Da Silva C. A., et al. (2009). Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J. Exp. Med. 206, 1149–1166 10.1084/jem.20081271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek R. D., Hunt N. C., Landers R. J., Lewis C. E., Royds J. A., Harris A. L. (2000). Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J. Pathol. 190, 430–436 [DOI] [PubMed] [Google Scholar]
- Leek R. D., Lewis C. E., Whitehouse R., Greenall M., Clarke J., Harris A. L. (1996). Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 56, 4625–4629 [PubMed] [Google Scholar]
- Lewis C. E., Pollard J. W. (2006). Distinct role of macrophages in different tumor microenvironments. Cancer Res. 66, 605–612 10.1158/0008-5472.CAN-05-4005 [DOI] [PubMed] [Google Scholar]
- Libreros S., Garcia-Areas R., Shibata Y., Carrio R., Torroella-Kouri M., Iragavarapu-Charyulu V. (2012). Induction of proinflammatory mediators by CHI3L1 is reduced by chitin treatment: decreased tumor metastasis in a breast cancer model. Int. J. Cancer 131, 377–386 10.1002/ijc.26379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinda K. M., Ponce L., Kleinman H. K., Shackelton L. M., Millis A. J. (1999). Gp38k, a protein synthesized by vascular smooth muscle cells, stimulates directional migration of human umbilical vein endothelial cells. Exp. Cell Res. 250, 168–173 10.1006/excr.1999.4511 [DOI] [PubMed] [Google Scholar]
- McQuade K. J., Beauvais D. M., Burbach B. J., Rapraeger A. C. (2006). Syndecan-1 regulates alphavbeta5 integrin activity in B82L fibroblasts. J. Cell Sci. 119, 2445–2456 10.1242/jcs.02970 [DOI] [PubMed] [Google Scholar]
- Nielsen K. R., Steffensen R., Boegsted M., Baech J., Lundbye-Christensen S., Hetland M. L., et al. (2011). Promoter polymorphisms in the chitinase 3-like 1 gene influence the serum concentration of YKL-40 in Danish patients with rheumatoid arthritis and in healthy subjects. Arth. Res. Therapy 13, R109 10.1186/ar3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K. C., Millis A. J. (2003). gp38k (CHI3L1) is a novel adhesion and migration factor for vascular cells. Exp. Cell Res. 287, 79–87 10.1016/S0014-4827(03)00069-7 [DOI] [PubMed] [Google Scholar]
- Nyirkos P., Golds E. E. (1990). Human synovial cells secrete a 39 kDa protein similar to a bovine mammary protein expressed during the non-lactating period. Biochem. J. 269, 265–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Ribes M., Allen E., Hudock J., Takeda T., Okuyama H., Vinals F., et al. (2009). Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis.[see comment]. Cancer Cell 15, 220–231 10.1016/j.ccr.2009.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. A., Drazen J. M., Tschumperlin D. J. (2010). The chitinase-like protein YKL-40 is secreted by airway epithelial cells at base line and in response to compressive mechanical stress. J. Biol. Chem. 285, 29817–29825 10.1074/jbc.M110.103416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P. W., Reizes O., Bernfield M. (2000). Cell surface heparan sulfate proteoglycans: selective regulators of ligand-receptor encounters. J. Biol. Chem. 275, 29923–29926 10.1074/jbc.R000008200 [DOI] [PubMed] [Google Scholar]
- Pelloski C. E., Mahajan A., Maor M., Chang E. L., Woo S., Gilbert M., et al. (2005). YKL-40 expression is associated with poorer response to radiation and shorter overall survival in glioblastoma. Clin. Cancer Res. 11, 3326–3334 pp.3326-34 10.1158/1078-0432.CCR-04-1765 [DOI] [PubMed] [Google Scholar]
- Perrin R. J., Craig-Schapiro R., Malone J. P., Shah A. R., Gilmore P., Davis A. E., et al. (2011). Identification and validation of novel cerebrospinal fluid biomarkers for staging early Alzheimer's disease. PLoS ONE 6:e16032 10.1371/journal.pone.0016032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson F., Rathcke C. N., Gall M. A., Parving H. H., Vestergaard H., Rossing P. (2012). High YKL-40 levels predict mortality in patients with type 2 diabetes. Diabetes Res. Clin. Pract. 96, 84–89 10.1016/j.diabres.2011.12.008 [DOI] [PubMed] [Google Scholar]
- Pini M., Rhodes D. H., Castellanos K. J., Hall A. R., Cabay R. J., Chennuri R., et al. (2012). Role of IL-6 in the resolution of pancreatitis in obese mice. J. Leukoc. Biol. 91, 957–966 10.1189/jlb.1211627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizano-Martinez O., Yanez-Sanchez I., Alatorre-Carranza P., Miranda-Diaz A., Ortiz-Lazareno P. C., Garcia-Iglesias T., et al. (2011). YKL-40 expression in CD14 liver cells in acute and chronic injury. World J. Gastroenterol. 17, 3830–3835 10.3748/wjg.v17.i33.3830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard J. W. (2004). Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 4, 71–78 10.1038/nrc1256 [DOI] [PubMed] [Google Scholar]
- Prince R. N., Schreiter E. R., Zou P., Wiley H. S., Ting A. Y., Lee R. T., et al. (2010). The heparin-binding domain of HB-EGF mediates localization to sites of cell-cell contact and prevents HB-EGF proteolytic release. J. Cell Sci. 123, 2308–2318 10.1242/jcs.058321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W., Zhu W., Schlatter L., Miick R., Loy T. S., Atasoy U., et al. (2007). Increased expression of the inflammatory protein YKL-40 in precancers of the breast. Int. J. Cancer 121, 1536–1542 10.1002/ijc.22881 [DOI] [PubMed] [Google Scholar]
- Rathcke C. N., Vestergaard H. (2006). YKL-40, a new inflammatory marker with relation to insulin resistance and with a role in endothelial dysfunction and atherosclerosis. Inflamm. Res. 55, 221–227 10.1007/s00011-006-0076-y [DOI] [PubMed] [Google Scholar]
- Recklies A. D., White C., Ling H. (2002). The chitinase 3-like protein human cartilage glycoprotein 39 (HC-gp39) stimulates proliferation of human connective-tissue cells and activates both extracellular signal-regulated kinase- and protein kinase B-mediated signalling pathways. Biochem. J. 365, 119–126 10.1042/BJ20020075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehli M., Krause S. W., Andreesen R. (1997). Molecular characterization of the gene for human cartilage gp-39 (CHI3L1), a member of the chitinase protein family and marker for late stages of macrophage differentiation. Genomics 43, 221–225 10.1006/geno.1997.4778 [DOI] [PubMed] [Google Scholar]
- Rehli M., Niller H. H., Ammon C., Langmann S., Schwarzfischer L., Andreesen R., et al. (2003). Transcriptional regulation of CHI3L1, a marker gene for late stages of macrophage differentiation. J. Biol. Chem. 278, 44058–44067 10.1074/jbc.M306792200 [DOI] [PubMed] [Google Scholar]
- Renkema G. H., Boot R. G., Au F. L., Donker-Koopman W. E., Strijland A., Muijsers A. O., et al. (1998). Chitotriosidase, a chitinase, and the 39-kDa human cartilage glycoprotein, a chitin-binding lectin, are homologues of family 18 glycosyl hydrolases secreted by human macrophages. Eur. J. Biochem. 251, 504–509 10.1046/j.1432-1327.1998.2510504.x [DOI] [PubMed] [Google Scholar]
- Roslind A., Johansen J. S., Junker N., Nielsen D. L., Dzaferi H., Price P. A., et al. (2007a). YKL-40 expression in benign and malignant lesions of the breast: a methodologic study. Appl. Immunohistochem. Mole. Morphol. 15, 371–381 10.1097/01.pai.0000213146.77772.6a [DOI] [PubMed] [Google Scholar]
- Roslind A., Knoop A. S., Jensen M. B., Johansen J. S., Nielsen D. L., Price P. A., et al. (2007b). YKL-40 protein expression is not a prognostic marker in patients with primary breast cancer. Breast Cancer Res. Treat. 112, 275–285 10.1007/s10549-007-9870-7 [DOI] [PubMed] [Google Scholar]
- Saidi A., Javerzat S., Bellahcene A., De Vos J., Bello L., Castronovo V., et al. (2008). Experimental anti-angiogenesis causes upregulation of genes associated with poor survival in glioblastoma. Int. J. Cancer 122, 2187–2198 10.1002/ijc.23313 [DOI] [PubMed] [Google Scholar]
- Scully S., Yan W., Bentley B., Cao Q. J., Shao R. (2011). Inhibitory activity of YKL-40 in mammary epithelial cell differentiation and polarization induced by lactogenic hormones: a role in mammary tissue involution. PLoS ONE 6:e25819 10.1371/journal.pone.0025819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelton L. M., Mann D. M., Millis A. J. (1995). Identification of a 38-kDa heparin-binding glycoprotein (gp38k) in differentiating vascular smooth muscle cells as a member of a group of proteins associated with tissue remodeling. J. Biol. Chem. 270, 13076–13083 10.1074/jbc.270.22.13076 [DOI] [PubMed] [Google Scholar]
- Shao R., Cao Q. J., Arenas R. B., Bigelow C., Bentley B., Yan W. (2011). Breast cancer expression of YKL-40 correlates with tumour grade, poor differentiation, and other cancer markers. Br. J. Cancer 105, 1203–1209 10.1038/bjc.2011.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R., Hamel K., Petersen L., Cao Q. J., Arenas R. B., Bigelow C., et al. (2009). YKL-40, a secreted glycoprotein, promotes tumor angiogenesis. Oncogene 28, 4456–4468 10.1038/onc.2009.292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y., Metzger W. J., Myrvik Q. N. (1997). Chitin particle-induced cell-mediated immunity is inhibited by soluble mannan: mannose receptor-mediated phagocytosis initiates IL-12 production. J. Immunol. 159, 2462–2467 [PubMed] [Google Scholar]
- Stearman R. S., Dwyer-Nield L., Grady M. C., Malkinson A. M., Geraci M. W. (2008). A macrophage gene expression signature defines a field effect in the lung tumor microenvironment. Cancer Res. 68, 34–43 10.1158/0008-5472.CAN-07-0988 [DOI] [PubMed] [Google Scholar]
- Suzuki T., Hashimoto S., Toyoda N., Nagai S., Yamazaki N., Dong H. Y., et al. (2000). Comprehensive gene expression profile of LPS-stimulated human monocytes by SAGE. Blood 96, 2584–2591 [PubMed] [Google Scholar]
- Tsutsui S., Yasuda K., Suzuki K., Tahara K., Higashi H., Era S. (2005). Macrophage infiltration and its prognostic implications in breast cancer: the relationship with VEGF expression and microvessel density. Oncol. Rep. 14, 425–431 [PubMed] [Google Scholar]
- Verhoeff J. J., van Tellingen O., Claes A., Stalpers L. J., van Linde M. E., Richel D. J., et al. (2009). Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer 9:444 10.1186/1471-2407-9-444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vind I., Johansen J. S., Price P. A., Munkholm P. (2003). Serum YKL-40, a potential new marker of disease activity in patients with inflammatory bowel disease. Scand. J. Gastroenterol. 38, 599–605 [DOI] [PubMed] [Google Scholar]
- Volck B., Johansen J. S., Stoltenberg M., Garbarsch C., Price P. A., Ostergaard M., et al. (2001). Studies on YKL-40 in knee joints of patients with rheumatoid arthritis and osteoarthritis. Involvement of YKL-40 in the joint pathology. Osteoarth. Cart. 9, 203–214 10.1053/joca.2000.0377 [DOI] [PubMed] [Google Scholar]
- Wang H., Lathia J. D., Wu Q., Wang J., Li Z., Heddleston J. M., et al. (2009). Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 27, 2393–2404 10.1002/stem.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen P. Y., Kesari S. (2008). Malignant gliomas in adults. New Eng. J. Med. 359, 492–507 10.1056/NEJMra0708126 [DOI] [PubMed] [Google Scholar]
- Wick W., Weller M., van den Bent M., Stupp R. (2010). Bevacizumab and recurrent malignant gliomas: a European perspective. J. Clin. Oncol. 28, e188–189; author reply e190–182. 10.1200/JCO.2009.26.9027 [DOI] [PubMed] [Google Scholar]