


Abstract
Human artificial chromosome (HAC)-based vectors represent an alternative technology for gene delivery and expression with a potential to overcome the problems caused by the use of viral-based vectors. The recently developed alphoidtetO-HAC has an advantage over other HAC vectors because it can be easily eliminated from cells by inactivation of the HAC kinetochore via binding of tTS chromatin modifiers to its centromeric tetO sequences. This provides unique control for phenotypes induced by genes loaded into the alphoidtetO-HAC. However, inactivation of the HAC kinetochore requires transfection of cells by a retrovirus vector, a step that is potentially mutagenic. Here, we describe an approach to re-engineering the alphoidtetO-HAC that allows verification of phenotypic changes attributed to expression of genes from the HAC without a transfection step. In the new HAC vector, a tTS-EYFP cassette is inserted into a gene-loading site along with a gene of interest. Expression of the tTS generates a self-regulating fluctuating heterochromatin on the alphoidtetO-HAC that induces fast silencing of the genes on the HAC without significant effects on HAC segregation. This silencing of the HAC-encoded genes can be readily recovered by adding doxycycline. The newly modified alphoidtetO-HAC-based system has multiple applications in gene function studies.
INTRODUCTION
Human artificial chromosomes or HAC-based vectors represent a novel system for gene delivery and expression that has several advantages over previous gene and cell therapy strategies (1–7). All HACs by definition contain a functional centromere and, therefore, represent a non-essential 47th chromosome that replicates and segregates like a normal chromosome in human cells, thereby avoiding integration into the host genome. HAC vectors have essentially unlimited cloning capacity; therefore, they are able to carry genes or genomic fragments up to several mega base pairs. Thus, HAC-based gene delivery vectors may provide long-term expression of complete genetic loci, while reducing the risk of an immune response and transgene silencing characteristic of viral-based vectors (8–12). Moreover, the presence of internal promoters, splicing sites and polyadenylation [poly(A)] sites allows tissue-specific expression of the therapeutic genes, making HACs suitable for gene delivery to different types of target cells. Several laboratories succeeded in complementation of gene deficiency in human recipient cell lines and in creation of transgenic mice using HAC vectors containing genomic copies of the genes with all their regulatory elements, demonstrating their potential as therapeutic gene expression vectors (13–24). A particularly impressive example is stem-cell–mediated gene replacement therapy in dystrophic mice using a HAC expressing the human 2.4-Mb dystrophin (DYS) gene (25,26). Tedesco et al. (27,28) reported transfer of this HAC carrying the entire human dystrophin genetic locus into blood vessel-associated stem cells (mesoangioblasts). Transfer of these genetically corrected cells to the mdx mouse resulted in the amelioration of dystrophic phenotypes in the mouse model of Duchenne muscular dystrophy. This example of HAC-mediated gene transfer shows efficacy in a pre-clinical model of Duchenne muscular dystrophy and offers the potential for future clinical translation.
HACs are engineered by ‘top-down’ or ‘bottom-up’ (de novo formation) approaches (1–7). The top-down approach is based on a telomere-associated human chromosome truncation technique. The bottom-up approach includes transfection of human cells with either natural high-order repeat or synthetic α-satellite (alphoid) DNA with a size bigger than 30 kb. In this case, HAC formation is accompanied by multimerization of the input DNA in the cell up to 1–5 Mb.
Recently, a new generation de novo HAC, the alphoidtetO-HAC, was engineered using a 40-kb synthetic alphoid DNA array (29). This array contains 42-bp tetracycline operator (tetO) sequences incorporated into every second alphoid DNA monomer. After amplification of the input DNA (40 kb) in human HT1080 cells and HAC formation, the resulting alphoidtetO-HAC contains <6000 copies of the tetO sequence in the 1.1 mega-base size alphoid DNA array. Because tetO sequences are bound with very high affinity and specificity by the tet repressor (tetR), the tetO sequences in the HAC can be targeted efficiently in vivo with tetR fusion proteins. The power of this system is that it allows the specific manipulation of the chromatin composition of a HAC kinetochore in vivo, whereas it leaves all kinetochores of the natural chromosomes unperturbed. Targeting of chromatin-modifying proteins into the HAC kinetochore demonstrated that a balance between open and condensed chromatin is critical for kinetochore function (29–34). The frequency of HAC loss was very high when repressive chromatin was induced in the HAC via the tTS (tet-repressor transcriptional silencer) (which contains the KRAB-AB domain of the SDkid-1 protein). tTS binding caused the loss of CENP-A, CENP-B, CENP-C and H3K4me2 from the HAC kinetochore accompanied by an accumulation of histone H3K9me3 on the alphoid DNA array. As a consequence, the functional HAC kinetochore was inactivated (29–34). This alphoidtetO-HAC elimination was highly efficient using either retrovirus or plasmid-induced expression to provide a high level of chromatin modifiers as tetR fusion proteins (Figure 1a).
Figure 1.

A scheme illustrating epigenetic changes in the alphoidtetO-HAC caused by tTS expression. (a) The condition when tTS is highly expressed from a retroviral vector in trans. Multiple tTS molecules bind to a great fraction of tetO sequences in the HAC and as previously shown lead to the formation of heterochromatin, spreading of which disrupts centrochromatin. As a result, the HAC kinetochore is inactivated, and the HAC is lost. (b and c) Conditions where a single copy of tTS is integrated into the HAC. The main difference of this system compared with viral expression is that fewer targets (tetO sequences) are bound by the effectors (tTS protein), as less protein is synthesized. Two scenarios are possible. (b) Despite the limited number of tTS molecules binding to tetO sequences in the HAC, active spreading of heterochromation induced by tTS results in the disruption of centrochromatin domains. (c) Binding of the limited number of tTS molecules to alphoid DNA array induces formation of the local chromatin domains that do not compromise activity of the HAC kinetochore but completely blocks transcription of the sequences within these domains. One of these domains is located near the gene-loading site leading to self-silencing of the tTS gene and transgenes nearby.
The alphoidtetO-HAC with a regulated kinetochore represents a novel system for gene function analysis and potentially for gene therapy because it provides a unique possibility to compare the phenotypes of target human cells with and without a functional copy of a gene inserted into the HAC. Such a rigorous control is required for proper interpretation of gene complementation studies. To adopt the existing alphoidtetO-HAC for gene delivery and expression studies, the HAC containing a loxP gene-loading site was transferred to Chinese hamster ovary (CHO) cells via micro-cell–mediated chromosome transfer (MMCT) (35). In those cells, a gene of interest can be easily inserted into the loxP site of the HAC by Cre-mediated recombination. In addition, the HAC can be transferred from these cells into different recipient cells via MMCT. Because physical characterization of the alphoidtetO-HAC was carried out (36), this HAC is an excellent candidate for gene function studies. Recently, we reported the capacity of the alphoidtetO-HAC to deliver genomic copies of two human genes, VHL (∼25 kb) and NBS1 (∼60 kb), and complement genetic deficiencies in patient-derived cell lines (37). Functional expression of pVHL and pNBS1 in recipient cells was proven by a set of specific tests based on the known functions of those proteins. Corresponding controls were conducted following elimination of the HAC from the cells by transfection with a retrovirus vector carrying a tTS chromatin silencer to inactivate HAC centrochromatin.
It is well known that transfection by both plasmid and retroviral vectors is potentially mutagenic. In this report, we describe re-engineering of an alphoidtetO-HAC vector to allow verification of phenotypic changes attributed to the expression of genes from the HAC without the necessity of viral transfection of the HAC-bearing cells. As a proof of principle, a gene of interest was inserted into the HAC along with a tTS-EYFP expression cassette. Such a configuration of effector (tTS) and target (tetO) apparently generates a self-regulating fluctuating repressive heterochromatin-like system that can be used to induce silencing of genes loaded into the HAC without a significant effect on the HAC segregation. This feature of the alphoidtetO-HAC carrying tTS is consistent with its potential broad use in gene function study experiments.
MATERIALS AND METHODS
Cell lines and media
The hypoxanthine phosphoribosyltransferase (HPRT)-deficient HT1080 cell line was obtained from Dr Zoia Larin Monaco (Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK). The cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% (v/v) tet system-approved fetal bovine serum (Clontech Laboratories, Inc.) at 37°C in 5% CO2. HPRT-deficient Chinese hamster ovary (CHO) cells (JCRB0218) carrying the alphoidtetO-HAC were maintained in Ham’s F-12 nutrient mixture (Invitrogen) plus 10% FBS with 8 µg/ml of blasticidin (BS) (Invitrogen). After loading of the transgene cassettes into the alphoidtetO-HAC, the HT1080 and CHO cells were cultured in 1× HAT medium (Invitrogen). Concentration of doxycycline in the medium was 1 µg/ml.
Micro-cell–mediated chromosome transfer
The alphoidtetO-HAC containing a single loxP gene-loading site was transferred from CHO cells to HPRT-deficient HT1080 cells using a standard MMCT protocol (35,38). The whole MMCT procedure takes 4 days (the growth of cells in the presence of colcemid for 3 days and the MMCT procedure itself 1 day). BS was used to select colonies with the HAC. Selection of BSR clones usually takes 2–3 weeks. Typically, 3–10 BSR colonies were obtained in one MMCT experiment. The BSR colonies were analyzed by fluorescence in situ hybridization (FISH) for the presence of the autonomous form of the HAC. The potential co-transfer of CHO chromosomes was excluded using a sensitive PCR test for rodent-specific SINE elements (Supplementary Table S1).
FISH analysis
FISH analysis was performed as previously described (29,37). HAC-containing cells were cultured in medium with 10 μg/ml of colcemid (Invitrogen) for 3 h at 37°C. Metaphase cells were trypsinized and collected by centrifugation for 5 min at 483g, treated in hypotonic solution 50 mM KCl for 20 min at 37°C and washed in methanol:acetic acid (3:1) solution three times. Cells were diluted to the appropriate density with fixative solution, spread onto pre-cleaned slides (Fisher Scientific) above steam (boiling water) and allowed to age 2 days at room temperature. Metaphase chromosomes on the slide were denatured by 70% formamide/2× SSC for 2 min at 72°C. Samples were dehydrated through a 70, 90 and 100% ethanol series for 4 min each and left to air-dry. Orange 552 dUTP (5-TAMRA-dUTP) (Abbott Molecular) labeled probes were denatured in hybridization solution at 78°C for 10 min and left at 37°C for 30 min. The hybridization mix probe was applied to the sample and incubated at 37°C overnight. Slides were washed with 0.4× SSC, 0.3% Tween 20 for 2 min at 72°C, briefly rinsed with 2× SSC, 0.1% Tween 20 (5 s to 1 min) and air dried in darkness. The samples were counterstained with Vectashield mounting medium with DAPI (Vector Laboratories). Slides were analyzed by fluorescence microscopy. Images were captured using a DeltaVision microscopy imaging system in the CRC, LRBGE Fluorescence Imaging Facility (NIH) and analyzed using image J software (NIH).
The probe used for FISH analysis
The probe used for FISH was BAC32-2mer(tetO) DNA containing 40 kb of alphoid-tetO array cloned into a BAC vector as described previously (29). BAC DNA was labeled using a nick-translation kit with Orange 552 dUTP (5-TAMRA-dUTP) (Abbott Molecular).
Analysis of HAC mitotic stability by FISH
Fluorescence images of human and hamster cells carrying the HAC with tTS-containing cassettes were analyzed. Cells were processed for FISH using a BAC-based probe after 7, 20 and 36 days of culturing without HAT selection in the presence or absence of doxycycline. Metaphase spreads were screened for the presence or absence of the HAC. At least 60 metaphases were screened for each clone. The loss rate (R) was calculated using the formula N1 = N0 × (1 − R)n, where N0 = percentage of metaphase spreads with alphoidtetO-HAC in cells cultured under selection, N1 = percentage of alphoidtetO-HAC-containing metaphase spreads after n days of culture in the absence of selection (20).
Construction of the tTS-containing cassettes used for loading into the HAC
The construction of the control X3.1-I-EGFP-I cassette (also referred as 264-EGFP) (construct #1) was described previously (35). The transgene cassettes tetR-IRES-DsRed2 (construct #2), tTS-IRES-DsRed2 (construct #3) and tTS-EYFP (construct #4) were assembled as follows. The X3.1-I-EGFP-I DNA was digested with BsrGI/SpeI. The 7241-bp fragment without the EYFP transgene was extracted from the gel and ligated with a synthetic polylinker (Supplementary Table S1) (Integrated DNA Technologies), producing the intermediate plasmid 265. The CAG promoter was isolated as a 1734-bp fragment by digestion of X3.1-I-EGFP-I using KpnI/EcoRI. This fragment was cloned into the pBluescript KS vector (Fermentas). Next, the CAG promoter-containing fragment was digested by SpeI and re-cloned into a single SpeI site of plasmid 265, producing the plasmid 266. This plasmid was used as a basic construct for further insertion of different cassettes. The tTS-EYFP cassette was amplified from the pFB-tTS-EYFP vector (29) using a pair of specific primers PacI-rtTS-GFP-F/AvrII-rtTS-GFP-R (Supplementary Table S1) and inserted into AvrII/PacI sites of the plasmid 266, producing the transgene construct #4 (tTS-EYFP). To construct the transgene cassette tTS-IRES-DsRed2 (construct #3), the tTS fragment of 849 bp was amplified from pFB-tTS-EYFP plasmid (29) with a pair of primers PacI-rtTS-IRES-Red-F/XmaI-rtTS-R (Supplementary Table S1). The IRES/DsRed2 fragment of 1261 bp was amplified from the 264-CAG-OKS-EF1-tTKRAB-DsRed-pA plasmid (kindly provided by Dr Tomilin, Institute of Cytology, RAS, Russia) with a pair of primers XmaI-IRES-Red-F/NheI-rtTS-IRES-Red-R (Supplementary Table S1). The fragments were digested by PacI/XmaI and XmaI/NheI, correspondingly, and cloned into the basic plasmid 266 digested by PacI/NheI by one ligation step, producing the transgene cassette tetR-tTS-IRES-DsRed2 (construct #3). To construct the transgene cassette tetR-IRES-DsRed2 (construct #2), the tetR-NLC fragment of 654 bp was amplified from the plasmid pTetR-TS (Clontech) with a pair of primers PacI-rtTS-IRES-Red-F/XmaI-tetR-NLS-R (Supplementary Table S1) and then digested by PacI/XmaI. The fragments tetR-NLS and IRES/DsRed2 were cloned into the basic plasmid 266 digested by PacI/NheI by one ligation step, producing the transgene cassette tetR-IRES-DsRed2 (construct #2). Detailed physical maps of the cassettes are shown in Supplementary Figure S1.
Loading of the transgene cassettes into the loxP site of the alphoidtetO-HAC
A total of 1–3 μg of transgene construct DNA and 1 μg of the Cre expression pCpG-iCre vector DNA were co-transformed into HPRT-deficient human HT1080 cells containing the alphoidtetO-HAC by lipofection with X-tremeGENE 9 DNA transfection reagent (Roche) or Lipofectamine 2000 (Invitrogen). HPRT-positive colonies were selected after 2–3 weeks growth in HAT medium. For each transgene cassette, from five to seven clones were selected. Correct loading of the transgene cassettes into the HAC was confirmed by genomic PCR with a specific pair of primers that diagnose reconstitution of the HPRT gene (Supplementary Table S1). Conditions of a transgene loading into the alphoidtetO-HAC propagated in CHO hamster cells were described previously (35,37).
RT–PCR analysis
Expression of the Hygro, HS-tk, HPRT, BRCA1 and EGFP genes was checked by RT–PCR. Pairs of specific primers were designed to amplify each gene as 265-, 267-, 449-, 893- and 483-bp fragments, respectively (Supplementary Table S1). cDNA was made from 1 µg of total RNA isolated from dox+ and dox− cultured HT1080 cells using the RevertAid™ First Strand cDNA Synthesis Kit (Fermentas) and priming with oligo dT as recommended by the kit supplier. RT–PCR reactions were performed with 1 µl of cDNA in a 50-µl volume. Standard reaction conditions were 95°C for 30 s, 60°C for 30 s, 72°C for 45 s × 35 cycles. The PCR products were sequenced on a PE-Applied Biosystem 3100 Automated Capillary DNA Sequencer.
Trichostatin A and suberoylanilide hydroxamic acid treatment
Aliquots of ∼2.4 × 106 HT1080 cells carrying the alphoidtetO-HAC with the silenced tTS-EYFP were incubated in 2 ml of non-selective medium containing either 100 ng/ml of Trichostatin A (TSA) (Wako) or in 10 mM of suberoylanilide hydroxamic acid (SAHA) (Vorinostat) for 24 h in the presence of doxycycline. Then cell samples were collected and analyzed by FACS.
FACS analysis
FACS analysis of EYFP and DsRed2 expression was performed on a FACSCalibur instrument (BD Biosciences) controlled using CellQuest acquisition software and analyzed statistically with FlowJo software (39,40). A minimum of 4 × 104 cells were analyzed for each cell sample.
Southern-blot hybridization analysis
Southern-blot hybridization was performed with a 32P-labeled probe. Genomic DNA was prepared in agarose plugs and restriction digested by PmeI or SpeI. The digested DNA was CHEF separated, and blot-hybridized with a 276-bp probe specific to a tetO-containing alphoid sequence (36). The probe sequence was PCR amplified from BAC32-2-mer/tetO (29) using the primers listed in Supplementary Table S1. Blots were incubated for 2 h at 65°C in pre-hybridization Church’s buffer (0.5 M Na-phosphate buffer containing 7% SDS and 100 µg/ml of salmon sperm carrier DNA). The labeled probe was heat denatured in boiling water for 5 min and snap cooled on ice. The probe was added to the hybridization buffer and allowed to hybridize overnight at 65°C. Blots were washed twice for 30 min at room temperature in 2× SSC, 0.1% SDS and then three times for 30 min at 65°C in 0.1× SSC, 0.1% SDS. Blots were exposed to X-ray film for 24 h at −70°C.
Chromatin immunoprecipitation assay and real-time PCR
Chromatin immunoprecipitation (ChIP) with antibodies against trimethyl H3Lys4 (Upstate) and CENP-A was carried out according to a previously described method (29,36). Briefly, cultured cells were cross-linked in 1% formaldehyde for 10 min at 37°C. After addition of 1/10 volume of 1.25 M glycine and incubation for 5 min, fixed cells were washed twice with cold phosphate-buffered saline. Soluble chromatin was prepared by sonication (Bioruptor sonicator; Cosmo Bio) to an average DNA size of 500 bp in sonication buffer and immnnoprecipitated in IP buffer (20 mM Tris–HCl, pH 8.0, 600 mM NaCl, 1 mM EDTA, 0.05% SDS, 1.0% Triton X-100, 20% glycerol, 1.5 μM aprotinin, 10 μM leupeptin, 1 mM DTT and 40 μM MG132). Protein G sepharose (Amersham, USA) blocked with bovine serum albumin was added, and the antibody–chromatin complex was recovered by centrifugation. The recovery ratio of the immunoprecipitated DNA relative to input DNA was measured by real-time PCR using a CFX96 real-time PCR detection system (Bio-Rad, USA) and iQ SYBR Green Supermix (Bio-Rad, USA). Primers for EYFP, Bsr, Hygro, HS-tk transgenes, alphoidtetO repeat and chromosome 21 alphoid repeat sequences, as well as for control loci, 5S ribosomal DNA and pericentromeric satellite 2 repeats, are listed in Supplementary Table S1. At least three independent ChIP experiments were performed to estimate the level of enrichment.
Construction of BRV-tTS-EYFP retrofitting vector
The BRV-tTS-EYFP vector to retrofit transformation-associated recombination (TAR)-isolated genes with a loxP site and a gene-silencing cassette was constructed as follows. First, the four following fragments with the appropriate restriction sites at their ends were obtained by PCR amplification. The bovine globulin terminator (ter)-cHS4-Amp-pBR322 fragment was PCR amplified from plasmid pFB-tTS-EYFP (29) using specific primers B346 and B345 (Supplementary Table S1). This fragment was digested with AsiSI and AscI. The cytomegalovirus promoter-DsRed2 fragment was PCR amplified from plasmid pDsRed2-C1 (Clontech) using specific primers B350 and B349 (Supplementary Table S1). Before ligation, this fragment was digested with BglII and AsiSI. The SV40pA terminator sequence was PCR amplified from plasmid phiC31 attP neo (41) using specific primers B348 and B347 (Supplementary Table S1). Before ligation, this fragment was digested with BamHI and SpeI. The cHS4-CAG-tTS-YFP fragment was excised from the plasmid pFB-tTS-EYFP (29) with endonucleases AvrII and AscI. Each of the aforementioned fragments was gel-purified, and the four fragments were assembled together in a single ligation reaction. The intermediate plasmid thus obtained was named 264-tTS-DsRed2. That plasmid was checked by PCR, restriction digestion and sequencing. Next, plasmids 264-tTS-DsRed2 and BRV1 (42), modified by insertion of unique BsiWI site, were digested with AvrII and AscI. The two fragments were ligated together, producing the vector BRV-tTS-EYFP. A detailed diagram of the BRV-tTS-EYFP retrofitting vector is shown in Supplementary Figure S2. The vector contains the 3′-end of the HPRT gene, a loxP gene acceptor sequence and two short targeting hooks (<300 bp each) homologous to the TAR cloning vector pVC604 used for gene isolation (42) separated by the unique BsiWI site.
RESULTS
Chromatin changes in the alphoidtetO-HAC kinetochore that may occur when a tTS cassette is expressed from the HAC
To omit the necessity for viral vector transfection to regulate the activity of the alphoidtetO kinetochore, we investigated whether a single copy of a tTS-containing cassette integrated into the HAC could induce inactivation of the HAC kinetochore. We deduced that cells with a single copy of tTS should have much lower level of the chromatin modifier than cells transfected under conditions where potentially hundreds of vector molecules might penetrate into the cell. More important, the presence of the tTS cassette, with its multiple targets (thousands of tetO sites) on the same DNA molecule (in cis configuration) might produce a negative autoregulation of the expression of the tTS module itself. Indeed, such an in cis configuration for effector (tTS) and target (tetO) could potentially generate a self-regulating repressive fluctuating heterochromatin-like system. This is because when the tTS silencer reaches a certain level of saturation on alphoidtetO array, the synthetic chromatin created will turn off its own expression. Subsequently, when the level of the tTS silencer falls, and then the tTS could potentially re-emerge from its repressed state.
Based on these considerations, we have considered several scenarios (Figure 1b and c). First, despite a relatively low level of expression, the chromatin modifier might disrupt the CENP-A kinetochore domain and induce HAC loss. Second, because of the self-induced suppression of tTS expression (and as a result of this a relatively low level of heterochromatinization of the alphoidtetO array), the HAC kinetochore might escape the inactivation seen in our previous studies (29), perhaps because of the plasticity of human kinetochore chromatin. However, because the HAC contains thousands of tetO sites, induction of synthetic heterochromatin-like repressed regions on the megabase-size alphoidtetO array could potentially induce transcriptional silencing of adjacent genes because of heterochromatin spreading. In this case, the tTS module might provide a novel approach to regulate the expression of HAC-encoded genes while maintaining a functional HAC kinetochore.
In this study, several tTS-containing cassettes were inserted into the alphoidtetO-HAC to determine the consequences of the silencer expression on HAC stability and the expression of genes loaded onto the HAC.
Characterization of the gene-loading system in the alphoidtetO-HAC propagated in human cells
The loxP site in the HAC was combined with the HPRT sequence fragment such that gene-loading events could be selected by reconstitution of the HPRT gene. All previous experiments on gene loading into the alphoidtetO-HAC were carried out in HPRT-deficient CHO hamster cells (35,37), as these are efficient donors for HAC transfer into different human cell lines. In this study, we performed the gene loading directly in human cells.
For this purpose, the alphoidtetO-HAC containing a single loxP gene-loading site was first moved from CHO cells into HPRT-deficient HT1080 cells (43) via MMCT under selection for Blasticidin (or BS) (Figure 2a). Eight drug-resistant clones were isolated. The presence of the autonomous form of the HAC was examined by FISH analysis. This confirmed that the HAC was maintained autonomously in seven of the eight clones analyzed (Figure 2c). Second, because the MMCT procedure can lead to deletions and rearrangements of the alphoid DNA array, four HAC-containing clones were checked for integrity by SpeI digestion and Southern-blot hybridization as previously described (36). As seen, all clones have SpeI profiles indistinguishable from the original alphoidtetO-HAC generated in human cells (clone AB2.2.18.21) (29,36) and from the HAC in CHO cells (Figure 3a).
Figure 2.

Schematic diagram of a gene loading into the alphoidtetO-HAC propagated in human cells. (a) Three steps of MMCT to transfer the HAC into HPRT-deficient HT1080 cells. In chicken DT40 cells, a loxP gene-loading site was introduced into the HAC (35). This modified HAC was transferred to hamster CHO cells and then to HPRT-deficient human HT1080 cells. (b) Loading of the X3.1-I-EGFP-I construct into the HAC. X3.1-I-EGFP-I contains a 3′ HPRT sequence, loxP site and EGFP transgene. (c) FISH analysis of the HAC-containing HT1080 clones. The HAC was visualized using a vector probe (red). Chromosomal DNA was counterstained with DAPI (blue). The HAC is indicated by an arrow. (d) Fluorescence image of cells carrying the HAC with expressing X3.1-I-EGFP-I construct.
Figure 3.
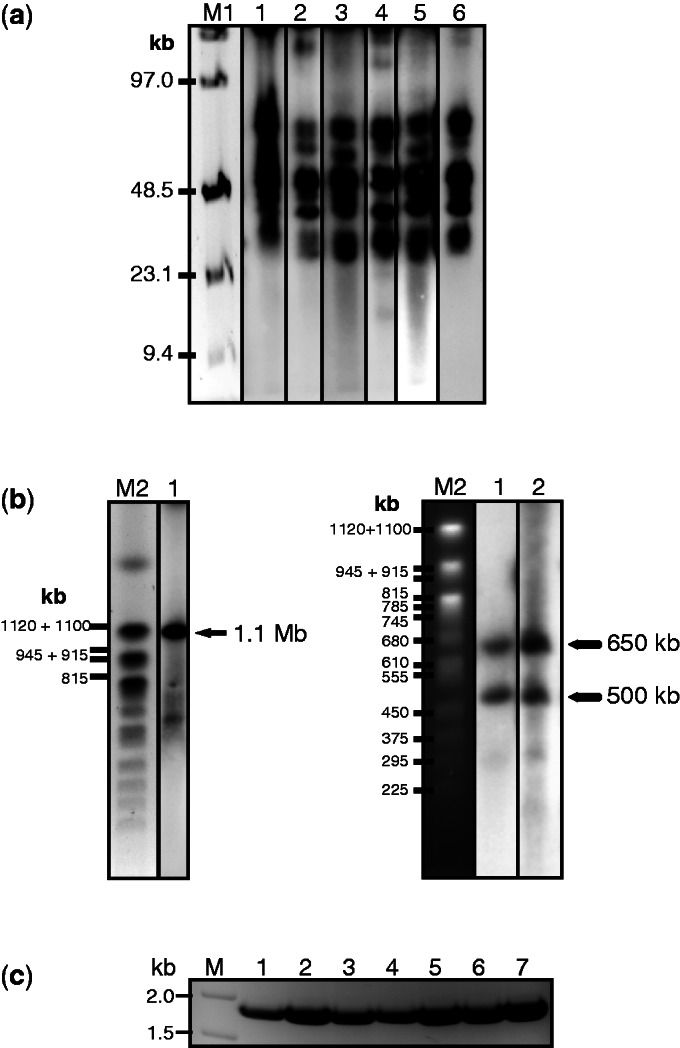
Physical analysis of the alphoidtetO-HAC used for loading of tTS-containing cassettes. (a) Analysis of integrity of the alphoidtetO-HAC synthetic array after its transfer into HPRT-deficient HT1080 cells. Genomic DNA from the cells with the HAC was digested with SpeI endonuclease, separated by CHEF gel electrophoresis (range 10–70 kb) and the transferred membrane was hybridized with the tetO-alphoid probe. Lane 1: the HAC with the loxP site in hamster CHO cells; lanes 2, 3, 4 and 5: four HAC clones with the loxP site in HPRT-deficient HT1080 cells; lane 6: the original HAC (clone AB2.218.21) generated in human cells (29). M1- Pulse Marker™ 0.1–200 kb (Sigma-Aldrich). (b) Mapping of the loxP site in a mega-base size alphoid DNA array. Genomic DNA from the cells possessing the original HAC was digested with PmeI endonuclease, separated by CHEF gel electrophoresis (range 200–1500 kb) and the transferred membrane was hybridized with the tetO-alphoid probe. A single 1.1-Mb fragment was detected for the original HAC (left, lane 1). Two bands of 650 and 500 kb were detected for genomic DNA from cells with the HAC bearing an inserted NBS1 gene (right, lanes 1 and 2). The size of the fragments was determined by comparison with the DNA size standard, Saccharomyces cerevisiae chromosomes. Lane M2: Yeast Chromosome PFG Marker BioLabs. (c) PCR analysis of clones with insertion of X3.1-I-EGFP-I into the HAC confirming restoration of the full-length HPRT gene.
A unique loxP site was previously inserted within a mega-size alphoid DNA array of the alphoidtetO-HAC (35). However, its position was not determined. For this study, the location of the loxP site within the array was mapped. Earlier we have shown that the entire 1.1-Mb alphoid DNA array may be excised from the HAC by PmeI digestion (36). For mapping, we used a previously constructed alphoidtetO-HAC carrying the full-length NBS1 gene sequence that contains a unique PmeI site (in the NBS1 sequence) (37). Chromosome-size genomic DNA of two independent NBS1-containing clones was prepared in agarose plugs from human cells harboring the alphoidtetO-HAC/NBS1, cut with PmeI endonuclease, separated by CHEF gel electrophoresis and hybridized with an alphoidtetO probe (Supplementary Table S1). Southern-blot hybridization revealed two bands of ∼650 and 500 kb corresponding in sum to the continuous ∼1.1-Mb alphoid DNA array in the original HAC (Figure 3b). Thus, the loxP site is located near the middle of the alphoid DNA array of the alphoidtetO-HAC. This means that genes loaded into the HAC loxP site will be equally flanked on either side by thousands of tetO sequences.
To evaluate the efficiency of gene loading into the loxP site of the alphoidtetO-HAC in human cells, a previously constructed vector, 264-EGFP (or X3.1-I-EGFP-I), (Supplementary Figure S1) and a Cre-recombinase expression vector were co-transfected into cells carrying the HAC (Figure 2b). Seven recombinant clones were selected by growth on HAT medium after 12–15 days. PCR analysis with specific primers (Supplementary Table S1) confirmed that the HPRT gene was reconstituted in all selected drug-resistant clones (Figure 3c). Fluorescence microscopy revealed expression of the EGFP transgene in all clones. A fluorescence image of one representative clone bearing the HAC is shown in Figure 2d. The efficiency of gene loading into the HAC in HPRT− HT1080 cells was comparable with that observed in CHO cells (<1–2 × 10−4) (35). In human cells, EGFP expression was stable for at least 6 months under selective conditions (data not shown). Thus, we conclude that genes of interest or transgene constructs can be efficiently inserted into the alphoidtetO-HAC propagated in human cells, and that they are subsequently stably expressed.
One copy of tTS loaded into the alphoidtetO-HAC does not inactivate the HAC kinetochore
To investigate the effects of a single copy of the tTS on the mitotic stability of the HAC, three different transgene cassettes were constructed (Figure 4a), and isogenic human HT1080 cell lines containing the different constructs loaded into the alphoidtetO-HAC were generated. Two cassettes contained the tTS and color markers, either EYFP or DsRed2 (constructs #3 and #4). The tTS is fused to EYFP in construct #4. In construct #3, the DsRed2 and tTS sequences are transcribed from the same promoter but are separated by an internal ribosome entry site (IRES) sequence(Supplementary Figure S1). A control cassette containing the tetR that is co-expressed with DsRed2 via an intervening IRES was also constructed (construct #2) (Supplementary Figure S1). All cassettes encode a fusion protein with a high affinity for the multiple tetO sequences in the alphoid array of the HAC. This binding to tetO sequences is negatively regulated by doxycycline. The loxP-targeted region in the HAC contains the drug-resistance marker hygromycin (Hygro), the 5′-end of the HPRT gene and the thymidine kinase gene of herpes simplex virus 1 (HS-tk) (35). After insertion of the transgene cassettes into the HAC and reconstitution of the HPRT gene, the order and orientation of the genes is as shown in Figure 4b. Importantly, this targeting site was not flanked by insulator elements. All cell lines were isolated in the presence of doxycycline to prevent tTS binding with the alphoidtetO array.
Figure 4.

Loading of tTS-containing cassettes into the alphoidtetO-HAC propagated in human cells. (a) Diagram of the transgene cassettes used in this study. All cassettes contain a 3′ HPRT sequence and loxP site. Control constructs are marked by either EGFP alone (construct #1) or DsRed2 that is co-transcribed with tetR (constructs #2). Experimental constructs contain the tTS (tetR with the KRAB-AB) either co-transcribed with DsRed2 (construct #3) or fused to EYFP (constructs #4). All constructs were inserted into the loxP site of the alphoidtetO-HAC propagated in HPRT-deficient HT1080 cells. (b) A map of the resulting transgene cluster in the HAC containing TK, Hygro and HPRT genes and a color marker. Arrows indicate direction of transcription of the transgenes. (c) Mitotic stability of the HAC carrying different transgene cassettes after growing the cells in the presence or absence of doxycycline. Two control clones containing the alphoidtetO-HAC in human HT1080 cells were checked for HAC loss after transfection with a retroviral vector carrying the tTS. Two HAC-containing clones for each construct (#2, #3 and #4) were checked in human cells. Two clones for construct #4 and one clone for construct #1 were also checked in CHO cells. Each clone was grown medium either with (open bars) or without (black bars) doxycycline.
To evaluate the effect of a single copy of the tTS (either fused to EYFP or co-expressed with DsRed2) on HAC stability, cells bearing HACs carrying different constructs were transferred into non-selective medium (HAT− and BS−) either with doxycycline (dox+) to inhibit binding of the tTS to the alphoidtetO array sequences or without doxycycline (dox−) to permit tTS binding. Cells were maintained in these media for 7, 20 and 36 days, and then FISH analysis was carried out with the probe specific for the alphoidtetO-HAC. For each construct, two randomly chosen clones were analyzed, and for each clone, at least 60 metaphases were examined. Figure 4c shows the estimated rates of HAC loss (black bars correspond to dox− versus white bars correspond to dox+ medium). As expected, the absence or presence of doxycycline had essentially no effect on stability of the HAC lacking the KRAB-AB sequence (construct #2). Expression of a tTS fusion protein from constructs #3 and #4 caused little, if any, increase in the population of cells lacking the HAC when the cells were grown in the absence of doxycycline compared with the HAC loss induced by expression of tTS from a retrovirus vector. Similar data were also obtained for hamster cells carrying the tTS-containing HAC (Figure 4c). Altogether, these data indicate that the expression of a single copy of the tTS-containing cassette loaded into the alphoidtetO-HAC does not induce significant destabilization of the HAC.
Expression of a single copy of tTS induces silencing of transgenes in the alphoidtetO-HAC
In accordance with the prediction that the tTS could potentially induce an autoregulated fluctuating repressive heterochromatin-like state on the alphoidtetO-HAC, we observed a quick disappearance of the fluorescence signal expressed from the tTS-EYFP or tTS-DsRed2 cassette when the cells were grown in the absence of doxycycline (Figure 5a and b). This silencing occurred in ∼93% of cells after culturing for 7–10 days without the drug. A similar phenomenon of transgene silencing in dox− medium without noticeable loss of the HAC was observed in CHO cells (Figure 5c). Because in both cassettes, the sequence encoding a fluorescent protein is either fused or co-expressed with the tTS sequence, this observation reveals a progressive decrease in the tTS protein under conditions when this protein binds to tetO sequences.
Figure 5.

(a) A schematic representation of the EYFP signal in the cells cultured in different media. (b) Fluorescence images of HT1080 cells with the HAC carrying the tTS-EYFP cassette (construct #4) cultured in dox− media and after adding doxycycline followed by 12 days of culture. Cells were cultured without HAC selection (in HAT− and BS− medium). (c) Fluorescence images of CHO cells with the alphoidtetO-HAC carrying the tTS-IRES-DsRed2 cassette (construct #3) cultured in dox+ and dox− media. Cells were cultured without HAC selection (in HAT− and BS− medium) for 30 days.
To further characterize the autoregulation of tTS expression, the dynamics of tTS-EYFP expression (construct #4) from the alphoidtetO -HAC in human cells were analyzed in dox+ and dox− media by measuring the mean green fluorescence (FACS) in the cell population. Each measurement was the average of values for three independent experiments. Figure 6a describes the protocol for time-course analysis of tTS-EYFP protein expression in the cells grown in different media. tTS-EYFP transgene silencing was relatively stable because even after 22 days of culture in dox− medium, the tTS-EYFP expression remained suppressed (Figure 6a and b; sample 4). After growing for one additional month in the presence of blastocidine (i.e. under selection for the HAC), no recovery of transgene expression was observed (Figure 6a and b; sample 5). At this point, FISH analysis was carried out again, revealing the presence of an autonomous alphoidtetO-HAC in 92% of cells. Remarkably, this tTS-induced silencing of the EYFP transgene was reversible. This was observed when the cells with no detectable EYFP signal were transferred to medium with doxycycline. After the cells were cultured without selection for 24 h, the EYFP signal was partially regained (Figure 6a and b; sample 7). The fraction of cells expressing tTS-EYFP gradually increased after 6 and 12 days in dox+ culture (Figure 6a and b; samples 8 and 9).
Figure 6.

Doxycycline regulated expression of the tTS-EYFP transgene loaded into the HAC. (a) A diagram illustrating time-course analysis of tTS-EYFP expression in the cells grown in different media. Samples 1–11 correspond to cultures used for FACS analysis. The cultures marked by red stars were analyzed by ChIP (see below). (b) Relative mean EYFP fluorescence determined by FACS of cells carrying the tTS-EYFP cassette (construct #4) grown in different media. Sample 1 corresponds to control HT1080 cells without the EYFP transgene (fluorescence background). Sample 2 corresponds to the cells with the HAC carrying the tTS-EYFP cassette grown in HAT and dox+ medium for 30 days. Sample 3 corresponds to the cells grown for 22 days in dox+ medium without selection. Sample 4 corresponds to the cells grown for 22 days without doxycycline. Sample 5 corresponds to the cells grown for a month in dox− medium with BS selection. Sample 6 corresponds to the cells grown in bsr−dox− medium. Samples 7, 8 and 9 correspond to the cells grown in dox+ BS+ medium for 24 h, 6 and 12 days, correspondingly. Samples 10 and 11 correspond to the cells treated either TSA or SAHA in dox+ medium. Error bars, SD (n = 3). (c) Transcription of Hygro, TK, EGFP and HPRT genes from the HAC. The level of the transgene transcripts from cells cultured in dox+ or dox− medium was analyzed by RT–PCR. The housekeeping gene, BRCA1, was used as an internal control. Lane 1 (dox−) and lane 2 (dox+) correspond to transcripts for BRCA1. Lane 3 (dox−) and lane 4 (dox+) correspond to transcripts for Hygro. Lane 5 (dox−) and lane 7 (dox+) correspond to transcripts for TK. Lane 6 (dox−) and lane 8 (dox+) correspond to transcripts for HPRT. Lane 9 (dox−) and lane 10 (dox+) correspond to transcripts for EGFP. Lane M- GeneRuler™ 1-kb DNA ladder. (d) ChIP analysis of H3K4me3 chromatin in the transgene cassette of the HAC in the presence and absence of doxycycline. The cell samples used for ChIP correspond to samples 2 and 6 in Figure 6a and b. Enrichment is shown relative to the 5S rRNA control locus. Satellite 2 sequence, Sat2, corresponding endogenous pericentromeric repeats was included as a negative control. (e) ChIP analysis of CENP-A chromatin in the HAC in the presence and absence of doxycycline. Enrichment is shown relative to the chromosome 21 centromere.
We also examined whether other genes, Hygro, HS-tk and HPRT, located in the same region of the HAC (Figure 4b) were also silenced when the cells were cultured in the absence of doxycycline. Using pairs of primers specific to Hygro, HS-tk and HPRT genes (Supplementary Table S1), RT–PCR analysis revealed bright bands of the expected size (265, 267 and 449 bp, respectively) in dox+ medium (Figure 6c; lanes 4, 7 and 8). Either no or faint bands (on the level of background) were observed in dox− medium (Figure 6c; lanes 3, 5 and 6). As a control, expression of the genomic BRCA1 gene was included. Using a pair of specific primers for exons 15–21 of BRCA1 (Supplementary Table S1), almost equally bright bands of 893 bp were detected in dox+ and dox− media (Figure 6c; lanes 1 and 2). As expected, for EGFP, the faint and bright 483-bp bands were observed in dox− and dox+ media, respectively (Figure 6c; lanes 9 and 10). Thus, we concluded that binding of the tTS-EYFP to tetO sequences silences genes located in regions neighboring the loxP site of the alphoidtetO -HAC.
Based on known mechanisms of gene silencing induced by the tTS (44,45), downregulation of the entire gene cluster in the alphoidtetO-HAC is likely to be caused by heterochromatinization of this region initiated from nearby tetO sequences. Indirect support for this conclusion came from experiments in which cells with silenced transgenes were treated with the histone deacetylase inhibitors, TSA and SAHA (46). tTS-EYFP expression was re-activated when these inhibitors were added to the culture (Figure 6a and b; samples 10 and 11 compared with sample 7), suggesting that an increase of the acetylation level of histone H3 in the HAC favors transcription of the transgene.
Chromatin state of different regions of the alphoidtetO-HAC in the presence and absence of doxycycline
ChIP analysis was used to detect chromatin architecture changes in the alphoidtetO-HAC DNA in the presence and absence of tTS binding. Specifically, we analyzed the chromatin structure of the HAC region containing the tTS-EYFP, Hygro and HS-tk genes, as well as the Bsr gene (repeated ∼30–40 times within the mega-size alphoid DNA array). For this experiment, cells carrying the HAC with construct #4 (tTS-EYFP) were grown either in HAT and dox+ or in dox− medium (Figure 6a; samples 2 and 6). ChIP analysis revealed that H3K4me3, a marker for active gene promoters, was associated with all three transgene sequences when the cells were cultured in dox+ medium (Figure 6d). The level of H3K4me3 was higher at tTS-EYFP than that at HS-tk and Hygro. This may be due to the presence of two copies of the cHS4 insulator upstream and downstream of tTS-EYFP. At the same time, the level of H3K4me3 on the transgene sequences dropped dramatically after removal of doxycycline (Figure 6d), suggesting that the insulator sequences do not protect expression of tTS-EYFP. A similar dynamic was observed for the Bsr gene sequences scattered throughout the megabase-size alphoid array. The Bsr gene sequences were more enriched in H3K4me3 when doxycycline was added into the medium compared with when doxycycline was omitted. These results clearly indicate that when tTS-EYFP cannot bind to alphoidtetO repeats, the megabase-size synthetic array in the HAC seems to form a more open chromatin structure. Thus, based on these results, we hypothesize that when the tTS-EYFP protein binds to tetO sequences after the removal of doxycycline, the heterochromatin formed on the alphoidtetO DNA array of the HAC spreads, leading to transcriptional repression of transgenes located adjacent to the gene-loading site. ChIP analysis also demonstrated that tTS-induced silencing of the transgenes does not affect the structure of centrochromatin in the HAC. Specifically, no significant difference in the level of CENP-A was observed when cells were grown in the presence or absence of doxycycline. Immunoprecipitation with antibodies against CENP-A yielded an enrichment for the alphoidtetO repeats in both cultures comparable with that seen at the control endogenous centromere of the chromosome 21 (Figure 6e). These results are in agreement with mitotic stability of the HAC when the cells are grown in the absence of doxycycline.
Development of an improved system to verify phenotypic changes attributed to expression of HAC-encoded genes
The phenomenon of transgene silencing by a single copy of the tTS can be used to develop a novel system to verify phenotypes induced by genes inserted into the alphoidtetO-HAC. This could be achieved by either modification of the original HAC vector (insertion of the tTS-containing module) or loading of the chromatin modifier cassette into the HAC along with a gene of interest. In this work, we exploited the second approach.
Previously the retrofitting vector pJBRV1 was constructed for loading full-length genes isolated by TAR cloning (47–49) into the alphoidtetO-HAC (37). Here, we have constructed a new retrofitting vector, BRV-tTS-EYFP, that is applicable for the regulated expression of TAR-isolated genes in the HAC (Figure 7a). The BRV-tTS-EYFP vector contains a 3′ HPRT-loxP cassette, allowing gene loading into a unique loxP site of the alphoidtetO-HAC, an independently expressed color marker, DsRed2, that reports either the inhibition or expression of a gene of interest loaded onto the HAC, and a single copy of the tTS fused to EYFP. In addition, similar to pJBRV1, the new vector contains two targeting sequences (hooks) that have homology to the TAR vector pVC604 used to clone full-length genes from complex genomes (42,49). After target gene retrofitting and insertion into the HAC, the tTS-EYFP fusion is constitutively expressed from the HAC. In the presence of doxycycline, the tTS-EYFP does not bind to the tetO sequences in the alphoidtetO-HAC. Under these conditions, the HAC has open chromatin permissive for expression of the DsRed2 color marker and a gene of interest. After doxycycline removal, the tTS-EYFP fusion binds to tetO sequences of the HAC, resulting in heterochromatin formation and silencing of the two color markers and a target gene (Figure 7b). As a proof of principle, the BRV-tTS-EYFP vector was inserted into the loxP site of the alphoidtetO-HAC in human HPRT-deficient HT1080 cells, and expression of two color markers was analyzed. The predicted loss of the fluorescence signals of tTS-EYFP and DsRed2 without detectable loss of the HAC was observed in three of the three selected clones cultured in the absence of doxycycline after 7–10 days (Supplementary Figure S3). Data for one representative clone are shown in Figure 7c–e.
Figure 7.

Use of the retrofitting vector BRV-tTS-EYFP to regulate expression of a gene loaded into the HAC by doxycycline. (a) Diagram of the retrofitting vector BRV-tTS-EYFP. The vector contains two targeting sequences (hooks) that are released by BsiWI endonuclease digestion. In vivo recombination in yeast between the vector and a YAC containing the gene of interest (isolated by TAR cloning) leads to assembly of a circular YAC/BAC molecule containing the gene of interest plus the tTS cassette all of which can be inserted into the HAC by Cre-lox recombination. (b) Inactivation of a gene expression by modulating the epigenetic status of the underlying HAC kinetochore chromatin. The BRV-tTS-EYFP vector along with a gene of interest is inserted into the single loxP site of the alphoidtetO-HAC. In the presence of doxycycline, the tTS cannot bind to tetO sequences in the HAC, and this region of the HAC is transcriptionally active. Removal of doxycycline leads to tTS-induced heterochromatinization of this region followed by gene silencing. (c) Fluorescence images of HT1080 cells with the HAC carrying the BRV-tTS-EYFP vector cultured in dox+ and dox− media followed by 10 days of culture without HAC selection (in HAT−, BS− medium). (d) Relative mean EYFP fluorescence of cells carrying the BRV-tTS-EYFP vector (FACS). (e) Mitotic stability of the HAC measured by FISH after growing the cells in the presence or absence of doxycycline. Open bars correspond to the frequency of HAC loss when the cells were grown in the presence of doxycycline. Black bars correspond to the frequency of HAC loss when doxycycline was excluded from the medium.
To summarize, gene loading into the alphoidtetO-HAC using the BRV-tTS-EYFP vector allows us to control gene expression from the HAC and thereby to verify gene-induced phenotypes without detectable HAC loss from the cell.
DISCUSSION
The alphoidtetO-HAC carrying a megabase-size synthetic, high-order alpha-satellite repeat array with a tetO in every other alphoid monomer represents an advanced HAC-based system for regulated gene delivery. The main advantage of this HAC is that its kinetochore is conditional and can be inactivated by tethering of the tTS silencer or other repressive activities (29–32,35). This feature provides a level of control for phenotypes induced by a gene loaded into the HAC that is important for gene function studies.
In our previous studies, induction of alphoidtetO-HAC loss was initiated by transfection of cells by viral or plasmid vectors expressing transcriptional repressors that efficiently induce heterochromatin formation. This transfection step is potentially mutagenic and might want to be avoided under some circumstances. In the present study, we demonstrated that the expression of a single copy of the tTS-containing cassette from the HAC silences HAC-encoded genes without inactivating the HAC kinetochore. This process likely involves the formation of a limited number of local heterochromatin domains as a consequence of the presence of effector (tTS) and target sites (tetO) on the same DNA molecule. Such a configuration generates a self-regulating fluctuating repressive heterochromatin-like state that is apparently not sufficiently deep to inactivate the HAC kinetochore. However, the level of heterochromatinization reached on the alphoidtetO array is sufficient to induce transcriptional silencing of transgenes loaded onto the HAC. It is worth noting that the observed phenomenon of gene silencing seems to be caused by a unique combination of the location of the gene-loading site (in the middle of a megabase-size alphoid DNA array), and the positions of tetO-recognition sites in the array that are accessible for binding with the tTS silencer.
The presence of the color markers, DsRed2 and EYFP (the second of which is present as a chimeric fusion to the tTS suppresser itself), in the tTS-containing cassettes allowed us to examine several parameters of heterochromatin seeding. Expression of the tTS-EYFP or DsRed2 protein was detectably suppressed after growing the cells for 4–5 days without doxycycline (i.e. under conditions when tTS-EYFP binds to tetO sequences in the HAC). Taking into account that tTS-EYFP is a relatively stable protein (50), this suggests that removal of doxycycline results in relatively quick silencing of the tTS-EYFP transcription. Such rapid gene silencing has been previously reported for constructs with tetO-binding sites located immediately upstream of a promoter sequence (44–45). In our case, the nearest tetO sequences are ∼15 kb up and 15 kb down from the tTS-EYFP transgene and are separated from tTS-EYFP by three other functional transgenes, HS-tk, Hygro and HPRT (Figure 4b). Indeed, most of the tetO sequences are located hundreds of kilobases away from the transgene. We found that the other transgenes were also inactivated by tTS-induced heterochromatinization initiated on the synthetic tetO-alphoid DNA array. Importantly, inactivation of the tTS-EYFP and other neighboring transgenes on the HAC was reversed by the addition of doxycycline. Thus, these observations indicate that tTS binding induces a repressive heterochromatin-like state that can be converted to transcriptionally permissive chromatin when tTS binding is prevented. It should be noted that the observed transgene silencing and its re-activation in the HAC is in sharp contrast to previously described situations, where transgenes become silenced in cell lines and transgenic animals (44,45). In all published works, tTS was constitutively expressed, and transgenes regulation was accomplished by the changing of affinity of an effector to a target. In our case, tTS regulates its own expression and, as a consequence, the expression of neighboring genes loaded onto the HAC.
Altogether, our findings demonstrate that expression of a single autoregulated copy of the tTS from the alphoidtetO-HAC provides an efficient method to switch off expression of the transgenes loaded onto the HAC, thus allowing verification of gene phenotypes by regulated removal of the relevant gene product. The additional advantage of this system is the possibility to coordinately silence groups of expressed genes, presumably because of robust silencing spreading from the alphoidtetO array. Another potential advantage of gene silencing in the alphoidtetO-HAC carrying a single copy of tTS is that the procedure does not lead to changes in the number of chromosomes in the cell.
One advantage of HAC-based technologies is the opportunity to work with full-length genes. TAR cloning allows selective isolation of any desired allele of a gene of interest as a pre-determined chromosomal fragment in yeast (47–49). The new retrofitting vector, BRV-tTS-EYFP, described here links this gene isolation strategy with the regulated gene expression in the alphoidtetO-HAC. A hypothetical scheme illustrating the entire process, including verification of gene-induced phenotypes, is diagrammed in Supplementary Figure S4.
In addition to gene expression experiments, the HAC-based system described here may be useful for comparative studies of chromatin barrier elements. The level of protection of the tTS-EYFP transgene by flanking sequences may be used to evaluate the activity of several barrier elements. This approach is currently being used to compare new barrier elements identified in our laboratory (39,40). Potentially, the phenomenon of tTS-EYFP silencing may be also exploited to screen drugs affecting de-condensation of heterochromatin. This is supported by our pilot experiments with inhibitors of histone deacetylases, SAHA and TSA (manuscript in preparation).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–4.
FUNDING
Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, USA (to V.L.); Wellcome Trust of which W.C.E. is a Principal Research Fellow [073915]. Funding for open access charge: Intramural funds of the US Department of Health and Human Services (to National Library of Medicine).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr A. Samoshkin for help in Southern-blot hybridization experiments, Dr Hee-Sheung Lee for help in cell culturing and Dr E. Pak and Dr E. Leo for valuable advice regarding FISH and FACS analyses, correspondingly. The authors also thank the CRC, LRBGE Fluorescence Imaging Facility (NIH) and personally Dr Karpova and Dr McNally for instructions, consultations and help with the usage of a DeltaVision microscopy imaging system.
REFERENCES
- 1.Saffery R, Choo KH. Strategies for engineering human chromosomes with therapeutic potential. J. Gene. Med. 2002;4:5–13. doi: 10.1002/jgm.236. [DOI] [PubMed] [Google Scholar]
- 2.Basu J, Willard HF. Human artificial chromosomes: potential applications and clinical considerations. Pediatr. Clin. North. Am. 2006;53:843–53. doi: 10.1016/j.pcl.2006.08.013. viii. [DOI] [PubMed] [Google Scholar]
- 3.Monaco ZL, Moralli D. Progress in artificial chromosome technology. Biochem. Soc. Trans. 2006;34(Pt. 2):324–327. doi: 10.1042/BST20060324. [DOI] [PubMed] [Google Scholar]
- 4.Ren X, Tahimic CG, Katoh M, Kurimasa A, Inoue T, Oshimura M. Human artificial chromosome vectors meet stem cells: new prospects for gene delivery. Stem Cell Rev. 2006;2:43–50. doi: 10.1007/s12015-006-0008-9. [DOI] [PubMed] [Google Scholar]
- 5.Oshimura M, Katoh M. Transfer of human artificial chromosome vectors into stem cells. Reprod. Biomed. 2008;16:57–69. doi: 10.1016/s1472-6483(10)60557-3. [DOI] [PubMed] [Google Scholar]
- 6.Kazuki Y, Oshimura M. Human artificial chromosomes for gene delivery and the development of animal models. Mol. Ther. 2011;19:1591–1601. doi: 10.1038/mt.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikeno M, Suzuki N. Construction and use of a bottom-up HAC vector for transgene expression. Methods Mol. Biol. 2011;738:101–110. doi: 10.1007/978-1-61779-099-7_7. [DOI] [PubMed] [Google Scholar]
- 8.Li CM, Park JH, Simonaro CM, He X, Gordon RE, Friedman AH, Ehleiter D, Paris F, Manova K, Hepbildikler S, et al. Insertional mutagenesis of the mouse acid ceramidase gene leads to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes. Genomics. 2002;79:218–224. doi: 10.1006/geno.2002.6686. [DOI] [PubMed] [Google Scholar]
- 9.Li Z, Düllmann J, Schiedlmeier B, Schmidt M, von Kalle C, Meyer J, Forster M, Stocking C, Wahlers A, Frank O, et al. Murine leukemia induced by retroviral gene marking. Science. 2002;296:497. doi: 10.1126/science.1068893. [DOI] [PubMed] [Google Scholar]
- 10.Raper SE, Chirmule N, Lee FS, Wivel NA, Bagg A, Gao GP, Wilson JM, Batshaw ML. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003;80:148–158. doi: 10.1016/j.ymgme.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 11.Odom GL, Gregorevic P, Chamberlain JS. Viral-mediated gene therapy for the muscular dystrophies: successes, limitations and recent advances. Biochim. Biophys. Acta. 2007;1772:243–262. doi: 10.1016/j.bbadis.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, Down J, Denaro M, Brady T, Westerman K, et al. Transfusion independence and HMGA2 activation after gene therapy of human ß-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basu J, Compitello G, Stromberg G, Willard HF, Van Bokkelen G. Efficient assembly of de novo human artificial chromosomes from large genomic loci. BMC Biotechnol. 2005;5:21. doi: 10.1186/1472-6750-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Auriche C, Carpani D, Conese M, Caci E, Zegarra-Moran O, Donini P, Ascenzioni F. Functional human CFTR pro,duced by a stable minichromosome. EMBO Rep. 2002;3:862–868. doi: 10.1093/embo-reports/kvf174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rocchi L, Braz C, Cattani S, Ramalho A, Christan S, Edlinger M, Ascenzioni F, Laner A, Kraner S, Amaral M, et al. Escherichia coli-cloned CFTR loci relevant for human artificial chromosome therapy. Hum. Gene Ther. 2010;21:1077–1092. doi: 10.1089/hum.2009.225. [DOI] [PubMed] [Google Scholar]
- 16.Breman AM, Steiner CM, Slee RB, Grimes BR. Input DNA ratio determines copy number of the 33 kb Factor IX gene on de novo human artificial chromosomes. Mol. Ther. 2008;1:315–323. doi: 10.1038/sj.mt.6300361. [DOI] [PubMed] [Google Scholar]
- 17.Yamada H, Li YC, Nishikawa M, Oshimura M, Inoue T. Introduction of a CD40L genomic fragment via a human artificial chromosome vector permits cell-type-specific gene expression and induces immunoglobulin secretion. J. Hum. Genet. 2008;53:447–453. doi: 10.1007/s10038-008-0268-0. [DOI] [PubMed] [Google Scholar]
- 18.Kazuki Y, Hoshiya H, Kai Y, Abe S, Takiguchi M, Osaki M, Kawazoe S, Katoh M, Kanatsu-Shinohara M, Inoue K, et al. Correction of a genetic defect in multipotent germline stem cells using a human artificial chromosome. Gene Ther. 2008;15:617–624. doi: 10.1038/sj.gt.3303091. [DOI] [PubMed] [Google Scholar]
- 19.Kuroiwa Y, Tomizuka K, Shinohara T, Kazuki Y, Yoshida H, Ohguma A, Yamamoto T, Tanaka S, Oshimura M, Ishida I. Manipulation of human minichromosomes to carry greater than megabase-sized chromosome inserts. Nat. Biotechnol. 2000;18:1086–1090. doi: 10.1038/80287. [DOI] [PubMed] [Google Scholar]
- 20.Ikeno M, Inagaki H, Nagata K, Morita M, Ichinose H, Okazaki T. Generation of human artificial chromosomes expressing naturally controlled guanosine triphosphate cyclohydrolase I gene. Genes Cells. 2002;7:1021–1032. doi: 10.1046/j.1365-2443.2002.00580.x. [DOI] [PubMed] [Google Scholar]
- 21.Kuroiwa Y, Kasinathan P, Sathiyaseelan T, Jiao JA, Matsushita H, Sathiyaseelan J, Wu H, Mellquist J, Hammitt M, Koster J, et al. Antigen-specific human polyclonal antibodies from hyperimmunized cattle. Nat. Biotechnol. 2009;27:173–181. doi: 10.1038/nbt.1521. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki N, Nishii K, Okazaki T, Ikeno M. Human artificial chromosomes constructed using the bottom-up strategy are stably maintained in mitosis and efficiently transmissible to progeny mice. J. Biol. Chem. 2006;281:26615–26623. doi: 10.1074/jbc.M603053200. [DOI] [PubMed] [Google Scholar]
- 23.Ito M, Ikeno M, Nagata H, Yamamoto T, Hiroguchi A, Fox IJ, Miyakawa S. Treatment of nonalbumin rats by transplantation of immortalized hepatocytes using artificial human chromosome. Transplant. Proc. 2009;41:422–424. doi: 10.1016/j.transproceed.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 24.Voet T, Schoenmakers E, Carpentier S, Labaere C, Marynen P. Controlled transgene dosage and PAC-mediated transgenesis in mice using a chromosomal vector. Genomics. 2003;82:596–605. doi: 10.1016/s0888-7543(03)00112-5. [DOI] [PubMed] [Google Scholar]
- 25.Hoshiya H, Kazuki Y, Abe S, Takiguchi M, Kajitani N, Watanabe Y, Yoshino T, Shirayoshi Y, Higaki K, Messina G, et al. A highly stable and nonintegrated human artificial chromosome (HAC) containing the 2.4 Mb entire human dystrophin gene. Mol. Ther. 2009;17:309–317. doi: 10.1038/mt.2008.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazuki Y, Hiratsuka M, Takiguchi M, Osaki M, Kajitani N, Hoshiya H, Hiramatsu K, Yoshino T, Kazuki K, Ishihara C, et al. Complete genetic correction of iPSs cells from Duchenne muscular dystrophy. Mol. Ther. 2010;18:386–393. doi: 10.1038/mt.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tedesco FS, Hoshiya H, D'Antona G, Gerli MF, Messina G, Antonini S, Tonlorenzi R, Benedetti S, Berghella L, Torrente Y, et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Transl. Med. 2011;3 doi: 10.1126/scitranslmed.3002342. 96ra78. [DOI] [PubMed] [Google Scholar]
- 28.Tedesco FS, Gerli MF, Perani L, Benedetti S, Ungaro F, Cassano M, Antonini S, Tagliafico E, Artusi V, Longa E, et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003541. 140ra89. [DOI] [PubMed] [Google Scholar]
- 29.Nakano M, Cardinale S, Noskov VN, Gassmann R, Vagnarelli P, Kandels-Lewis S, Larionov V, Earnshaw WC, Masumoto H. Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev. Cell. 2008;14:507–522. doi: 10.1016/j.devcel.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cardinale S, Bergmann JH, Kelly D, Nakano M, Valdivia MM, Kimura H, Masumoto H, Larionov V, Earnshaw WC. Hierarchical inactivation of a synthetic human kinetochore by a chromatin modifier. Mol. Biol. Cell. 2009;20:4194–4204. doi: 10.1091/mbc.E09-06-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergmann JH, Rodríguez MG, Martins NM, Kimura H, Kelly DA, Masumoto H, Larionov V, Jansen LE, Earnshaw WC. Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. EMBO J. 2011;30:328–340. doi: 10.1038/emboj.2010.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bergmann JH, Jakubsche JN, Martins NM, Kagansky A, Nakano M, Kimura H, Kelly DA, Turner BM, Masumoto H, Larionov V, et al. Epigenetic engineering: histone H3K9 acetylation is compatible with kinetochore structure and function. J. Cell Sci. 2012;125(Pt. 2):411–421. doi: 10.1242/jcs.090639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohzeki J, Bergmann JH, Kouprina N, Noskov VN, Nakano M, Kimura H, Earnshaw WC, Larionov V, Masumoto H. Breaking the HAC Barrier: Histone H3K9 acetyl/methyl balance regulates CENP-A assembly. EMBO J. 2012;31:2391–2402. doi: 10.1038/emboj.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bergmann JH, Martins NMC, Larionov V, Masumoto H, Earnshaw WC. HACking the centromere chromatin code: in sights from human artificial chromosomes. Chromosome Res. 2012;20:505–519. doi: 10.1007/s10577-012-9293-0. [DOI] [PubMed] [Google Scholar]
- 35.Iida Y, Kim JH, Kazuki Y, Hoshiya H, Takiguchi M, Hayashi M, Erliandri I, Lee HS, Samoshkin A, Masumoto H, et al. Human artificial chromosome with a conditional centromere for gene delivery and gene expression. DNA Res. 2010;17:293–301. doi: 10.1093/dnares/dsq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kouprina N, Samoshkin A, Erliandri I, Nakano M, Lee HS, Fu H, Iida Y, Aladjem M, Oshimura M, Masumoto M, et al. Organization of synthetic alphoid DNA array in human artificial chromosome (HAC) with a conditional centromere. ACS Synth. Biol. 2012;1:590–601. doi: 10.1021/sb3000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JH, Kononenko A, Erliandri I, Kim TA, Nakano M, Iida Y, Barrett JC, Oshimura M, Masumoto H, Earnshaw WC, et al. Human artificial chromosome (HAC) vector with a conditional centromere for correction of genetic deficiencies in human cells. Proc. Natl Acad. Sci. USA. 2011;108:20048–20053. doi: 10.1073/pnas.1114483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koi M, Shimizu M, Morita H, Yamada H, Oshimura M. Construction of mouse A9 clones containing a single human chromosome tagged with neomycin-resistance gene via microcell fusion. Jpn. J. Cancer Res. 1989;80:413–418. doi: 10.1111/j.1349-7006.1989.tb02329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim JH, Ebersole T, Kouprina N, Noskov VN, Ohzeki JI, Noskov VN, Ohzeki J, Masumoto H, Mravinac B, Sullivan BA, et al. Human pericentromeric gamma-satellite DNA maintains open chromatin structure and protects a transgene from epigenetic silencing at an ectopic site. Genome Res. 2009;19:533–544. doi: 10.1101/gr.086496.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ebersole T, Kim JH, Samoshkin A, Kouprina N, Pavlicek A, White R, Larionov V. tRNA genes protect a reporter gene from epigenetic silencing in mouse cells. Cell Cycle. 2011;10:2729–2791. doi: 10.4161/cc.10.16.17092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dafhnis-Calas F, Xu Z, Haines S, Malla SK, Smith MC, Brown WR. Iterative in vivo assembly of large and complex transgenes by combining the activities of phiC31 integrase and Cre recombinase. Nucleic Acids Res. 2005;33:e189. doi: 10.1093/nar/gni192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kouprina N, Annab L, Graves J, Afshari C, Barrett JC, Resnick MA, Larionov V. Functional copies of a human gene can be directly isolated by transformation-associated recombination cloning with a small 3' end target sequence. Proc. Natl Acad. Sci. USA. 1998;95:4469–4474. doi: 10.1073/pnas.95.8.4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lugo TG, Baker RM. Chromosome-mediated gene transfer of HPRT and APRT in an intraspecific human cell system. Somatic Cell Genet. 1983;9:175–188. doi: 10.1007/BF01543176. [DOI] [PubMed] [Google Scholar]
- 44.Freundlieb S, Schirra-Müller C, Bujard H. A tetRacycline controlled activation/repression system with increased potential for gene transfer into mammalian cells. J. Gene Med. 1999;1:4–12. doi: 10.1002/(SICI)1521-2254(199901/02)1:1<4::AID-JGM4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 45.Groner AC, Meylan S, Ciuffi A, Zangger N, Ambrosini G, Dénervaud N, Bucher P, Trono D. KRAB–Zinc Finger Proteins and KAP1 Can Mediate Long-Range Transcriptional Repression through Heterochromatin Spreading. PLoS Genet. 2010;6:e1000869. doi: 10.1371/journal.pgen.1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chavan AV, Somani RR. HDAC inhibitors - new generation of target specific treatment. Mini Rev. Med. Chem. 2010;10:1263–1276. doi: 10.2174/13895575110091263. [DOI] [PubMed] [Google Scholar]
- 47.Larionov V, Kouprina N, Graves J, Chen XN, Korenberg JR, Resnick MA. Specific cloning of human DNA as yeast artificial chromosomes by transformation-associated recombination. Proc. Natl Acad. Sci. USA. 1996;93:491–496. doi: 10.1073/pnas.93.1.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kouprina N, Larionov V. TAR cloning: insights into gene function, long-range haplotypes and genome structure and evolution. Nat. Rev. Genet. 2006;7:805–812. doi: 10.1038/nrg1943. [DOI] [PubMed] [Google Scholar]
- 49.Kouprina N, Larionov V. Selective isolation of genomic loci from complex genomes bytransformation-associated recombination cloning in the yeast Saccharomyces cerevisiae. Nat. Protoc. 2008;3:371–377. doi: 10.1038/nprot.2008.5. [DOI] [PubMed] [Google Scholar]
- 50.Elizabeth M, Burns EM, Christopoulou L, Corish P, Tyler-Smith C. Quantitative measurement of mammalian chromosome mitotic loss rates using the green fluorescent protein. J. Cell Sci. 1999;112:2705–2714. doi: 10.1242/jcs.112.16.2705. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.