


Abstract
Bacillus subtilis has two replicative DNA polymerases. PolC is a processive high-fidelity replicative polymerase, while the error-prone DnaEBs extends RNA primers before hand-off to PolC at the lagging strand. We show that DnaEBs interacts with the replicative helicase DnaC and primase DnaG in a ternary complex. We characterize their activities and analyse the functional significance of their interactions using primase, helicase and primer extension assays, and a ‘stripped down’ reconstituted coupled assay to investigate the coordinated displacement of the parental duplex DNA at a replication fork, synthesis of RNA primers along the lagging strand and hand-off to DnaEBs. The DnaG–DnaEBs hand-off takes place after de novo polymerization of only two ribonucleotides by DnaG, and does not require other replication proteins. Furthermore, the fidelity of DnaEBs is improved by DnaC and DnaG, likely via allosteric effects induced by direct protein–protein interactions that lower the efficiency of nucleotide mis-incorporations and/or the efficiency of extension of mis-aligned primers in the catalytic site of DnaEBs. We conclude that de novo RNA primer synthesis by DnaG and initial primer extension by DnaEBs are carried out by a lagging strand–specific subcomplex comprising DnaG, DnaEBs and DnaC, which stimulates chromosomal replication with enhanced fidelity.
INTRODUCTION
The bacterial DNA polymerase III holoenzyme comprises several proteins and is an integral part of the DNA replication machinery known as the replisome. At the core of the replisome is a DNA polymerase activity, which copies the parental strands to synthesize nascent DNA. In the Gram-negative model organism Escherichia coli, a DnaE-type polymerase (known as the α subunit) forms a core heterotrimer with the 3′–5′ proofreading exonuclease ε (1) and the polypeptide θ the function of which is not known (2). These proteins are collectively responsible for the accurate synthesis of nascent DNA (3,4). The Gram-positive model organism Bacillus subtilis and the rest of the Firmicutes (low-G+C-content Gram-positive bacteria) use two essential DNA polymerases, DnaEBs and PolC, during replication (5–7). In vivo, PolC is the main replicative polymerase, while the DnaE polymerase acts in lagging strand synthesis. In a fully reconstituted replication assay, the leading strand is copied by PolC and the lagging strand by PolC and DnaEBs, with DnaEBs acting to extend RNA primers before handing them off to PolC, in a manner analogous to the eukaryotic pol α, which extends RNA primers at the lagging strand before handing them off to the more processive pol δ for bulk DNA synthesis (3,7–9). In addition to DNA replication, DnaEBs is involved in DNA repair/mutagenesis (10).
In E. coli, the DnaE-type Pol α is attached to the replisome via direct interactions with a C-terminal domain (CTD, Cτ) of the clamp loader protein τ, the product of the dnaX gene (11). A shorter variant of τ is produced in Gram-negative bacteria, either by a translational frameshift (12) or a transcriptional slippage (13), resulting in the γ polypeptide, which is identical to τ but lacks the Cτ and cannot bind to Pol α. Both polypeptides are found in the trimeric τ2γ core of the clamp loader that bridges two Pol α polymerases synthesizing nascent DNA on the leading and lagging strands. In Gram-positive bacteria, only τ is present forming the τ3 core of the clamp loader (14). It is, therefore, conceivable that three polymerases are associated with the replisome in Gram-positive organisms. This notion is supported by E. coli studies where a functional triple polymerase replisome with a τ3 core has been observed in vivo (15), and reconstituted in vitro with purified proteins, including τ but in the absence of γ (4). However, the B. subtilis τ interacts only with the PolC and not with the DnaEBs (14,16). It is, therefore, not known how DnaEBs is associated with the replisome and compartmentalized to the lagging strand. Unlike other replicative polymerases, the intrinsic processive replication activity of DnaEBs is too weak to fulfil DNA replication in vivo (7,10,14). This enzyme is also error-prone, causing frameshifting at a high rate (10,17). It is unclear which replisomal interactants stimulate the DnaEBs activity and fidelity to fulfil the needs of a progressing replication fork.
Here we show that DnaEBs physically interacts with four replisomal proteins in addition to DnaN and single-stranded DNA binding proteins (SSB) (18,19): the replicative helicase DnaC, the primase DnaG, the HolA subunit of the DnaX complex and PolC. We reveal that DnaC, DnaG and DnaEBs form a ternary complex when DnaC is in the ring-shaped hexameric form, indicating that this complex may operate dynamically along the lagging strand template; as the DnaC replicative helicase translocates along this strand in the 5′–3′ direction and forms a functional complex with the DnaG primase, the latter synthesizes de novo RNA primers, which emerge behind the forward moving helicase–primase complex (20–24). The RNA primers are then extended by DnaEBs (7).
Furthermore, we have characterized the individual DnaC, DnaG and DnaEBs activities and showed that their physical interactions are functionally relevant. We confirmed that DnaC requires the loader protein DnaI to assemble into a functional hexameric ring around its DNA substrate, in agreement with previous studies (25–27). We established that DnaG initiates primer synthesis preferentially at 5′-d(CTA), 5′-d(TTA) and 5′-d(TTT) sites, and its activity is stimulated by DnaC without altering the length of the primers. In addition, we showed that primer hand-off can take place within the ternary complex at a very early stage after de novo polymerization of very small RNA primers, only 2–4 ribonucleotides in size, without the requirement of other auxiliary replication proteins. Based on these data, we suggest that during lagging strand replication, the DnaEBs is recruited by the DnaC–DnaG complex to further extend the primers.
Finally, we showed that although the native DnaEBs polymerase activity is not affected within binary complexes by DnaC and/or DnaG, its fidelity is improved by both DnaC and DnaG, but not by DnaI. This modulation is nucleotide specific, suggesting that it is the result of allosteric effects induced by direct protein–protein interactions of the DnaC helicase and/or the DnaG primase with the DnaEBs polymerase. These interactions likely increase the stringency of the polymerase catalytic site, thereby lowering the frequency of nucleotide mis-incorporations and mis-aligned primer extension. Hence, the ternary DnaC–DnaG–DnaEBs complex forms a spatial adaptor that compartmentalizes DnaEBs to act along the lagging strand and enhances the stimulation of chromosomal replication with higher fidelity.
MATERIALS AND METHODS
Yeast two-hybrid constructions and assay
Sequences indicated in red in the Supplementary Table S1 (yeast two-hybrid matrix assay, see Supplementary Data) were amplified by PCR from the wild-type strain 168 genomic DNA and cloned as translational fusions with the Gal4 DNA-binding (BD) and Gal4 activation (AD) domains of vectors pGBDU-C1 (bait, Ura+) and pGAD-C1 (prey, Leu+), respectively. Constructions were then introduced by transformation into the Saccharomyces cerevisiae haploid strains PJ69-4 a (bait) and PJ69-4α (prey) and checked by DNA sequencing. Other fusions were from the laboratory collection. The two-hybrid assay was carried out according to a previously described mating strategy (16). Interactions were scored by replica-plating diploids on plates selecting for the His+ or Ade+ phenotype. Empty pGBDU-C1 and pGAD-C1 vectors were used as negative controls and to detect self-activating peptides.
Protein purifications
DnaE
DnaE (125 kDa) was expressed as an intein fusion from the pTYB3 plasmid in E. coli B834 (DE3) in the presence of 1 mM IPTG and 100 µg/ml ampicillin at 30°C for 4 h, as described before (10). Cell paste from 3 l of Luria-Bertani broth (LB) culture was suspended in 37.5 ml of TEN1000 buffer [50 mM Tris–HCl (pH 7.5), 2 mM Ethylenediaminetetraacetic acid (EDTA), 1 M NaCl]. Protease inhibitors, 1 mM phenyl methyl sulphonyl fluoride (PMSF) and 300 μL of protease inhibitor cocktail (SIGMA) were added to the cell suspension, and the cells were lysed by sonication. The bacterial extract was clarified by centrifugation (42 000g, 30 min, 4°C), and, after filtering through a 0.22-µm filter, the supernatant was applied to a 12-ml chitin column equilibrated in TEN1000. After washing the column extensively, first with TEN2000 [50 mM Tris–HCl (pH 7.5), 2 mM EDTA, 2 M NaCl] and then with TEN1000, intein-mediated self-cleavage was carried out by an on-column incubation of the immobilized fusion protein for 42 h with 100 mM DTT in TEN1000. The eluted protein was buffer exchanged into TEN50 [50 mM Tris–HCl (pH 7.5), 2 mM EDTA, 50 mM NaCl] and applied to a 26/60 Superdex S200 (GE Healthcare) gel filtration column equilibrated in TEN50. The DnaE containing fractions were pooled and applied to a Q-sepharose (GE Healthcare) equilibrated in TEN50 and eluted with a NaCl gradient up to 1 M. The DnaE containing fractions were pooled diluted in TEN0 to adjust the conductivity to 50 mM NaCl and then applied to a heparin column equlibrated in TEN50 and eluted with a NaCl gradient up to 2 M NaCl. The DnaE-containing fractions were pooled, extensively dialyzed against 50 mM Tris-HCl pH 7.5, 300 mM NaCl, 2 mM EDTA and 20% v/v glycerol, spectrophotometrically quantified (DnaE extinction coefficient, 86 530 M−1 cm−1), aliquoted and stored at −80°C.
DnaG
DnaG (68 kDa) was expressed from the pSMG11 plasmid in E. coli MCC26 in the presence of 0.5 mM IPTG and 100 μg/ml ampicillin at 25°C for 7 h. Cell paste from 2 l of LB culture was suspended in 25 ml of lysis buffer [50 mM Tris–HCl (pH 8), 1 mM EDTA, 0.5 M NaCl]. Protease inhibitors, 1 mM PMSF and 200 μL of protease inhibitor cocktail (SIGMA) were added to the cell suspension, and the cells were lysed by sonication. The bacterial extract was clarified by centrifugation (42 000g, 30 min, 4°C), and, after filtering through a 0.22-μm filter, the supernatant was applied to a 12-ml chitin column equilibrated in buffer RC [50 mM Tris–HCl (pH 8), 1 mM EDTA, 0.5 M NaCl, 1 mM DTT]. After washing the column extensively in wash buffer [50 mM Tris–HCl (pH 8), 1 mM EDTA, 1 M NaCl], intein-mediated self-cleavage was carried out by an on-column incubation of the immobilized fusion protein for 24 h with 100 mM DTT in buffer RC. The eluted protein was collected, and the total volume was reduced to 6 ml using Amicon spin concentrators (Millipore 10 000 MWCO) and applied to a 26/60 Superdex S75 (GE Healthcare) gel filtration column equilibrated in TN50D [50 mM Tris–HCl (pH 8), 50 mM NaCl, 1 mM DTT]. The DnaG-containing fractions were pooled, and applied to a 5 ml Q-sepharose (GE Healthcare) equilibrated in TN50D. DnaG was eluted with TEN500D (50 mM Tris HCl ph 8, 500 mM NaCl, 1 mM DTT) and dialyzed extensively against the storage buffer (50 mM Tris HCl pH8, 50 mM NaCl, 1 mM DTT and 30% v/v glycerol, quantified (DnaG extinction coefficient, 45 270 M−1 cm−1), aliquoted and stored at −80°C.
DnaC–DnaI complex
DnaC (50.4 kDa) and DnaI (35.9 kDa) were co-expressed from the pSMG33 plasmid in E. coli ER2556 in the presence of 1 mM IPTG and 100 µg/ml ampicillin at 30°C for 3 h, as described before (25). Cell paste from 0.5 l (for DnaC purifications) or 2 l (for DnaC–DnaI purifications) of LB culture were suspended in 8 ml or 32 ml, respectively, of lysis buffer [25 mM Tris–HCl (pH 8), 2 mM EDTA, 0.2 M NaCl, 10 mM DTT, 2.5 mM ATP, 5 mM MgCl2] supplemented with 0.5 mg/ml lysozyme. Protease inhibitors, 1 mM PMSF and 200 μl of protease inhibitor cocktail (no EDTA, SIGMA) were added to the cell suspension, and the cells were incubated on ice for 30 min and then lysed by freezing thawing at 80°C. The bacterial extract was clarified by centrifugation (49 000g, 60 min, 4°C), and, after filtering through a 0.22-µm filter, total protein in the supernatant was precipitated by the addition of 30% w/v ammonium sulphate at 4°C. The pellet was collected by centrifugation (40 000g, 30 min, 4°C), suspended in 5 ml of buffer A [50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 1 mM DTT, 1 mM ATP, 2 mM MgCl2], desalted through a 5-ml desalting column (GE Healthcare) and loaded onto a 26/60 Superdex S200 gel filtration column equilibrated in buffer A. To purify the intact DnaC–DnaI complex, the relevant fractions from the Superdex S200 gel filtration column (see above) were pooled, concentrated to 20 ml using Amicon spin concentrators (Millipore 10 000 MWCO), made up to 10% v/v glycerol, quantified spectrophotometrically (DnaC–DnaI extinction coefficient, 52 720 M−1 cm−1 for a monomer–monomer or 316 320 M−1 cm−1 for the native DnaC6–DnaI6 complex) and stored at −80°C.
Hexameric DnaC
To separate hexameric DnaC from DnaI, fractions containing the DnaC–DnaI complex were pooled (total volume, 18 ml), and the salt concentration was adjusted to 50 mM NaCl with 2 M urea added before loading onto a 26/60 Superdex S200 (GE Healthcare) gel filtration column equilibrated in buffer A* [50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 1 mM DTT, 2 M urea]. The DnaC-containing fractions were pooled, desalted through a 5-ml desalting column and loaded onto a 5-ml Hi-Trap Q column (GE Healthcare) equilibrated in buffer B [50 mM Tris–HCl (pH 7.8), 50 mM NaCl, 1 mM DTT]. DnaC was eluted with a salt gradient at ∼300 mM NaCl. Relevant fractions were pooled, buffer exchanged into buffer A* without ATP, made up to 10% v/v glycerol, quantified spectrophotometrically (DnaC extinction coefficient, 26 360 M−1 cm−1) and stored at −80°C.
Monomeric DnaC
To separate DnaC from DnaI and to disrupt the hexameric form of DnaC into monomers, the co-expressed DnaC and DnaI proteins were purified through a Superdex 200 column (Hi-load 26/60, GE Healthcare), equilibrated in buffer A [50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 1 mM DTT, 1 mM ATP, 2 mM MgCl2], as a single peak corresponding to the dodecameric complex DnaC6:DnaI6. The corresponding fractions were pooled, and the proteins precipitated with 30% w/v ammonium sulphate. Protein was collected by centrifugation at 40 000g for 20 min at 4°C, and the pellet suspended in buffer B [50 mM Tris (pH 8), 50 mM NaCl, 1 mM DTT] supplemented with 2 M guanidine hydrochloride to disrupt the dodecameric DnaC6–DnaI6 complex. The protein sample was then loaded onto a Superdex 75 column (Hi-load 26/60, GE Healthcare), equilibrated with buffer B supplemented with 2 M guanidine hydrochloride. DnaC and DnaI separate fractions were collected and pooled separately. The DnaI protein was further loaded onto a 5-ml Q sepharose column equilibrated with buffer B, and eluted with buffer B2 [50 mM Tris (pH 8), 200 mM NaCl, 1 mM DTT]. DnaI was then dialysed against the storage buffer (buffer B plus 50% v/v glycerol). To disrupt the hexameric form of DnaC, the DnaC sample was treated with buffer B supplemented with 2 M guanidine hydrochloride, and loaded onto a Superdex 200 column (Hi-load 26/60, GE Healthcare), equilibrated with buffer B containing 2 M guanidine hydrochloride. The DnaC corresponding fractions were pooled and dialysed extensively against the storage buffer (buffer B containing 50% v/v glycerol).
Assaying protein–protein interactions by analytical gel filtration
Analytical gel filtration experiments were carried out with 4.6 µM DnaC–DnaI, 2.3 µM DnaG and 2.3 µM DnaEBs in binding buffer [50 mM Tris (pH 7.4), 1 mM EDTA, 1 mM DTT, 100 mM NaCl, 1 mM ATP, 2 mM MgCl2] using a Superose 6 HR (10/30) pre-packed column (GE Healthcare) run at 0.3 ml/min and collecting 0.5 ml fractions. Proteins were mixed in 1 ml of the binding buffer and incubated for 10 min at room temperature before loading onto the column. The elution profiles from each experiment were monitored at 280 nm and plotted as a function of the fraction numbers, as indicated. Samples from fractions 9–18 were analysed by SDS–PAGE and Coomassie Blue staining to verify the identity of the proteins.
Surface plasmon resonance analysis
Surface plasmon resonance (SPR) experiments to investigate the DnaG–DnaE–DnaC/DnaI interactions were carried out with a four-channel sensor chip C1 and a Biacore™ 3000 system (GE Healthcare). The system was primed with running buffer (20 mM sodium acetate, pH 5), which was degassed and filtered through a 0.22-µm filter. The surface of the chip was washed with three injections of 10 µl of 0.1 M glycine–NaOH, pH 12, containing 0.3% v/v Triton X-100, at 10 µl/min followed by a prime run with running buffer to remove the Triton X-100. The sensor surface was activated by treating with a mixture of 0.2 M EDC [1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride] and 0.05 M N-hydroxysuccinimide (NHS), according to the manufacturer’s instructions.
Typically for DnaG immobilization on the sensor chip for binding kinetic studies, serial injections of 10 µl of DnaG 1 nM, 5 nM and finally two injections of 1 nM each in (20 mM sodium acetate, pH 5) were loaded on the activated sensor chip at 5 µl/min (2-min contact time) in one of the two channels, while in the second control channel, the same buffer without protein was loaded for direct comparison. Unreacted NHS esters on the sensor surface were deactivated with 70 µl of ethanolamine hydrochloride loaded in both channels at 10 µl/min (7-min contact time). Finally, the system was primed with the running buffer [0.01 M 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) (pH 7.4), 0.15 M NaCl, 0.005% v/v surfactant P20, 1 mM ATP, 2 mM MgCl2]. Under these conditions, ∼150–165 RU (response units) were obtained. Binding of DnaC–DnaI and DnaE to DnaG was examined by single-cycle analysis where 10-µl samples of increasing concentrations of DnaC–DnaI [1.24, 2.48, 4.95 and 9.91 µM in 40 mM Tris (pH 8.0), 150 mM NaCl, 1 mM ATP, 2 mM MgCl2 and 0.005% v/v Tween 20] or DnaE (0.115, 0.23, 0.46, 0.92 and 1.84 µM) were injected at 2 µl/min in a single cycle serially first over a control surface and then over a surface with immobilized DnaG. The running buffers for each experiment were the same buffers as those of the equivalent proteins. Control data were subtracted from the experimental data, and the final normalized data were exported to GraphPad and plotted as RU over time to give standard sensograms. Kinetic analysis was carried out using the BIAevaluation Software version 4.0 software (Biacore).
Helicase assays
Helicase activity was assayed by monitoring the displacement of radiolabelled oligonucleotides (5′-TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCAGTGCCAAGCTTGCATGCCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3′ or 5′-TTGCGGTCCCAAAAGGGTCAGTGATGCAACATTTTGATGCCGCAGTGCCAAGCTTGCATGCCACGTCCAGCTTGATCTCATAGGGGCCCATGGCTCGAGTTGAC-3′) annealed to the single-stranded circular DNA M13mp18 (ssM13) of 7 249 bases. The 104-mer oligonucleotides were radiolabelled at their 5′ ends using γ32P-ATP and T4 polynucleotide kinase (New England Biolabs), purified through an S-200 mini-spin column (GE Healthcare) and then annealed onto M13mp18 (the bold-underlined bases indicate the region of complementarity with the ssM13 DNA, whereas the four consecutive underlined Ts in italic indicate a specific DnaG priming site in the second oligonucleotide). They produced two different double-fork substrates: one with poly(dT) tails and multiple 5′-d(TTT) prime sites along both tails, and the other with random sequence tails and a defined 5′-d(TTTT) site in the 5′ tail. All reactions were initiated by the addition of 2.5 mM ATP and carried out at 37.5°C in helicase buffer, 20 mM Tris (pH 7.5), 50 mM NaCl, 0.658 nM radiolabelled DNA substrate, 12 mM MgCl2 and 2 mM DTT for various times, as indicated.
Activation of B. subtilis DnaC by DnaI was established by mixing 0.5 μM DnaC with increasing concentrations of DnaI (0.125, 0. 250, 0.5, 1, 2 and 4 μM) and incubating for 10 min at 37°C before being added to the reaction buffer. Reactions were carried out for 1 h at 37°C and terminated with the addition of stop buffer (1% w/v SDS, 40 mM EDTA, 8% v/v glycerol, 0.1% w/v bromophenol blue). To assess the effects of DnaG and DnaEBs on the DnaC helicase activity, DnaC (0.5 μM, referring to monomers) was mixed with increasing concentrations of DnaEBs (0.04, 0.08, 0.16, 0.32, 0.64 and 1.3 μM) and DnaG (0.125, 0.25, 0.5, 1, 2 and 4 μM) and incubated for 10 min at 37°C before being added to the reaction buffer. Reactions were carried out for 1 h at 37°C and terminated with the addition of stop buffer. In all cases, reaction samples (10 μl) were loaded onto a 10% w/v non-denaturing polyacrylamide mini-gel, and electrophoresis was carried out in TBE at constant voltage (130 V). Gels were then dried and analysed using a molecular imager and associated software (Biorad). All experiments were carried out in triplicates, and average values of percentage displacement of the radiolabelled oligonucleotide from the ssM13 DNA were plotted as a function of the relevant effector protein. Experimental errors were within ±5%.
Coupled helicase–primase–polymerase assays
To set up coupled assays, DnaC and DnaI (0.5 μM each, referring to monomers) were mixed with 0.25 μM DnaG and/or 83.3 nM DnaEBs and incubated for 10 min at 37°C before being added to the helicase reaction buffer. These molar ratios are consistent with an assumed active complex from the literature comprising a DnaI6:DnaC6:DnaG3:DnaEBs stoichiometry. Reactions were initiated by the addition of 2.5 mM ATP and carried out for 20 min at 37°C in the presence or absence of rNTPs and/or dNTPs (0.5 mM each), as appropriate. Samples (10 μl) at various time intervals (0, 1, 3, 5, 10, 15 and 20 min) were analysed, as described above. All experiments were performed in triplicates, and average values of percentage displacement of the radiolabelled oligonucleotide from the ssM13 DNA were plotted as a function of time. Experimental errors were within ±5%.
RNA primer synthesis assay and thermally denaturing high performance liquid chromatography analysis
RNA priming assays and denaturing high performance liquid chromatography (HPLC) analyses were conducted as previously determined for other mesophilic DnaG primases from Firmicutes that belong to the Bacilli class (28). Briefly, all RNA primer synthesis reactions were carried out in 100 µl of nuclease-free water containing 50 mM HEPES (pH 7.5), 100 mM potassium glutamate, 10 mM DTT, 2 µM ssDNA template, 40 mM magnesium acetate and 0.8 mM of each NTP and/or dNTP, unless otherwise specified. The two oligonucleotides that comprised all 64 trinucleotide sequences were initially used to assess template specificity, as described previously (29). The trinucleotide-specific template 5′-CAGA(CA)5XYZ(CA)3-1,3-propanediol, whereby XYZ was the trinucleotide of interest, was added to the priming reactions as indicated. Priming reactions were incubated at 30°C for 1 h, desalted in a Sephadex G-25 spin column and dried using a speed vacuum. The pellet was suspended in water to one-tenth the original volume of the sample, and 8 μl of that sample was analysed by HPLC under thermally denaturing conditions at 80°C.
For the denaturing HPLC analyses, a gradient 0–8.8% v/v acetonitrile over 16 min was used to obtain optimal separation of primer products and ssDNA template peaks on a DNA Sep column. Primer products ssDNA templates were detected by UV absorbance at 260 nm. Retention times of the ss primer products were correlated to the retention times of the appropriate oligonucleotide standard to confirm composition and length. Supplementary Table S2 shows the RNA, DNA and RNA:DNA oligonucleotides standards used in this study. To normalize variability introduced during sample preparation and injection into the HPLC column, primer abundance was determined by using the ssDNA template as an internal standard. The moles of RNA primers synthesized were quantified using the relative extinction coefficient for the oligonucleotides and reported as the sum of moles for all primer lengths, as previously described (30).
Polymerase assays
DnaEBs (10 nM) polymerase activity was monitored by a standard primer extension assay at 37°C in a buffer containing 50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 50 mM NaCl, 1 mM DTT, 25 μM dNTPs, 25 μM rNTPs and 2 nM DNA substrate. The DNA substrate was prepared by radiolabelling a 45-mer oligonucleotide (5′-CAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTAC-3′) using γ32P-ATP and T4 polynucleotide kinase (New England Biolabs). The radiolabelled substrate was then purified through an S-200 mini-spin column (GE Healthcare) and annealed to M13mp18. DnaEBs polymerase extension of the 45-mer oligonucleotide was monitored over time (0, 1, 2, 4, 8 and 16 min). Reactions were terminated by the addition of 2 μl of 0.5 M EDTA and 5 μl of 3× gel-loading dye (0.1% w/v bromophenol blue, 0.1% w/v xylene cyanol and 25% v/v glycerol) and incubation for 5 min at 90°C before transferring them to ice. The reaction products were resolved by electrophoresis through 2% w/v high-resolution alkaline agarose gels. Gels were fixed in 7% v/v trichloroacetic acid, dried onto Whatman paper and analysed using a molecular imager and associated software (Biorad).
The dNTP mis-incorporating activity of DnaEBs (10 nM) was assayed by primer extension assays at 37°C for 16 min using different oligonucleotide templates in the absence of one of the dNTPs in different reactions, as appropriate. The reaction products were resolved by electrophoresis through a 10% w/v urea-polyacrylamide denaturing sequencing gels. All gels were dried and analysed using a molecular imager and associated software (Biorad). All experiments were carried out in triplicates. In all cases, the effects of DnaG and DnaC/DnaI were assessed by pre-incubating DnaEBs (10 nM) with the relevant protein (30 nM DnaG, 60 nM DnaC, 60 nM DnaI) for 5 min before starting the reaction.
Molecular modelling
Swissmodel web service (31) was used to construct homology models for B. subtilis DnaG based on the crystal structure of the E. coli protein [(32); PDB code 3B29], and for DnaEBs based on the crystal structure for Thermus aquaticus polIIα [(33), PDB code 3E0D]. Alignments and quality metrics are included in the supplementary information (Supplementary Table S3). Using Chimera (34), the nucleic acid component of both models was elaborated to include a fuller length of the template strand both upstream and downstream of each catalytic site, while a 5-nt length of double-stranded (ds) nucleic acid was included to represent the situation where a short primer has been synthesized. For the DnaG model, this required extending the observed ssDNA in a 5′ direction, while modelling the hybrid duplex into the basic groove, as described before (32). For the DnaEBs model, this involved adding a length of ssDNA that exited the catalytic site in the direction of the C-terminal domain, in line with the electron density described before (33). A crude model for the open state of DnaEBs, as required for direct interaction with DnaG and nucleic acid hand-off (see ‘Discussion’ section), was built using Chimera, by modelling simple rigid-body motions of the thumb and CTD domains in a similar (for the thumb) or more extreme (for the CTD) manner to that inferred from the displacements seen crystallographically between the apo- and DNA-bound conformations of PolIII (33,34).
RESULTS
DnaEBs interacts directly and indirectly with the helicase and primase
To coordinate DNA unwinding and nucleotide incorporation at replication forks, the helicase and the DNA polymerases are physically connected by direct (like in bacteriophage T7) or indirect (like in E. coli, via DnaX) contacts in the replisome (35). Because DnaEBs binds DnaN and SSB, but not DnaX (14,16,36,37), the way this polymerase is connected to the helicase (if any) remains mysterious. To answer this point and improve our view of the interactions in the B. subtilis replisome, a rather large yeast two-hybrid screen was conducted. In a first analysis, the full-size DnaEBs protein and seven peptides derived thereof were crossed against known replisomal proteins (full-size with the exception of PolC, which was tested as two overlapping N-terminal and C-terminal fragments) in both directions (bait and prey). Of 240 interactions tested, only 8 (3%) were positives (Figure 1A). They confirmed that DnaEBs interacts with SSB, and identified three new potential partners: DnaC, DnaG and HolA, a subunit of the DnaX complex. To expand this study to the whole replisome, two other replication proteins (PolC and DnaG) were fragmented, and all fragments constructed here, and available in the Philippe Noirot’s collection, were crossed against each other in both directions. In all, 1015 crosses were tested, and 57 (∼6%) were positive (Figure 1B, see Supplementary Table S1 for details). This identified 18 different replisomal interactions. Of these, 13 had been previously reported (blue lines Figure 1B), and the remaining five (green lines) correspond to the three DnaEBs interactions detected above and to two new interactions: DnaEBs–PolC and DnaG–DnaX (Figure 1A). These results thus suggest that DnaEBs is physically connected to the helicase in the B. subtilis replisome by both direct and indirect interactions. The interacting domains of DnaEBs, DnaC, DnaG and PolC are shown Figure1C.
Figure 1.

DnaEBs interacts with the replicative helicase DnaC and primase DnaG. (A) Yeast two-hybrid matrix interaction assays illustrating interactions of DnaEBs with DnaC, DnaG, PolC, HolA and SSB. The indicated proteins were expressed as baits (Gal4 Binding Domain fusion) and/or as preys (Gal4 Activating Domain fusion). Pairs of independent diploid yeast cells expressing combinations of fusions were subjected to selection for the Adenosine+ phenotype as described in ‘Materials and Methods’ section and Supplementary Data. The empty bait and prey vectors (BD and AD) were used as negative controls. Plates were incubated at 30°C for the time indicated at the top left of each panel (in days). 1: AD-DnaEBs805–1115 and BD-DnaC; 2: AD-DnaEBs and BD-DnaG; 3: AD-DnaEBs1–826 and BD-HolA; 4: AD-PolC875–1437 and BD-DnaEBs618–1115; 5: AD-SSB and BD-DnaEBs618–1115. (B) Overview of replisomal interactions. Arrows are oriented from the bait to the prey. Blue: known interactions; green: new interactions; grey: interaction showed previously biochemically (14,19). (C) Schematic representation of protein-binding domains. Coloured boxes: protein binding domains; dashed grey lines: identified interactions. PHP: polymerase and histidinol phosphatase domain; activity: polymerase domain; OB: OB-fold; Zn: zinc finger binding domain; primase: primases conserved region; hel. bind.: helicase binding domain; Nter and Cter: DnaB-like helicases N- and C-terminal conserved domains.
Figure 10.

Effects of the DnaG primase and DnaC/DnaI helicase on the error-prone DnaEBs polymerase activity. (A) The sequences of the oligonucleotides used to prepare the primer extension substrates are shown. Each of the bottom four oligonucleotides was annealed to the top oligonucleotide to produce substrates 1–4. The sequences shown in pale shading were the templates used by the DnaEBs polymerase during primer extensions with these substrates. (B–E) Representative urea–polyacrylamide denaturing gels showing the effects of DnaG (30 nM) (B), DnaC/DnaI (60 nM each protein) (C), DnaG+DnaC/DnaI (D) and DnaI (60 nM) (E) on the error-prone polymerase activity of DnaEBs (10 nM). Lanes labelled C1 and C2 represent primer extension reactions with (showing full extension of the DNA substrate) and without (showing no extension) DnaEBs, respectively, in the presence of all four dNTPs. Lanes labelled with − and + signs represent DnaEBs extension reactions in the absence and presence of the relevant effector proteins in the different substrates (1–4) shown underneath the gels. The missing dNTP nucleotide for each reaction is indicated underneath each gel. All reactions were carried out at 37°C for 16 min.
To confirm and analyse the biological functions of the interactions connecting DnaEBs to the DnaC helicase and DnaG primase, the proteins were purified and analytical gel filtration experiments were carried out. In an initial study, no interaction was detected between DnaC and either DnaG or DnaEBs (not shown). We hypothesized that this apparent lack of interaction may be a consequence of the inability of the purified DnaC to form the stable ring-shaped hexamer required for helicase and ATPase activities at the replication fork [(25) and our data]. Therefore, we co-purified DnaC and DnaI, the DnaC loader, as this allows the formation of a stable complex DnaC6–DnaI6 with an apparent ring-shaped structure and ATPase and helicase activities (25). With this purified DnaC6–DnaI6 complex, the search for protein interactions by gel filtration was repeated. Results presented in Figure 2A confirmed the stable DnaC–DnaI interaction and showed that this complex binds to DnaG and DnaEBs, a significant amount of DnaG or DnaEBs eluting much earlier (fractions 9–12) when mixed with DnaC–DnaI than alone (DnaG fractions 15–18, DnaEBs fractions 13–18). This shift in the DnaEBs and DnaG elution profiles was also observed with a mixture containing DnaC–DnaI, DnaEBs and DnaG. In this mixture, DnaI appeared to dissociate from the complex, and a significant fraction of this protein eluted in fractions 16–18. An interaction was also detected between DnaEBs and DnaG (the elution profile of DnaEBs remained unchanged because the resolving power of Superose 6 was not sufficient to separate the DnaEBs–DnaG complex from DnaEBs alone, but that of DnaG shifted significantly from fractions 16–18 to 14–18). Detection of protein complexes by gel filtration is an indication of relatively strong protein–protein interactions. It is worth noting that although the E. coli helicase DnaBEc and primase DnaGEc are known to physically and functionally interact, no DnaBEc–DnaGEc complex was ever detected by gel filtration because of its weak and transient nature. Hence, the detection of complexes between the B. subtilis replisomal proteins by gel filtration suggests relatively strong protein–protein interactions. However, during gel filtration, only the most stable protein–protein complexes survive until the end of the experiment, while the weaker more transient complexes tend to partially dissociate and separate within the column. The results suggest that the interactions DnaC–DnaI–DnaG, DnaC–DnaI–DnaEBs, DnaEBs–DnaG and possibly DnaC–DnaI–DnaEBs–DnaG are partially stable with the off rate high enough to cause apparent spreading of DnaEBs and DnaG across several fractions, as the complexes move along the column and dissociate.
Figure 2.

DnaEBs, DnaC and DnaG form a ternary complex. (A) Gel filtration. Purified DnaC–DnaI, DnaEBs and DnaG proteins were mixed, incubated and then analysed for complex formation by gel filtration as described in ‘MATERIALS AND METHODS’ section. The composition of the mixture and the chromatograms obtained are indicated on the right. Column fractions 9–18 were analysed by SDS–PAGE stained by Coomassie Blue. Fraction numbers are indicated above the gels, protein names to the right and molecular weight of protein standards to the left. (B) SPR. Analysis of protein–protein interactions was carried out with immobilized DnaG and with DnaC/DnaI and DnaEBs in the mobile phase by single-cycle analysis with serial injections of increasing concentrations of DnaC/DnaI or DnaEBs, as described in ‘MATERIALS AND METHODS’ section. Control background data were subtracted from the experimental data, and the relevant sensograms are shown. Kinetic parameters were obtained using the BIAevaluation Software version 4.0. The derived parameters for the DnaG–DnaC/DnaI interaction were ka = 1.08 × 103 M−1 s−1, T(ka) = 174, kd = 2.87 × 10−4 s−1, T(kd) = 44.5 [Rmax (RU) = 22.5, T(Rmax) = 317], Ka = 3.77 × 106 (M−1), Kd = 2.95 × 10−7, (M) Chi2 = 1.62; and for the DnaG–DnaEBs interaction were ka = 2.4 × 103 M−1 s−1, T(ka) = 33.2, kd = 1.81 × 10−3 s−1, T(kd) = 42.8 [Rmax (RU) = 88.8, T(Rmax) = 47.6], Ka = 1.33 × 108 (M−1), Kd = 7.52 × 10−7, (M) Chi2 = 15.1.
Protein–protein interactions were kinetically assessed by SPR. Results with the immobilized DnaG protein confirmed the DnaG–DnaC–DnaI and DnaG–DnaEBs interactions (Figure 2B). The observed kinetic parameters for the DnaG–DnaC–DnaI and DnaG–DnaEBs interactions were comparable with each other and consistent with the analytical gel filtration results. Because of the strict requirement of the DnaC–DnaI complex for ATP, it was technically impossible to immobilize this complex on the chip surface, as the adenosine amino groups of the ATP strongly interacted with the NHS esters on the chip surface and blocked protein immobilization. Immobilization of DnaEBs was also not effective, as this protein precipitated on the surface of the chip when introduced in the immobilization buffer (20 mM sodium acetate, pH 5). Therefore, it was not possible to study the DnaEBs–DnaC–DnaI complex by SPR. Collectively, the results reported here suggest that DnaEBs is physically attached to the DnaC helicase in the B. subtilis replisome by five different sets of protein–protein interactions. Among these, the DnaEBs–DnaC and DnaEBs–DnaG–DnaC interactions are the most direct. Interestingly, DnaC interacts with DnaEBs and DnaG only in the form of a functional hexameric helicase, and the three proteins may form a ternary complex. It is thus likely that DnaEBs and DnaG operates in association with the DnaC helicase translocating along the lagging strand template in the B. subtilis replisome. To assess whether these protein–protein interactions are functionally relevant, we characterized the individual activities and the interacting effects by in vitro biochemical assays.
Characterization of the B. subtilis DnaG primase activity
Bacillus subtilis DnaG initiation specificity was predicted to be similar to that of primases from other Firmicutes belonging to the Bacilli class (28). To test this hypothesis, two oligonucleotides that comprised all 64 possible trinucleotide sequences were initially used to assess template specificity, followed by trinucleotide-specific ssDNA oligonucleotides to verify template specificity. These results confirmed that B. subtilis DnaG initiated primer synthesis preferentially on 5′-d(CTA) and, to a slightly lesser degree, on 5′-d(TTA; Figure 3A and data not shown), similar to the primases from Bacillus anthracis, Geobacillus stearothermophilus and Staphylococcus aureus (29,38,39). In addition, the trinucleotide 5′-d(TTT) was determined to support de novo primer synthesis. The highest concentration of DnaG (3.6 µM) examined produced predominantly full-length 16-mer primers with the 23-mer 5′-d(CTA)-specific template. Both full-length and shorter RNA primers, ranging from 5 - to 16-mers, with the majority being 10-mers, were synthesized at the lower more physiological enzyme concentrations (0.45–1.8 µM; Figure 3A). At high DnaG concentrations, a 16-mer RNA polymer is predominantly produced, presumably indicating more efficient primase recycling. These results demonstrate that B. subtilis DnaG, at lower more physiologically relevant concentrations, preferentially synthesizes RNA primers that are 10 nt in length. RNA primer synthesis by B. subtilis DnaG increased with increasing enzyme, NTP abundance and magnesium ion concentrations of 30–50 mM (Figure 3B–D).
Figure 3.

Characterization of the B. subtilis DnaG primase activity. (A) Representative chromatograms showing RNA primers synthesized by B. subtilis DnaG at the enzyme concentration indicated on the right. RNA polymers synthesized in the priming reactions with 2 µM trinucleotide-specific template containing the preferred 5′-d(CTA) initiation sequence and 0.4 mM each NTP were visualized by thermally denaturing HPLC. The lengths of the primers were determined by comparison with RNA standards. The 10-mer and 16-mer RNA polymers are denoted. (B) Bar graph showing B. subtilis DnaG concentration–dependent primer synthesis and total amount of RNA primers synthesized in the priming assays described and shown in panel A. (C) B. subtilis primase activity as a function of magnesium ion concentration. Shown are the relative levels of RNA primers synthesized by B. subtilis DnaG (1.8 µM) in reactions with the 23-mer 5′-d(CTA)-containing ssDNA template and the indicated concentration of magnesium acetate. (D) B. subtilis primase activity as a function of NTP concentration. Shown are the relative levels of RNA primers synthesized by B. subtilis DnaG (1.8 µM) in reactions with 2 µM of the 5′-d(CTA)-containing ssDNA template and the indicated concentration of each NTP.
The DnaC helicase stimulates the DnaG primase activity
The effect of preformed hexameric B. subtilis DnaC helicase on DnaG activity was assessed. When the activity of DnaG alone was evaluated in the presence of the 5′-d(CTA)-containing template at 2, 4 or 8 µM, there was no change in the lengths of the RNA primers synthesized (Figure 4A). Addition of preformed hexameric B. subtilis DnaC helicase stimulated primer synthesis by B. subtilis DnaG relative to primase alone without altering the length of the RNA polymers (Figure 4A). However, the abundance of primers 5–15-mers in length increased, whereas the quantity of the full-length 16-mer RNA polymers was unchanged at all three template concentrations tested.
Figure 4.

The effect of B. subtilis DnaC on DnaG primase activity as a function of ssDNA concentration. (A) Representative chromatograms showing the RNA primers produced by B. subtilis DnaG with 2, 4 or 8 µM d(CTA)-containing ssDNA template in the absence or presence of DnaC and DnaEBs. Concentration of the protein(s) and ssDNA template used in the priming assays are indicated in the right panels, with the respective chromatogram on the left. The 6-mer, 10-mer and 16-mer RNA polymers are denoted and were based on the elution of RNA standards. Priming reactions contained 0.8 mM each NTP. (B) Quantification and comparison of RNA primers synthesized by B. subtilis DnaG in the absence or presence of DnaC and DnaEBs with 2, 4 or 8 µM of the d(CTA)-containing ssDNA template. Priming reactions were carried out as described in A with various combinations of DnaG (G) and DnaC (C).
The quantity of total primers synthesized by B. subtilis DnaG was evaluated in the absence and presence of hexameric DnaC in priming reactions containing 2, 4 or 8 µM of the ssDNA template. In the priming reactions with DnaG alone, no substantial enhancement of primer production was observed with increasing template concentrations (Figure 4B). In the presence of helicase at stoichiometric concentrations of DnaC6:DnaG3, primer abundance increased ∼2-fold relative to primer levels synthesized by DnaG alone at all three template concentrations (Figure 4B), showing that there was no adverse sequestration of DnaC by the template. Overall, our data reveal that DnaC stimulates the activity of DnaG without generally affecting the length of primers synthesized.
Characterization of the B. subtilis DnaC helicase activity
Helicase activity was assayed by monitoring the displacement of a 104-mer oligonucleotide annealed onto ssM13 and forming a double-fork substrate with poly(dT) tails. Purified monomeric or hexameric B. subtilis DnaC helicase did not exhibit detectable helicase activity in vitro under a variety of conditions tested (data not shown). The B. subtilis DnaG primase or the DnaEBs polymerase did not activate the monomeric or hexameric DnaC helicase (Figure 5A and data not shown). The helicase activity was only detectable when monomeric DnaC was mixed with the putative B. subtilis helicase loader protein DnaI, and increased with increasing concentrations of DnaI, reaching a maximum at 0.5 μM DnaI (monomer), corresponding to 1:1 DnaI:DnaC molar ratio (Figure 5B). This is in agreement with the previously reported activation of DnaC by DnaI in vitro (25). DnaI is required to assemble a functional DnaC hexamer onto the DNA substrate, but it is not known whether it remains associated after loading with the translocating helicase (25–27). In E. coli, the helicase loader must detach from the helicase after loading before helicase translocation can begin (40), and it is likely that this is also the case with the B. subtilis DnaI helicase loader. The DnaC helicase activity was dependent on the presence of a DNA fork, as an oligonucleotide that fully annealed onto ssM13 DNA without forming a fork was not displaced (Figure 5C), consistent with a steric exclusion model of translocation first observed for the E. coli DnaB helicase (41). Our data confirm that monomeric DnaC is functionally loaded onto the DNA template, and that strand separation is the result of steric exclusion during translocation.
Figure 5.

Characterization of the B. subtilis DnaC helicase activity. (A) The DnaC helicase activity was assayed by monitoring the displacement of a 104-mer oligonucleotide annealed onto ssM13 and forming a double-fork substrate with poly(dT) tails. DnaC did not exhibit any detectable helicase activity on its own or in the presence of the DnaG primase and the DnaEBs polymerase. Control reactions with DnaG and DnaEBs confirmed, as expected, that these proteins have no helicase activities. Control samples in lanes a and b represent annealed substrate and fully displaced (boiled) controls. Reactions were carried out with 500 nM monomeric DnaC incubated for 1 h at 37°C as described in ‘Materials and Methods’ section, with increasing concentrations of DnaE (40–1300 nM) or DnaG (125–4000 nM). (B) The DnaC helicase activity was detectable only in the presence of the putative DnaI helicase loader. Similar helicase reactions were carried out as above in the presence of DnaI (125, 250, 500, 1000, 2000–4000 nM monomeric). Data were plotted as a percentage of the radiolabelled oligonucleotide displacement versus DnaI concentration. Maximal stimulation of the DnaC helicase activity was observed at 500 nM DnaI, indicating optimal 1:1 stoichiometry consistent with a DnaC6:DnaI6 complex. (C) The helicase activity requires a fork DNA substrate. Helicase reactions were carried out as above for 20 min at 37°C with DnaC (500 nM) and DnaI (500 nM) in the presence or absence of DnaG (250 nM) with two different DNA substrates (with or without fork), as indicated. The double-fork substrate forms 5′ and 3′ poly(dT) tails, and the non-fork DNA substrate was the same used in the polymerase assay with a synthetic oligonucleotide annealed onto ssM13 (see ‘Materials and Methods’ section). Control samples in lanes a and b represent annealed substrate and fully displaced (boiled) controls.
Lagging strand primer synthesis in a minimal coupled primase–helicase assay
Having established that B. subtilis DnaG can initiate primer synthesis from 5′-d(TTT) sites, we used the radiolabelled poly(dT)-tailed helicase substrate in a reconstituted lagging strand–coupled primase–helicase assay to establish whether DnaG can synthesize RNA primers, using the multiple 5′-d(TTT) sites along the lagging strand, while being carried by a forward-moving helicase at a replication fork. We argued that the ATP in the reaction mixture will provide the necessary fuel for DnaC translocation and also a substrate for DnaG to synthesize and extend poly(A) primers on the poly(dT) tails during helicase translocation (Supplementary Figure 1SA). This coupled primase–helicase assay effectively reconstitutes in vivo lagging strand primer synthesis, but in a ‘stripped down’ version. DnaC and DnaI in equimolar concentrations (0.5 μM each, referring to monomers) were incubated with DnaG (0.25 μM), equivalent to a molar ratio of DnaC6:DnaI6:DnaG3, for 10 min at 37°C before being added to the reaction mixture containing the DNA substrate. Significant stimulation of the helicase activity was observed with concomitant poly(A) primer synthesis (Figure 6A and B). The poly(A) RNA primers synthesized de novo by DnaG annealed onto the poly(dT) tails of the displaced radiolabelled oligonucleotide and caused distinct diffuse gel shifts, indicating the formation of DNA:RNA heteroduplexes with variable length RNAs (Figure 6B). To confirm that the observed gel shifts were produced by the formation of DNA:RNA heteroduplexes and not by the formation of protein:DNA complexes, the reaction products were treated with RNase H before electrophoresis. After RNase H treatment, the shifted bands disappeared, with only the displaced radiolabelled oligonucleotide showing in the gel, confirming the formation of DNA:RNA duplexes (Figure 6C). Treatment of the reaction products before electrophoresis with proteinase K (for 30 min or 2 h), urea (for 30 min), guanidinium hydrochloride (for 30 min) and a range of SDS concentrations (1, 2 and 5% w/v) did not eliminate the gel shifts, indicating that this effect was not due to the formation of protein:DNA complexes after the oligonucleotide was displaced from the ssM13 DNA (data not shown).
Figure 6.

A reconstituted coupled helicase–primase assay. (A) The helicase activity of DnaC (500 nM) in the presence of DnaI (500 nM) is shown as a function of time. The DNA substrate used was a radiolabelled synthetic oligonucleotide annealed onto ssM13 DNA, producing a double-fork substrate with poly(dT) tails. (B) Helicase reactions were carried out exactly as in panel A but in the presence of DnaG (250 nM). Stimulation of the helicase activity (compare the graphs in panels A and B) as well as concomitant de novo RNA primer synthesis from multiple 5′-d(TTT) sites along the tails of the annealed oligonucleotide (shifted bands) can be clearly seen. (C) Helicase reactions (20 min) in the presence of DnaC/DnaI (500 nM each protein) and DnaG (250 nM) with the double-fork poly(dT) DNA substrate. After 20-min incubation at 37°C, the reaction products were incubated with RNase H (2 units, New England Biolabs) for 1 h at 37°C before addition of the stop buffer and further incubation at 37°C for 20 min, followed by electrophoresis. Lanes a and b represent annealed and boiled substrates, respectively. Lane c represents a standard 20-min helicase reaction. Lanes marked with + and − signs represent RNase H–treated and non-treated (RNase H storage buffer was added and the reaction was processed in a manner identical to the RNase H treated reaction) helicase reactions, respectively. (D) Identical helicase reactions as described in panel B were carried out with a radiolabelled oligonucleotide annealed onto ssM13, producing a double-fork substrate the tails of which had random sequences. No gel shifts were apparent, suggesting that no RNA primers were synthesized. In all helicase experiments, data from triplicate experiments were quantified and plotted as % displacement of the annealed oligonucleotide as a function of time. Experimental errors were within a ±5 range. Lanes labelled a and b represent annealed and fully displaced (boiled) controls, while control reactions with individual DnaC, DnaI and DnaG proteins are shown in the relevant lanes in panels A, B and D.
To confirm further the formation of DNA:RNA heteroduplexes, we used a different 104-mer oligonucleotide, which annealed onto ssM13 at exactly the same sequence, forming a double-fork substrate, but with the poly(dT) sequence of the tails replaced with random sequences containing a single 5′-TTTT-3′ site in the 5′ tail proximal to the ss–ds stranded junction of the fork (see ‘MATERIALS AND METHODS’ section; helicase assays). We hypothesized that because there is only ATP in the reaction mixture, the DnaG primase, in the absence of the full NTP complement, will not be able to synthesize long RNA primers and no DNA:RNA duplexes would form, therefore abolishing the observed shift of the displaced oligonucleotide. Identical primase–helicase coupled assays were carried out with this substrate, and no shifted bands of the displaced 104-mer were observed (Figure 6D), confirming that the observed gel shifts are the result of RNA–DNA heteroduplexes formed with the poly(dT) double-forked substrate. In summary, we established a minimal coupled primase–polymerase assay to detect DnaG primer synthesis and DnaEBs primer extension on the lagging strand.
Primase-to-polymerase hand-off in a reconstituted lagging strand–coupled primer synthesis–primer extension–translocation assay
To determine whether B. subtilis DnaG can directly hand-off the nascent RNA primer to DnaEBs in the absence of auxiliary DNA replication proteins, priming assays were performed with these two proteins using the 23-mer 5′-d(CTA)-specific template in the presence of NTPs and/or dNTPs. Treatment of the reaction products with RNase H confirmed RNA content in the primers synthesized (compare Figure 7A and B). The presence of only dNTPs eliminated primer production, demonstrating the requirement for complementary ribonucleotides to initiate de novo nucleotide polymerization (compare Figure 7A and C). In the presence of DnaEBs and dNTPs, the reaction products were longer than the nucleotide polymers produced with only DnaG (compare Figure 7A and D), and in the presence of both DnaEBs and DnaC, the abundance of these longer nucleic acid products was increased (compare Figure 7D and E), consistent with the stimulation of DnaG by DnaC observed before (Figure 4A). Comparisons with the oligonucleotide standards shown in Supplementary Table S2 suggested that in the presence of both DnaG and DnaEBs, a mixture of RNA–DNA hybrid molecules were synthesized. The major RNA–DNA hybrid products were 2-mer RNA–14-mer DNA and 4-mer RNA–12-mer DNA oligonucleotides, with both products having the same length of 16 nt but eluting differently because of their different compositions (Figure 7D). To further confirm the presence of RNA–DNA hybrid molecules, the products of this reaction were treated with RNase H, which specifically digests only RNA. After digestion with RNase H, the products were resolved into more distinct peaks, with major peaks indicating 12-mer and 14-mer DNA molecules (Figure 7F). These experiments demonstrated the formation of RNA–DNA hybrid nucleic acids. Furthermore, they confirmed that DnaG and DnaEBs interact directly and functionally, resulting in efficient primer hand-off and extension in the absence of auxiliary proteins. More importantly, these findings showed that primer hand-off occurred very early after the polymerization of only two ribonucleotides, suggesting that the RNA primer is handed off to DnaEBs directly via a DnaG–DnaEBs interaction.
Figure 7.

DnaEBS extends RNA primers after de novo di-ribonucleotide polymerization by DnaG. (A) Representative chromatograms from primase assays showing RNA primers synthesized by B. subtilis DnaG (1.8 µM) in reactions containing the 23-mer 5′-d(CTA)-specific ssDNA template and 0.8 mM each NTP before RNase H treatment. (B) RNA primers synthesized as described in panel A were incubated with RNase H to confirm content. (C) Priming reactions included dNTPs without the presence of ribonucleotides. (D) Priming reactions contained DnaEBs, as well as both NTPs and dNTPs. (E) B. subtilis DnaC helicase was added to the priming reactions, along with DnaEBs and both NTPs and dNTPs. (F) RNA:DNA hybrid primers synthesized in the reaction described in panel D were treated with RNase H to determine primer composition. Elution times for the various primer products synthesized were determined by using RNA, DNA and RNA:DNA size markers shown in Supplementary Table S2. A schematic of de novo primer synthesis and extension is shown below the chromatograms.
We next investigated whether DnaG to DnaEBs hand-off can take place in a coupled translocation, primer synthesis and primer extension assay. This is effectively a partially reconstituted lagging strand replication system with de novo RNA primer synthesis by DnaG and extension of RNA primers by DnaEBs coupled to DnaC helicase translocation. First, we examined the effect of DnaEBs in the absence of dNTPs. DnaC and DnaI in equimolar concentrations (0.5 μM each, referring to monomers) were incubated with DnaG (0.25 μM) and DnaEBs (0.083 μM), equivalent to a DnaC6:DnaI6:DnaG3:DnaEBs1, for 10 min at 37°C before being added to the reaction mixture containing the poly(dT) DNA substrate (0.658 nM) and 2.5 mM ATP. Gel shifts were clearly apparent, indicating de novo RNA primer synthesis from multiple 5′-d(TTT) sites along the lagging strand and the formation of DNA:RNA duplexes. The shifted bands appeared earlier than in reactions with only DnaG present and were better defined and somewhat smaller compared with the rather diffuse bands observed in reactions with only DnaG present (compare Figures 6B and 8A). This indicates that DnaEBs stimulates de novo primer synthesis by DnaG.
Figure 8.
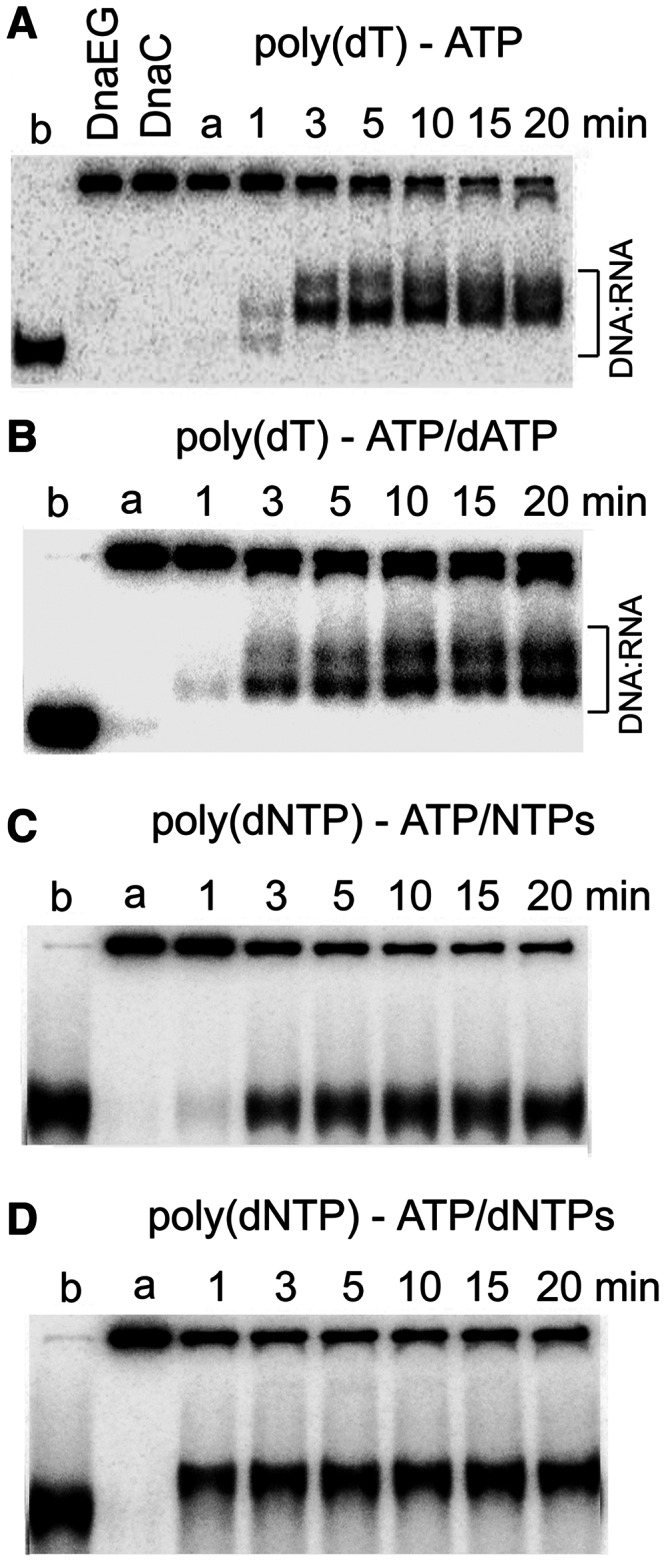
Primer formation during helicase assays. (A) Coupled helicase–primase–polymerase assays were carried out with the poly(dT)-tailed DNA substrate as described in Figure 6B, but in the presence of DnaEBs (83.3 nM). The shift of the displaced oligonucleotide appears earlier than in equivalent reactions in the absence of DnaEBs (compare with Figure 6B), indicating the DnaEBs stimulates primer synthesis. (B) Similar reactions as in panel A were carried out, but this time in the presence of dATP (0.5 mM). Similar shifts of the displaced oligonucleotide were observed compared with panel A. (C) Coupled helicase–primase assays were carried out with the random sequence tail DNA substrate, which has a single 5′-d(TTTT) site on the 5′ tail, in the presence of rNTPs (0.5 mM each). No shift of the displaced oligonucleotide was observed. (D) Coupled helicase–primase–polymerase assays were carried out as described in panel C, but this time in the presence of dNTPs (0.5 mM each; no rNTPs). A clear well-defined shift of the displaced oligonucleotide was apparent from the first time point (1 min), suggesting the formation of very small RNA primers that were extended further by DnaEBs.
Then we examined the effect of DnaEBs in identical reactions with the poly(dT) substrate but in the presence of ATP (2.5 mM) and dATP (0.5 mM). We predicted that in the presence of dATP, DnaEBs will be able to extend the RNA primers synthesized by DnaG along the poly(dT) tails (Supplementary Figure 1SB). Again, similar shifts of the displaced radiolabelled oligonucleotide were apparent (Figure 8B). However, by comparing the shifts in Figure 8A resulting from RNA primers annealing to the displaced oligonucleotide, and the shifts in Figure 8B presumably resulting from RNA–DNA hybrids annealing to the displaced oligonucleotide, it was difficult to distinguish between them.
To unequivocally prove that DnaG–DnaEBs hand-off was taking place, we repeated these assays using the substrate with random sequence tails, which contained random tail sequences and a single 5′-d(TTTT) prime site on the 5′ -tail. The reaction with DnaC6:DnaI6:DnaG3–DnaEBs, 2.5 mM ATP and a full set of rNTPs did not produce shifted bands (Figure 8C), but the reaction with 2.5 mM ATP and a full set of dNTPs produced a well-defined shift, which appeared clearly from the first time point (1 min; Figure 8D). We interpret from these data that with one 5′-d(TTTT) site available in this substrate, DnaG can only produce very small RNA primers (2–3 nt), which are unable to cause a noticeable shift when annealed to the displaced oligonucleotide (Supplementary Figure 1SC). However, in the presence of dNTPs, DnaEBs efficiently extends these small RNA primers to synthesize larger RNA–DNA hybrids, which anneal to the displaced radiolabelled oligonucleotide and are sufficiently large to cause a noticeable shift (Supplementary Figure 1SD). Another important point obtained from these data is that although in the reaction with ATP and dNTPs, DnaG was capable of forming only very small primers (2 or 3 nt) from the single 5′-d(TTTT) site, despite their small size, they were efficiently extended by DnaEBs. Collectively, these data show that in this partially reconstituted lagging strand replication system, there is de novo RNA primer synthesis by DnaG and extension of RNA primers by DnaEBs coupled to DnaC helicase translocation. More importantly, the DnaG–DnaEBs hand-off is efficient even with the synthesis of very small RNA primers only 2–3 nt long, suggesting again that the RNA primer is handed off very early to DnaEBs directly via a DnaG–DnaEBs interaction.
DnaEBs native polymerase activity is not significantly affected by the DnaC helicase and/or the DnaG primase
The DnaEBs native polymerase activity was monitored by a primer extension assay using a synthetic oligonucleotide annealed onto ssM13 DNA. DnaEBs was able to extend the annealed oligonucleotide with a characteristic periodic pattern of products as a function of time (Figure 9). The DnaG primase and/or the DnaC helicase in the presence or absence of DnaI did not noticeably affect the DnaEBs primer extension activity (Figure 9). The same results were obtained using a substrate prepared from synthetic oligonucleotides (42-mer oligonucleotide annealed onto a 65-mer oligonucleotide, data not shown). We conclude that the DnaEBs native polymerase activity is not affected by its interaction with the DnaG primase and/or the DnaC helicase.
Figure 9.

The effects of DnaG primase and DnaC helicase on the native DnaEBs polymerase activity. Representative alkaline agarose gels showing DnaEBs primer extension time courses in the presence or absence of effector proteins, DnaG, DnaC, DnaC/DnaI, DnaC–DnaG and DnaC/DnaI–DnaG, as indicated. 10 nM DnaEBs was premixed with different proteins: 30 nM DnaG, 60 nM monomeric DnaC and 60 nM monomeric DnaI, as appropriate. Reactions were carried out as described in ‘Materials and Methods’ section. Lanes marked 0 represent the radioactive template before protein addition; lanes G, C, I, CI and CG represent primer extension control reactions (16 min) in the presence of DnaG, DnaC, DnaI, DnaC/DnaI and DnaC–DnaG, respectively. Molecular weight markers are labelled appropriately.
DnaEBs error-prone polymerase activity is affected by the DnaG primase and the DnaC helicase
Previous studies have shown that DnaEBs lacks proofreading activity, is error-prone and is efficient at lesion bypass by frameshifting (10,14,17,42). The fidelity of DnaEBs activity was assayed with primer extension assays using synthetic oligonucleotide substrates and a pool of three of four dNTPs (Figure 10A). The four tested template sequences, 24 bases in length, contained at least 10 bases complementary to the missing nucleotide (Figure 10A sequences marked in green). Consistent with previous studies, our purified DnaEBs was highly error-prone. It efficiently and repeatedly incorporated wrong deoxynucleotides at positions along the DNA templates where the complementary dNTP was missing in the pool (Figure 10B–E lanes labelled with a minus sign and data not shown).
Under our assay conditions, the error efficiency was dependent on the nature of the missing deoxynucleotide. DnaEBs was efficient at mis-incorporating at T and C positions along the template in the absence of the correct complementary bases A (-dATP) and G (-dGTP), respectively producing almost fully extended products (Figure 10B–E lanes labelled with a minus sign in substrates 1 and 4). The efficiency of mis-incorporations was somewhat decreased at G template positions in the absence of the correct complementary base C (-dCTP), which was evident by the predominance of a range of partially extended products (Figure 10B–E lanes labelled with a minus sign in substrate 3). It decreased further at template positions with the base A in the absence of T (-dTTP), as was evident by the presence of small products that were not extended beyond position 8 (Figure 10B–E lanes labelled with a minus sign in substrate 2).
Inclusion of the DnaG primase in these reactions resulted in a marked decrease in the error rate with substrates 1, 3 and 4 missing dATP, dCTP and dGTP, respectively (Figure 10B compare lanes labelled with minus and plus signs in substrates 1, 3 and 4). However, there was no apparent difference in the error rate with substrate 2 missing dTTP (Figure 10B compare lanes labelled with minus and plus signs in substrate 2). Furthermore, the extent of the DnaG-dependent decrease in the error rate in the substrates 1, 3 and 4 was somewhat varied. A bigger decrease was apparent with substrate 4 in the absence of dGTP, a smaller decrease with substrate 3 in the absence of dCTP and an even smaller decrease with substrate 1 in the absence of dATP. These data indicate that the error rate of DnaEBs depends on the nature of the nucleotide template when the complementary dNTP is missing; DnaEBs is more efficient at mis-incorporating opposite pyrimidines (T and C templates) than purines (A and G templates). Secondly, the strength of the inhibitory effect of DnaG on nucleotide mis-incorporation by DnaEBs also depends on the nature of the template; the DnaG-mediated inhibitory effect on errors is largest opposite C template bases and then decreases progressively opposite G and T template bases, with no observed inhibition at mis-incorporations opposite A bases.
A similar decrease in the efficiency of DnaEBs errors was observed in the presence of the DnaC–DnaI complex (Figure 10C). In this case, a small inhibitory effect on mis-incorporations was also apparent opposite A bases (Figure 10C compare lanes labelled with minus and plus signs in substrate 2), an effect that was not observed with DnaG as explained above. Addition of both DnaG and the DnaC–DnaI complex also resulted in a reduction of the DnaEBs error rate, but not in an additive way (Figure 10D). Interestingly, DnaI on its own did not change the DnaEBs error-prone synthesis (Figure 10E), suggesting first, that the observed effect is due to specific protein–protein interactions and not an artefact, and second, that the effects on the DnaEBs errors observed in the presence of the DnaC–DnaI complex is the result of a DnaEBs–DnaC interaction. This was confirmed further with similar experiments carried out in the presence of hexameric DnaC, but in the absence of DnaI. DnaC on its own and in the absence of DnaI also exerted the same improvement of the DnaEBs fidelity in a similar manner to the DnaC–DnaI complex (data not shown).
The combined data show that the effects of the replicative helicase DnaC and the primase DnaG on the error-prone polymerase activity of DnaEBs are substrate dependent, suggesting that they are mediated by direct physical protein–protein interactions, which likely improve the fidelity of DnaEBs by allosterically altering the stringency of its catalytic site.
A structural model for the primase–polymerase hand-off
Reasonable-quality homology models for both B. subtilis DnaG and DnaEBs could be obtained from crystal structure templates using the Swissmodel Web Service (see ‘Materials and Methods’ section). In preparation for the analysis of the possibility of direct hand-off from DnaG to DnaEBs, the models of both proteins were elaborated to include a long length of template DNA strand both upstream and downstream of the two enzymes’ catalytic sites (for details, see ‘Materials and Methods’ section). The resulting model for the DnaG complex is shown in Figure 11A. The short length of ssDNA present in the crystal structure of the template protein (PDB entry 3B29) was extended in a 5′ direction, while a section of partially dsDNA/RNA was modelled into the basic groove, following the hypotheses discussed by Corn et al. (32). Note that this results in a sharp bend in the template DNA at the catalytic site. The resulting model for the DnaEBs complex is shown in Figure 11B. Evidently, in contrast to the rather open catalytic site in DnaG, in the presence DnaEBs, this region is buried. Only the extremity of the extruding hybrid duplex is visible, in addition to the modelled 5′ piece of template DNA strand, which enters the catalytic site at the point of contact between the fingers, β-binding and CTDs. As suggested by Wing et al. (33), we modelled the template strand interacting closely with the CTD. As a result, there is again a sharp bend in this strand at the catalytic site. Remarkably, the degree and direction of this bending are almost identical in the two models. This suggests that, if the two proteins can be brought into close enough physical contact, direct hand-off from DnaG to DnaEBs could take place with almost no significant change in the conformation of the nucleic acid.
Figure 11.

A structural model for the primase–polymerase (DnaG–DnaEBs) hand-off. (A) Model for DnaG (grey-blue) bound to a section of template DNA (grey), at the stage where 5 nt of primer (pink) have been synthesized. (B) Model for DnaE (PHP nuclease domain: red; palm: orange; thumb: yellow; fingers: green; beta binding: cyan; CTD: blue) bound to a section of template DNA (grey) hybridized to 10 nt of newly synthesized RNA/DNA (pink). (C) Model for the DnaG–DnaE complex, handing off the template DNA after synthesis of 5 nt of primer (colour coding as in panels A and B). Note that DnaE is oriented as shown in panel B, while DnaG is rotated 180° around the y-axis compared with the view in panel A. (D) View of the DnaG–DnaE complex from the direction of the duplex exit channel, emphasising the minor motion required to transfer the duplex from the basic groove of DnaG into its binding site in DnaE (arrow).
It is also clear from these models that some reorganization in the domains of DnaEBs will be required to expose the catalytic cleft for the transfer process. In particular, major motion in the CTD domain and a smaller adjustment in the thumb domain would be needed. Encouragingly, the crystal structure of the unliganded form of the template protein (34) suggests that such motions are quite reasonable. Rotating the CTD out of the way, and withdrawing the thumb domain slightly, we created a docking surface for DnaE that looks plausible (Figure 11C and D). First, the two catalytic sites are brought into proximity, such that hand-off would only require a movement of a few angstroms in the nucleic acid. Second, the entrance and exit directions for the template strand are perfectly aligned. Third, DnaG is predicted to make major interactions with the fingers and β-binding domains of DnaE, in agreement with our yeast two-hybrid experimental data that broadly specify the interaction interfaces.
DISCUSSION
Detailed studies of the E. coli replisome provided fundamental knowledge in our understanding of the structure and function of replisomes. Although basic structural/functional principles are common, it is now becoming apparent that replisomes exhibit architectural and functional diversity. Our combined data show that the replisome of the Gram-positive model bacterium B. subtilis is drastically different from the E. coli replisome and more similar to the eukaryotic and T7 phage replisomes because first, it uses two distinct DNA polymerases like the eukaryotic replisome (3,7,8,43), and secondly, one of the replicative polymerases (DnaEBs) forms a ternary complex with the replicative helicase DnaC and the primase DnaG when the helicase is a ring-shaped hexamer like the T7 replisome. As this hexamer assembles during initiation (and replication restart) and encircles the lagging strand template, the DnaEBs–DnaC–DnaG proteins form a subcomplex in the replisome that is pivotal during lagging strand synthesis. We have carried out biochemical characterizations of the three proteins and report the first functional analysis of this ternary complex to uncover basic principles of lagging strand synthesis.
Consistent with previous studies, the B. subtilis DnaC helicase activity was detected only in the presence of the putative helicase loader DnaI and required the presence of fork substrate confirming an assembly loading mechanism and a steric exclusion mode of DNA unwinding (25,41). DnaI is required to assemble a functional hexameric DnaC ring around the DNA substrate, but it has not formally been established whether it remains associated with the translocating helicase after loading or whether it is detached as is the case in the E. coli system with the functionally analogous DnaC helicase loader (25,27,40). It is likely that DnaI detaches from the functional DnaC during translocation, and some supporting evidence for this is provided by the observation in our gel filtration experiments that DnaI is displaced from the complex when DnaG and DnaEBs interact with DnaC to form the ternary complex. The helicase activity of DnaC was significantly stimulated by the primase DnaG, consistent with similar observations in other species (20–24,28,44–48).
Bacillus subtilis DnaG was found to initiate primer synthesis from 5′-d(CTA), 5′-d(TTA) and 5′-d(TTT). The interaction of DnaG with pre-formed hexameric DnaC helicase stimulated primer synthesis without altering the length of the RNA primers synthesized in priming assays. Helicase stimulation increased the abundance of smaller primers (5–15 ribonucleotides in length), while the quantity of the full-length 16-mer RNA polymers was unchanged. This feature differs considerably from the S. aureus system, whereby the DnaC helicase stimulated the production of predominantly full-length primers (39), and from the E. coli and G.stearothermophilus systems where the replicative DnaB helicases modulated their cognate primases to synthesize predominantly shorter primers (44,48). Priming reactions with B. subtilis DnaG3:DnaEBs demonstrated a direct interaction between these enzymes, with primer hand-off and extension occurring after di-ribonucleotide synthesis.
For the first time, we were able to detect lagging strand primer synthesis in a ‘stripped down’ in vitro reconstituted replication assay coupling RNA primer synthesis by DnaG, extension of the RNA primers by DnaEBs and fork translocation by DnaC. Using this assay, we showed that DnaEBs was able to efficiently extend RNA primers as short as 2 nt synthesized by DnaG, indicating that the DnaG to DnaEBs hand-off can take place very early during the process of de novo RNA primer synthesis by DnaG. It does not require maturation of RNA primers to relatively long sizes or the intervention of any other replisomal proteins. This is distinctly different from the process in E. coli, where the intervention of other proteins is required for the DnaG to DnaEEc primer hand-off to occur; the E. coli DnaG aided by its interaction with SSB remains stably bound to a primed site, and the χ subunit of the clamp loader interacts with SSB to facilitate primase displacement from the primed site before the DnaEEc polymerase can extend it (49). Furthermore, the ability of DnaEBs to extend extremely small RNA primers is an important result because it is consistent with direct hand-off of RNA primers from the DnaG to DnaEBs likely via their direct physical interaction. The fact that DnaEBs stimulated primer synthesis during translocation is another indication that the two proteins directly interact while operating on the DNA template. It may well be the case that at least the de novo primer synthesis by DnaG and initial extension by DnaEBs are not distributive and may be carried out by a distinct lagging strand–specific replisomal subcomplex comprising DnaG, DnaC and DnaEBs before the hand-off to the more processive and distributive PolC for Okazaki fragment synthesis. This is reminiscent of the eukaryotic pol α–primase action, where after the synthesis of small RNA primers (∼8–12 nt), an intrinsic counting mechanism shifts the primer from the primase active site in the p48 subunit to the polymerase active site in the p180 subunit within the same pol α–primase complex (50–53). Structural modelling and docking studies, guided by the information gained by our yeast-two hybrid data on the DnaG–DnaEBs interaction patches, revealed a structural model with the two catalytic sites in proximity, such that hand-off would only require a movement of a few angstroms in the nucleic acid with the entrance and exit directions for the template strand perfectly aligned. This is perfectly consistent with a direct hand-off mechanism mediated by a DnaG–DnaEBs interaction.
DnaEBs was poorly processive and relatively slow, consistent with previous studies (7). It produced well-defined DNA fragments with a characteristic periodic pattern as a function of time, which may indicate an inherent ability to extend primers to well-defined relatively short lengths before handing them off to PolC.
The DnaC helicase, with or without DnaI, and the DnaG primase did not significantly affect DnaEBs activity. These observations are consistent with the fully reconstituted replication assay, which suggests that DnaEBs adds small patches of DNA to primers before handing the hybrid primer product off to PolC. However, previous studies suggest a more potent DnaEBs polymerase activity that is further stimulated on SSB-coated templates (10,17). Moreover, the processivity of DnaEBs increases significantly (to more than the length of an Okazaki fragment) when the β sliding clamp is loaded on the template by the τδδ′ clamp loader (14). Hence, if DnaEBs were to add small patches of DNA to primers before hand-off to PolC, the hand-off would likely be an active process rather than passive falling off DnaEBs from the template before replacement by PolC (54). Such a passive mechanism would require DnaEBs to work independently of the β clamp. The two mechanisms (passive versus active) are not mutually exclusive. It may be that two hand-off mechanisms can occur during lagging strand synthesis: an active mechanism when DnaEBs operates in association with the β clamp to synthesize relatively long stretches of nascent DNA or a passive mechanism when DnaEBs operates independently of the β clamp to synthesize relatively short regular stretches of nascent DNA. The complexity of dynamic protein–protein interactions within the B. subtilis replisome may differentially regulate the activity of the DnaEBs in vivo by dynamically switching it from poorly active to highly active modes in response to the physiological needs of the cell. For example, different levels of activity may be required when using damaged or non-damaged DNA templates during DNA synthesis.
Although the native wild-type polymerase activity of DnaEBs was not significantly inhibited within the ternary helicase–primase–polymerase complex, its error rate was significantly reduced by the helicase and the primase. DnaEBs was able to efficiently extend primers in the absence of any one of the four dNTPs in primer extension assays. The efficiency of mis-incorporations was nucleotide dependent: higher error rates occurred opposite template pyrimidines (T and C) compared with purines (A and G). Both the DnaG primase and DnaC helicase decreased the mis-incorporation efficiency of DnaEBs in a nucleotide-dependent manner. DnaC was able to exert its inhibitory effect on DnaEBs either on its own or when in complex with the helicase loader DnaI, while DnaI did not affect DnaEBs mis-incorporation activity. The fact that the efficiency of mis-incorporations by DnaEBs and the effects of the DnaC and DnaG proteins were nucleotide dependent suggests that their effects are mediated by direct physical protein–protein interactions that allosterically affect the catalytic site of DnaEBs and increase its stringency. As DnaEBs lacks proofreading activity, and is error-prone and efficient at lesion bypass by frameshifting (10,14,17,42), the enhanced fidelity of this enzyme within the helicase–primase–polymerase ternary complex may contribute towards the reduction of DnaEBs errors during lagging strand DNA synthesis. DnaEBs also appears to physically interact with the MutS–MutL mismatch repair (MMR) system. Localized DnaEBs-GFP foci decrease in a MutS-dependent manner after mismatch incorporation (55). This suggests that DnaEBs errors may be corrected by the MMR mismatch repair system. Here we show that the interactions of DnaEBs with the helicase–primase complex not only temporally restrict DnaEBs to the lagging strand but also increase its fidelity independently of the MMR repair system.
The helicase–primase–polymerase lagging strand replisomal subcomplex confers a different architecture to the B. subtilis replisome than its E. coli counterpart. Such architecture could be typical of other related Firmicutes, closely related to B. subtilis, and possibly other two polymerase (PolC–DnaE) replication systems. This replisome structure may represent the archetypal bacterial replication architecture, as Gram-negative bacteria diverged from the Firmicutes at a later stage in evolution (56,57). The two polymerases may have evolved through distinct pathways providing different solutions to the problem of asymmetric DNA synthesis. Whereas DnaE co-evolved with core bacterial functions (i.e. the transcription machinery, the intermediary metabolic enzymes and the ribosome), PolC has co-evolved with RNA degradation enzymes present exclusively in the low-G+C-content Firmicutes (57). The DnaC–DnaG–DnaEBs lagging strand complex may also be involved in metabolic control of replication, as all three proteins were shown to be sensitive to metabolic signals originating from the three-carbon part of glycolysis (58).
In conclusion, we show that DnaC helicase, DnaG primase and DnaEBs polymerase form a functional replisomal subcomplex that is regulated by a highly complex network of protein–protein interactions. Within this subcomplex, DnaEBs is temporally restricted at the lagging strand, and its fidelity is improved. RNA primers synthesized de novo by DnaG are handed-off directly to DnaEBs for further extension without the involvement of other replisomal proteins. Collectively, these structural/functional properties render the B. subtilis replisome distinctly different from its E. coli counterpart.
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online: Supplementary Tables 1–3 and Supplementary Figure 1.
FUNDING
The Wellcome Trust [091968/Z/10/Z to P.S.]; a PhD fellowship from the School of Chemistry, University of Nottingham (to R.O); and the Ministère de l'Enseignement Supérieur et de la Recherche (MESR) (ED GGC, Université Paris Sud; to H.N.). L.J. is on the CNRS staff. Funding for open access charge: The Wellcome Trust.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Patrice Polard and Steven McGovern for providing the plasmids pSMG6, pSMG11 and the E.coli strain MCC26. The authors especially thank Geoffrey Briggs for technical advice on the DnaC/DnaI purification scheme and Matthew Green for help with the DnaEBs structure modelling.
REFERENCES
- 1.Hamdan S, Carr PD, Brown SE, Ollis D, Dixon NE. Structural basis for proofreading during replication of the Escherichia coli chromosome. Structure. 2002;10:535–546. doi: 10.1016/s0969-2126(02)00738-4. [DOI] [PubMed] [Google Scholar]
- 2.Mueller GA, Kirby TW, DeRose EF, Li D, Schaaper RM, London RE. Nuclear magnetic resonance solution structure of the Escherichia coli DNA polymerase III theta subunit. J. Bacteriol. 2005;187:7081–7089. doi: 10.1128/JB.187.20.7081-7089.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McHenry CS. Bacterial replicases and related polymerases. Curr. Opin. Chem. Biol. 2011;15:587–594. doi: 10.1016/j.cbpa.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McInerney P, Johnson A, Katz F, O’Donnell M. Characterization of a triple DNA polymerase replisome. Cell. 2007;27:527–538. doi: 10.1016/j.molcel.2007.06.019. [DOI] [PubMed] [Google Scholar]
- 5.Dervyn E, Suski C, Daniel R, Bruand C, Chapuis J, Errington J, Jannière L, Ehrlich SD. Two essential DNA polymerases at the bacterial replication fork. Science. 2001;294:1716–1719. doi: 10.1126/science.1066351. [DOI] [PubMed] [Google Scholar]
- 6.Titok M, Suski C, Dalmais B, Ehrlich SD, Jannière L. The replicative polymerases PolC and DnaE are required for theta replication of the Bacillus subtilis plasmid pBS72. Microbiology. 2006;152:1471–1478. doi: 10.1099/mic.0.28693-0. [DOI] [PubMed] [Google Scholar]
- 7.Sanders GM, Dallmann G, McHenry CS. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol. Cell. 2010;37:273–281. doi: 10.1016/j.molcel.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 8.Kunkel TA, Burgers PM. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 2008;18:521–527. doi: 10.1016/j.tcb.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pellegrini L. The polα-primase complex. Subcell. Biochem. 2012;62:157–169. doi: 10.1007/978-94-007-4572-8_9. [DOI] [PubMed] [Google Scholar]
- 10.Le Chatelier E, Bécherel OJ, d’Alençon E, Caneill D, Ehrlich SD, Fuchs RP, Jannière L. Involvement of DnaE, the second replicative DNA polymerase from Bacillus subtilis, in DNA mutagenesis. J. Biol. Chem. 2004;279:1757–1767. doi: 10.1074/jbc.M310719200. [DOI] [PubMed] [Google Scholar]
- 11.Su XC, Jergic S, Keniry MA, Dixon NE, Otting G. Solution structure of domains IVa and V of the τ subunit of Escherichia coli DNA polymerase III and interaction with the α subunit. Nucleic Acids Res. 2007;9:2825–2832. doi: 10.1093/nar/gkm080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flower AM, McHenry CS. The γ subunit of DNA polymerase III holoenzyme of Escherichia coli is produced by ribosomal frameshifting. Proc. Natl Acad. Sci. USA. 1990;87:3713–3717. doi: 10.1073/pnas.87.10.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yurieva O, Skangalis M, Kuriyan J, O’Donnell M. Thermus thermophilus dnaX homolog encoding γ- and τ-like proteins of the chromosomal replicase. J. Biol. Chem. 1997;272:27131–27139. doi: 10.1074/jbc.272.43.27131. [DOI] [PubMed] [Google Scholar]
- 14.Bruck I, O’Donnell M. The DNA replication machine of a Gram-positive organism. J. Biol. Chem. 2000;275:28971–28983. doi: 10.1074/jbc.M003565200. [DOI] [PubMed] [Google Scholar]
- 15.Reyes-Lamothe R, Sherratt DJ, Leake MC. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science. 2010;328:498–501. doi: 10.1126/science.1185757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noirot-Gross MF, Dervyn E, Wu LJ, Mervelet P, Errington J, Ehrlich SD, Noirot P. An expanded view of bacterial DNA replication. Proc. Natl Acad. Sci. USA. 2002;99:8342–8347. doi: 10.1073/pnas.122040799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruck I, Goodman MF, O’Donnell M. The essential C family DnaE polymerase is error-prone and efficient at lesion bypass. J. Biol. Chem. 2003;278:44361–44368. doi: 10.1074/jbc.M308307200. [DOI] [PubMed] [Google Scholar]
- 18.Costes A, Lecointe F, McGovern S, Quevillon-Cheruel S, Polard P. The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet. 2010;6:e1001238. doi: 10.1371/journal.pgen.1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simmons LA, Davies BW, Grossman AD, Walker GC. Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol. Cell. 2008;29:291–301. doi: 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bailey S, Eliason WK, Steitz TA. Helicase and its complex with a domain of DnaG primase. Science. 2007;318:459–463. doi: 10.1126/science.1147353. [DOI] [PubMed] [Google Scholar]
- 21.Chintakayala K, Larson MA, Griep MA, Hinrichs SH, Soultanas P. Conserved residues of the C-terminal p16 domain of primase are involved in modulating the activity of the bacterial primosome. Mol. Microbiol. 2008;68:360–371. doi: 10.1111/j.1365-2958.2008.06155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thirlway J, Turner IJ, Gibson CT, Gardiner L, Brady K, Allen S, Roberts CJ, Soultanas P. DnaG interacts with a linker region that joins the N- and C-domains of DnaB and induces the formation of 3-fold symmetric rings. Nucleic Acids Res. 2004;32:2977–2986. doi: 10.1093/nar/gkh628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Syson K, Thirlway J, Hounslow AM, Soultanas P, Waltho JP. The solution structure of the helicase-interaction domain of the primase DnaG: a model for helicase activation. Structure. 2005;13:609–616. doi: 10.1016/j.str.2005.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corn JE, Pease PJ, Hura GL, Berger JM. Crosstalk between primase subunits can act to regulate primer synthesis in trans. Mol. Cell. 2005;20:391–401. doi: 10.1016/j.molcel.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Velten M, McGovern S, Marsin S, Ehrlich SD, Noirot P, Polard P. A two-protein strategy for the functional loading of a cellular replicative DNA helicase. Mol. Cell. 2003;11:1009–1020. doi: 10.1016/s1097-2765(03)00130-8. [DOI] [PubMed] [Google Scholar]
- 26.Ioannou C, Schaeffer PM, Dixon NE, Soultanas P. Helicase binding to DnaI exposes a cryptic DNA-binding site during helicase loading in Bacillus subtilis. Nucleic Acids Res. 2006;34:5247–5258. doi: 10.1093/nar/gkl690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soultanas P. Loading mechanisms of ring helicases at replication origins. Mol. Microbiol. 2012;84:6–16. doi: 10.1111/j.1365-2958.2012.08012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larson MA, Griep MA, Bressani R, Chintakayala K, Soultanas P, Hinrichs SH. Class-specific restrictions define primase interactions with DNA template and replicative helicase. Nucleic Acids Res. 2010;38:7167–7178. doi: 10.1093/nar/gkq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larson MA, Bressani R, Sayood K, Corn JE, Berger JM, Griep MA, Hinrichs SH. Hyperthermophilic Aquifex aeolicus initiates primer synthesis on a limited set of trinucleotides comprised of cytosines and guanines. Nucleic Acids Res. 2008;36:5260–5269. doi: 10.1093/nar/gkn461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koepsell SA, Larson MA, Frey CA, Hinrichs SH, Griep MA. Staphylococcus aureus primase has higher initiation specificity, interacts with single-stranded DNA stronger, but is less stimulated by its helicse than Escherichia coli primase. Mol. Microbiol. 2008;68:1570–1582. doi: 10.1111/j.1365-2958.2008.06255.x. [DOI] [PubMed] [Google Scholar]
- 31.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein-homology modelling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corn JE, Pelton JG, Berger JM. Identification of a DNA primase template tracking site redefines the geometry of primer synthesis. Nat. Struct. Mol. Biol. 2008;15:163–169. doi: 10.1038/nsmb.1373. [DOI] [PubMed] [Google Scholar]
- 33.Wing RA, Bailey S, Steitz TA. Insigths into the replisome from the structure of a ternary complex of the DNA polymerase IIIα-subunit. J. Mol. Biol. 2008;382:859–869. doi: 10.1016/j.jmb.2008.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey S, Wing RA, Steitz TA. The structure of T. acquaticus DNA polymerase III is distinct from eukaryotic replicative DNA polymerases. Cell. 2006;126:893–904. doi: 10.1016/j.cell.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 35.Pattersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 36.Patel SS, Pandey M, Nandakumar D. Dynamic coupling between the motors of DNA replication: hexameric helicase, DNA polymerase, and primase. Curr. Opin. Chem. Biol. 2011;15:595–605. doi: 10.1016/j.cbpa.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costes A, Lecointe F, McGovern S, Quevillon-Cheruel S, Polard P. The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001238. e1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thirlway J, Soultanas P. In the Bacillus stearothermophilus DnaB-DnaG complex the activities of the two proteins are modulated by distinct but overlapping networks of residues. J. Bacteriol. 2006;188:1534–1539. doi: 10.1128/JB.188.4.1534-1539.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koepsell SA, Larson MA, Griep MA, Hinrichs SH. Staphylococcus aureus helicase but not Escherichia coli helicase stimulates S. aureus primase activity and maintains initiation specificity. J. Bacteriol. 2006;188:4673–4680. doi: 10.1128/JB.00316-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makowska-Grzyska M, Kaguni JM. Primase directs the release of DnaC from DnaB. Mol. Cell. 2010;37:90–101. doi: 10.1016/j.molcel.2009.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaplan DL. The 3 ‘-tail of a forked-duplex sterically determines whether one or two DNA strands pass through the central channel of a replication-fork helicase. J. Mol Biol. 2000;301:285–299. doi: 10.1006/jmbi.2000.3965. [DOI] [PubMed] [Google Scholar]
- 42.Low RL, Rashbaum SA, Cozzarelli NR. Purification and characterization of DNA polymerase III from Bacillus subtilis. J. Biol. Chem. 1976;251:1311–1325. [PubMed] [Google Scholar]
- 43.Simmons LA, Davies BW, Grossman AD, Walker GC. Beta clamp directs localization of mismatch repair Bacillus subtilis. Mol. Cell. 2008;29:291–301. doi: 10.1016/j.molcel.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamdan SM, Richardson CC. Motors, switches, and contacts in the replisome. Annu. Rev. Biochem. 2009;78:205–243. doi: 10.1146/annurev.biochem.78.072407.103248. [DOI] [PubMed] [Google Scholar]
- 45.Bird LE, Pan H, Soultanas P, Wigley DB. Mapping protein-protein interactions within a stable complex of DNA primase and DnaB helicase from Bacillus stearothermophilus. Biochemistry. 2000;39:171–182. doi: 10.1021/bi9918801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitkova AV, Khopde SM, Biswas SB. Mechanisms and stoichiometry of interaction of DnaG primase with DnaB helicase of Escherichia coli in RNA primer synthesis. J. Biol. Chem. 2003;278:52253–52261. doi: 10.1074/jbc.M308956200. [DOI] [PubMed] [Google Scholar]
- 47.Biswas SB, Wydra E, Biswas EE. Mechanisms of DNA binding and regulation of Bacillus anthracis DNA primase. Biochemistry. 2009;48:7373–7382. doi: 10.1021/bi900086z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson SK, Bhattacharyya S, Griep MA. DnaB helicase stimulates primer synthesis activity on short oligonucleotide templates. Biochemistry. 2000;39:736–744. doi: 10.1021/bi991554l. [DOI] [PubMed] [Google Scholar]
- 49.Yuzhakov A, Kelman Z, O’Donnell M. Trading places on DNA; a three-point switch undelies primer handoff from primase to the replicative DNA polymerase. Cell. 1999;96:153–163. doi: 10.1016/s0092-8674(00)80968-x. [DOI] [PubMed] [Google Scholar]
- 50.Copeland WC, Wang TS. Enzymatic characterization of the individual mammalian primase subunits reveals a biphasic mechanism for initiation of DNA replication. J. Biol. Chem. 1993;268:26179–26189. [PubMed] [Google Scholar]
- 51.Arezi B, Kirk BW, Copeland WC, Kuchta RD. Interactions of DNA woth human DNA primase monitored with photoactivatable cross-linking agents: implications for the role of the p58 subunit. Biochemistry. 1999;38:12899–12907. doi: 10.1021/bi9908991. [DOI] [PubMed] [Google Scholar]
- 52.Zerbe LK, Kuchta RD. The p58 subunit of human DNA primase is important for primer initiation, elongation, and counting. Biochemistry. 2002;41:4891–4900. doi: 10.1021/bi016030b. [DOI] [PubMed] [Google Scholar]
- 53.Nunez-Ramirez R, Klinge S, Sauguet L, Melero R, Recuero-Checa MA, Kilkenny M, Perera RL, Garcia-Alvarez B, Hall RJ, Nogales E, et al. Flexible tethering of primase and DNA pol α in the eukaryotic primosome. Nucleic Acids Res. 2011;39:8187–8199. doi: 10.1093/nar/gkr534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McHenry CS. Breaking the rules: bacteria that use several DNA polymerase IIIs. EMBO Rep. 2011b;12:408–414. doi: 10.1038/embor.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klocko AD, Schroeder JW, Walsh BW, Lenhart JS, Evans ML, Simmons LA. Mismatch repair causes the dynamic release of an essential DNA polymerase from the replication fork. Mol. Microbiol. 2011;82:648–663. doi: 10.1111/j.1365-2958.2011.07841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Briggs GS, Smits WK, Soultanas P. The chromosomal replication machinery of low G+C content Firmicutes. J. Bacteriol. 2012;194:5162–5170. doi: 10.1128/JB.00865-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Engelen S, Vallenet D, Médigue C, Danchin A. Distinct co-evolution patterns of genes associated to DNA polymerase III DnaE and PolC. BMC Genomics. 2012;13:69. doi: 10.1186/1471-2164-13-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jannière L, Canceill D, Suski C, Kanga S, Dalmais B, Lestini R, Monnier AF, Chapuis J, Bolotin A, Titok M, et al. Genetic evidence for a link between glycolysis and DNA replication. PloS One. 2007;2:e447. doi: 10.1371/journal.pone.0000447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.