


Abstract
Read alignment is an ongoing challenge for the analysis of data from sequencing technologies. This article proposes an elegantly simple multi-seed strategy, called seed-and-vote, for mapping reads to a reference genome. The new strategy chooses the mapped genomic location for the read directly from the seeds. It uses a relatively large number of short seeds (called subreads) extracted from each read and allows all the seeds to vote on the optimal location. When the read length is <160 bp, overlapping subreads are used. More conventional alignment algorithms are then used to fill in detailed mismatch and indel information between the subreads that make up the winning voting block. The strategy is fast because the overall genomic location has already been chosen before the detailed alignment is done. It is sensitive because no individual subread is required to map exactly, nor are individual subreads constrained to map close by other subreads. It is accurate because the final location must be supported by several different subreads. The strategy extends easily to find exon junctions, by locating reads that contain sets of subreads mapping to different exons of the same gene. It scales up efficiently for longer reads.
INTRODUCTION
Developments in next-generation (next-gen) sequencing technologies that parallelize the sequencing process have dramatically increased world-wide sequencing capacity in the past few years. Individual projects, such as the 1000 Genomes project (1) or The Cancer Genome Atlas (http://cancergenome.nih.gov, March 2013), can produce tens or hundreds of terabytes of sequence. A single Illumina HiSeq system now has the capacity to generate >4 billion bases of sequence per hour. Meanwhile the typical length of an individual sequence read has increased from ∼30 to 100 bp, and is likely to increase further.
Next-gen sequencing is revolutionalizing many areas of biological research. It may be used to detect variation in genomic DNA, to measure gene expression, to identify RNA transcripts and for many other purposes. Read mapping, the alignment of sequence reads to a reference genome, is the first step for many of these analyses and is often the most computationally intensive part of the analysis.
All read aligners have to take algorithmic shortcuts because the computational cost of exhaustively comparing every read to every possible position in the genome is prohibitively expensive. The first step is almost always to map a shorter part of the read (a seed) to the genome. Typically, only a small number of mismatches are permitted, and indels are disallowed entirely. This is partly for specificity, but also because too many mismatches may cause later steps like backtracking to fail. Most aligners then work out from the location that the seed mapped to, trying to match the remainder of the read to the genome surrounding the original location, a process often called the extension step (2). Typically a short seed will map to multiple locations in the genome, so the seed must be extended at multiple locations before it can be settled which of the original locations has the best overall match to the complete read. At each location, the extension step must contend with the possibility of sequencing errors, polymorphisms or indel events. If the read was generated from RNA, then each extension step must moreover deal with the possibility that the read might span two or more exons that might be well separated in the genome. The extension steps are far more expensive than mapping the original seed, especially for longer reads. Much of the computational cost is incurred because the final mapping location cannot be decided until all the extension steps are largely complete. If the original seed contains too many sequencing errors or mutations relative to the reference genome, or spans an unexpected exon junction, then the read alignment may be doomed from the start.
Popular aligners that extend from a seed in various ways include Bowtie (3), Bowtie2 (4), BWA (5), Novoalign (http://www.novocraft.com, 2013), Maq (6) and MrsFast (7). The extension step usually involves backtracking (3,5), Smith–Waterman dynamic programming (4–6,8) or Needleman–Wunsch dynamic programming (9) (Novoalign). A survey of read aligners can be found in (10). In general, the running time of dynamic programming increases quadratically with read length (11,12). Many clever algorithms have been proposed to make the extension step more efficient, including bounded backtracking (5) and banded and bit-vector versions of dynamic programming (13,14). Bowtie2 has abandoned backtracking in favour of an Single Instruction Multiple Data-accelerated dynamic programming procedure (4). Despite all efforts, seed extension remains intrinsically expensive for longer reads.
A recent trend to avoid problems with the choice of seed is to try multiple spaced seeds (8,9,13,15–18). This multiplies the candidate locations, which must then be prioritized by some form of filtering to improve specificity. A recent technique to do this is q-gram filtering. This procedure extracts a number of q-grams (substrings or seeds of length q) from a sliding window moved along the read (13,19–21) or from the entire read (8,9,17). A measure of local similarity or a count of matched q-grams is then used to determine whether the candidate regions should be included for further examination. Local similarity has been measured efficiently using parallelograms (13).
In this article, we propose a new multi-seed strategy that differs from previous algorithms by choosing the mapped genomic location for the read directly from the seeds. The strategy consists of a seed-and-vote step, which achieves local alignment simultaneously in multiple parts of the read, followed by an in-fill step to complete the alignment. The new strategy uses a relatively large number of short equi-spaced seeds from each read, which we call subreads. Instead of trying to prioritize the seeds, the strategy allows all the subreads to vote on the optimal location for the read. The voting procedure has similarities with q-gram counting, but is used instead to determine a unique location. The new strategy differs from previous procedures in a number of ways: the subreads are shorter and more numerous than conventional seeds; they are mapped without mismatches; and the local alignment is determined directly by counting subreads without further intermediate steps. The subread procedure then uses conventional algorithms including dynamic programming to complete the alignment, filling in the detailed mismatch and indel information between the subreads that make up the winning voting block. The alignment is extremely fast because the overall genomic location has already been chosen before the detailed alignment is done, and because the in-fill is required for very short local regions only, with known flanking locations already provided by the matched subreads. The strategy has been implemented in two software tools: Subread for general purpose alignment and Subjunc for detecting exon–exon junctions from RNA reads.
Seed-and-vote local alignment may at first impression seem too naive, as it does not require any conventional concept of sequence similarity like edit distance to be specified explicitly. Instead, a suitable balance between sensitivity and specificity is achieved implicitly by choosing a considerable number of relatively short subreads. In extensive testing, the strategy proves to be not only fast but more than competitive with existing aligners in terms of sensitivity and accuracy. The strategy is sensitive because no individual subread is required to map exactly, nor are individual subreads constrained to map close by other subreads. The strategy is accurate because the final location must be supported by several different subreads. Crucially, the strategy scales up efficiently for longer reads.
Insertions and deletions are genomic variants that have been linked to the onset and progression of a number of diseases. For example, a 6 bp indel in the promoter region of gene Casp8 was identified to be associated with susceptibility to multiple cancers (22). A 14 bp indel in the gene Ncx1 was found to modulate the age at onset in late-onset Alzheimer’s disease (23). Indel detection is an important part of read alignment when mapping genomic DNA, but presents special problems for many aligners. The need to detect indels makes dynamic programming and backtracking very time-consuming, and the presence of indels can make similarity measures like Hamming distance misleading. Q-gram filtering methods are seldom designed for detecting insertions and deletions. SWIFT, for example, can detect insertions and deletions within the sliding windows, but not in the entire region of the read (20). By contrast, our Subread software finds indels rapidly anywhere in the read, mainly leveraging the fact that indels can be restricted to very small regions bounded by flanking local alignments.
RNA-seq presents particular challenges for aligners because RNA transcripts typically comprise multiple exons that might be thousands of bases apart in genomic location. Elucidating the splicing mechanism is important for understanding various biological processes, which might make use of different isoforms from the same genes to exert their functions. Ordinary DNA mapping techniques designed for contiguous reads cannot be applied successfully to map sequences that span exon–exon junctions. RNA-seq mapping has therefore concentrated on the detection of exon–exon junctions in the read. Junction detectors need to split the read into smaller segments, typically non-overlapping segments of about 25 bp (24–26). Each segment is then mapped separately to the reference genome, and an exon–exon junction is detected when segments from the same read map to different exons. Our subread strategy can be viewed as a more flexible and higher resolution version of segmentation that uses shorter more numerous overlapping segments. Subjunc is a specialized version of our subread software that performs complete alignment of RNA-seq reads including detection of exon–exon junctions. Compared with segmentation, the use of overlapping subreads allows shorter subsequences to be matched to exons while taking full advantage of longer single-exon subsequences when they exist. Junctions can be detected closer to the ends of the reads. At the same time, the seed-and-vote strategy provides speed improvements both at the full read level and within each single-exon subsequence.
Not all analyses of RNA-seq data require detection of splice junctions. A popular type of gene-level differential expression analysis uses read counts that are summarized at the gene level (27–30). For this type of analysis, the seed-and-vote paradigm provides a special efficiency, because each read can be anchored to a particular exon in a particular gene, even before the exon–exon junctions have been detected. This means that Subread can be used to generate gene-level count summaries, without the need to run Subjunc. This provides spectacular speed improvements for this particular type of analysis over alternative alignment pipelines.
This article presents results from an extensive suite of test scenarios to compare Subread with other popular aligners. We present results both from simulations and from a range of calibration data sets, including the 1000 Genomes project, sequencing data with spike-in controls, and benchmark RNA-seq data from the Sequencing Quality Control (SEQC) project. The tests include indel detection for genomic DNA mapping scenarios and exon-junction detection for RNA-seq. Special attention is given in our comparisons to accuracy, i.e. to incorrectly mapped reads, as well as to the more commonly examined questions of sensitivity and speed.
MATERIALS AND METHODS
Data sets
We used a 1000 Genomes data set, a SEQC data set and simulation data sets to compare alternative methods for read mapping and exon–exon junction detection. The 1000 Genomes data set includes 27.5 million pairs of 100 bp reads, which were generated from an exome sequencing of a Puerto Rico individual (SRR070481). The sequencing was performed by Washington University Genome Sequencing Center in October 2010, using an Illumina Genome Analyzer II sequencer.
The SEQC project, which is the third stage of the well-known MAQC project (31), is producing benchmark next-gen sequencing data. It aims to use these data to evaluate current analysis methods and to provide a guideline for analysing the sequencing data. Four types of samples are being sequenced in this project, including A, B, C and D. Sample A is the Universal Human Reference RNA (UHRR). Sample B is the Human Brain Reference RNA (HBRR). Samples C and D are mixed from A and B at mixing percentages of 75%A:25%B and 25%A:75%B, respectively. We chose one library for each sample and included them in this study. Each library has ∼6 million pairs of 101 bp reads. This data set was generated by City of Hope, USA, in August 2011, using an Illumina HiSeq sequencer.
One hundred and one base pair simulation data were generated from a modified human reference genome GRCh37(hg19), in which 80 bp or longer repetitive sequences were removed so as to make each simulated read have a unique known mapping location. SNPs and indels were randomly introduced to the human genome GRCh37, at rates of 0.0009 and 0.0001, respectively, to simulate genomic variation. This setting is the same as that used in the work of Li and Durbin (5). Real quality scores, extracted from a 101 bp SEQC Sample A read data set, were used for simulation reads. Sequencing errors were generated according to the quality score each read base has. The lower the quality score, the more likely a sequencing error was introduced. So the distribution of sequencing errors is similar to that of the real base calling errors. This makes the simulation read data very similar to the real read data. Supplemental Figure S1 shows the mean error rates at each base location in SEQC reads and in simulation reads.
Two 101 bp simulation data sets were generated. One contained indels and the other did not. Indels were not introduced to the reference genome when generating the data set containing no indels. Each data set included 10 million single-end reads. Two 202 bp simulation data sets (one contained indels and the other did not) were generated in the similar means, except that quality score of each base in each SEQC read was duplicated before being assigned to the longer reads.
In addition to the simulation data sets generated from the filtered human genome, we generated a 101 bp simulation data set from the unfiltered human genome, in which repetitive regions were kept. This data set contained indels in it. We also used Mason (32) and Art (33) to generate two extra simulation data sets. The unfiltered human genome was used for them as well. We generated 100 000 100-bp-long reads from using each read simulator. For Mason, we used an SNP rate of 0.0009, an indel rate of 0.0001 and the default sequencing error rate (0.004). For Art, we provided it with a quality profile, which was created from the SEQC data set used in this study, to make it introduce sequencing errors using the real base calling errors. The indel rate used was 0.001. For all other parameters of Maons and Art, default values were used. Versions of Mason and Art used are 0.1 and 1.5.0, respectively.
ERCC spike-in control data
The Ambion(textregistered) External RNA Controls Consortium (ERCC) spike-in control includes 92 spike-in transcripts, which are spiked in difference concentrations in each of the two mixes (Mix 1 and Mix 2) (http://www.lifetechnologies.com, 2013). The transcripts in these two mixes are present at defined Mix 1:Mix 2 molar concentration ratios, described by four subgroups (log fold changes of 2, 0, −0.58 and −1, respectively). Each group contains 23 transcripts spanning a 106-fold concentration range, with approximately the same transcript size and GC content. The median length of the spike-in transcript sequence is 994 bp.
The ERCC spike-in control sequencing data used in this study were created as part of the SEQC study. Mix 1 and Mix 2 were pooled with SEQC sample A (UHRR) and sample B (HBRR), respectively, before library preparation was performed. Spike-in transcript sequences were combined with human genome so that a hybrid index can be built by each aligner. Spike-in reads and human reads were then mapped to the hybrid index.
To compute fold changes for each spike-in transcript, read counts were normalized by total number of mapped spike-in reads and by the transcript length (reads per 1 kb transcript per 10 000 mapped spike-in reads). An offset count of 0.5 was added to the raw read counts to avoid taking the log of zero.
Exon–exon junctions derived from NCBI RefSeq annotation
When comparing different methods for detecting exon–exon junctions, we assessed their ability to discover junctions that originate from annotated exons. We obtained chromosomal coordinates of annotated exons from NCBI RefSeq human gene annotation (Build 37.2). We call a reported junction as a ‘known’ junction, if it connects two annotated exons from the same gene, i.e. the 5′ splicing point of the junction is located at the last base position of the 5′ exon and the 3′ splicing point is located one base before the first base of the 3′ exon.
Mapping quality scores
Subread and Subjunc output a mapping quality score (MQS) for each mapped read, defined by
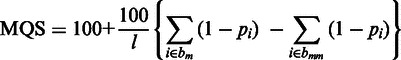 |
where l is the read length, pi is the base-calling P value for the ith base in the read, bm is the set of locations of matched bases and bmm is the set of locations of mismatched bases.
Base-calling P values can be readily computed from the base quality scores available in the FASTQ file (raw read data file). High-quality bases have low base-calling P values. Read bases that were found to be insertions are treated as matched bases in the MQS calculation. The MQS is a read-length normalized value, which is in the range of 0–200. If a read can be best mapped to more than one location, its MQS will be divided by the number of such locations.
Building index for reference genome
To build an index, 16 bp sequences were extracted from the reference genome in every three bases, i.e. there is a 2 bp gap between each pair of neighbouring 16 bp sequences. Correspondingly, each read has to be scanned three times for the mapping, i.e. three sets of subreads are extracted, which start from the first, second and third base of the read, respectively.
We build a hash table for a reference genome to enable fast access to the chromosomal locations of subreads extracted from each read. The hash table includes all the informative 16 bp sequences extracted from the reference genome (keys) and also their chromosomal locations (values). Each base in each 16 bp sequence is encoded by 2 bits. Therefore, each 16 bp sequence occupies 4 bytes of space. For mouse or human genomes, their index sizes are 6.2 and 6.6 GB, respectively. The actual peak memory usage will be slightly higher than these, because sequences of the entire genome are loaded into memory as well when performing alignment. The index-building function provides the option of breaking the index into multiple parts so as to reduce the memory footprint (only one part is present in the memory at any time).
Aligners and junction detectors under comparison
Versions of aligners included in this study are as follows: Subread (1.3.1), Bowtie2 (2.0.0-Beta3), BWA (0.5.9), Maq (0.7.1), MrsFast (2.3.0.2) and Novoalign (2.07.11). All aligners were run using their default settings except Novoalign and MrsFast, which were run with the options -rRandom and -n 1, respectively, to report at most one hit for each read so that they can be compared with other aligners. Versions of junction detectors included in this study are: Subjunc (1.3.1), TopHat (1.3.0), TopHat 2 (2.0.0) and MapSplice (1.15.2). All the programs were tested on a HP Blade supercomputer, which includes 16 Xeon 2.93 GHz CPU cores and 128 GB of memory.
Subread and Subjunc can be downloaded from http://subread.sourceforge.net or http://www.bioconductor.org (Rsubread package).
RESULTS
The seed-and-vote paradigm
We describe a new multi-seed alignment strategy that chooses the mapped genomic location for the read directly from the seeds (Figure 1). The new strategy uses a number of overlapping seeds from each read, called subreads. Instead of trying to pick the best seed, the strategy allows all the seeds to vote on the optimal location for the read. The algorithm then uses more conventional alignment algorithms to fill in detailed mismatch and indel information between the subreads that make up the winning voting block. Figure 1B illustrates the proposed seed-and-vote mapping approach with an artificial example.
Figure 1.

Seed-and-vote mapping paradigm. (A) Schematic of the proposed mapping paradigm. Subreads (or seeds) are short continuous sequences extracted from each read. Substrings in green are uninformative subreads, and they are excluded from voting. Little red bars denote mismatched bases. Mapping location of the read is determined by the largest consensus set. The thin solid arrows point to the mapping location of each subread included in the largest consensus set. Mapping location of the read, as indicated by the black up-pointing triangle, is voted for by all the subreads in the largest consensus set. The dashed arrows indicate other mapping locations for the subreads, and these locations were disregarded due to insufficient number of votes. (B) Using an artificial example to illustrate the paradigm. Six subreads are extracted from the artificial read. Each square bracket denotes an extracted subread, which contains five continuous bases, and the number embedded in the blue cycle indicates the subread number. Base sequence of each subread is encoded into a string of 0’s and 1’s (each base is encoded into a 2-bit binary number). Encoded value for each subread is used as its key in the hash table. The key’s value gives the chromosomal location/s in the genome to which the corresponding subread is perfectly matched (no mismatches allowed). Four candidate mapping locations are found for this artificial read, which receive 2, 5, 1 and 2 votes (number of consensus subreads), respectively. The location that receives the largest number of votes, in this case the location with five votes, is selected as the final mapping location for this artificial read. (C) Indel detection performed under the seed-and-vote paradigm. (C1) shows the mapping results of subreads when there are no indels found in the reads (assuming no mismatches exist in the read for simplicity). (C2) and (C3) show respectively the schematic for detecting an insertion (Ins) and a deletion (Del) in the situation where insertion or deletion is found in the read and flanking subreads are found at both sides of insertion or deletion. (C4) gives the schematic for detecting indels when they occur at the locations close to the end of the reads where flanking subreads can be found at only one side. In (C2) and (C3), chromosomal locations pointed to by red arrows are the true mapping locations of subreads 8, 9 and 10, respectively, and chromosomal locations pointed to by dotted black arrows indicate the chromosomal locations to which they will be mapped if no indels exist before them. d is the indel length, equal to the difference between the location pointed to by the red arrow and the location pointed to by the dotted black arrow from the same subread. Regions encompassed by the dotted green lines are found to contain indels [(C2) and (C3)] or are candidate regions for searching indels (C4). Bases in these regions are not covered by subreads that have made successful votes, and their mapping locations will be determined by aligning to the corresponding regions (within the dotted green lines) in the reference genome. In (C4), a 4 bp window is moved along the uncovered bases to look for potential indels. When three or more bases in the window are found to be mismatches, the indel detection process is triggered for the search of indels.
Optimal subread length
A set of equally spaced overlapping substrings, the subreads, are extracted from the read, and each is mapped to the reference genome. No mismatches are permitting when mapping each subread, so this step can be accomplished with superb speed and efficiency via a hash index of the genome. Instead of allowing mismatches, we keep the subreads relatively short to achieve a good balance between sensitivity and accuracy. Tests show that a range of subread lengths, from 10–25 bp, work well from this point of view (data not shown). Subread uses subreads of length 16 because that is in the optimal range for sensitivity and accuracy and because sequences of this length will fit exactly into a machine word on a 32-bit computer system or into half a word on a 64-bit computer system. This uses computer memory in the most efficient way and reduces data access time (Supplemental Methods, Supplemental Figure S2).
For the subread strategy to work effectively, it is necessary that each subread has reasonable specificity, so subreads corresponding to highly repetitive or overly common sequences are removed from the subread set. Examination of the human genome shows that 81% of all possible 16 bp sequences occur 24 or fewer times in the genome (Supplemental Methods, Supplemental Figure S3). With this motivation, we define as uninformative any subread whose sequence occurs >24 times in the reference genome. The informative subreads are therefore those subreads that occur ≤24 times in the reference genome. Simulations show that higher thresholds lead to higher mapping sensitivity but lower accuracy (Table 5). Our goal is to achieve a high mapping accuracy and a high mapping speed; therefore, we decided to use a more stringent threshold to filter out uninformative subreads. A cut-off of 24 repeats was used for Subread when comparing it with other aligners in this study unless otherwise stated. Subread provides an option (‘-f’) in the index building program so that users can adjust this threshold if appropriate.
Table 5.
Performance of aligners in mapping simulation reads generated from the unfiltered human genome (repetitive regions were kept)
| Aligner | Art |
Mason |
Our simulator |
Time (min) | |||
|---|---|---|---|---|---|---|---|
| Rec (%) | Acc (%) | Rec (%) | Acc (%) | Rec (%) | Acc (%) | ||
| Subread | 81.5 | 97.9 | 88.8 | 96.1 | 88.5 | 98.0 | 17 |
| Subread -f 100 | 84.4 | 97.7 | 91.5 | 96.0 | 91.3 | 97.9 | 19 |
| Subread -f 200 | 85.5 | 97.6 | 92.5 | 95.9 | 92.4 | 97.8 | 21 |
| Subread -f 300 | 86.1 | 97.5 | 93.1 | 95.8 | 92.9 | 97.7 | 22 |
| Bowtie2 | 87.6 | 95.2 | 95.2 | 95.3 | 95.7 | 96.0 | 83 |
| BWA | 87.1 | 97.2 | 95.5 | 95.7 | 78.6 | 96.4 | 497 |
| Novoalign | 89.8 | 97.3 | 93.5 | 97.1 | 140 | ||
One hundred thousand 100 bp reads were each generated from Art and Mason simulators, and 10 million 101 bp reads were generated from our simulator. ‘-f’ option of Subread specifies the threshold for removing uninformative subread. For example, ‘-f 100’ means those subreads that occur 100 or more times in the reference genome were removed. Running time was measured using our simulation data.
Any set of informative subreads that vote for the same mapping location for the read is called a consensus set. In general, a read will have more than one consensus set. This is partly because of ambiguity, because a subread can be mapped to more than one location, but also because different regions of the read could genuinely originate from disjoint regions of the reference sequence, for example because an RNA read could span one or more exon–exon junctions.
The largest consensus set for each read determines its mapping location. When there is no unique largest consensus set, because two or more consensus sets mapping to different locations have the same number of votes, the one covering more bases in the genome is chosen. If there is still a tie, it is broken on the basis of either MQSs or by the Hamming distance between the read and each candidate region.
How many subreads and how many votes?
The remaining parameters that determine the mapping algorithm are the number of subreads selected from each read and the consensus threshold. The consensus threshold is the minimum number of subreads (votes) required for reporting a mapping location. An extensive simulation study was undertaken to establish optimal values for these parameters (Supplemental Materials). Numbers of subreads ranging from 7 to 28, and consensus thresholds ranging from 10 to 70% of the number of subreads, were examined for the mapping of 10 million 101 bp reads. Not surprisingly, sensitivity decreased and accuracy improved with the consensus threshold increase for any fixed subread number (Supplemental Figure S4). However, setting the consensus threshold at ∼30% of the subread number gave good performance with respect to both accuracy and sensitivity across a wide range of subread numbers and cut-offs for removing uninformative subreads (Supplemental Figure S5). Smaller numbers of subreads are preferred from a computational cost point of view. By taking all the evaluation results into account, we decided to select 10 subreads from each read and use a consensus threshold of three for mapping.
Detecting indels around the subreads
Detecting deletions and insertions is an especially difficult aspect of read mapping that typically incurs considerable computational cost. Our seed-and-vote strategy, however, facilitates an efficient and accurate approach to indel detection with only very modest computation overhead. First consider indels that are flanked by consensus subreads. In that case, the genomic positions of the flanking subreads determine the indel length and bound the locations of the indel bases. Indels near the ends of the reads will not have flanking subreads on both sides. In this case, we move a window along the unmapped regions to identify indels. The subread approach only needs to align read bases not covered by the mapped subreads, a considerable computational saving compared with full alignment of the entire read.
Figure 1C illustrates how we identify indels and determine their lengths and locations. Figure 1(C1) shows the mapping locations of subreads when there are no indels found in a read. For simplicity, here every extracted subread is mapped to a unique location. It can be seen that the distances between mapping locations of subreads in the reference genome are the same as their distances in the read. We make use of this distance concordance to infer indel lengths. When a read contains an insertion [Figure 1(C2)], mapping locations of the subreads on the right side of the insertion will be shifted to the left by a distance d, which is equal to the length of insertion. Similarly, when a read contains a deletion [Figure 1(C3)], mapping locations of subreads on its right side will be shifted to the right by a distance equal to the length of deletion. Because there are no mismatches allowed in the flanking subreads, the indels are called with high confidence. The uncovered bases, which are not covered by mapped subreads due to indel occurrences, are then aligned to the genomic interval between the mapped locations of the flanking subreads (encompassed by the green dotted line) using a Smith–Waterman dynamic programming procedure. Because the indel length has been determined already by the flanking subreads, the Smith–Waterman algorithm can be instructed to find an alignment of the correct indel length.
As it can be seen, the dynamic programming procedure is only required for aligning uncovered bases, rather than aligning the entire read sequence using this procedure as carried out by other aligners such as Novoalign. The running time of Subread increased by only 3% when using this procedure to discovered indels in the 1000 Genomes data set included in this study. The dynamic programming procedure also reported correct indel lengths for 98% of the reads that were found to contain indels in this data set.
However, indels might not have flanking subreads at both their sides, especially when their locations are near the ends of the reads [Figure 1(C4)]. In this case, a 4 bp window is moved from the first (or last) mapped base to the start (or end) of the read to identify indels. We require at least three mismatches in the window to consider a potential indel. Any potential indel that improves the similarity between uncovered bases and the corresponding reference region is reported.
Discovering exon–exon junctions between the subreads
A unique feature of RNA-seq is the ability to measure distinct isoforms of a gene including alternative splicing events. Here we use the seed-and-vote paradigm to develop a novel approach for detecting exon–exon junctions and producing complete mapping results from RNA-seq reads.
Figure 2 shows a schematic of the approach. The entire set of reads is scanned twice. In the first scan, a number of subreads are extracted from each read, which are then used to vote for the mapping locations of reads in the reference genome. Any locations which receive at least one vote will be considered. We select the two most voted mapping locations for each read and examine whether there are any splicing sites existing between the two selected locations. We require that a donor site (‘GT’) and a receptor site (‘AG’) exist between the two locations before considering them to have splicing points in between. We also require that the length of region in the reference genome that is spanned by the two sets of subreads that voted for the two best locations, excluding the region between the identified donor site and receptor site, must be equal to the length of the region in the read that is spanned by two same sets of subreads, when no indels are allowed. This is illustrated in the mapping of Read 1 in Figure 2 (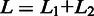 ). When indels are allowed, the length difference will be equal to or less than the specified maximal indel length. This first scan is very sensitive in finding potential exon–exon junctions because any mapping locations with as low as one vote are considered. On the other hand, the requirements on the length of mapped regions and on the donor/receptor sites ensure a high accuracy to be achieved.
). When indels are allowed, the length difference will be equal to or less than the specified maximal indel length. This first scan is very sensitive in finding potential exon–exon junctions because any mapping locations with as low as one vote are considered. On the other hand, the requirements on the length of mapped regions and on the donor/receptor sites ensure a high accuracy to be achieved.
Figure 2.

Schematic for detecting exon–exon junctions under seed-and-vote paradigm. A two-scan procedure is used to detect exon–exon junctions and to determine the mapping of each read. Three artificial reads are used to illustrate this procedure (Read 1, Read 2 and Read 50). In the first scan, a set of subreads are extracted from each read and mapped to the reference genome. The best two mapping locations from each read, which receives the two largest numbers of votes, are selected for further consideration. If donor and receptor sites are found between these two locations and total size (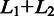 ) of the two mapped regions in the reference is equal to the size (L) of the read region that is spanned by the subreads that vote for the best two mapping locations, the determined splicing points will be recorded in the putative exon–exon junction table. Anchor locations of each read in the genome and in the read are also recorded, which gives the mapping location to which the read is best mapped and the location of the leftmost base of the set of extracted subreads that vote for that location, respectively. Anchor locations will be used for retrieving putative splicing points and for the validation performed by the second scan. The first scan is applied to all the reads, and two tables are produced on completion. These two tables include chromosomal locations of putative splicing points found for each exon–exon junction and anchor information for each read, respectively. The input to the second scan includes these two tables and also the read data. For each read, the second scan uses its anchor location to search for the putative splicing points falling within the read from the junction table output from the first scan and then examines all mapping possibilities (including mapping the read as an exonic read) to eventually determine how the read should be mapped. The similarity between the read sequence and the mapped regions when it is mapped as a junction read has to be greater than that from being mapped as an exonic read (i.e.
) of the two mapped regions in the reference is equal to the size (L) of the read region that is spanned by the subreads that vote for the best two mapping locations, the determined splicing points will be recorded in the putative exon–exon junction table. Anchor locations of each read in the genome and in the read are also recorded, which gives the mapping location to which the read is best mapped and the location of the leftmost base of the set of extracted subreads that vote for that location, respectively. Anchor locations will be used for retrieving putative splicing points and for the validation performed by the second scan. The first scan is applied to all the reads, and two tables are produced on completion. These two tables include chromosomal locations of putative splicing points found for each exon–exon junction and anchor information for each read, respectively. The input to the second scan includes these two tables and also the read data. For each read, the second scan uses its anchor location to search for the putative splicing points falling within the read from the junction table output from the first scan and then examines all mapping possibilities (including mapping the read as an exonic read) to eventually determine how the read should be mapped. The similarity between the read sequence and the mapped regions when it is mapped as a junction read has to be greater than that from being mapped as an exonic read (i.e. 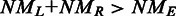 ), if it is called a junction read. The cyan dashed line indicates the mapping location of the first base or the last base of the read when it is assumed that the read does not contain junctions. Putative splicing points are removed from the final results if they are found to not have any supporting reads after the second scan is completed. The final output from this two-scan procedure is a table of validated exon–exon junctions with the number of supporting reads included, and also the complete mapping results for each read including CIGAR strings, which describes how each base in each read is mapped.
), if it is called a junction read. The cyan dashed line indicates the mapping location of the first base or the last base of the read when it is assumed that the read does not contain junctions. Putative splicing points are removed from the final results if they are found to not have any supporting reads after the second scan is completed. The final output from this two-scan procedure is a table of validated exon–exon junctions with the number of supporting reads included, and also the complete mapping results for each read including CIGAR strings, which describes how each base in each read is mapped.
The second scan will perform full read alignment for each read (including those reads mapped as exonic reads, which have only one candidate mapping location), using the output from the first scan. The second scan also serves as a validation procedure that will examine all mapping possibilities for each read and choose the best possible ones for them. It also assigns reads to the exon–exon junctions discovered from the first scan and removes those junctions that failed to get any supporting reads.
Output from the first scan includes discovered putative exon–exon junctions and anchor information for reads. For each read, its anchor location in the genome is the mapping location of leftmost base of the leftmost subread in the set of subreads that voted for the best mapping location of the read, and its anchor location in the read is the location of the same base in the read. The anchor region in each read is the region that is spanned by the set of subreads that voted for the best mapping location, and the region in the genome that is mapped to by the anchor region in the read is its anchor region in the genome. The anchor location saved for each read allows the second scan to retrieve all putative exon–exon junction locations falling within the read discovered from the first scan. The second scan considers all possibilities of how each read should be finally mapped, including locations where the read is mapped as an exonic read (no junction break points were found in the read), locations where the read is mapped as a junction read that has one junction break point or locations where the read is mapped as a junction read that includes more than one junction break point.
We illustrate the proposed algorithm with example involving a couple of reads, shown graphically in Figure 2. Read 1 is found to contain a putative junction break point located at the right side of the anchor, when using its anchor location to search for splicing points from the exon–exon junction table for this read discovered by the first scan (Figure 2). To confirm if this is a true junction, we examine whether including this junction in the mapping result will improve its sequence similarity to the reference genome. If this junction is included, its position in Read 1 can be worked out from calculating the distance between the anchor location of this read in the genome (950 on chromosome 1) and location Splicing Point 1 in the genome (1000 on chromosome 1), as this distance is equal to the distance between this junction location in the read and anchor location in the read (when no indels exist). Each exon–exon junction has two splicing points in the reference genome, Splicing Point 1 and Splicing Point 2. If there are indels, the junction position in the read will be shifted to the right, denoting insertion in the read, or left, denoting deletion in the read, by the number of indel bases. NML denotes the number of matched bases found in the region between the determined junction location and anchor location in the read. We further map the region, which is located between the junction location and the 3′ end of the read, to a genomic region starting from location Splice Point 2. Again, we allow indels in this mapping. The number of matched bases found in this region is denoted by NMR. Sum of NML and NMR gives the total number of matched bases for the entire read region located at the right side of the anchor, when this region is being considered to contain a junction. We then compare this region directly to a continuous reference region starting from the anchor location and ending at the location indicated by the cyan dash line, and count the number of matched bases, denoted by NME. This comparison checks the possibility that this region can be mapped as an exonic region, i.e. no junction exists in this region. Indels are allowed in this comparison. If the sum of NML and NMR is greater than NME, the discovered junction will then be confirmed and this read will also be counted as one of the supporting reads for this junction. Otherwise, this region will be mapped as an exonic region. For the artificial read Read 1, this junction is confirmed and added to the table of validated exon–exon junctions.
After determining the mapping of read region on the right side of the anchor, the second scan moves on to map the region at the left side of the anchor. There is no putative junction break points found in this region; therefore, the mapping of this region is quite straightforward. The voting subreads have already determined the mapping locations of those bases in the anchor, and only indels need to be figured out in those bases, which are located outside of the anchor region if there is any. This is done by testing if adding indels to every base could increase the matched bases.
Read 50, however, is found to contain a putative junction break point in the region on the left side of the anchor, in addition to its confirmed junction break point on the right side of the anchor. The second scan performs a test, similar to what it has done for confirming the right side junction, to validate the junction in this left region.
After both scans are completed, every read will be fully aligned and a list of validated junction locations will be generated. Those putative junction locations, which were reported by the first scan but failed to get any supporting reads in the second scan, were removed from the result (e.g. the junction indicated by a red cross was removed). The number of supporting reads is provided for each reported junction. Mapping results for each read are reported, in addition to the chromosomal locations of discovered exon–exon junctions. For each junction read, the mapping location of each of its bases is recorded in a CIGAR string (34).
Subread outputs the same mapping results for the mapping of exonic reads as those given by Subjunc. For the mapping of each junction read, the mapping region given by Subread will overlap with one of the mapping regions given by Subjunc, as they use the same set of consensus subreads to determine the mapping location (Subread) or anchor location (Subjunc). Therefore, mapping locations of reads are essentially the same between Subread and Subjunc, meaning that Subread has the same mapping accuracy as Subjunc.
Subread is faster than previous aligners
First, we compared alternative aligners on a recent 1000 Genomes data set of 27.5 million pairs of 100 bp DNA reads. Bowtie2, Maq and Subread all succeeded in mapping almost all the reads to the human genome, and they also had the highest percentages of normalized found intervals given by the Rabema program (35). The metric ‘normalized found intervals’ developed in Rabema is similar to the recall used in this study, except that it down-weights those reads that are mapped to multiple locations.
Subread was nearly four times as fast as the nearest competitor, Bowtie2 (Table 1). There was a 30-fold difference in speed between Subread and the slowest aligners. Subread remained more than twice as fast as any other aligner even when tuned to use a small memory footprint. MrsFast and Maq used considerable memory for this data set, because their memory use is dependent on the number of reads being mapped. The 1000 Genomes data set used in this evaluation gives a typical size of read data used in the field of genomic variation detection by using next-gen sequencing technology. The speed of Subread makes it suitable for production use.
Table 1.
Performance of aligners in mapping genomic DNA reads from the 1000 Genomes project
| Aligner | Mapped (%) | Rabema intervals (%) | Time (h) | Memory (Gb) |
|---|---|---|---|---|
| Subread (default) | 97.7 | 86.7 | 1.6 | 7.6 |
| Subread (low memory) | 97.7 | 86.7 | 2.9 | 4.3 |
| Bowtie2 | 99.1 | 87.2 | 6.0 | 3.3 |
| BWA | 95.6 | 82.6 | 15.2 | 3.3 |
| Maq | 98.1 | 86.3 | 48.3 | 19.1 |
| Novoalign | 93.9 | 68.9 | 18.7 | 8.2 |
| MrsFast | 70.3 | 73.8 | 48.2 | 25.8 |
Columns give the percentage of reads that are successfully mapped, the percentage of normalized found intervals given by the Rabema program (in ‘all’ category, maximal error rate of 8%), the time taken and the peak memory usage. Results are given for Subread with default settings and when set to use less memory.
The speed advantage of Subread increases as reads become longer. Subread is seven times as fast as the next fastest for mapping 202 bp reads (Supplemental Table S1). With reads of this length, only Subread, Bowtie2 and BWA were able to complete the task successfully.
Next, we compared the aligners on the SEQC RNA-seq data. On this RNA data, Subread mapped by far the highest percentage of reads of any of the aligners while maintaining the same relative speed advantage as observed for DNA reads (Table 2). Although junction detectors such as TopHat or MapSplice can be used here to achieve a mapping percentage comparable with Subread, this requires >15 times the computing time (Table 7), making this route less attractive for routine whole genome expression profiling.
Table 2.
Performance of aligners in mapping RNA-seq reads from the SEQC project
| Aligner | Mapped (%) | Time (min) | Memory (Gb) |
|---|---|---|---|
| Subread (default) | 96.9 | 23 | 7.6 |
| Subread (low memory) | 96.9 | 40 | 4.3 |
| Bowtie2 | 85.7 | 90 | 3.3 |
| BWA | 78.6 | 284 | 3.3 |
| Maq | 66.4 | 685 | 5.2 |
| Novoalign | 78.4 | 361 | 8.1 |
| MrsFast | 46.2 | 398 | 7.4 |
Columns give the percentage of reads that are successfully mapped, the time taken and the peak memory usage. Results are given for Subread with default settings and when set to use less memory.
Table 7.
Performance of exon–exon junction detectors in junction detection and read mapping from using SEQC RNA-seq data
| Method | Number of junctions (’000) |
Known junctions (%) |
Supporting junction reads (%) |
Time (h) | Memory (Gb) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | A | B | C | D | A | B | C | D | |||
| Subjunc | 152 | 142 | 155 | 157 | 84.4 | 86.6 | 85.6 | 85.8 | 95.8 | 95.1 | 95.7 | 95.3 | 1.4 (1.9) | 8.4 (4.7) |
| MapSplice | 171 | 157 | 173 | 175 | 78.3 | 81.4 | 80.1 | 80.2 | 94.4 | 93.5 | 94.2 | 93.8 | 5.6 | 4.3 |
| TopHat | 156 | 145 | 159 | 161 | 82.5 | 84.9 | 83.8 | 84.0 | 93.8 | 93.5 | 93.8 | 93.6 | 9.2 | 2.9 |
| TopHat 2 | 152 | 141 | 155 | 157 | 83.8 | 85.9 | 85.0 | 85.2 | 94.1 | 93.5 | 93.9 | 93.7 | 9.9 | 3.5 |
Columns give the number of reported exon–exon junctions, percentage of reported junctions that span known exons, percentage of reported junction reads that support those known junctions, for each of the four samples included in the SEQC project (A, B, C and D). Sample A is Universal Human Reference RNA made up from 10 cancer cell lines and sample B is Human Brain Reference RNA. C and D are the mixtures of A and B. Chromosomal coordinates of annotated exons from NCBI RefSeq mouse annotation build 37.2 are used to determine whether or not a junction spans known exons. Running time and peak memory usage of Subjunc when set to use less memory are given in parenthesis.
Subread is more accurate than previous aligners
Recovering spiked-in expression levels
We examined accuracy first by sequencing spiked-in RNA transcripts, and comparing the read count coverage for each transcript with the known expression level for that transcript. The SEQC (MAQC III) project is now using Ambion(textregistered] ERCC spike-in control (36) to evaluate inter-laboratory concordance in using next-gen sequencing technologies. The SEQC RNA-seq data set included in this study contained reads sequenced from these spike-in transcripts, in addition to the reads sequenced from UHRR and HBRR samples. Each spike-in transcript sequence contains a string of continuous bases, and there are no exon–exon junctions in these sequences. The set of ERCC spike-in transcripts span a large concentration range, making them useful for evaluating methods developed for processing next-gen sequencing data.
A set of 92 spike-in transcripts were pooled with UHRR and HBRR RNA to make Mix 1 and Mix 2 sample. The spike-ins produce a set of transcripts 250–2000 nt in length that mimic natural eukaryotic mRNAs. The two mixes contain the same set of spike-in transcripts, but at different known concentrations in the two mixes, so that the nominal fold change between Mix 1 and Mix 2 for each transcript is known. The true fold changes vary from 0.5 to 4. Each aligner was used to map reads from the Mix 1 and Mix 2 samples to a mixed reference genome consisting of the human reference genome (GRCh37) plus the spike-in transcript sequences. Each spike-in transcript was treated as a separate chromosome. Reads mapped to each spike-in transcript were counted and used to compute a 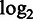 fold change for that transcript between the two samples.
fold change for that transcript between the two samples.
Subread returned fold changes that were closer to the true fold changes than any other aligner (Table 3). Subread also mapped more reads than any other aligner except Bowtie2, but Bowtie2 had the worst accuracy, suggesting that its alignment is somewhat too aggressive.
Table 3.
Performance of aligners in mapping ERCC spike-in reads from the SEQC project
| Aligner | Number of mapped spike-in reads (%) |
MSE of log2FC | |
|---|---|---|---|
| Mix 1 | Mix 2 | ||
| Subread | 86 906 (0.64%) | 133 589 (1.2%) | 1.10 |
| Bowtie2 | 87 983 (0.65%) | 135 105 (1.2%) | 1.34 |
| BWA | 85 835 (0.64%) | 131 821 (1.2%) | 1.28 |
| Maq | 81 772 (0.61%) | 125 698 (1.1%) | 1.33 |
| Novoalign | 84 556 (0.63%) | 129 711 (1.2%) | 1.32 |
| MrsFast | 70 294 (0.52%) | 109 144 (1.0%) | 1.15 |
Columns 2–3 give the number (and percentage) of reads that were correctly mapped to spike-in transcripts when sequencing the Mix 1 and Mix 2 samples. Column 4 shows the mean squared error (MSE) with which the log fold changes (log2FC) computed from the transcript-wise read counts estimate the known log fold changes between the Mix 1 and Mix 2 samples.
Detecting indels
Next, we evaluated aligners according to their ability to detect known indels. To contruct a genome with known deletions, we extracted a long sequence including one million bases from chromosome 1 of human reference genome and introduced deletions (at a rate of 0.02%) and SNPs (at a rate of 0.09%) to it. We then extracted 101 bp reads from locations in this sequence containing deletions, and recorded the position and length of the deletions in each read. Deletions could be located at any base position of reads, except their first and last four bases. Each read contained only one deletion event. To assess the ability of aligners in detecting indels with different lengths, we generated 16 data sets spanning every possible deletion length from 1 to 16 bp. The first such data set includes reads with 1 bp deletions, the second includes reads with 2 bp deletions and so on. Base quality scores in each read were taken from reads in a 101 bp SEQC data set. Bases in each read were mutated according to their quality scores to simulate sequencing errors, i.e. the lower the quality score a base has, the more likely it will be changed to a different nucleotide.
Figure 3 shows the recall rate and accuracy of each aligner in detecting deletions at each cumulative deletion size. Maq and MrsFast do not support indel detection and therefore excluded from this evaluation (Maq only supports indel detection for paired-end reads). We added BWA-SW (37) in this evaluation.
Figure 3.
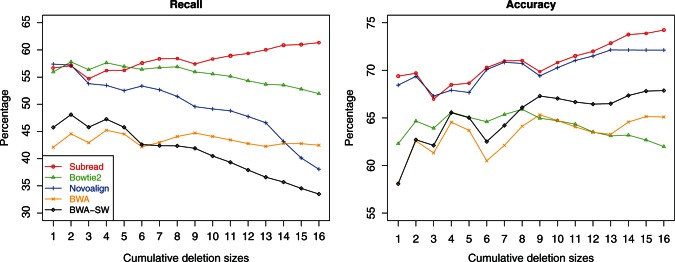
Performance of aligners in detecting deletions of different sizes. The horizontal axis gives the cumulative deletion sizes. For each size, the data sets with equal or smaller deletion sizes were combined and used for calculating the recall and accuracy for that size for each aligner. Aligners were run at their best possible settings for the detection of deletions with different sizes.
Subread was found to clearly outperform the other aligners in both accuracy and recall. It is also the only aligner that has achieved increasingly higher performance in both accuracy and recall with the increase of deletion size. The superior performance of Subread in detecting indels should be due to the power of using perfectly matched flanking subreads to discover indels. Novoalign has the second best accuracy. However, it has a rapidly decreasing recall rate with the increase of deletion length, and its recall is worse than Bowtie2. Bowtie2 has the second best recall; however, its accuracy is one of the worst. BWA-SW has a higher accuracy but lower recall than BWA. BWA-SW and BWA were found to have the worst performance among all aligners in this evaluation. Although only deletions were included in this evaluation, similar results should be observed when comparing for the detection of insertions.
Correctly mapping simulated reads
Next, we examined the ability of aligners to map reads to correct locations. We firstly used the two 101 bp simulation data sets generated from the modified human genome in which repetitive regions have been removed (MATERIALS AND METHODS). One data set contained indels and the other did not. Reads in each data set had a unique known mapping location.
The non-indel data set enables us to perform a fair comparison for the aligners that do not support indel detection, including Maq and MrsFast. Aligners supporting indel detection were configured to disable indel detection if possible so that all aligners can be compared at equivalent terms, when using this data set for comparison. Table 4 shows that Subread has the highest accuracy in all aligners. The mapping accuracy was measured as the fraction of correctly mapped reads in all mapped reads. The accuracy achieved by Novoalign and Maq is slightly lower than that of Subread. Novoalign has a slightly higher recall rate than Subread; however, Maq has a much lower recall rate. The recall rate was calculated as the fraction of correctly mapped reads in all reads. Bowtie2 has the highest recall rate among all aligners; however, its mapping accuracy is one of the worst. BWA and MrsFast were found have both low recall rate and low accuracy among all aligners.
Table 4.
Performance of aligners in mapping simulation reads generated from the filtered genome (repetitve regions were removed)
| Aligner | Without INDELs |
With INDELs |
Time (min) | Memory (Gb) | ||
|---|---|---|---|---|---|---|
| Rec (%) | Acc (%) | Rec (%) | Acc (%) | |||
| Subread | 95.96 | 99.72 | 95.58 | 99.31 | 16 (29) | 7.6 (4.3) |
| Bowtie2 | 99.04 | 99.41 | 98.65 | 99.03 | 66 | 3.3 |
| BWA | 81.06 | 99.22 | 80.24 | 98.50 | 205 | 2.4 |
| Maq | 90.56 | 99.69 | 622 | 5.9 | ||
| Novoalign | 95.99 | 99.69 | 95.57 | 99.29 | 91 | 8.0 |
| MrsFast | 72.78 | 99.45 | 256 | 4.6 | ||
Two data sets were used. One data set contains indels and the other does not. Column ‘Rec (%)’ gives the percentage of correctly mapped reads in all simulation reads included in the data set, and column ‘Acc (%)’ gives the percentage of correctly mapped reads in all mapped reads. Maq and MrsFast do not support indel detection, and therefore they do not have recall and accuracy values for the indel-containing data set. Running time and peak memory usage of Subread, Bowtie2, BWA and Novoalign were measured using the indel-containing data set. Running time and peak memory usage of Subread when set to use less memory are given in parentheses.
We then used the data set including indels to compare those aligners that support indel detection. Here, a correctly mapped read must have a correct CIGAR string, in addition to having the correct mapping coordinate on the reference genome as given by its leftmost base. The CIGAR string describes the location and length of indels in the read if there is any. Again, Subread was found to achieve the highest mapping accuracy (Table 4). The accuracy and recall rate of Novoalign were found to be slightly lower than those of Subread. Similar to its performance in mapping the data set that does not contain indels, Bowtie2 had a high recall rate but low accuracy. The accuracy and recall rate of BWA were both found to be the worst for this data set.
We further compared running time and peak memory used by these aligners. The running time and peak memory used by Subread, Bowtie2, BWA and Novoalign were measured on the data set including indels, and other aligners were measured on the data set that does not include indels. Subread is the only aligner that allows the tuning of amount of memory used for read mapping. Subread was found to have a mapping speed 4–39 times as fast as other aligners when using 7.6 GB of memory, and 2–21 times as fast when using 4.3 GB of memory (Table 4). Subread achieves this enormous speed advantage mainly due to its efficient voting mechanism, which does not require the expensive operation of extending a seed sequence to the entire read which is being carried out by all other aligners.
Subread maintained improved accuracy over competitor aligners when mapping longer 202 bp reads (Supplemental Table S1).
We also performed simulations using the unfiltered human genome, in which repetitive regions were not removed, to complement the above simulation that used the unique regions of the human genome. Simulation reads were generated from three simulators including Art, Mason and our own simulator (MATERIALS AND METHODS). Reads that were called correctly mapped must have a correct CIGAR string. In this simulation, we also tried using different cut-offs for removing uninformative subreads for Subread. As before, Subread continues to achieve better mapping accuracy and much higher mapping speed, with small cost to sensitivity (Table 5). Bowtie2 was found to have the worst accuracy in all the comparisons here, although it had a relatively good recall.
It can also be seen that when a higher threshold was used for removing uninformative subreads, Subread has a lower accuracy but a higher recall. The decrease of accuracy should be because more uninformative subreads remained when using a higher threshold, and these uninformative subreads introduced more ambiguity to the mapping. We prefer a low threshold (20–100) in favour of mapping accuracy, although users can tune it to achieve the balance between accuracy and recall they desire. More importantly, the speed advantage of Subread is almost unaffected by the choice of threshold values.
Next, we examined how well the MQS can be used to measure the confidence of read alignment, using simulation data. MQS has been found useful in downstream analyses, for example they have been used by genotypers to improve the performance of variant calling etc. (38). Figure 4a and c show that for each aligner, as expected, reads mapped with high MQS contained fewer incorrect alignments. This supports the use of MQS as a means to find high-confidence read alignments. Subread reports more correct alignments (higher recall) than Bowtie2 for the reads with high to medium MQS, and fewer incorrect alignments for the reads with medium to low MQS. Note that multi-mapping reads were assigned low MQS by every aligner. Novoalign seems to have a lower recall than Subread for the reads with high MQS, but a slightly better recall for the reads with low to medium MQS. Note that the overwhelming majority of mapped reads were given high MQS by every aligner. BWA has the best recall in Mason data set, but the worst recall in our simulation data set.
Figure 4.

Recall and accuracy of aligners with respect to MQSs. (a) and (c) give the cumulative number of correctly mapped reads and incorrectly mapped reads from high to low mapping quality. (b) and (d) show the cumulative accuracy and error fractions from high to low mapping quality. (a) and (b) use the indel data set included in Table 4, and (c) and (d) use the Mason data set included in Table 5. Each point in each plot corresponds to an MQS given by an aligner. Subread was run with default setting in (a) and (b) and with -f 100 in (c) and (d).
When assessing the accuracy of alternative aligners relative to each MQS, we use error fraction instead of absolute number of incorrect alignments, to take into account of the fact that different aligners reported different total number of incorrect alignments. The error fraction relative to an MQS s is calculated as the number of those incorrectly mapped reads, which had MQS equal to or higher than s, divided by the total number of incorrectly mapped reads reported by the aligner. The mapping accuracy relative to s is then calculated as the number of correctly mapped reads with MQS equal to or higher than s divided by the total number of reads that had their MQS equal to or higher than s. Figure 4b and d show that Subread outperforms all other aligners in mapping accuracy at each MQS value. Taken together, Subread was found to have comparable recall but higher accuracy across the entire MQS range, compared with other aligners.
Subjunc outperforms other junction detectors
The proposed seed-and-vote mapping paradigm has been demonstrated to be more accurate and efficient in read mapping. Here, we show that it is also useful in detecting exon–exon junctions. We now compare Subjunc, our new junction detector developed under the seed-and-vote paradigm, with other methods including MapSplice, TopHat and TopHat 2, using both simulation data and real data (SEQC data).
We randomly selected ∼600 genes from human genome and generated junction reads and exonic reads from them. Indels and sequencing errors were introduced to the read data in the same way as that used for generating simulation data for comparing aligners. It is known that distribution of gene expression levels is subject to exponential distribution. We therefore assign to genes the expression levels taken from an exponential distribution to make the simulation data more similar to real RNA-seq data. This also enables us to examine the performance of each junction detection method in detecting exon–exon junctions for both highly expressed genes and lowly expressed genes. We quantify expression levels of genes using number of reads per kilobases total exon model to take into account gene length differences.
We generated three simulation data sets with different sequencing coverages including 30 times (30×), 70 times (70×) and 100 times (100×). Length of generated reads is 101 bp. These data sets roughly correspond to RNA-seq data sets containing 18 million, 42 million and 60 million reads, respectively (size of the transcriptome is estimated to be 2% of genome size). Thus, they represent typical sizes of sequencing data sets currently being generated.
Table 6 shows the results of comparing junction detectors using these three data sets. It can be seen that Subjunc achieves the highest accuracy in exon–exon junction detection among all detectors. MapSplice has a higher recall rate than Subjunc, but its accuracy is clearly lower than Subjunc. TopHat 2 has a slightly better performance than TopHat, but both of them were found to have the worst accuracy, and TopHat was found to have the worst recall rate.
Table 6.
Performance of exon–exon junction detectors in junction detection and read mapping from using simulation data
| Coverage | Method | Junctions |
Junction reads |
All reads |
Time (min) | |||
|---|---|---|---|---|---|---|---|---|
| Rec (%) | Acc (%) | Rec (%) | Acc (%) | Rec (%) | Acc (%) | |||
| 30× | Subjunc | 92.4 | 98.3 | 90.5 | 98.0 | 93.2 | 96.1 | 2 |
| MapSplice | 93.1 | 97.3 | 86.3 | 95.4 | 95.8 | 88.9 | 15 | |
| TopHat | 92.0 | 92.3 | 86.7 | 91.9 | 95.7 | 92.0 | 16 | |
| TopHat 2 | 92.4 | 93.4 | 87.6 | 93.5 | 96.4 | 89.0 | 15 | |
| 70× | Subjunc | 93.2 | 98.0 | 90.7 | 98.0 | 93.3 | 96.1 | 3 |
| MapSplice | 94.0 | 97.0 | 86.3 | 95.4 | 95.8 | 88.8 | 17 | |
| TopHat | 93.0 | 91.7 | 87.2 | 91.9 | 95.9 | 92.0 | 26 | |
| TopHat 2 | 93.5 | 93.0 | 88.1 | 93.4 | 96.6 | 88.9 | 24 | |
| 100× | Subjunc | 93.3 | 98.0 | 90.8 | 98.0 | 93.3 | 96.2 | 5 |
| MapSplice | 94.3 | 96.9 | 86.3 | 95.5 | 95.9 | 89.0 | 18 | |
| TopHat | 93.0 | 91.0 | 87.2 | 91.9 | 95.9 | 92.0 | 32 | |
| TopHat 2 | 93.7 | 93.0 | 88.1 | 93.6 | 96.6 | 89.0 | 30 | |
Three simulation data sets were used, which have different sequencing coverages (30×, 70× and 100×). Column ‘Junction’ gives the recall rate and accuracy for the detection of exon–exon junctions by each method. Recall rate is the percentage of correctly reported junctions in all junctions generated in the simulation data, and accuracy is the percentage of correctly reported junctions in all reported junctions. Column ‘Junction reads’ (or ‘All reads’) gives the recall rate and accuracy of mapping of junction reads (or any reads). Recall rate is the percentage of correctly reported junction reads (or any reads) in all generated junction reads (or all reads) in the simulation data, and accuracy is the percentage of correctly mapped junction reads (or any reads) in all reported junction reads (or any reads). Results are given for Subjunc with default settings.
It is important to compare junction detectors for their performance in mapping reads, especially junction reads, in addition to comparing them for calling exon–exon junctions. This, however, has been overlooked in the literature. The precise mapping of RNA-seq reads (especially junction reads) is crucial for some downstream analyses, such as detection of functional variations (indels, SNPs, etc), allele-specific gene expression analysis and so on. A serious problem with variant calling is known to be the high false-positive rate. Here, we also compared the four junction detectors for their performance in mapping the reads.
Subjunc was found to outperform other detectors in read mapping by a clear margin, especially in the mapping of junction reads, in which it achieves both best accuracy and best recall rate (Table 6). The superior read mapping accuracy achieved by Subjunc gives it a lot more power in calling exon–exon junctions. Moreover, Subjunc is substantially faster in read mapping and junction calling. More comparison results for speed and memory usage can be seen from the comparison using the SEQC data set.
We used the SEQC RNA-seq data to further compare junction detectors. Table 7 shows the comparison results. We compared locations of exon–exon junctions in the genome reported by each method with the chromosomal regions of annotated human exons obtained from NCBI RefSeq annotation (build 37.2), to examine the difference between alternative methods in detecting exon–exon junctions originated from the splicing of known exons. Subjunc was found to have the highest percentage of ‘known’ junctions in all its reported junctions for every sample type (column ‘% known junction’), although its absolute numbers of discovered junctions are less than MapSplice and TopHat. This suggests that Subjunc has a higher accuracy in calling junctions than other methods, which is concordant with the simulation results. MapSplice now has the lowest percentage of ‘known’ junctions, although it calls more junctions than any other methods, suggesting that its accuracy is the worst among all the methods. TopHat 2 called less junction than TopHat, but its percentage of ‘known’ junctions is higher than TopHat. Compared with Subjunc, TopHat 2 had a comparable number of reported junctions, but its percentage of ‘known’ junctions is lower. It is worth noting that overall most of the reported junctions by each method (∼80% or more) were found to originate from the well-annotated RefSeq exons. Also, every method found more exon–exon junction in samples C and D than in A and B, which is expected because samples C and D are the mixture of samples A and B.
We then compared these junction detectors by examining the percentage of reported junction reads that support their reported ‘known’ junctions. Subjunc was found to have the highest percentage of supporting junction reads in each sample type (column ‘Supporting junction reads (%)’), indicating higher accuracy of Subjunc for mapping junction reads. TopHat and TopHat 2 were found to have the lowest percentages of supporting junction reads. This comparison result is consistent with the accuracy comparison results for junction reads from simulation.
Consistent with the speed advantage of Subread shown in the read mapping comparisons, Subjunc was found to achieve a large speed advantage as well. Subjunc was found to be four to seven times as fast as other methods when using 8.4 GB of memory, and three to five times as fast when using 4.7 GB of memory. This greatly reduces the computational burden on discovering genome-wide splicing events.
These results show improved speed and accuracy performance for Subjunc in exon–exon junction detection over existing methods.
DISCUSSION
Next-generation sequencing technologies entered mainstream genomic research only a few years ago, and the best ways to solve mapping and alignment problems are still being developed. Sequencing technologies continue to evolve at an astonishing rate, and read alignment is certain to be a significant bottleneck in the analysis of genomic data in the future at all levels of medical and biological research. Current read alignment tools are being challenged by increased data volumes and show deteriorating performance with longer reads. In this study, we propose a new multi-seed read alignment paradigm, called seed-and-vote, that abandons the computationally heavy extend operation of existing aligners in favour of a voting strategy to quickly and accurately locate the locations of reads in the reference genome.
The seed-and-vote paradigm has been found to be effective not only for quickly identifying mapping locations but also for detecting indels and for detecting exon–exon junctions for RNA-seq data. Using the subreads flanking the indel regions to locate indels and to determine their sizes enables highly accurate detection of indels. The overhead computational cost for indel detection is small, because the indel detection is only required for those regions that are not covered by the subreads that have made successful votes. To detect exon junctions, the algorithm uses the best two mapping locations voted by subreads extracted from junction reads to generate a set of candidate exon–exon junctions, and then performs rigorous validation for them to achieve a high detection accuracy.
We used a variety of data sets to demonstrate the performance of this paradigm against existing aligners. In particular, we used ERCC spike-in controls for the evaluation. Spike-in data sets have proved effective for evaluating methods developed for the analysis of microarray data (31,39–43). To the best of our knowledge, our study is the first to use spike-in controls to evaluate the performance of read aligners for mapping next-gen sequencing data. The unbiased design of ERCC spike-in transcripts and their known concentration and fold changes make them ideal for assessing the accuracy of read aligners. Our Subread aligner was found to clearly outperform other aligners in this comparison. Furthermore, we used SEQC RNA-seq data, 1000 Genomes exome sequencing data and simulation data to demonstrate the performance of Subread and Subjunc. Consistently across all evaluation data sets, the seed-and-vote paradigm showed higher accuracy and much higher mapping speed than the seed-and-extend methods, with little cost to recall. In particular, the superior performance of Subread in indel detection will bring a lot of benefit to the downstream analyses such as genomic variation detection etc. The similar indel detection approach was implemented in Subjunc as well, making it a valuable tool for detecting genomic variation in functional genomic regions (eg. exons).
Subread and Subjunc allow the tuning of memory used in read mapping. This gives them great flexibility in running on computers with different configurations. Subread and Subjunc achieve their top mapping speed when the entire hash table index is loaded into memory in one go, which takes 7.6 and 8.4 GB of memory, respectively, when mapping reads to human or mouse genome. The amount of memory used by Subread is comparable with or better than those of Novoalign, Maq and MrsFast, but is higher than Bowtie2 and BWA. Given that the contemporary computers are all equipped with large memories, for example the HP Blade supercomputers include hundreds of gigabytes of memory and laptops now can easily have 8 GB of memory, the memory use is not of concern compared with the mapping speed, which is increasingly becoming a bottleneck for the read mapping. Moreover, Subread and Subjunc still have a significant speed advantage when using memory comparable with Bowtie2 and BWA, and with MapSplice, TopHat and TopHat 2, respectively.
The main scoring scheme used by our seed-and-vote paradigm is the vote number. The mapping location that receives the highest number of votes is chosen for the read. It is guaranteed that the best mapping location is found when the maximum possible number of votes is achieved. It would, however, be interesting to measure how this scheme is correlated to other scoring schemes such as edit distance etc. We used the simulation data to measure this. As expected, voting number is inversely correlated to edit distance, i.e. large voting number correspond to small edit distance and vice versa (Supplemental Figure S6).
A crucial advantage of the Subread seed-and-vote strategy is that it scales up to map longer reads with negligible increase in computational time. Reads of 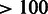 bp are already available, and much longer reads (1000 bp say) may be not far away. We believe that seed-and-vote will continue to yield good mapping results using 10 extracted subreads, even for longer reads, because 160 bases should be more than sufficient to determine the correct location for each read. This implies that local alignment can be achieved for a very long read virtually as quickly as for a shorter read. The time taken for the in-fill step will increase, but not enough to materially affect the overall time taken. By contrast, the running times of other existing aligners increase rapidly with read length. Subread is already faster than other aligners for 50–100 bp, and this advantage should become more pronounced as read lengths increase. Simulations in this article confirm that Subread maintains an accuracy advantage as well as being faster for 202 bp. More comprehensive evaluations should be performed as long read benchmark data sets become available.
bp are already available, and much longer reads (1000 bp say) may be not far away. We believe that seed-and-vote will continue to yield good mapping results using 10 extracted subreads, even for longer reads, because 160 bases should be more than sufficient to determine the correct location for each read. This implies that local alignment can be achieved for a very long read virtually as quickly as for a shorter read. The time taken for the in-fill step will increase, but not enough to materially affect the overall time taken. By contrast, the running times of other existing aligners increase rapidly with read length. Subread is already faster than other aligners for 50–100 bp, and this advantage should become more pronounced as read lengths increase. Simulations in this article confirm that Subread maintains an accuracy advantage as well as being faster for 202 bp. More comprehensive evaluations should be performed as long read benchmark data sets become available.
The success of the proposed paradigm in read mapping and its potentially high scalability make it become a promising new tool for the general-purpose sequence search, i.e. finding sequences from a collection of sequences (often stored in a database) that have a high overall similarity to or share common subsequences with the query sequences. The query sequences could be tens or thousands of bases long in the general biological sequence search. Blast (Basic Local Alignment Search Tool) is one of the most widely used algorithms for this kind of sequence search (44). It also makes use of the seed-and-extend paradigm, which means it has the limitations of this paradigm shown in this study, especially the long running time. Our proposed seed-and-vote paradigm can be readily extended to search for a sequence of thousands of bases long from a large sequence database, by extracting more subreads from the query sequence. We speculate that using 30% of total number of extracted subreads as the consensus threshold for calling hit sequences, which is the consensus threshold used in this study for read mapping, may still give a reasonably good accuracy and recall rate, not to mention its super fast searching speed. However, further studies will be needed to investigate how to use this paradigm to perform the sequence search in the most efficient and accurate way.
Some of the most commonly used statistical analyses of RNA-seq or ChIP-seq data do not actually require detailed alignment information, but are instead based purely on tables of read counts for each gene, or other pre-determined genomic feature, in each biological sample (30,45–48). In our own biological research, we frequently conduct differential expression analyses using total read counts mapped to the exome of each gene, or differential marking analyses of epigenetic modification using total read counts summarized by gene promoter regions or by gene bodies (49). This type of analysis focuses on the total expression level or total read coverage of each gene. Subread is particularly efficient for this type of analysis, because it can identify the gene or feature to which a read maps directly from the seed-and-vote step. When mapping RNA-seq reads, Subread has the capacity to use part of the read sequence to vote for the mapping location for the entire read, and this capacity enables Subread to call mapping locations for reads spanning exon–exon junctions using the longest matched region in the read. For this genewise counts of RNA-seq reads, Subread yields equal or better results than other aligners >15 times as quickly, turning weeks of computing time for large problems into an overnight run. We have created a Bioconductor package Rsubread to give access to Subread capabilities from the R command line, making a pipeline from FASTQ files to read count tables and statistical analyses using packages such as edgeR (50), baySeq (51) or diffBind (www.bioconductor.org, 2013) particularly convenient. Rsubread includes functions to summarize counts at the gene or exon level given annotation for the reference gene. The latest NCBI RefSeq annotation for the human and mouse genomes are included in the package by default, and annotation for other genomes can be uploaded by the user.
This study presents a new paradigm for aligning next-gen sequencing data that opens new directions for read mapping algorithms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1, Supplementary Figures 1–6 and Supplementary Methods.
FUNDING
Project Grant [1023454] and a Fellowship from the Australian National Health and Medical Research Council (NHMRC) (to GKS); Victorian State Government Operational Infrastructure Support; Australian Government [NHMRC IRIIS]. Funding for open access charge: NHMRC Project Grant [1023454].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Terry Speed, Rafael Irizarry and Aaron Lun for critical reading of the manuscript, and Leming Shi and Charles Wang for providing the SEQC pilot data.
REFERENCES
- 1.Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marco-Sola S, Sammeth M, Guig R, Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods. 2012;9:1185–1188. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- 3.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hach F, Hormozdiari F, Alkan C, Hormozdiari F, Birol I, Eichler EE, Sahinalp SC. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods. 2010;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.David M, Dzamba M, Lister D, Ilie L, Brudno M. SHRiMP2: sensitive yet practical SHort Read Maping. Bioinformatics. 2011;27:1011–102. doi: 10.1093/bioinformatics/btr046. [DOI] [PubMed] [Google Scholar]
- 9.Misra S, Agrawal A, Liao WK, Choudhary A. Anatomy of a hash-based long read sequence mapping algorithm for next generation DNA sequencing. Bioinformatics. 2011;27:189–195. doi: 10.1093/bioinformatics/btq648. [DOI] [PubMed] [Google Scholar]
- 10.Li H, Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief. Bioinform. 2010;11:473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 12.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 13.Weese D, Holtgrewe M, Reinert K. RazerS 3: faster, fully sensitive read mapping. Bioinformatics. 2012;28:2592–2599. doi: 10.1093/bioinformatics/bts505. [DOI] [PubMed] [Google Scholar]
- 14.Myers EW. A fast bit-vector algorithm for approximate string matching based on dynamic programming. JACM. 1999;46:395–415. [Google Scholar]
- 15.Lin H, Zhang Z, Zhang MQ, Ma B, Li M. ZOOM! Zillions of oligos mapped. Bioinformatics. 2008;24:2431–2437. doi: 10.1093/bioinformatics/btn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizk G, Lavenier D. GASSST: global alignment short sequence search tool. Bioinformatics. 2010;26:2534–2540. doi: 10.1093/bioinformatics/btq485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS One. 2009;4:e7767, 2009. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kehr B, Weese D, Reinert K. STELLAR: fast and exact local alignments. BMC Bioinformatics. 2011;12(Suppl. 9):S15. doi: 10.1186/1471-2105-12-S9-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasmussen KR, Stoye J, Myers EW. Efficient q-gram filters for finding all ϵ-matches over a given length. J. Comput. Biol. 2006;13:296–308. doi: 10.1089/cmb.2006.13.296. [DOI] [PubMed] [Google Scholar]
- 21.Burkhardt S, Crauser A, Ferragina P, Lenhof HP, Rivals E, Vingron M. Proceedings of RECOMB’99. New York: ACM; 1999. q-gram based database searching using a suffix array (QUASAR) pp. 77–83. [Google Scholar]
- 22.Sun T, Gao Y, Tan W, Ma S, Shi Y, Yao J, Guo Y, Yang M, Zhang X, Zhang Q, et al. A six-nucleotide insertion-deletion polymorphism in the CASP8 promoter is associated with susceptibility to multiple cancers. Nat. Genet. 2007;39:605–613. doi: 10.1038/ng2030. [DOI] [PubMed] [Google Scholar]
- 23.Bi XH, Lu CM, Liu Q, Zhang ZX, Zhao HL, Yu J, Zhang JW. A 14 bp indel variation in the NCX1 gene modulates the age at onset in late-onset Alzheimer’s disease. J. Neural. Transm. 2012;119:383–386. doi: 10.1007/s00702-011-0696-4. [DOI] [PubMed] [Google Scholar]
- 24.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trapnell C, Pachter L, Salzberg SL. Tophat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. Mapsplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–2887. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- 28.Langmead B, Hansen KD, Leek JT. Cloud-scale RNA-sequencing differential expression analysis with Myrna. Genome Biol. 2010;11:R83. doi: 10.1186/gb-2010-11-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MAQC Consortium. Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, et al. The Microarray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holtgrewe M. 2010. Mason—a read simulator for second generation sequencing data. Technical Report, Mathematics Department, Freie Universität Berlin, TR-B-10-06. [Google Scholar]
- 33.Huang W, Li L, Myers JR, Marth GT. ART: a next-generation sequencing read simulator. Bioinformatics. 2012;28:593–594. doi: 10.1093/bioinformatics/btr708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holtgrewe M, Emde AK, Weese D, Reinert K. A novel and well-defined benchmarking method for second generation read mapping. BMC Bioinformatics. 2011;12:210. doi: 10.1186/1471-2105-12-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker SC, Bauer SR, Beyer RP, Brenton JD, Bromley B, Burrill J, Causton H, Conley MP, Elespuru R, Fero M, et al. The external RNA controls consortium: a progress report. Nat. Methods. 2005;2:731–734. doi: 10.1038/nmeth1005-731. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCall MN, Irizarry RA. Consolidated strategy for the analysis of microarray spike-in data. Nucleic Acids Res. 2008;36:e108. doi: 10.1093/nar/gkn430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunning MJ, Ritchie ME, Barbosa-Morais NL, Tavare S, Lynch AG. Spike-in validation of an Illumina-specific variance-stabilizing transformation. BMC Res. Notes. 2008;1:18. doi: 10.1186/1756-0500-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 42.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 43.Shi W, Oshlack A, Smyth GK. Optimizing the noise versus bias trade-off for Illumina whole genome expression beadchips. Nucleic Acids Res. 2010;38:e204. doi: 10.1093/nar/gkq871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 45.Robinson MD, Strbenac D, Stirzaker C, Statham AL, Song JZ, Speed TP, Clark SJ. Copy-number-aware differential analysis of quantitative DNA sequencing data. Genome Res. 2012;22:2489–2496. doi: 10.1101/gr.139055.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao L, Glazov EA, Pattabiraman DR, Al-Owaidi F, Zhang P, Brown MA, Leo PJ, Gonda TJ. Integrated genome-wide chromatin occupancy and expression analyses identify key myeloid pro-differentiation transcription factors repressed by Myb. Nucleic Acids Res. 2011;39:4664–4679. doi: 10.1093/nar/gkr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vrba L, Garbe JC, Stampfer MR, Futscher BW. Epigenetic regulation of normal human mammary cell type-specific miRNAs. Genome Res. 2011;21:2026–2037. doi: 10.1101/gr.123935.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Connell RJ, Thon MR, Hacquard S, Amyotte SG, Kleemann J, Torres MF, Damm U, Buiate EA, Epstein L, Alkan N, et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012;44:1060–1065. doi: 10.1038/ng.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pal B, Bouras T, Shi W, Vaillant F, Sheridan J, Fu N, Breslin K, Jiang K, Ritchie ME, Young M, et al. Ezh2 coordinates global changes in the mammary epigenome induced by hormonal cues and controls mammary progenitor activity. Cell Rep. 2012;3:411–426. doi: 10.1016/j.celrep.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 50.Robinson M, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hardcastle TJ, Kelly KA. bayseq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics. 2010;11:422. doi: 10.1186/1471-2105-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.