


Abstract
A crucial step in analyzing mRNA-Seq data is to accurately and efficiently map hundreds of millions of reads to the reference genome and exon junctions. Here we present OLego, an algorithm specifically designed for de novo mapping of spliced mRNA-Seq reads. OLego adopts a multiple-seed-and-extend scheme, and does not rely on a separate external aligner. It achieves high sensitivity of junction detection by strategic searches with small seeds (∼14 nt for mammalian genomes). To improve accuracy and resolve ambiguous mapping at junctions, OLego uses a built-in statistical model to score exon junctions by splice-site strength and intron size. Burrows–Wheeler transform is used in multiple steps of the algorithm to efficiently map seeds, locate junctions and identify small exons. OLego is implemented in C++ with fully multithreaded execution, and allows fast processing of large-scale data. We systematically evaluated the performance of OLego in comparison with published tools using both simulated and real data. OLego demonstrated better sensitivity, higher or comparable accuracy and substantially improved speed. OLego also identified hundreds of novel micro-exons (<30 nt) in the mouse transcriptome, many of which are phylogenetically conserved and can be validated experimentally in vivo. OLego is freely available at http://zhanglab.c2b2.columbia.edu/index.php/OLego.
INTRODUCTION
In eukaryotes, alternative splicing (AS) is critical for amplifying genomic complexity by generating multiple mRNA isoforms from a single gene (1,2). More than 90% of human multi-exon genes express transcripts that potentially undergo AS (3,4). Besides the extent of AS, decades of research have revealed the key roles of this process in post-transcriptional gene-expression regulation, and how its disruption can cause various genetic diseases (5,6).
Global insights into AS were initially achieved largely from analysis of expressed sequence tag (EST) data, which provide a means of cataloguing AS events at the genome-wide scale (7). In general, EST data have low coverage and limited capability for quantifying exon-inclusion level, especially in specific conditions, such as in different tissues. This issue was later addressed by splicing-sensitive microarrays, such as exon-junction arrays (8,9) or exon arrays (10), which were designed based on gene structures and AS events observed in ESTs and other sequenced transcript data. However, microarrays are largely restricted to studies of annotated AS events, and their signal-to-noise ratio is also limited by issues such as cross-hybridization. Recently, ultra-high-throughput mRNA sequencing (mRNA-Seq) provided a powerful alternative to profile the transcriptome at unprecedented depth and resolution, with the advantages of being highly quantitative, sensitive and able to discover novel splice junctions and exons (11).
A key step in analyzing mRNA-Seq data is to map hundreds of millions of reads, currently of size 50–150 nucleotides (nt), back to the reference genome, and to detect known or novel splice junctions. Various algorithms have been developed in the past few years for this purpose, with specific consideration to mapping speed and to short read lengths (12–18). The early versions of TopHat (16) first align all exon-body reads to the genome using an external aligner, Bowtie (19), and all aligned reads are clustered and counted to locate potential exons based on read coverage (exon islands). Potential splice sites are then searched locally, and nearby exons are paired in silico to generate a database of candidate exon junctions, followed by alignment of unmapped reads in the first stage against this junction database. This procedure is relatively fast and reliable because exon identification before junction search largely limits the search space, despite the caveat that junctions spanning exons at low levels might be missed. To overcome this limitation, several other programs turned to more exhaustive searches by using double- or multiple-seed-and-extend approaches to find exon junctions de novo. For example, SpliceMap (17) splits each read of ∼50 nt in the middle and maps each part (seed) to the genome separately, again relying on an external aligner for genomic mapping, and then it extends the alignments to find junctions. To handle longer reads that can span multiple junctions—obtained with more recent technologies—MapSplice (18) and later versions of TopHat (16) segment each read into multiple seeds to detect splice junctions.
Although different heuristics are used in each algorithm, an important limitation shared by these tools is their use of relatively long seeds (∼25 nt). This is due in part to their dependence on an external aligner for seed mapping, whose output is then parsed to detect exon junctions. As a consequence, the number of hits for each seed has to be small, which constrains the choice of seed size and limits the resolution in locating potential exon positions. This constraint increases the chance that one or more seeds will fail to align, because they span exon junctions, reducing the sensitivity of junction detection. This issue becomes more severe for reads spanning small exons, which are frequently alternatively spliced and regulated to have variable inclusion levels in specific conditions.
As sequencing technologies keep evolving, the throughput and read length are increasing rapidly, which imposes even greater challenges for mRNA-Seq data processing. For example, a single sequencing lane from the Illumina HiSeq 2000 can currently produce >200 million paired-end reads, with read lengths up to 150 nt. Therefore, mapping speed, without sacrificing accuracy, becomes more critical. In addition, longer reads tend to span more exon junctions and have more complex structures, especially when they cover small exons or exons expressed at a low level. Here we address these challenges and present a new program named OLego, which is designed for fast de novo mapping of spliced mRNA-Seq reads with both high specificity and sensitivity.
MATERIALS AND METHODS
Overview of mRNA-Seq read mapping
Analysis of mRNA-Seq data typically starts from mapping a set of N relatively short reads of length L nt to the reference genome. For higher eukaryotes, and mammals in particular, the vast majority of genes consist of multiple exons and introns. Therefore, a read can be mapped continuously to a single exon (exonic alignment), or to multiple exons that span one or more exon junctions (junction alignment). Due to sequencing errors or polymorphisms in the sequenced sample, compared with the reference genome, a read alignment has to tolerate a certain number of substitutions or small insertions and deletions (indels)—collectively denoted as mismatches here—as measured by an editing distance of M nt between the query reads and the target reference genome sequences.
OLego finds junction alignments using a multiple-seed-and-extend approach, which is also used by several other programs, such as MapSplice (18), but with several distinct and important features (Figure 1). In essence, each read is processed independently in a series of steps, without relying on an external aligner. Reads can therefore be processed in parallel when multiple threading is enabled. OLego performs more exhaustive and yet efficient searches using small seeds (12–14 nt; 14 nt for this study), whose hits are clustered, ranked and refined to find the best alignment. This greatly improves the sensitivity for de novo discovery of splice junctions and small exons. In addition, particular attention is paid when a small unaligned segment of a read is flanked by aligned regions on both ends in the presence of large genomic gaps, typically due to the presence of a small exon (<30 nt) or micro-exon (20). To ensure the efficiency of this exhaustive procedure in terms of both time and memory usage, Burrows–Wheeler transform (BWT) and full-text minute-space (FM)-index (21) are used in multiple steps to map seeds and discover junctions and small exons with a small memory footprint (<4 GB in general for mammalian genomes). More details of the algorithm are described below.
Figure 1.

Overview of OLego. Each read is processed independently by OLego. (1) Continuous mapping to the genome or exonic alignment is attempted first. If no hits are found within the allowed number of mismatches, junction alignment is searched through steps starting from (2) seeding (3) seed mapping and hit clustering into candidate alignments, and (4) candidate-exon identification and extension. (5) Junctions are then searched between two consecutive candidate exons and at the end of the read, and small exons are searched when necessary. (6) Finally, exons and junctions are connected and ranked to identify the optimal alignment for the whole read.
The workflow of OLego
Exonic mapping of reads
For each read, continuous mapping to the genome with BWT and FM-index is first attempted, using essentially the same approach as in BWA (22), with minor modifications. At most M’ nt (currently M’ = min{2,M}, where M is the number of mismatches allowed for the whole read), mismatches are allowed in this step. If an exonic alignment is found, the read will be reported as an exon-body read, and the algorithm turns to the next read. Otherwise, it will be processed in the following steps to search for a junction alignment. Note that a smaller number of mismatches are allowed here to avoid promiscuous exonic alignment with mismatches near the end of a read, when it actually spans an exon junction with a small anchor at the end. Exonic alignment with >M’ mismatches (but ≤ M mismatches) will be recovered later (step 4 below).
Seeding
Each unmapped read subject to junction search is segmented into multiple seeds of a specified size w. Spaces are allowed between seeds if the read length is not a multiple of the seed size, such that the read can be evenly covered by seeds. Because the boundaries of seeds relative to exon junctions are random, an exon of size ≥2w is guaranteed to have at least one seed inside the exon, assuming a sufficient sequencing depth. The default seed size w in OLego is 14 nt, considering the balance between sensitivity and speed to deal with mammalian-sized genomes. The use of a smaller seed size in OLego greatly increases the chance of finding hits of one or more seeds in each exon, especially for small exons ≤50 nt.
Seed mapping and hit clustering
Each seed is mapped independently to the genome by querying BWT and FM-index, allowing ≤m mismatches (default: m=0). Due to the small seed size, each seed is expected to have a substantial number of hits. For example, at a seed size of 14 nt, the average number of hits for each seed is estimated to be W = 11 for a mammalian genome (3 × 109/414), although this number varies for different seeds. If a seed has an exceedingly large number of hits (W > 1000), it is considered as repetitive and all its hits are discarded; otherwise, we keep all W hits of a seed, and recover their original genomic coordinates from the BWT index. The hits of all kept seeds are then clustered head-to-tail to locate potential alignments of the complete read according to their genomic coordinates, so that the distances between any two neighbor hits in each potential alignment are less than twice the specified maximum intron size I (default: I = 500 000 nt). We require 2I in the clustering of hits, because there might be a missing internal exon between two neighbor hits (see below). Each potential alignment is scored and ranked according to an ‘E-value’ estimated from the number of aligned seeds and their uniqueness 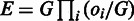 , where oi is the total occurrence in the whole genome for seed i in the potential alignment, and G is the size of the genome. Only the top 100 potential alignments with E < 10 are examined further.
, where oi is the total occurrence in the whole genome for seed i in the potential alignment, and G is the size of the genome. Only the top 100 potential alignments with E < 10 are examined further.
To maximize the speed, we do not allow mismatches in seed mapping by default, given the small seed size and low sequencing errors that minimize the chance of failure in seed mapping. In addition, even if no hits are found or kept for some seeds, owing to sequencing errors, polymorphisms or their repetitive nature, such parts can still be recovered in the following hole-filling and candidate-exon-extension step. Each potential alignment is treated separately in the following steps.
Candidate-exon identification and extension
In each potential alignment, the hits are further grouped into individual candidate exons, using more stringent criteria. This is done using the diagonal coordinates of the hits, which are calculated by subtracting the start coordinates of the corresponding seeds in the query read from the genomic start coordinates of the hits (23). The hits whose diagonal coordinates are within M’ nt differences are considered to be in the same candidate exon, which tolerates potential indels in mRNA-Seq reads. After this step, holes between hits within each candidate exon are filled in by realigning the orthologous sequences in the query read and the reference genome using banded dynamic programming, which allows substitutions and small indels. In addition, each candidate exon is also extended on both ends by allowing ≤M’ mismatches to find potential exon boundaries. If a candidate exon already covers the whole read with ≤M mismatches at this point, a candidate exonic alignment is recorded.
Junction and small-exon search
There are two types of junction searches: double-anchor and single-anchor. Double-anchor search is performed between each pair of neighboring candidate exons. Candidate splice sites are searched locally around the exon boundaries (default: ±6 nt). At the same time, the match of sequences between the reference genome and the query read around exon boundaries are examined. If nucleotides near exon boundaries are aligned properly (≤M’ mismatches), a candidate exon junction is recorded. Otherwise, if a gap remains in the query read after local search of exon boundaries according to the candidate splice sites, this typically suggests a missing internal exon without any hits of seed sequences in the exon, as discussed above. In this case, further searching for the missing internal exon is carried out. The sequence in the gap region of the read, flanked by the dinucleotides (AG/GT) of the two splice sites, is queried against the reference genome using BWT and FM-index to find the missing internal exon, requiring a minimum exon size (default: 9 nt) and proper intron size (default: ∼20–500 000 nt). This gives a chance of ∼5.8 × 10−5 (2 × 500 000/49 + 8, 8 nt are dinucleotides for four splice sites) to find a random match.
Single-anchor search is performed at the ends of the first and last candidate exons if they do not reach the boundaries of the read. Candidate 5′ or 3′ splice sites are searched locally (default: ±6 nt) near the exon boundary, and the unaligned part of the read after this local adjustment, flanked by the 3′ or 5′ splice site dinucleotide, is searched against the reference genome with BWT and FM-index. The size of the match at the end has to be larger than the minimum size (default: a = 8 nt), and the intron size is restricted in the proper range as well. This gives a chance of ∼0.03 (500 000/48 + 4, 4 nt are splice site dinucleotides) to find a random match.
Connecting junctions and reporting the final alignment
All candidate junctions are connected along the read to find the optimal path that represents the complete alignment of the whole read. If multiple candidate alignments can be found within the desired number of mismatches, all candidate alignments are first ranked according to the number of mismatches. If the top two or more alignments have the same number of mismatches, they are further ranked to resolve ambiguity by an additional criterion that takes into consideration splice-site strength and intron size. This criterion is also used to filter out potential false positives in de novo junction search (details are given below).
A regression model to score exon junctions
Splice sites show extended consensus sequences beyond the strictly required GT/AG dinucleotides (for canonical splice sites), which are crucial for accurate and efficient exon recognition by the splicing machinery (24,25). These motifs have been used previously for bioinformatic splice-site prediction in several methods, such as GeneSplicer (26) and SplicePort (27). In addition, intron size also affects the efficiency of splicing, and shows a distinct distribution in the mammalian genome (28).
When multiple alignments with the same number of mismatches exist, they are further ranked to prioritize the most reliable alignments according to the strength of exon junctions, using a regression model that combines splice-site motif score and intron size. To this end, we collected true splice sites from annotated gene models. For example, for mouse data, NCBI37/mm9 Ensembl (29) gene annotations were downloaded from the UCSC genome browser (30); all the splice-site pairs (242 141 pairs) were then retrieved as a true-positive training dataset. We also randomly selected the same number of pairs of GT/AG sites separated by 20–500 000 nt from the mouse genome to generate the training dataset of false splice sites.
The splice-site score for each exon junction is calculated using ±15 nt sequences around the 5′ and 3′ splice sites, respectively (31). Therefore, for each pair of splice sites corresponding to an exon junction, 60 nt are taken into account. We define the splice-site score S of an exon junction as
| (1) |
where Bi is the nucleotide (A, C, G or T) at position i, 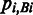 is the probability of observing
is the probability of observing 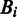 at position i of the 60-nt splice-site motif derived from the true dataset and
at position i of the 60-nt splice-site motif derived from the true dataset and 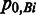 is the probability of observing Bi in the background intronic sequences. Junction splice-site scores are calculated for all the entries in both true and false training datasets. Meanwhile, the corresponding intron sizes are recorded.
is the probability of observing Bi in the background intronic sequences. Junction splice-site scores are calculated for all the entries in both true and false training datasets. Meanwhile, the corresponding intron sizes are recorded.
Splice-site score and intron size are combined by a logistic function:
| (2) |
and
| (3) |
Here x1 and x2 are splice-site score and intron size, respectively, and the coefficients are determined by fitting the true and false training datasets. We have provided the regression models for mouse and human in the package, and the parameters of these models, including the coefficients and their statistical significance, are summarized in Supplementary Table S1). Scripts are also included to allow users to generate their own models for other species and gene annotations.
For every candidate junction identified by single- or double-anchor de novo junction search, we calculate the logistic probability with Equation 2. At the ‘junction connection’ stage, logistic probabilities of all junctions in a candidate alignment are averaged for final ranking. Those de novo alignments with low logistic probabilities (default: ≤0.5) are regarded as low confidence and filtered out.
Practical considerations in implementation
OLego can either perform de novo junction searches or work with a database of annotated splice junctions. For de novo junction search, we currently require the canonical GT/AG splice sites because they account for ∼99% of all known introns in mammals (32). To further reduce false-positive detection of splice sites, we also require an average logistic probability of >0.5 for each alignment. When OLego is provided with a database of annotated splice junctions, several special considerations are given for alignment to known splice sites because they define a much smaller search space. Specifically, we allow non-canonical splice sites, a less stringent threshold on the minimum anchor size (5 nt vs. 8 nt for single-anchor search), and no constraint on intron size.
OLego takes FASTA or FASTQ files as input, and outputs alignments in SAM format. The junctions from the best alignments are collected and reported in BED format. It loads mRNA-Seq reads in batches, and in each batch the reads are assigned randomly to different threads, when multiple threading is enabled. Therefore, OLego supports multiple threading in the whole alignment workflow. This is distinct from many available tools, for which multiple threading is only supported at the stages when an external aligner is involved. For paired-end mRNA-Seq data, each end is first mapped independently, and the results for both ends are then combined according to their distance and orientations on the reference genome, to help resolve possible ambiguity in alignments of single-end reads. Different types of mRNA-Seq libraries with or without strand information can be handled properly.
OLego is an open source code project. It is released under GPLv3 and is freely available online at http://zhanglab.c2b2.columbia.edu/index.php/OLego. It was implemented in C++ and relied heavily on the source code library of BWA (version 0.5.9rc1) (22).
Evaluation on simulated datasets
We generated simulated mRNA-Seq reads using the program BEERS from the RUM package (14). For mouse (mm9), BEERS uses gene models derived from 11 annotation tracks (AceView, Ensembl, Geneid, Genscan, NSCAN, Other RefSeq, RefSeq, SGP, Transcriptome, UCSC and Vega) in the UCSC genome browser to avoid bias toward or against any particular set of gene annotations. It is also capable of simulating polymorphisms and random sequencing errors (at a default rate of 0.5%) with positional biases (e.g. higher error rate toward the end of reads) that mimic real mRNA-Seq data produced by the Illumina platform. We carried out two sets of simulations with default parameters, each consisting of 10 million paired-end reads, but with different read lengths (100 and 150 nt, respectively); three replicates were generated for each set.
We compared OLego (v1.0.0) with three other published programs: TopHat (version 1.4.0), MapSplice (version 1.15.2) and PASSion (version 1.2.1). TopHat and MapSplice use seed-and-extend approaches, as described above. Alternatively, PASSion (12) uses a different strategy, called pattern growth, which does not require segmentation of reads, to find exon junction reads in paired-end mRNA-Seq data. For all these programs, default parameters were used for mapping, except that the size of introns was restricted in the range of 20–500 000 nt for OLego, TopHat and MapSplice, and 20–409 600 nt for PASSion owing to its discrete choices for the maximum intron size. In addition, up to four mismatches were allowed by OLego (-M 4). In this setting, OLego searches with a seed size of 14 nt (-w 14), allowing no mismatches in the seed; de novo single-anchor junction search is enabled and a minimum anchor size of 8 nt is required (-a 8). For MapSplice, the configuration file paired.cfg included in the package was used to maximize the sensitivity (see ‘Discussion’ section). Both MapSplice and Tophat used a seed size of 25 nt and minimum anchor size of 8 nt, and they tolerated one and two mismatches in the seed, respectively. The reads were mapped onto the reference mouse genome (mm9) without any exon junction annotations provided. Up to 16 Intel Xeon CPU cores (2.0 GHz) on a Linux server were used for mapping. The BED format junction output files from these programs were used to evaluate discovery of unique exon junctions, and the alignment outputs (in SAM or BAM format) were used to evaluate the accuracy of junction alignment and small-exon discovery.
Evaluation on real mRNA-Seq data
We downloaded mRNA-Seq data (accession: SRX088978) used in a previous study (14) from the NCBI Sequence Read Archive (33). This mouse retina mRNA-Seq library was originally prepared with a 350 nt (±25 nt) average insert size, and sequenced on an Illumina Genome Analyzer IIx, with 120 nt paired-end reads (14). One lane of reads (∼26 million reads) was extracted and used in our study. Parameters used in OLego for read alignment were the same as those used in simulation, as described above.
RT-PCR validation of novel micro-exons
Retinal tissues from three 2-month-old female C57BL/6 J mice were purchased from The Jackson Laboratory. Total RNA was extracted using Trizol (Invitrogen), followed by DNAse I digestion (Promega), phenol-chloroform extraction and ethanol precipitation. RNA (1 µg) was reverse-transcribed with Improm-II reverse transcriptase (Promega) and oligo dT primers.
Radioactive touchdown PCR with [α-32 P]-dCTP and Taq Gold polymerase (Invitrogen) was used to amplify endogenous transcripts with primers described in Supplementary Table S4 and Figure 7A. PCR products were separated by 8% native PAGE, visualized by autoradiography and quantified on a phosphorimager (Fuji Image Reader FLA-5100) using Multi Gauge software Version 2.3. The inclusion ratio of each exon was then calculated by normalizing the signal intensity of the inclusion isoform to the total intensity of both isoforms, and expressed as a percentage.
Figure 7.
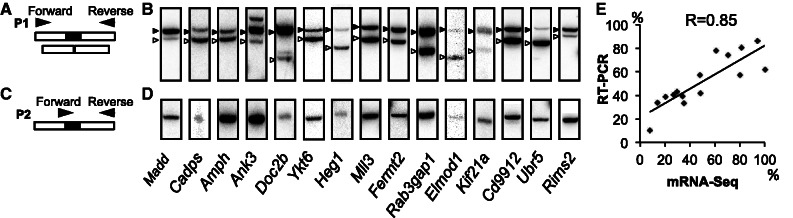
Experimental in vivo validation of micro-exons discovered by OLego. (A, C) Primers were designed either in the flanking exons to detect both micro-exon inclusion and skipping isoforms (A), or at the exon junction to specifically detect micro-exon expression (C). Primers positions and structure of each isoform are indicated (not to scale). (B, D) RT-PCR analysis of micro-exon expression in mouse retina using primers described in (A, C). Micro-exon included and skipped isoforms are indicated next to the corresponding bands by solid and empty arrowheads, respectively. (E) Correlation of micro-exon inclusion ratios estimated from mRNA-Seq data and those measured by radioactive PCR, as described in (B) (n = 3).
RESULTS
Exon junction discovery in simulated datasets
We first evaluated the performance of OLego for de novo exon junction discovery using two sets of simulated mRNA-Seq data. We also carried out a comparison of OLego with two other widely used seed-and-extend programs, TopHat (16) and MapSplice (18), and a recently published program, PASSion, which is based on a pattern-growth algorithm to search exon junctions in paired-end data (12). All of the compared programs were previously benchmarked and demonstrated good performance (12,14). In each simulation set, we generated 10 million 100-nt or 150-nt paired-end reads, which were aligned by the four programs. Unique exon junctions reported by each program were used to estimate the positive predictive value (PPV) as a measure of accuracy, and the false-negative rate (FNR) as a measure of sensitivity, and this process was repeated in three replicates to average the results (Table 1). In these tests, a slightly higher PPV was achieved by Tophat and OLego (97.7–98.4%), compared with PASSion (96.3% for both 100-nt and 150-nt reads) and MapSplice (95.1% for 100-nt reads; 97.1% for 150-nt reads). In terms of sensitivity, OLego discovered substantially more true junctions than the other programs. OLego’s FNR (8.2% for 100-nt reads and 6.8% for 150-nt reads) almost halved the FNRs of TopHat (15.4% for 100-nt reads and 12.8% for 150-nt reads) and PASSion (14.8% for 100-nt reads and 15.5% for 150-nt reads), whereas MapSplice had an intermediate FNR (10.3% for 100-nt reads and 9% for 150-nt reads). Therefore, OLego achieved both high sensitivity and accuracy, suggesting the benefit of more exhaustive searches using small seeds, combined with quantitative modeling of exon-junction strength and alignment quality. As expected, all seed-and-extend-based tools achieved better sensitivity when the read size increased; interestingly, PASSion’s sensitivity decreased slightly with longer reads.
Table 1.
The number of exon junctions identified by OLego, MapSplice, TopHat and PASSion on simulated data
| Measurement | 100-nt reads (178 449 junctions) |
150-nt reads (189 106 junctions) |
||||||
|---|---|---|---|---|---|---|---|---|
| OLego | MapSplice | TopHat | PASSion | OLego | MapSplice | TopHat | PASSion | |
| Found junctions total | 166 954 | 168 219 | 153 446 | 157 967 | 180 410 | 177 142 | 167 707 | 165 871 |
| Found true junctions | 163 740 | 159 984 | 151 013 | 152 094 | 176 172 | 172 052 | 164 847 | 159 773 |
| Missed true junctions | 14 708 | 18 464 | 27 436 | 26 355 | 12 934 | 17 054 | 24 259 | 29 333 |
| PPV | 0.981 | 0.951 | 0.984 | 0.963 | 0.977 | 0.971 | 0.983 | 0.963 |
| FNR | 0.082 | 0.103 | 0.154 | 0.148 | 0.068 | 0.090 | 0.128 | 0.155 |
PPV, positive predictive value or precision; FNR, false-negative rate.
We then assessed the extent of overlap among the four programs with regard to the true junctions they identified. In all pairwise comparisons, the number of common junctions identified by the programs was higher than expected by chance (Table 2, upper diagonal vs. lower diagonal). For example, in 100-nt reads, OLego identified most true junctions found by MapSplice (97.9% or 156 654/159 984), TopHat (97.8% or 147 731/151 013) and PASSion (96.8% or 147 266/152 094), whereas only 91.1–91.8% (146 798/159 984, 138 566/151 013 and 138 558/152 094, respectively) were expected (P < 2.2 × 10−16, Fisher’s exact test). The striking statistical significance of the overlap suggests that some junction reads are easier to align, whereas others are more difficult for all four programs. This observation can be interpreted in several ways, including multiple hits of read sequences in the transcriptome (e.g. introduced by paralogous genes), ambiguity of sequence alignment at exon junctions, short anchors on either side of some exon junctions or complications introduced by simulated sequencing errors in some mRNA-Seq reads.
Table 2.
Pairwise comparison of exon junctions discovered by OLego, MapSplice, TopHat and PASSion on simulated data
 |
The observed (upper diagonal; shaded) and expected (lower diagonal) overlap of discovered true exon junctions between each pair of programs is shown.
We also compared how read coverage affected each program in sensitivity of exon-junction detection. For this purpose, we binned the simulated junctions according to their ground-truth read coverage, and for each program, we estimated the sensitivity of junction discovery separately for each bin (Figure 2). As expected, all programs had higher sensitivity when the coverage increased. OLego achieved higher or comparable sensitivity in all bins, relative to the other three programs. For example, for 100-nt reads, OLego had a sensitivity of 95.7% for junctions supported by >4 reads, which was comparable with MapSplice (95.8%), despite more specific junction identifications by OLego. OLego performed best in all other bins, with sensitivity between 74.8% (for junctions supported by only 1 read) and 93.9% (for junctions supported by four reads). On the other hand, TopHat and PASSion had relatively lower sensitivity, as observed from all bins. Importantly, the advantage of OLego in sensitivity was particularly clear for exon junctions with low coverage, compared with the other three programs (63.7%, 48.3% and 54.1% for junctions supported by only one read for MapSplice, TopHat and PASSion, respectively; Figure 2), again suggesting the benefit of more exhaustive searches using short seeds.
Figure 2.
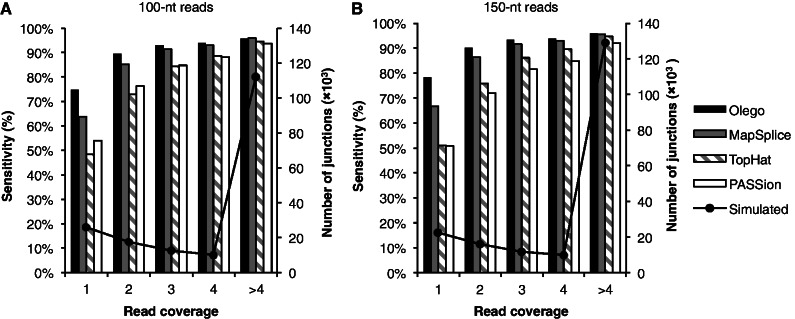
Sensitivity of junction detection at different coverages. (A) Tests on 10 million 2 × 100-nt simulated reads; (B) Tests on 10 million 2 × 150-nt simulated reads. For each panel, the simulated junctions were binned according to their coverage, from 1 read per junction to >4 reads per junction. The true numbers of junctions in the simulation are shown by lines with markers on the right axis, and the sensitivity of OLego, MapSplice, TopHat and PASSion are indicated by bars on the left axis.
Mapping speed
We next compared OLego, MapSplice, TopHat and PASSion in terms of mapping speed because this becomes increasingly critical as the throughput of mRNA-Seq technologies increases dramatically. OLego supports multiple threading in the whole cycle of mapping individual reads, whereas the other three programs support multiple threading in a limited number of steps. Therefore, we first ran all programs with multiple threading enabled using 16 CPU cores (2.0 GHz per core). It took OLego, TopHat, MapSplice and PASSion 0.8, 2, 5.5 and 6.8 h, respectively, to align the 10 million 2 × 100-nt reads, and 1.4, 3.3, 10.5 and 9.3 h, respectively, to align the 10 million 2 × 150-nt reads. OLego was faster than TopHat by more than 2-fold, whereas MapSplice and PASSion were substantially slower than OLego by ∼7-fold. To further compare read-mapping speed and the benefit of multiple threading, we ran OLego and TopHat using different numbers of CPU cores (1, 4, 8 and 16) on both sets of simulated data (Figure 3). When a single CPU core was used, OLego and TopHat had similar mapping speeds, despite the fact that OLego performed more exhaustive searches using much smaller seeds. When more CPU cores were used, the mapping speed of OLego increased linearly as a function of the number of CPU cores. The mapping speed of TopHat also increased, but at a slower rate. This is presumably because TopHat supports multiple threading only in steps that involve the external aligner Bowtie. When ≥8 CPU cores were used for alignment, OLego used half as much or even less time, compared with TopHat. These comparisons suggest that OLego not only achieved high sensitivity and specificity, but also substantially improved the mapping speed.
Figure 3.

Comparison of mapping speed. (A) Tests on 2 × 100-nt simulated reads; (B) Tests on 2 × 150-nt simulated reads. Running time (wall time) of TopHat (square) and OLego (triangle) on 10 million simulated paired-end reads with different numbers of CPU cores is shown. The values were averaged across three replicates for each test, with error bars indicating standard deviations.
Small or micro-exon discovery in simulated datasets
Alternatively spliced exons are generally shorter than exons that undergo constitutive splicing (34), and some are extremely small [e.g. 6 nt (35)]. Owing to the limited information content encoded in such short sequences, micro-exons (<30 nt) (20) and their AS are intriguing with respect to their functional significance and underlying regulatory mechanisms. However, these exons are more likely to be missed in de novo searches because they are much less likely to have seed sequences completely within the exon. Therefore, we evaluated the performance of OLego, TopHat, MapSplice and PASSion in finding small exons or micro-exons (9–39 nt). OLego consistently performed best in both sensitivity and accuracy, compared with the other three programs (Figure 4). In terms of specificity, OLego achieved a PPV of 96.5% for 100-nt reads and 95.7% for 150-nt reads, respectively, compared with ∼91.7–93.6% for TopHat, ∼88.3–93.3% for MapSplice and ∼90.7–91.8% for PASSion. OLego achieved an overall sensitivity of 86.6% for 100-nt reads and 90.8% for 150-nt reads, respectively, which was much higher than TopHat (∼63.9–68.5%), MapSplice (∼75.9–80.6%) and PASSion (∼67.7–78.7%). We further grouped exons according to their sizes to evaluate the sensitivity of each program. For exons of size 27–39 nt, MapSplice discovered a smaller number of exons than OLego (83.3% vs. 88.4% for 100 nt reads and 87.2% vs. 92.3% for 150 nt reads), and gave a lower PPV (91.3% vs. 96.6% for 100 nt reads, and 94.5% vs. 95.9% for 150 nt reads). TopHat and PASSion had the lowest sensitivity among the four programs (∼75.8–80.6% for TopHat, and ∼69.8–80.1% for PASSion). Again, the advantage of OLego was most prominent in detecting extremely short micro-exons. For example, in detecting micro-exons of size 9–15 nt, OLego had a sensitivity of 75.7% for 100-nt reads and 78.4% for 150-nt reads, respectively, which was substantially higher compared with TopHat (6.1% and 9.3%, respectively), MapSplice (27.8% and 33.8%, respectively) and PASSion (47.4% and 38.4%, respectively).
Figure 4.

Discovery of small and micro-exons in simulated mRNA-Seq data. (A) 2 × 100-nt simulated reads; (B) 2 × 150-nt simulated reads. In each panel, internal exons within mapped reads were counted. The numbers of true (open columns) and false (solid columns) exons of different sizes, compared with the ground truth (horizontal bar) are shown for OLego, TopHat, MapSplice and PASSion, respectively. The overall sensitivity (SN9–39) and the sensitivity for exons of size 9–15 nt (SN9–15) are indicated on each plot.
Exon junction discovery in real data
After systematic evaluation of OLego using simulated data, we proceeded to analyze an mRNA-Seq dataset prepared from mouse retina RNA, which consisted of ∼26 million 120-nt paired-end reads (33). We first examined known and novel exon junctions identified de novo by OLego. In total, mapping of these reads identified 234 440 unique exon junctions. Among them, 159 938 junctions (68.2%) were previously annotated, based on a comprehensive database of gene models derived from multiple sources (denoted as inclusive gene models; see ‘Materials and Methods’ section for more details) (14); more strictly, 137 606 (58.7%) junctions were annotated in RefSeq genes. We next binned all identified junctions according to the number of supporting reads, and categorized junctions in each bin into annotated junctions and three classes of novel junctions: novel junctions in which both splice sites are annotated separately, but the intron itself is not annotated (class I); novel junctions with only one site annotated (class II); and novel junctions with neither site annotated (class III) (Figure 5A). This analysis suggested that for exon junctions supported by >4 reads, 96.9% junctions were previously annotated, and an additional 0.69% exon junctions were class I novel junctions. On the other hand, for exon junctions supported by a single read, only 24.4% were previously annotated, and 39.8% were class III novel junctions. This trend is not surprising because more abundant exon junctions are more likely to be known from previous data. As sequencing depth increases, it becomes more likely to observe novel rare splicing events, which are, however, complicated by sequencing and alignment errors.
Figure 5.
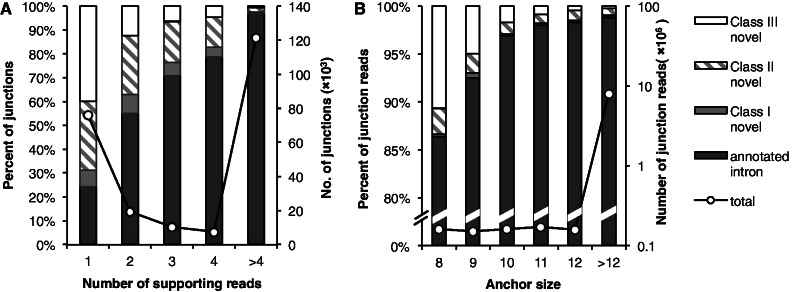
Distributions of exon junctions discovered in mouse retina mRNA-Seq data. (A) The junctions found by OLego were binned according to the numbers of supporting reads. Different patterns indicate categories of junctions in the bar plot: annotated junctions; junctions with both splice sites annotated (Class I novel); junctions with only one splice site annotated (Class II novel) and junctions without any splice site annotation (Class III novel). The total number of junctions discovered in each bin is shown by the solid line with axis on the right. (B) The junction alignments were grouped according to their anchor sizes. The categories of the junctions are shown in the same way as in panel (A), and the numbers of junction alignments are shown by the solid line with the y-axis on the right.
Distinguishing novel exon junctions from artifacts introduced by alignment errors in real mRNA-Seq data is difficult. Nevertheless, we reasoned that there are two major sources of mapping errors that can introduce false exon junction detection. The first type is ambiguous determination of splice sites, due to repetitive sequences in double-anchor junction search; the second type is errors introduced in single-anchor search when the number of matched nucleotides at the other end of the junction (anchor size) is limited. Manual examination of unannotated junctions suggested that the latter might be dominant. To study the relationship between anchor size and false junction detection, we binned all the aligned junction reads by the anchor size (Figure 5B). The rationale here is that the boundaries of mRNA/cDNA fragments in library preparation are random relative to the position of the splice sites, which is what we actually observed (Figure 5B, black curve). If all reads were aligned perfectly, the percentage of junction reads sampled from annotated (real) junctions should not vary as a function of anchor size. On the other hand, it is clear that a higher error rate is expected to occur when the anchor size is small, owing to an increase in the chance of random matches. Therefore, examining the percentage of reads sampled from known junctions as a function of anchor size provides an independent method of estimating the bound of mapping errors. Specifically, with an anchor size >12 nt, 98.8% of splicing events in reads were mapped to annotated exon junctions, and an additional 0.23% were mapped to class I novel junctions. On the other hand, for junction reads with an anchor size of 8 nt, 86.4% of splices were mapped to annotated junctions, and an additional 0.3% were mapped to class I novel junctions. Therefore, we estimated that the false mapping rate of an exon junction read with anchor size of 8 nt could be as high as 12.3–13.3%, although these alignments represented a minor proportion of all junction alignments (1.86%). Similarly, the false mapping rate of an exon junction read with anchor size of 10 nt was estimated to be 1.9–2.9%. By requiring a more stringent anchor size of 10 nt, we identified 208 567 unique exon junctions, among which 76.4% were annotated previously in inclusive gene models. The proportion was 97.1% and 35.3% for junctions supported by >4 reads and by a single read, respectively.
Micro-exon discovery in real data
Our evaluation on simulated datasets suggested that OLego is particularly sensitive and accurate for micro-exon discovery. In this real mRNA-Seq dataset, we identified 1665 micro-exons between 9 nt and 27 nt (Figure 6A and Supplementary Table S2), after requiring a minimal match of 10 nt at both ends for junctions flanking the micro-exon. Among these, 1035 exons (62.2%) were annotated in inclusive gene models (14), and more restrictively, 715 (42.9%) were annotated in RefSeq genes. Among the remaining 630 exons that lack any evidence in current gene models, we examined the 5′ splice site of the upstream intron and the 3′ splice site of the downstream intron flanking each micro-exon. We found that 417 exons (66.2% out of 630 or 25% out of 1665) had both the upstream and downstream constitutive splice sites annotated in the current gene models, as well as supporting reads that connect them to the micro-exon on both sides. This subset is expected to have a higher reliability, and we refer to it as high-confidence novel micro-exons. As a comparison, TopHat, MapSplice and PASSion found 790, 713 and 1242 micro-exons, respectively, among which 81, 85 and 163 exons are of high confidence with the same criteria (Supplementary Figure S1). Compared with OLego, short micro-exons are under-represented in the results of all three programs, consistent with our observations from simulation data.
Figure 6.

Discovery of micro-exons in mouse retina mRNA-Seq data. (A) Number of micro-exons identified by OLego. Exons are binned by their sizes (∼9–27 nt), and in each bin, they are classified into three groups: annotated micro-exons in previous gene models (black), high-confidence novel micro-exons (exons with both flanking constitutive splice sites annotated; gray) and other (blank). (B) Cumulative distribution of exon inclusion level for annotated and high-confidence novel micro-exons; only those cassette exons with ≥10 reads that support either isoform were included for this analysis. (C) The distribution of total splice-site score (3′ + 5′ splice sites) for each group of micro-exons is shown as a boxplot. (D) The pyrimidine (C/U) content in the upstream 100-nt intronic sequences, calculated using 10-nt sliding windows. (E) Cross-species conservation around the micro-exons. The medians of phastCons scores across 30 vertebrate species in the intronic regions immediately upstream and downstream of the annotated and high-confidence novel micro-exons are shown. (F) An example of a 9-nt novel micro-exon in the Kcnn2 gene is shown. This exon is missing in current gene models (e.g. RefSeq) or cDNA/EST data (not shown), but both isoforms are abundant in the mouse retina (the two tracks on the top). The micro-exon is embedded in a longer stretch of conserved sequences.
A large proportion (988/1665 or 59.3%) of the micro-exons have a size that is a multiple of three (Figure 6A). This is a prominent feature of regulated AS (36) and is consistent with our results on simulated data (Figure 4). Indeed, 42.2% (437/1035) of annotated exons and 67.9% (283/417) of high-confidence novel exons are cassette exons, for which both inclusion and skipping were observed in these mRNA-Seq data. For these cassette micro-exons, novel exons tend to have much lower inclusion levels, compared with annotated exons (Figure 6B). The difference between annotated and novel exons can be explained in part by their difference in splice signals. Compared with the annotated micro-exons, novel exons have weaker summed 3′ and 5′ splice site scores (9.12 vs. 11.89, median; Figure 6C) and polypyrimidine tract (Figure 6D), although their scores are still clearly above background. Another not mutually exclusive possibility is that inclusion of novel cassette micro-exons shifts the reading frame more frequently, compared with annotated cassette micro-exons (47.7% vs. 16%), which would likely introduce premature stop codons and thereby trigger nonsense-mediated mRNA decay to reduce the apparent inclusion level (37).
To assess the functional significance of the novel micro-exons, we examined their sequence conservation in vertebrate species (38). For both annotated and high-confidence novel micro-exons, we observed a high level of sequence conservation in flanking intronic regions (Figure 6E). For example, a 9-nt cassette exon in the Kcnn2 gene is located in a long stretch of highly conserved sequences, and both isoforms were abundantly detected by mRNA-Seq, but not in previous cDNA/EST data (Figure 6F). Presumably, these regions harbor conserved cis-regulatory elements, which might be important for regulated splicing of these micro-exons and are thus under evolutionary selection pressure.
In vivo validation of the micro-exons discovered by OLego
To assess the accuracy of OLego’s micro-exon predictions, we experimentally tested the expression and inclusion ratios of micro-exons in mouse retina. We ranked the high-confidence novel micro-exons by the number of supporting reads for the inclusion isoform, and selected 15 exons for PCR validations (Supplementary Table S3 and Supplementary Figure S2). Two sets of primers were designed to validate each micro-exon: (i) primers were positioned in the flanking exons of the micro-exon to detect both exon inclusion and exclusion; and (ii) one of the primers was positioned on the exon junction spanning the micro-exon and a flanking exon, while the other primer was positioned in a flanking exon (Figure 7A, C; Supplementary Table S4). This ensured that we would both quantify the inclusion ratios and specifically detect the micro-exon, respectively. For all tested exons, we detected two isoforms with a size corresponding to the inclusion and exclusion of the micro-exon, respectively (Figure 7B). In addition, amplification with primers specific to the micro-exon junction confirmed the identity of the included/skipped micro-exon (Figure 7D). Therefore, OLego performs well in micro-exon discovery, as we were able to validate15 out of 15 predicted novel micro-exons. This is further supported by the observation that the inclusion ratios estimated from the RNA-Seq data and those measured in the PCR validation are highly correlated (Pearson correlation coefficient R = 0.85; Figure 7B, E, Supplementary Table S3).
DISCUSSION
Here we present OLego, a program designed for fast mapping of hundreds of millions of mRNA-Seq reads to the reference genome with high accuracy and sensitivity, which allows identification of known and novel exon junctions. Since the first publication of mRNA-Seq studies (11), the technologies have evolved rapidly, with the most prominent features including increases in read length and throughput, and a reduction in sequencing errors.
The first generation of tools that align RNA-seq reads to the genome is based on the construction of a database of known or predicted exon junction sequences (3,39,40), so that junction reads can be mapped against this junction sequence database without alignment gaps. This strategy is fast and accurate for alignments to annotated exon junctions. However, it relies on the fact that reads are short (∼36 nt), so that they rarely span more than one junction, and another important caveat is that this approach does not allow the discovery of exon junctions de novo. As the read length increases, it becomes more and more frequent for a read to span three or more exons, but it is difficult to build a sequence database of alternative isoforms that span many exons, while preserving the ‘uniqueness’ of sequences that can potentially match mRNA-Seq reads. Algorithms designed specifically to map spliced mRNA-Seq reads were subsequently developed, with TopHat being one of the first (41), followed by several others, such as SpliceMap (17), GSNAP (13), MapSplice (18) and PASSion (12).
Although different heuristics were used in each of these programs, most of them use a seed-and-extend strategy, which was also used in programs developed earlier to map traditional cDNA/EST sequences to genomic DNA sequences, such as sim4 (42), BLAT (23) and exonerate (43). With this strategy, the size and position of the seeds are critical determinants of mapping sensitivity. In general, a match of at least one seed in each exon is critical for successful alignment of a read, although tricks like single-anchor junction search can be used to match sequences near the ends of a read. To achieve sensitivity, these earlier programs typically used short seeds of size 11–12 nt. BLAT is one of the first programs to allow fast mapping of cDNA/EST sequences to the whole genome, by hashing the whole genome using non-overlapping seeds or tiles (default = 11 nt). However, speed is a bottleneck in processing ultra-high-throughput mRNA-Seq data.
To improve the speed of genome-wide mapping of large numbers of short reads, various schemes have been used to index the reference genome sequences to enable faster querying. For example, GSNAP uses a hash table indexing all the k-mers (k typically in the range from 11 to 15 nt) every 3 nt in the reference genome. The overlapping 3-nt spaced seed hashing scheme is necessary to reduce the memory footprint to ∼4 GB. An alternative approach to hashing is the BWT- and FM-index-based method used by many programs, including Bowtie, which is integrated into TopHat and MapSplice. This invertible full-text indexing scheme is more memory-efficient, and allows fast query of sequences of varying length, in contrast to the fixed size of seed sequences in hashing-based methods. This flexibility makes it possible to align different types of reads with different granularity, e.g. fast alignment of exon-body reads without requiring seed partitioning, followed by alignment of spliced reads using short seeds and short or micro-exons of varying sizes.
Most currently available mRNA-Seq read splice-mapping tools typically segment reads into non-overlapping, relative long (∼25 nt) seeds, which are mapped to the genome without gaps by an external mapper. The relatively long seeds restrict the number of hits, so that the temporary results generated by the external mapper are manageable in post-processing steps to produce final junction alignments. However, even with relatively long seeds, the pipeline-based methods still generate temporary files of enormous size, which can be a significant concern regarding both space and speed when these files are parsed to produce final results. For example, with the basic configuration (paired.cfg), MapSplice required about 140 and 200 GB of disk space to store temporary files to align the 10 million paired-end 100 - or 150-nt reads, respectively, in our simulation. In the more exhaustive mode (Try_hard.cfg in the package), the disk usage of MapSplice increased to >500 GB for 100-nt reads and 800 GB for 150-nt reads. Interestingly, with this mode we did not observe an increase in sensitivity, but did observe a dramatic drop in accuracy (data not shown).
The relatively long seeds increase the chance that the seeds themselves span exon junctions, reducing the number of seeds mapped to exonic sequences, which are critical for final sensitivity. For example, the median size of mammalian exons is ∼120 nt. In this case, ∼21% (25/120) of 25-nt sliding windows overlap with exon junctions. This problem gets worse for alternative exons, especially those regulated to have variable inclusion levels in different conditions, such as different tissues. For example, the median size of cassette exons regulated by the neuron-specific splicing factor Nova is ∼80 nt (44), so that ∼31% (25/80) of 25-nt sliding windows overlap with exon junctions. Smaller seeds greatly reduce the chance of overlaps with exon junctions in seed sequences. With the ∼14-nt seeds used in OLego, a read covering an exon of ≥28 nt is guaranteed to have at least one seed inside the exon, which increases the sensitivity of mapping reads sampled from these relatively small exons. Instead of using a smaller seed size, PASSion (12) uses a different strategy, based on a pattern-growth algorithm. This method can only be applied in paired-end mRNA-Seq data when one exonic read is aligned in the first pass without allowing large gaps, while the other read, which spans one or two exon junctions, is missed in the first pass and is to be refined in later steps. In this scenario, the aligned read in a pair is used as an anchor, and local searches of the (maximum) unique substrings starting from the ends of the other read are performed using a pattern-growth algorithm, constrained by the maximum intron size. This process can continue iteratively until the substrings cover the whole read. The advantage of PASSion in eliminating the read-segmentation step is attractive, and this algorithm was reported to show competitive performance compared with several other programs. However, it is unclear how this algorithm handles sequencing errors, the repetitive nature of substrings and longer mRNA-Seq reads in which both reads in a pair span exon junctions. In practice, OLego achieved both higher sensitivity and accuracy in discovery of exon junctions and micro-exons using realistic simulated data.
OLego aligns each read independently in one pass, without filtering of junctions based on their summary statistics derived from all reads, such as the uniformity of the positions of reads mapped to the junction used by MapSplice and the read coverage around the junction used by TopHat and PASSion. To ensure the accuracy of junction mapping, OLego limits the de novo search to canonical GT/AG splice sites, and uses a built-in model of exon-junction strength that combines splice-site scores and intron size. This intentional choice is owing to the fact that canonical splice sites account for ∼99% mammalian introns (32). The exon-junction scoring is effective to find the real splice site in a single-anchor junction search, in which multiple hits flanked by splice-site dinucleotides can be found when the anchor sequence is short. It also improves the accuracy in a double-anchor junction search, when ambiguity exists owing to repetitive sequences near the splice sites. As a result, OLego achieved an accuracy comparable with that of TopHat and better than those of MapSplice and PASSion, while at the same time having a much lower FNR. The advantage of OLego compared with the other programs is particularly prominent for exon junctions of low abundance, as demonstrated in simulations. Nevertheless, the default parameters are chosen to balance accuracy and sensitivity, which is suitable for many applications of mRNA-Seq to quantify splicing levels. For efforts focusing on the discovery of novel junctions, including those of low abundance, filtering of junctions based on supporting evidence and anchor size can certainly improve the accuracy further.
While this article was under revision, another program named TrueSight was published (45). TrueSight also uses splice-site motifs together with other features to build a regression model to distinguish true- vs. false-positive exon junctions, and the authors reported improved PPV and sensitivity compared with TopHat, MapSplice and PASSion. One important difference between TrueSight and OLego is that the former builds exon junction models on the fly, using already-mapped junction reads, and it updates the model and the alignment iteratively using an expectation maximization (EM) algorithm. The benefit of the EM algorithm is unclear, given that a large number of exon junctions from reads mapped in previous steps (or in annotated gene models) is already available, and logistic regression in general is not sensitive to a certain level of noises. In our experiment, we were able to use 10% of the training data to derive our logistic regression model and obtain essentially the same results (data not shown). On the other hand, the iterative procedure in TrueSight appears to be computationally intensive, so that TrueSight is significantly slower than the other programs in the authors’ original comparison and it has a relatively large memory footprint (10 GB memory per 30 million reads). Furthermore, TrueSight also relies on an external mapper for seed mapping, sharing the same limitation on seed size as TopHat and MapSplice, which OLego aims to improve.
We used several strategies to achieve fast mapping with small seeds to the mammalian-sized genome. First, we require perfect matches in seed sequences, given the fact that sequencing errors in typical mRNA-Seq data are as low as 0.5%. Mismatches, including substitutions and indels, are handled when the alignments are refined for each exon. Second, after hits of seeds are clustered, candidate alignments are ranked and filtered by the number and uniqueness of matched seeds. Therefore, the later time-consuming steps to locate exon junctions are only applied to the most promising candidate alignments. Third, BWT- and FM-index-based querying in the genome is not only applied in the step of seed mapping, but also in the later steps to locate splice sites (single-anchor junction search) and micro-exons. The capability of fast querying of sequences of different sizes using BWT is particularly helpful. Finally, we do not need to filter the alignments according to the abundance of each junction, so that each read can be mapped independently in one pass. This makes it possible to support multiple threading in the whole cycle of alignment. Indeed, although OLego performed more exhaustive searches than TopHat, the speeds of these two programs were still comparable, even with a single CPU core. Furthermore, the speed of OLego increased faster than that of TopHat as the number of CPU cores increased, such that with ≥8 CPU cores, OLego used half or less time, compared with TopHat. The other two programs, MapSplice and PASSion, were substantially slower in our comparison. We estimate that on 8 CPU cores, OLego can map a typical lane of 200 million paired-end mRNA-Seq reads of 100 nt and 150 nt to the mammalian genome in ∼29 and ∼46 h, respectively. Combined with its small memory footprint, OLego can efficiently run on desktop workstations. It is also worth noting that increasing the seed size will further improve OLego’s mapping speed, despite the risk of potential decrease in sensitivity of exon junction detection, especially for those flanking small or micro-exons.
We paid special consideration to searches of small or micro-exons. Even with the small seeds used in this study, these exons might still lack internal seed sequences without overlap with exon junctions. However, these exons can be recovered when matches to sequences in the flanking upstream or downstream exons are found, and the micro-exon sequences can be determined accordingly, so that they can be effectively searched against the indexed genome together with the flanking splice sites. As demonstrated by simulation, OLego was successful in identifying most of the extremely small exons of size 9–15 nt (75.7–78.4%), whereas TopHat and MapSplice missed most of them (only 6.1–33.8% identified). PASSion identified more exons in this range (∼38.4–47.4%) than TopHat and MapSplice, but the numbers were still much smaller than OLego’s. TopHat provided an optional ‘micro-exon-search’, which is supposed to improve the sensitivity of micro-exon search. However, even with this option enabled, TopHat only found 12% of these extremely small exons in the 100-nt dataset, as compared with 75.7% by OLego. Finally, we were able to identify >400 high-confidence novel micro-exons in a single mRNA-Seq library of moderate depth (∼26 million paired-end reads) prepared from mouse retina RNA. The inclusion level of these exons is lower than that of annotated ones, which is likely why they were not previously identified, and this can be explained by their weak splicing signals. However, we were able to validate 100% of the novel micro-exons tested by RT-PCR, demonstrating OLego’s high sensitivity and accuracy. Some of these micro-exons likely have functional significance, as judged from their deep phylogenetic conservation (see also Supplementary Figure S2).
With its high sensitivity and accuracy and fast mapping speed, OLego can be used for efficient alignment of large-scale mRNA-Seq data being generated at unprecedented rate and depth. It can be combined with downstream analysis tools for transcript reconstruction and quantification to facilitate the process of revealing the transcriptomic complexity of mammals and other species.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1–4 and Supplementary Figures 1–2.
FUNDING
National Institutes of Health (NIH) [GM74688 to M.Q.Z. and A.R.K., K99GM95713 to C.Z.]; National Basic Research Program of China [2012CB316503 to M.Q.Z.]. Funding for open access charge: NIH [GM74688 to M.Q.Z. and A.R.K., K99GM95713 to C.Z.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Michael Schatz and Martin Akerman for critical reading of the manuscript, and members of the Krainer, Zhang and Robert Darnell labs for helpful discussion. C.Z. would also like to thank the Darnell lab for computing resources and support.
REFERENCES
- 1.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 2.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 4.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Licatalosi DD, Darnell RB. Splicing regulation in neurologic disease. Neuron. 2006;52:93–101. doi: 10.1016/j.neuron.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 7.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 8.Castle JC, Zhang C, Shah JK, Kulkarni AV, Kalsotra A, Cooper TA, Johnson JM. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008;40:1416–1425. doi: 10.1038/ng.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, et al. Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 2005;37:844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 10.Clark T, Schweitzer A, Chen T, Staples M, Lu G, Wang H, Williams A, Blume J. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biol. 2007;8:R64. doi: 10.1186/gb-2007-8-4-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Lameijer EW, t Hoen PA, Ning Z, Slagboom PE, Ye K. PASSion: a pattern growth algorithm-based pipeline for splice junction detection in paired-end RNA-Seq data. Bioinformatics. 2012;28:479–486. doi: 10.1093/bioinformatics/btr712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grant GR, Farkas MH, Pizarro AD, Lahens NF, Schug J, Brunk BP, Stoeckert CJ, Hogenesch JB, Pierce EA. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011;27:2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang S, Zhang J, Li R, Zhang W, He Z, Lam TW, Peng Z, Yiu SM. SOAPsplice: genome-wide ab initio detection of splice junctions from RNA-Seq data. Front. Genet. 2011;2:46. doi: 10.3389/fgene.2011.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Au KF, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Volfovsky N, Haas BJ, Salzberg SL. Computational discovery of internal micro-exons. Genome Res. 2003;13:1216–1221. doi: 10.1101/gr.677503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferragina P, Manzini G. Opportunistic data structures with applications. Proc FOCS 2000. 2000:390–398. [Google Scholar]
- 22.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hastings ML, Krainer AR. Pre-mRNA splicing in the new millennium. Curr. Opin. Cell Biol. 2001;13:302–309. doi: 10.1016/s0955-0674(00)00212-x. [DOI] [PubMed] [Google Scholar]
- 25.Zhang MQ. Statistical features of human exons and their flanking regions. Hum. Mol. Genet. 1998;7:919–932. doi: 10.1093/hmg/7.5.919. [DOI] [PubMed] [Google Scholar]
- 26.Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 2001;29:1185–1190. doi: 10.1093/nar/29.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dogan RI, Getoor L, Wilbur WJ, Mount SM. SplicePort—an interactive splice-site analysis tool. Nucleic Acids Res. 2007;35:W285–W291. doi: 10.1093/nar/gkm407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fox-Walsh KL, Dou Y, Lam BJ, Hung SP, Baldi PF, Hertel KJ. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc. Natl Acad. Sci. USA. 2005;102:16176–16181. doi: 10.1073/pnas.0508489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang C, Hastings ML, Krainer AR, Zhang MQ. Dual-specificity splice sites function alternatively as 5′ and 3′ splice sites. Proc. Natl Acad. Sci. USA. 2007;104:15028–15033. doi: 10.1073/pnas.0703773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burset M, Seledtsov IA, Solovyev VV. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000;28:4364–4375. doi: 10.1093/nar/28.21.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kodama Y, Shumway M, Leinonen R. The sequence read archive: explosive growth of sequencing data. Nucleic Acids Res. 2012;40:D54–D56. doi: 10.1093/nar/gkr854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stamm S, Zhang MQ, Marr TG, Helfman DM. A sequence compilation and comparison of exons that are alternatively spliced in neurons. Nucleic Acids Res. 1994;22:1515–1526. doi: 10.1093/nar/22.9.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carlo T, Sierra R, Berget SM. A 5′ Splice site-proximal enhancer binds SF1 and activates exon bridging of a microexon. Mol. Cell Biol. 2000;20:3988–3995. doi: 10.1128/mcb.20.11.3988-3995.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xing Y, Lee C. Alternative splicing and RNA selection pressure - evolutionary consequences for eukaryotic genomes. Nat. Rev. Genet. 2006;7:499–509. doi: 10.1038/nrg1896. [DOI] [PubMed] [Google Scholar]
- 37.Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- 38.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Meth. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 41.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slater G, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang C, Frias MA, Mele A, Ruggiu M, Eom T, Marney CB, Wang H, Licatalosi DD, Fak JJ, Darnell RB. Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science. 2010;329:439–443. doi: 10.1126/science.1191150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Li-Byarlay H, Burns P, Borodovsky M, Robinson GE, Ma J. TrueSight: a new algorithm for splice junction detection using RNA-seq. Nucleic Acids Res. 2013;41:e51. doi: 10.1093/nar/gks1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.