


Abstract
Transcription-blocking oxidative DNA damage is believed to contribute to aging and to underlie activation of oxidative stress responses and down-regulation of insulin-like signaling (ILS) in Nucleotide Excision Repair (NER) deficient mice. Here, we present the first quantitative proteomic description of the Caenorhabditis elegans NER-defective xpa-1 mutant and compare the proteome and transcriptome signatures. Both methods indicated activation of oxidative stress responses, which was substantiated biochemically by a bioenergetic shift involving increased steady-state reactive oxygen species (ROS) and Adenosine triphosphate (ATP) levels. We identify the lesion-detection enzymes of Base Excision Repair (NTH-1) and global genome NER (XPC-1 and DDB-1) as upstream requirements for transcriptomic reprogramming as RNA-interference mediated depletion of these enzymes prevented up-regulation of genes over-expressed in the xpa-1 mutant. The transcription factors SKN-1 and SLR-2, but not DAF-16, were identified as effectors of reprogramming. As shown in human XPA cells, the levels of transcription-blocking 8,5'-cyclo-2'-deoxyadenosine lesions were reduced in the xpa-1 mutant compared to the wild type. Hence, accumulation of cyclopurines is unlikely to be sufficient for reprogramming. Instead, our data support a model where the lesion-detection enzymes NTH-1, XPC-1 and DDB-1 play active roles to generate a genomic stress signal sufficiently strong to result in transcriptomic reprogramming in the xpa-1 mutant.
INTRODUCTION
Stochastic accumulation of oxidative DNA damage has been regarded as a major contributor to age-related functional loss ever since Harman formulated the original hypothesis of the oxidative damage theory of aging (1). A logical extension of this theory is that DNA repair processes should contribute to increased life expectancies. The existence of accelerated aging syndromes associated with DNA repair defects supports this model (2). Systematic gene expression profiling of segmental progeroid Nucleotide Excision Repair (NER)-defective mice has demonstrated that suppression of insulin-like signaling (ILS) pathways and activation of oxidative stress response pathways are associated with segmental progeroid phenotypes (1,3–6). Suppression of ILS per se is associated with lifespan extension (7,8). The transcriptomic modulation in segmental progeroid NER-mutants is therefore believed to reflect a ‘survival response’ to accumulation of transcription-blocking oxidative DNA damage (9,10), but important questions remain to be answered. Firstly, given that Base Excision Repair (BER) is more important than NER in repairing oxidative DNA damage, it is puzzling that similar accelerated aging syndromes are not seen in BER-deficient mouse models (11). Secondly, we do not know which types of lesions are responsible for age-related functional loss, although the fact that NER-but not BER mutants-show the more severe phenotypes would point to a role for 8,5′-cyclopurines as these are the only oxidized bases known to be a substrates for NER but not BER (12). Thirdly, there is little direct evidence to suggest whether passive accumulation of DNA damage is sufficient to cause these phenotypes or whether it is an active process that can be modulated genetically.
Caenorhabditis elegans (C. elegans) is frequently used to study genetic factors influencing longevity (13). C. elegans is also well suited to reveal phenotypes that may be masked in mammals due to extensive redundancy of BER enzymes since NTH-1 is the only known DNA glycosylase dedicated to removing oxidized bases in this animal (14). Moreover, the NER pathway is highly conserved (15), with global genome repair (GG-NER) primarily protecting germ cells and early embryos whereas transcription-coupled repair (TC-NER) becomes more important in later developmental stages (16). In mammalian cells GG-NER depends on UV-DDB and XPC/hHR23 for DNA-damage detection whereas TC-NER is initiated by stalling of RNA polymerase II on a lesion and depends on CSB (17). Both branches depend on XPA for damage verification and formation of the preincision complex (18). C. elegans xpa-1 mutants are UV-sensitive and have reduced capacity to repair UV-induced DNA damage (see (15) for a recent review). Contradictory reports exist as to whether NER-deficient xpa-1 mutant animals have shortened lifespan (discussed in (15)). We previously showed a small, but significant reduction of median lifespan in xpa-1 mutants that was accompanied with up-regulation of oxidative stress response genes (19). Moreover, we showed that deletion of the BER enzyme NTH-1 reversed the transcriptome changes and restored normal lifespan of the xpa-1 mutants (19) supporting a model where the NTH-1 enzyme itself generates a response that results in lifespan shortening in xpa-1 mutants. Here, we provide evidence for an active reprogramming response in xpa-1 mutants.
MATERIALS AND METHODS
For more detailed experimental procedures please refer to Supplementary Materials and Methods.
Caenorhabditis elegans and bacterial strains
C. elegans strains were cultured at 20°C on solid Nematode Growth Medium (NGM) agar plates using standard procedures (20). Wild type (WT) Bristol N2, RB877 nth-1(ok724) III, RB864 xpa-1(ok698) I and CL2166 [dvIs19[pAF15(gst-4::gfp::NLS)] III] (21) C. elegans, as well as Escherichia coli HT115(DE3) and OP50 were obtained from the Caenorhabditis Genetics Centre (CGC) (University of Minnesota, St. Paul, MN, USA), funded by the National Institutes of Health (NIH) National Center for Research Resources. RB864 xpa-1(ok698) I and RB877 nth-1(ok724) III were backcrossed five times into the N2 (WT) reference strain and are referred to below as the xpa-1 and nth-1, respectively. RNAi feeding constructs were obtained from Ahringer RNAi feeding library, except pL4440-xpc-1, which was provided by Wim Vermeulen (Erasmus Medical Center, Netherlands). pL4440-slr-2 was generated for this study. All RNAi constructs were confirmed by sequencing.
RNA isolation and microarray processing
Mixed-stage C. elegans populations were cultured on HT115(DE3) pL4440. Five biological replicates were analysed. RNA was isolated and processed essentially as previously described (19) and analysed at the NTNU Genomics Resource Center (Trondheim, Norway). Pre-processing of the raw-data (Affymetrix.cel-files) was done according to the standard analysis pipeline at the Bioinformatics and Expression Analysis (BEA) Core Facility at Karolinska Institutet, Huddinge, Sweden (www.bea.ki.se). Briefly, cel-files were imported into the Affymetrix software Expression Console, pre-processed and normalized using Global Median, following background correction (PM-GCBG) and the summarization method of Plier. No outlier effects were revealed by Quality Control (QC) plots. Raw-data, as well as unprocessed and processed data are found at NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), GSE39252.
Extended data- and statistical analysis
After pre-processing and normalization, the Student’s t-test was executed to reveal transcripts statistically significantly regulated at the 95% confidence level (P < 0.05) between xpa-1 and WT. A comparative 4-fold change filtering on the data was conducted. The Gene Set Enrichment Analysis (GSEA) was performed using Cytoscape (http://www.cytoscape.org/) and the BiNGO plug-in (22). The analysis was executed using the Hyper-geometric Test with Benjamini–Hochberg False Discovery Rate (FDR) correction. For Chi-square tests we used the web-based statistical tool http://www.graphpad.com/quickcalcs/index.cfm. Pearson correlation analysis was used to evaluate any over-representation between the xpa-1 sample group and any of the eight developmental stages in WT (oocytes to adults) C. elegans (data found in (23)).
Quantitative proteomic analysis by IPTL
Mixed-stage WT and xpa-1 nematode populations were cultured on HT115(DE3) pL4440. Four biological replicates were analysed. Crude extracts were prepared by bead beating from packed nematode pellets as previously described (24). Samples containing 100 µg protein were prepared for quantitative proteomic analyses by isobaric peptide termini labeling (IPTL) as previously described (25). Rapid-IPTL was performed as described (26) and labeled peptides were analysed on a Ultimate 3000 nanoLC system (Dionex, Sunnyvale CA, USA) connected to an LTQ Orbitrap XL mass spectrometer (ThermoScientific, Bremen, Germany) as previously described (25).
Quantitative western blot analysis
Standard western blot analysis was performed from whole-nematode crude protein extracts prepared by bead beating from mixed-stage cultures of WT and xpa-1 using the following primary antibodies: rabbit anti-alpha actin (Abcam) or rabbit anti-Sti-1/Hop (27) or rabbit anti-DHS-21 (28), provided by Klaus Richter (Technische Universität München, Garching, Germany) and Joohong Ahnn (Hanyang University, Republic of Korea), respectively. Evaluation of Pgst-4::gfp transgene expression was performed on whole-nematode protein extracts prepared from ∼200 adult CL2166 nematode, the Pgst-4::gfp transgene expression was measured relative to actin by western blot analysis using anti-GFP (Clontech) and anti-actin (Sigma) as primary antibodies followed by quantification using ImageJ.
Quantification of ATP and ROS content
Adenosine triphosphate (ATP) and reactive oxygen species (ROS) measurements were performed using standard assays. ATP content was determined on fresh 25 µg nematode extracts using the ATP Bioluminescence Assay Kit CLS II (Roche) according to the manufacturer’s protocol. All measurements were conducted in flat bottom 96-well dark plates with a Wallac 1420 Victor microplate reader. ROS were quantified using 50 µg nematode extracts incubated in 500 µl of a 5 µM H2-DCF-DA (2′-7′-dichlorofluorescein-diacetate) solution in S-Basal plus protease inhibitor cocktail. Each sample was read in triplicate (150 µl each) in flat bottom 96-well plates using a Wallac 1420 Victor microplate reader at excitation and emission wavelengths of 485 and 528 nm, respectively. Readings were performed over 2 h to ensure that the reaction was occurring as expected. Data collected at the 1 h time point are shown. ROS and ATP measurements were conducted on three independent biological replicates and are presented as means ± SEM.
Western blotting of the polyubiquitinated proteins
Age-synchronized young adult nematodes (4 days old) were collected in M9 prior to freezing at −80°C. Nematodes were lysed using a Dounce homogenizer and the amount of polyubiquitinated proteins was determined by western blotting using the FK1 antibody (Enzo Life Sciences) and anti-alpha-tubulin antibody (Sigma).
Body length measurement
WT and xpa-1 nematodes were cultured on solid NGM agar plates containing 2 mM IPTG and 50 µg/ml of carbenicillin seeded with HT115(DE3) carrying the RNAi feeding constructs indicated. Synchronized L1 larvae were transferred to NGM plates seeded with the appropriate RNAi food and cultured for ∼50 h until the late L4/young adult stage. The nematodes were collected, washed, stained with Bengal Rose (Sigma-Aldrich), killed by heat and stored at 4°C until body length measurement. Triplicate measurements were obtained using a Zeiss SteREO Lumar V12 microscope (45× magnification, Zeiss AxioVision 4.8 software) on at least 30 nematodes mounted on microscope slides. Measurements were combined into a single dataset to perform statistical analysis. Outliers were defined as values below the 1st quartile – (1.5 × interquartile range) or above the 3 rd quartile + (1.5 × interquartile range).
DNA lesion measurements
DNA was isolated from mixed-stage nematode populations grown on NGM agarose plates seeded with HT115(DE3) pL4440 using Qiagen 500/G Genomic tip columns per the manufacturer’s instructions. The isolated DNA was precipitated in isopropanol, washed twice with 70% (volume fraction) ethanol and then stored in absolute ethanol at −80°C until further processing. The DNA extracts were dried under vacuum, solubilized with Milli-Q water and split into equal aliquots for quantitative DNA lesion determinations using stable isotope-dilution liquid chromatography/tandem mass spectrometry (LC/MS/MS) and gas chromatography/tandem mass spectrometry (GC/MS/MS) methodologies. For the LC/MS/MS method, stable isotope-labeled internal standards, (5′R)-8,5′-cyclo-2′-deoxyadenosine-15N5 [R-cdA-15N5] and (5′S)-8,5′-cyclo-2′-deoxyadenosine-15N5 [S-cdA-15N5], were added to 50 µg aliquots of each DNA extract. Both R-cdA-15N5 and S-cdA-15N5 were prepared using dATP-15N5 (Medical Isotopes, Inc., Pelham, NH) as described (29). The samples were then processed and analysed as described previously (30), using a Thermo-Scientific Accela High Speed LC system coupled to a Thermo-Scientific Finnigan TSQ Quantum Ultra AM triple quadrupole MS/MS system operated in multiple-reaction-monitoring (MRM) mode. Final results were reported in terms of the number of lesions/106 DNA nucleosides. Additional details regarding sample preparation, LC/MS/MS methodology and instrumental parameters are in the Supplementary Material. For the GC/MS/MS method, stable isotope-labeled internal standards, 8-hydroxyguanine-15N5 [8-OH-Gua-15N5], 8-hydroxyadenine-15N5 [8-OH-Ade-15N5], 2,6-diamino-4-hydroxy-5-formamidopyrimidine-13C15N2 [FapyGua-13C15N2] and 2,4-diamino-5-formamidopyrimidine-13C15N2 [FapyAde-13C15N2] (Cambridge Isotope Laboratories, Andover, MA), were added to 50 µg aliquots of each DNA extract. The GC/MS/MS determination of the DNA lesions using MRM mode quantification was performed based on modifications to the selected ion monitoring (SIM) mode gas chromatography/mass spectrometry (GC/MS) methodology originally developed at the National Institute of Standards and Technology (NIST) (31–33). Each sample was analysed using an Agilent 7000 Series GC/MS/MS triple quadrupole instrument equipped with an ion source in positive electron-ionization mode, an Agilent 7890A GC and an Agilent 7693 autosampler. Final results were reported in terms of the number of lesions/106 DNA bases. Additional details regarding sample preparation, GC/MS/MS methodology and instrumental parameters are in the Supplementary Material.
Reverse transcription quantitative PCR
Synchronized WT and xpa-1 nematodes were cultured for three generations on solid NGM agarose plates containing 2 mM IPTG and 50 µg/ml of carbenicillin seeded with HT115(DE3) carrying the RNAi constructs indicated. RNAi feeding to deplete skn-1, ddb-1 (and the corresponding control) were performed for one generation since these foods resulted in embryonic lethality. N-acetylcysteine (NAC) treatment was performed as described (34). Briefly, L4 hermaphrodites were exposed to 10 mM NAC (Sigma-Aldrich) on NGM plates seeded with OP50 for 24 h. Nematodes were collected, washed twice in H2O, and disrupted by bead beating before isolating RNA using the RNAeasy Mini Kit (Qiagen). cDNA synthesis and RT-qPCR was performed as described previously (24) using tbg-1, gst-4, sod-3, hsp-1 and aqp-1 primers.
RESULTS
Proteomic reprogramming in xpa-1 mutants
A quantitative proteomic approach was used to query whether the transcriptome modulation we previously observed in xpa-1 mutant animals coincided with changes in protein levels. We used IPTL (26) combined with high-resolution mass spectrometry (35) (Figure 1A). IPTL permits relative quantification using quantification points distributed throughout the entire MS/MS spectrum. Isobaric masses from the pooled double-labeled peptides resulted in single peaks in MS acquisition mode. The relative quantitative abundance of the peptides are given by the ion intensities of peptide fragment ions pairs with mass shifts corresponding to tetradeuterium (4.0 Da) in the MS/MS spectrum. To determine the ratios of the relative peptide quantity, the software tool IsobariQ was used after automatic decoy database searches performed with Mascot.
Figure 1.
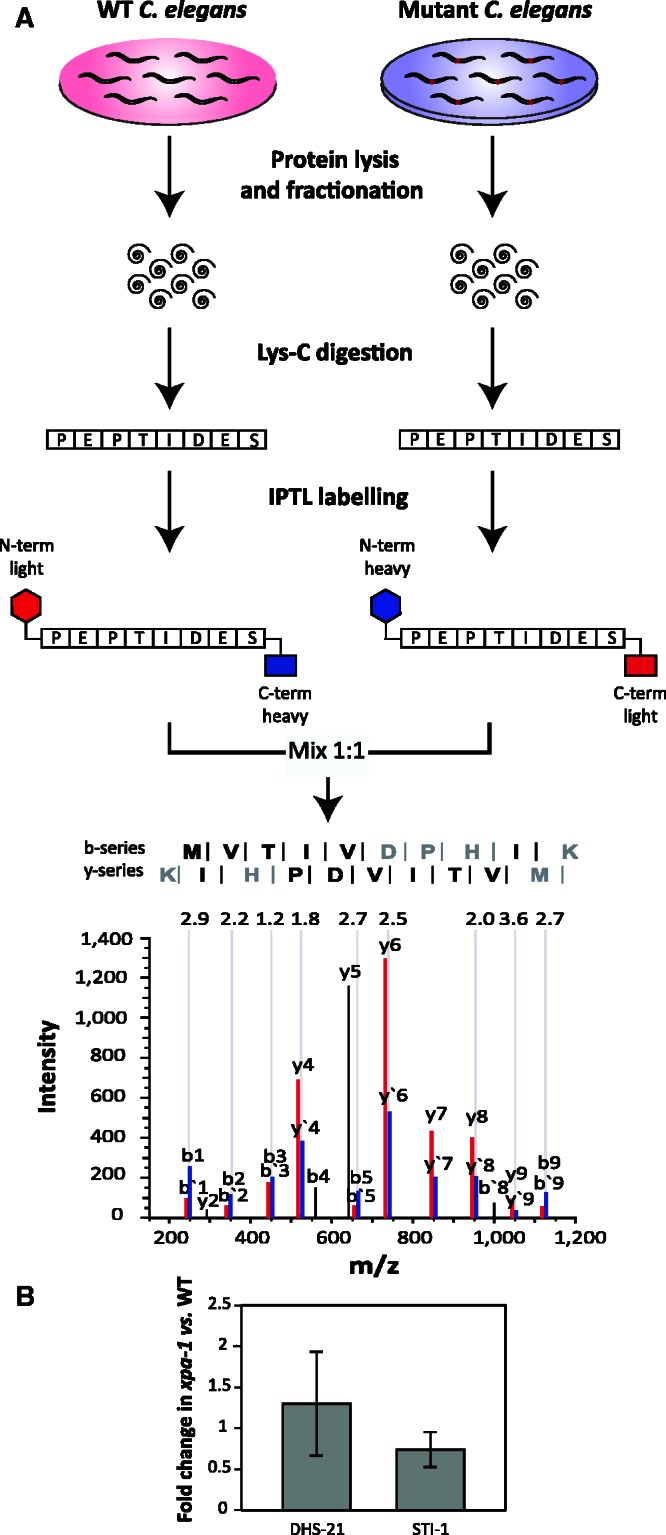
Comparative quantitative proteomics by IPTL. (A) Experimental design. C. elegans N2 and xpa-1 Lys-C resulting peptides were crosswise labeled using the IPTL technique, and combined 1:1 before submission to LC-MS/MS. As an example, N-terminal light/C-terminal heavy (red) and N-terminal heavy/C-terminal light (blue) pairs (10 ratio counts input) from the mass spectrum of a selected peptide identified as MVTIVDPHIK (inset with the detected y and b series ions represented), present in the protein F40F9.6, 2.3-fold up-regulated. (B) Quantitative analysis of DHS-21 and STI-1 protein levels by western blotting. Protein levels were normalized to alpha actin and presented as average fold changes ± SD from four independent replicates.
The comprehensive analysis of the identified peptides and proteins from the whole dataset, including peptide identities and normalized ratios is available in Supplementary Tables S2 and S3. In total 2271 proteins were reliably quantified of which 166 were quantified in at least 2 replicates showing ≤40% variation (coefficient of variation (CV) ≤40). In total, we identified 25 proteins to be regulated with a 2.0-fold change threshold for protein differentiation (Table 1). For the identified proteins for which antibodies were available, differential expression was validated by western blot (Figure 1B). Confirming the proteomic analysis, DHS-21, a dicarbonyl/L-xylulose reductase (DCXR) and STI-1, a heat-shock co-chaperone were up- and down-regulated, respectively. Gene Set Enrichment Analyses (GSEA) of the regulated proteins based on the gene ontology (GO) annotation, Biological Process (BP), revealed enrichment of BP clusters involved in metabolism and biosynthesis, and development and growth (Figure 2A).
Table 1.
Quantitative proteomic identification of differentially expressed proteins in xpa-1 mutants
| Protein group | Common name | Protein function | Meana norm. ratio | CV | P-value |
|---|---|---|---|---|---|
| JC8.3 | RPL-12 | Large ribosomal subunit L12 | 0.2 | 19.0 | 1.96E-12 |
| C13B4.2 | USP-14 | Homolog of ubiquitin-specific protease 14 | 0.3 | 20.1 | 2.05E-06 |
| Y82E9BR.14 | Unknown | 0.4 | 4.7 | 3.48E-04 | |
| R11A8.6 | IRS-1 | Cytoplasmic isoleucyl-tRNA synthetase | 0.4 | 38.6 | 4.51E-04 |
| F59D8.2 | VIT-4 | Yolk protein gene | 0.5 | 18.4 | 1.02E-03 |
| R09E12.3 | STI-1 | Heat-shock co-chaperone | 0.5 | 37.5 | 1.32E-03 |
| F48E8.5 | PAA-1 | Structural subunit of protein phosphatase 2A | 0.5 | 28.8 | 1.81E-03 |
| Y110A7A.6 | Phosphofructokinase-2/fructose-2,6-biphosphatase | 0.5 | 39.6 | 3.29E-03 | |
| K12H4.7 | Unknown | 0.5 | 12.0 | 3.31E-03 | |
| F57B9.10 | RPN-6.1 | Non-ATPase subunit of the 19S proteasome regulatory complex | 0.5 | 24.5 | 3.44E-03 |
| Y37E3.10 | Unknown | 0.5 | 32.1 | 4.71E-03 | |
| R07H5.8 | Unknown | 0.5 | 38.3 | 6.45E-03 | |
| ZK652.9 | COQ-5 | Putative 2-hexaprenyl-6-methoxy-1,4-benzoquinone methyltransferase | 2.0 | 1.7 | 4.62E-02 |
| W07E11.1 | Unknown | 2.0 | 5.2 | 4.47E-02 | |
| C47B2.6 | GALE-1 | Putative UDP-galactose-4-epimerase | 2.0 | 12.6 | 4.28E-02 |
| T24H7.2 | Ortholog of hypoxia-up-regulated vertebrate proteins | 2.3 | 12.9 | 2.23E-02 | |
| F40F9.6 | AAGR-3 | Acid Alpha Glucosidase Related | 2.3 | 9.7 | 2.03E-02 |
| Y47G6A.21 | Unknown | 2.3 | 26.1 | 1.95E-02 | |
| K10B3.10 | SPC-1 | Alpha spectrin ortholog | 2.4 | 4.9 | 1.73E-02 |
| ZC395.10 | Unknown | 2.4 | 19.0 | 1.48E-02 | |
| R11D1.11 | DHS-21 | Short-chain dehydrogenase/reductase | 2.9 | 15.1 | 4.15E-03 |
| H06O01.3 | CTG-1 | CRAL/TRIO and GOLD domain containing | 3.1 | 37.0 | 2.73E-03 |
| K07D4.3 | RPN-11 | Non-ATPase subunit of the 19S proteasome regulatory complex | 3.6 | 14.6 | 6.88E-04 |
| F10B5.1 | RPL-10 | Large ribosomal subunit L10 protein | 4.9 | 35.7 | 2.96E-05 |
| T21E3.3 | LPR-2 | Related to gp330/megalin | 7.2 | 32.9 | 2.92E-07 |
aThe ratios of the 2-fold-regulated proteins obtained by quantitative proteome analysis of xpa-1 mutants versus control are displayed at Mean Norm Ratio.
Figure 2.

GSEA reveals over-represented BPs regulated in xpa-1 mutants. Differential gene products from the proteomic (A) and transcriptomic (B) analysis were imported into Cytoscape and analysed using the BiNGO plug-in for BPs enrichment analysis. Up- and down-regulated genes are highlighted in blue and red, respectively. The nodes are colored yellow to orange, representing increasing statistical significance after Benjamini–Hochsberg correction (from P < 0.05 to P < 7 * 10−7, respectively).
Proteome changes in xpa-1 mutants correspond to transcriptome modulation
While no previous comparative quantitative proteomic study has been done to characterize xpa-1 mutants, two conflicting previous transcriptome descriptions exist (36,37). In order to assess whether the proteomic profile could be correlated to transcriptome modulation, we performed an independent gene expression profiling based on five biological replicates to be able to use more stringent statistical analysis. Predictably, this resulted in identification of a smaller number of regulated genes with 247 unique transcripts being significantly differentially expressed in xpa-1 mutants compared to the isogenic WT control (P < 0.05) (Supplementary Table S1).
As observed in the transcriptomic analysis performed by Boyd et al. (36), few genes were regulated more than 4-fold. Interestingly, GSEA revealed that the BP ‘determination of adult lifespan’ was over-represented among the regulated (P < 0.05) genes in xpa-1 mutants (Figure 2B) which, as previously observed (19), reflected regulation of stress response genes rather than a suppression of ILS. A Chi-square test with Yates’ correction revealed that electron transport chain genes (ETC) were over-represented in the present dataset (P = 0.02). Statistically significant over-representation was also found for probe sets regulated by the transcription factor SLR-2 (P = 0.028; (38)). Borderline statistical significance was found for genes regulated by the main C. elegans redox sensor SKN-1 (P = 0.096; (39–41)). Hence, the transcriptomic analyses reflect that xpa-1 mutant experiences oxidative stress.
As widely observed (42,43) we detected no direct overlap among the transcriptomic and proteomic datasets. However, many of the regulated proteins are annotated as, e.g. oxidative stress responsive (www.wormbase.org) and we hypothesized that the proteomic changes were secondary to the oxidative stress signature of the transcriptome. In support of this, a striking similarity between the datasets was revealed by GSEA upon expanding the primary lists with interactors obtained using the functional interaction browser FunCoup (44). Highly enriched clusters in both datasets included ‘nematode larval development’, ‘embryonic development ending in birth or egg hatching’, and ‘determination of adult lifespan’ (Supplementary Figure S1). To exclude whether these over-represented BPs resulted from the xpa-1 population being on average younger than the WT, we measured the distribution of the different developmental stages using a COPAS Biosorter. The distribution curves were superimposable illustrating that there was no systematic difference in developmental stages between the xpa-1 and WT populations (Supplementary Figure S2B). Estimating the fraction of the different developmental stages, the xpa-1 mutants gave a slightly lower fraction of L1 nematodes (P = 0.04) but higher representation of L4/adults (P = 0.08), but the variation between the replicates were considerable (Supplementary Figure S2C). However, the xpa-1 transcriptome did not reveal an under-representation of transcripts enriched in the L1 developmental stage as we did not find any strong correlation with any of the over-represented sets of transcripts extracted from the L1 through L4 developmental stages as determined by Hill et al. (23) (Pearson correlation coefficient r2 between 0.40 and 0.57 for all stages (data not shown)). By conducting the opposite analysis, comparing the xpa-1 dependent gene list to that of Hill’s et al. total transcriptomic gene list for each developmental stage, no significant correlation was found (data not shown). Hence, we conclude that the xpa-1 and the WT mixed-stage populations had similar distribution of developmental stages. The transcriptomic and proteome changes therefore reflect strain differences.
Antioxidant defenses are induced in xpa-1 mutant
To validate the transcriptomic changes, we used qRT-PCR to measure the expression levels of selected genes regulated in xpa-1 mutants, such as the two oxidative stress responsive genes, mitochondrial Fe/Mn-dependent superoxide dismutase sod-3 and glutathione-S-transferase gst-4. The expression of both genes were increased by about 1.5-fold in xpa-1 mutants relative to a constitutively expressed tubulin control whereas the expression of hsp-1, included as a negative control, was unchanged (Figure 3A). The Pgst-4::gfp transcriptional fusion expression was increased upon RNAi-mediated depletion of xpa-1 gene down-regulation (Figure 3B). The expression of sod-3 and gst-4 are regulated by the transcription factor SKN-1, the C. elegans ortholog of human Nrf2 (45). Thus, to test whether the transcriptional induction of SKN-1 target genes reflected oxidative stress, we measured the steady-state level of ROS using the ROS sensitive probe H2-DCF-DA. The xpa-1 mutant had a 1.3-fold increase of steady-state ROS levels compared to the WT (p < 0.05, Student’s t-test) (Figure 3B). In line with the mitochondrial respiratory chain being the major cellular source of ROS in the well-fed state, the increased ROS formation was accompanied by a 1.8-fold increase in the ATP levels (p < 0.05, Student’s t-test) (Figure 3B). Hence, the transcriptomic and proteomic reprogramming in xpa-1 mutants reflect a bioenergetic shift characterized by increased steady-state ROS and ATP levels.
Figure 3.
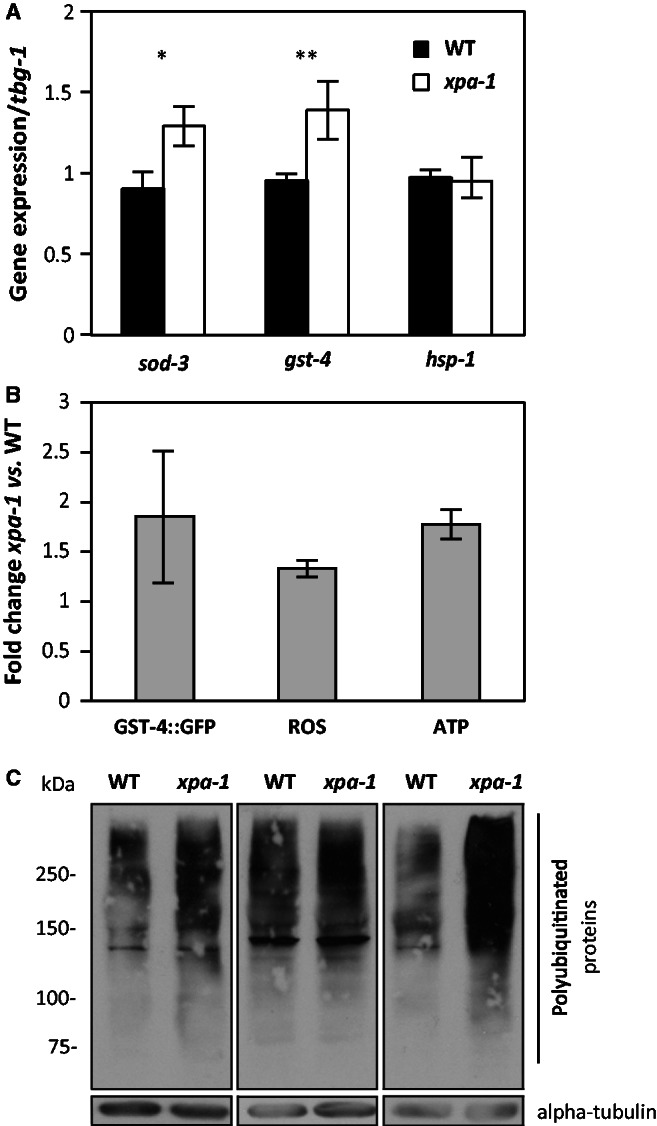
Bioenergetic imbalance and induction of antioxidant defense genes in xpa-1. (A) Transcript levels of sod-3, gst-4 and hsp-1 were measured by qRT-PCR. All transcript levels were normalized to that of gamma-tubulin (tbg-1) and presented as average ± SEM from at least three independent experiments. *P < 0.05, **P < 0.01; Student’s t-test. (B) Pgst-4::gfp transgene expression was measured by western blotting in CL2166 fed empty vector control or xpa-1 RNAi. Protein levels were normalized to alpha actin and presented as fold-changes relative to control as average ± SEM from two independent biological replicates. Steady-state ROS levels were measured using the ROS sensitive probe H2-DCF-DA and are presented as fold-change of relative fluorescence units (RFUs) normalized to WT as average ± SEM from three independent biological replicates. ATP levels were measured using the ATP Bioluminescence Assay Kit CLS II. The ATP levels are presented as fold-change of relative luminescence units (RLUs) normalized to the WT and given as average ± SEM from three independent replicates. (C) Polyubiquitinated proteins accumulate in xpa-1. Three separate nematode lysates were separated by 7.5% SDS-PAGE and blotted against polyubiquitinated proteins or alpha-tubulin as a loading control. Note that the exposure times of the film vary between the three independent experiments.
Polyubiquitinated proteins accumulate in xpa-1 mutants
Oxidative stress is expected to lead to accumulation of damaged and modified proteins. Regulation of proteasomal subunits, as seen in the transcriptomic as well as proteomic datasets (Table 1, Supplementary Figure S1), might therefore be expected. The proteasome is a 26S multi-subunit protease that contains a barrel-shaped 20S catalytic chamber, 20S alpha-rings (encoded by pas-1 - 7), and 20S beta-rings (encoded by pbs-1 - 7), with the 19S regulatory particle capping the barrel. To test the functional significance of this regulation, we analysed proteasomal function. The catalytic activity of the proteasome, as measured by the processing of synthetic substrate, and the levels of the 20S alpha-ring subunits indicating proteasome abundance, were indistinguishable in xpa-1 mutants and WT extracts (Supplementary Figure S3A and B). In contrast, polyubiquitinated proteins accumulated in the xpa-1 mutants (Figure 3C), possibly reflecting increased accumulation of damaged proteins as a consequence of oxidative stress. The accumulation of polyubiquitinated proteins would also be consistent with proteasomal dysfunction as a result of regulation of the 19S regulatory complex non-ATPase subunit RPN-6.1 and RPN-11 (Table 1). Differential regulation of individual proteasomal subunits as observed here is not unprecedented; proteasomal subunits are subject to a ‘bounce-back’ response where impaired proteasome activity induces expression of proteasomal proteins. In C. elegans, e.g. it was observed that depletion of PAS-5 and RPN-2 by RNAi induced up-regulation of pbs-4, rpt-5 and rpn-11 in a SKN-1-dependent manner (46). Moreover, down-regulation of RPN-6.1 in the short-lived xpa-1 mutants (37) is in agreement with recent observations that RPN-6.1 overexpression alone was sufficient to increase proteasome activity in human embryonic stem cells (47), as well as proteasome activity, stress resistance and longevity in C. elegans (48).
A survival response promoting growth and development is activated in xpa-1 mutants
The results presented above confirm that small changes identified by comparative quantitative transcriptomic and proteomic analyses reflect changes in biochemical phenotypes. As it is accepted that the transcriptome modulation reflect survival responses, we hypothesized that depleting the protein found up-regulated in xpa-1 mutants would confer a much stronger phenotypic effect in this mutant than in the WT. GSEA identified ‘growth and development’ as an over-represented BP cluster (Supplementary Figure S1A) where several up-regulated proteins were annotated as positive regulators of growth (e.g. SPC-1, RPL-10, W07E11.1, R07H5.8, VIT-4, IRS-1 and ZC395.1, Table 1). Since we observed that xpa-1 mutants had delayed progression through the L4 stage, we used growth as a read-out to test this hypothesis. Body length measurements confirmed that xpa-1 mutant nematodes were significantly shorter than WT nematodes (P < 0.001) (Figure 4A). As expected, RNAi-mediated depletion of two positive regulators of growth, alpha spectrin (SPC-1) (49) and beta-H spectrin (SMA-1), which was not detected as regulated in xpa-1 mutants, but is positively regulated by SPC-1 (50), resulted in statistically significant growth retardation in both xpa-1 and WT nematodes (P < 0.001). However, depletion of both SPC-1 and SMA-1 affected xpa-1 mutants more strongly with median lengths of xpa-1 mutants being half that of WT nematodes (0.84 mm versus 0.40 mm and 0.40 mm versus 0.20 mm after spc-1 and sma-1 RNAi, respectively). Depletion of SPC-1 and SMA-1 gave non-synchronous populations with broad distribution of larval stages (Supplementary Figure S4) and nematode lengths suggesting a high inter-individual variation in the dependence of growth-promoting survival responses. Depletion of STI-1, which was included as a negative control since it was down-regulated in xpa-1 mutants, had no greater effect on xpa-1 mutants than on WT nematodes (medians lengths of WT versus xpa-1 were 1.03 mm versus 0.95 mm and 1.06 mm versus 0.96 mm on L4440 and sti-1, respectively). Hence, the proteomic changes observed in the NER-defective xpa-1 mutant improves growth and development. However these responses were not sufficient to protect against lifespan shortening in xpa-1 mutants (Supplementary Figure S5A).
Figure 4.

Reprogramming serves as a survival response in xpa-1. (A) Body length of WT and xpa-1 nematodes fed with empty vector control (con), sti-1, spc-1 and sma-1 RNAi measured when WT nematodes on control food reached the L4/adult stage. Results are presented as medians (line) or means (square) inside the boxes showing the 25–75 percentile range with whiskers presenting SD. The lengths of 150 (spc-1 and sma-1 RNAi) or 90 (control and sti-1) were measured for each condition. (B) The measured levels of oxidized DNA bases in DNA isolated from WT and xpa-1 mutant nematodes. The DNA lesions were measured using GC/MS/MS and the results represent average lesion levels ± standard deviation from 5 to 7 independent biological replicates. Statistical analysis was performed using one-way ANOVA with Newman–Keuls post-test. *P < 0.05, **P < 0.01. (C) The measured levels of oxidized DNA nucleosides in DNA isolated from WT and xpa-1 mutant nematodes. The DNA lesions were measured using LC/MS/MS and the results represent average lesion levels ± standard deviation from 5 to 7 independent biological replicates. Statistical analysis was performed using one-way ANOVA with Newman–Keuls post-test. *P < 0.05, **P < 0.01.
Cyclopurine levels are reduced in xpa-1 mutants
As elevated ROS levels are expected to lead to increased oxidation of DNA bases, we utilized both GC/MS/MS (Figure 4B) and LC/MS/MS (Figure 4C) to determine the type and level of oxidatively induced DNA lesions in the WT and xpa-1 mutants. The levels of the ring-opened formamidopyrimidines (FapyGua and FapyAde) were significantly increased, with 36% and 45% higher levels, respectively, in the xpa-1 mutant compared to the WT (Figure 4B). A 57% increased 8-hydroxyadenine (8-OH Ade) content was measured in the xpa-1 mutant whereas no statistically significant difference was found for 8-hydroxyguanine (8-OH Gua) (Figure 4B). Accumulation of transcription-blocking cyclopurines, R-cdA and S-cdA, was suggested to cause the transcriptomic responses observed in progeroid NER-defective animals (9). Although a series of studies failed to detect increases in their steady-state levels in NER-mutants (51), cyclopurine accumulation was recently observed in progeroid ERCC1-deficient mice (10). In xpa-1 mutants, the steady-state levels of R-cdA and S-cdA was measured using LC/MS/MS and found to be reduced by 54% (P < 0.05) and 50% (P < 0.01), respectively, compared to the WT (Figure 4C). Therefore, increased steady-state ROS levels lead to accumulation of some oxidized DNA bases in xpa-1 mutants. As the xpa-1 mutants had lower accumulated levels of cyclopurines than the WT control, the transcriptional and proteomic changes do not result simply from global accumulation of cyclopurines.
Loss of NTH-1 reverses transcriptome modulation
We previously observed that deletion of the BER enzyme NTH-1 restored normal lifespan in the xpa-1 mutant (37). This phenotypic effect is difficult to reconcile with passive accumulation of DNA damage as a cause of lifespan shortening in the xpa-1 mutant. Instead it supports a model where NTH-1 actively generates a genomic stress signal sufficiently strong to result in lifespan shortening and transcriptome modulation.
We tested this directly using qRT-PCR to measure if RNAi-mediated depletion of NTH-1 cancelled the up-regulation of selected mRNAs in the xpa-1 mutant. Consistent with an active reprogramming model, depletion of NTH-1 completely reversed sod-3 over-expression and even induced further down-regulation of aqp-1 and gst-4 (Figure 5A). To test whether this was a general effect of DNA repair mediated DNA-damage detection, we depleted XPC-1, RAD-23 and DDB-1, which serve as lesion-detection factors in GG-NER. Depletion of XPC-1 suppressed the over-expression of all three genes tested, down-regulation of ddb-1 reversed over-expression of sod-3 and gst-4, with aqp-1 being borderline statistically significant (P = 0.092), and down-regulation of rad-23 induced down-regulation of gst-4. TC-NER had a limited effect since down-regulation of csb-1 only reversed sod-3 over-expression. Next, we tested which transcription factors mediated the response. As alluded to above, the xpa-1 mutant transcriptome signature suggested a role for the oxidative stress sensor SKN-1 (52). Surprisingly, SKN-1 down-regulation reversed over-expression of aqp-1, but not gst-4 and sod-3. However, gst-4 is a direct target of SKN-1 and it is possible that the strong suppression of gst-4 expression upon skn-1(RNAi), with ∼10-fold down-regulation of gst-4 mRNA expression in both WT and xpa-1 mutant (Supplementary Figure S6B), masks the more subtle up-regulation in xpa-1 mutant. Depletion of another stress response regulator SLR-2 (53), which controls expression of many genes regulated in the transcriptomic dataset (Chi-square test P < 0.05), suppressed sod-3 and gst-4 over-expression. In contrast, no effect was seen upon depletion of DAF-16 (54,55), which confirms our previous observation that, in the xpa-1 mutants, transcriptome modulation does not involve ILS suppression (37).
Figure 5.

NTH-1 and XPC-1 are upstream factors required for transcriptome modulation in xpa-1. (A) Transcript levels of sod-3, aqp-1 and gst-4 were measured by qRT-PCR in WT or xpa-1 nematodes fed with RNAi constructs targeting nth-1, xpc-1, rad-23, ddb-1, csb-1, skn-1 and daf-16. All transcript levels were normalized to that of gamma-tubulin (tbg-1) and presented as fold-changes relative to the WT. (B) Transcript levels measured in WT or xpa-1 mutants in the absence (-) or presence of the antioxidant NAC. (C) Cartoon showing the working model: in the WT situation (top), transcription-blocking lesions (small circle) do not accumulate and are not processed by BER. In the absence of xpa-1 (middle) the NER pathway is not functioning, leaving NTH-1 to bind unprocessed DNA lesions. This generates genomic stress signals and activates reprogramming, which induces ROS generation. When both xpa-1 and nth-1 are missing (bottom) there is no DNA repair enzyme that may bind the lesion site, the lesion is bypassed and no reprogramming is initiated. The results are presented as average ± SEM from at least three independent replicates. *P < 0.05, **P < 0.01; Student’s t-test.
Lowering the ROS levels by including the antioxidant NAC has been shown to reduce the expression of stress response genes (34). As expected both sod-3 and gst-4 mRNA levels were reduced upon NAC treatment in WT nematodes (Figure 5B). Surprisingly no effect of NAC treatment was seen in the xpa-1 mutant (Figure 5B). This is consistent with the idea that the gene expression changes in the xpa-1 mutant are not simply a result of elevated ROS levels but rather a result of reprogramming response to promote growth and development in the absence of this important DNA repair enzyme.
In conclusion, we present the first comparative quantitative proteomic study of the NER-defective mutant xpa-1 and show that reprogramming of xpa-1 mutant seems to be an active process that promotes growth and fitness of this mutant. We show that reprogramming requires NTH-1 and lesion-detection enzymes acting in GG-NER rather than simply being a consequence of passive accumulation of oxidatively induced DNA damage. In addition, our data suggest that the SKN-1 and SLR-2 transcription factors, but not DAF-16, are among the effectors of reprogramming.
DISCUSSION
Gene expression profiling of segmental progeroid NER-defective mice has demonstrated that suppression of ILS and activation of antioxidant defense pathways are associated with segmental progeroid phenotypes (5). It is believed that these complex phenotypes result from stochastic accumulation of oxidatively induced DNA damage (4,9). In this study we have addressed several outstanding questions related to this model: Firstly, by integrating quantitative proteomics and transcriptomic datasets from the NER-defective xpa-1 mutants we show that transcriptional reprogramming is accompanied by detectable changes on the proteome level. We show that the reprogramming reflects increased steady-state ROS and ATP levels and that it is a good predictor of mutant phenotypes. Secondly, we show that the proteomic reprogramming promotes growth and survival. Thirdly, we show that these responses do not result passively from global genome accumulation of cyclopurine DNA lesions as the levels of R-cdA and S-cdA are reduced by 50% in xpa-1 mutants compared to the WT strain. Finally, we offer evidence to suggest that reprogramming is an active process that requires lesion-detecting enzymes of BER (NTH-1) and GG-NER (XPC-1, DDB-1) as upstream factors.
In this study, we reconcile previous conflicting reports on gene expression profiling of C. elegans xpa-1 mutants and strengthen the relevance of transcriptomic profiling as a surrogate phenotype for genomic stress induced by DNA repair deficiency, by integrating the global gene expression signature with the first comparative quantitative proteomic analysis of a C. elegans DNA repair mutant. For quantitative proteome analysis, we used the IPTL technology, which is the only relative quantification method that currently produces several quantification points for each peptide. Both the xpa-1 mutant proteome and transcriptome suggested increased oxidative stress in the reprogramming response. This was reproduced biochemically with an increased steady-state ROS level which is in line with reports on extensive alterations of bioenergetic profiles in CSB- (56,57), XPA- (58), and DNA-glycosylase deficient human cells (59).
One important distinction between the reprogramming of C. elegans xpa-1 mutant and segmental progeroid NER-defective mice is, however, that there is no evidence for DAF-16 involvement, hence no bona fide ILS suppression, in xpa-1 mutant. The DAF-16—independent reprogramming was experimentally confirmed here by demonstrating that daf-16(RNAi) did not suppress up-regulation of sod-3, gst-4 and aqp-1 mRNA expression in xpa-1 mutants. Hence, the reprogramming in xpa-1 mutants only involves oxidative stress responses, whereas the severe segmental progeroid NER-syndromes also engage ILS suppression to improve fitness. One prediction that can be made from this model is that further suppression of ILS or caloric restriction would further extend lifespan in the xpa-1 mutant. We observed that the xpa-1 mean lifespan is reduced by 3 d compared to the WT when grown on OP50, however; the xpa-1 mean lifespan is less than 1 d shorter when grown on HT115(DE3) (Supplementary Figure S5). A precedent for mutant-specific diet-dependent reduction of lifespan has previously been reported for a mutant in the target of rapamycin complex 2, RICT-1, and may result from genetic difference between the parental bacterial strains that affect fat metabolism and TOR signaling (60). Moreover, the rict-1 mutant also consumed less food on HT115(DE3) compared to OP50. Hence, the extension of rict-1, and possibly also xpa-1, lifespan when cultured on HT115(DE3), could also be due to suppressed ILS as a result of mild starvation.
A major question is how the reprogramming is initiated. Garinis et al. (9) proposed that persistent transcription-blocking lesions were the source of transcriptome modulation and, therefore, the genomic damage resulting in age-dependent functional loss. Aging is associated with oxidative stress, but few ROS-induced lesions are known to present a powerful block to transcription and, moreover, cyclopurines are the only oxidatively induced base lesions preferentially repaired by NER. Although we cannot exclude that lesion levels in a subset of, e.g. neuronal cells might be the determinants of such responses, cyclopurines did not accumulate globally in xpa-1 mutants. This is in line with available literature showing decreased cdA levels in cultured human NER-defective cells (61,62). The cause for the decrease of cyclopurine levels upon NER deficiency is unknown, but it is reasonable to suspect that it might result from the oxidative stress responses in milder NER-mutants as presented here. Thus, our data are not in conflict with the current model but suggests another level of active control that also allows for the increasing number of reports describing transcriptome modulation and accelerated aging phenotypes in a wide range of mutants in DNA repair and genome maintenance enzymes. We propose a model for reprogramming in DNA repair mutants (Figure 5C). In the absence of xpa-1 (Figure 5C mid panel) the NER pathway is not functioning, giving other lesion-detecting DNA repair enzymes, such as NTH-1, the opportunity to bind a wide range of altered DNA bases and structures that they fail to effectively process. We propose that this generates a stronger genomic stress signal, or a stronger transcription block, than the original DNA lesion itself. However, when both xpa-1 and nth-1 are missing (Figure 5C, bottom panel) there is no DNA repair enzyme available to stall at the lesion site and promote reprogramming. We propose that upon xpa-1 deficiency, lesion detection by NTH-1 and GG-NER binding activate a signaling cascade, e.g. via the proposed DNA charge transport mechanism, which allows them to activate signaling cascades (63,64) leading to bioenergetic changes and activation of oxidative stress responses which serves to promote growth and development of this DNA repair mutant.
According to this model, one might postulate that NTH-1 should bind but not process NER-repairable lesions. In support of this we found that C. elegans NTH-1 binds cdA almost comparably to the preferred substrate thymine glycol (dissociation constant kD of 1.291 × 10−6 M for cdA and 1.015 × 10−6 M for thymine glycol, measured using BIAcore T100 and evaluated by the Langmuir model). This model is substantiated here as we show that the reprogramming has elements of an active process involving NTH-1 and the DNA-damage-detecting factors of GG-NER (XPC-1 and DDB-1) as upstream requirements. While xpc-1(RNAi) suppressed the up-regulation as efficiently as nth-1(RNAi), no significant suppression of gst-4 mRNA induction was seen after ddb-1 RNAi. This is probably related to the fact that SKN-1 activation upon ddb-1 RNAi induced sufficiently strong gst-4 over-expression (106- and 46-fold induction in WT and xpa-1, respectively, Supplementary Figure S6B) to mask the transcriptional changes induced by xpa-1 deficiency. Surprisingly, TC-NER seemed to have only a limited role in transcriptional changes observed upon xpa-1 deficiency, perhaps indicating that the germline is a central tissue for initiating the reprogramming response as GG-NER is more important than TC-NER in this organ (15). The individual contribution of transcription factors as effectors of the transcriptome modulation is less clear-cut. Nevertheless, roles for the two stress responsive transcription factors suggested from the transcriptomic and proteomic signatures, SKN-1 and SLR-2, were directly confirmed.
In conclusion, lesion-detection enzymes of BER and GG-NER are required for active reprogramming to trigger a survival response in C. elegans xpa-1 mutants.
NIST Disclaimer
Certain commercial equipment, instruments and materials are identified in this article to specify an experimental procedure as completely as possible. In no case does the identification of particular equipment or materials imply a recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials, instruments or equipment are necessarily the best available for the purpose.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–4, Supplementary Figures 1–6, Supplementary Methods and Supplementary References [63–69].
FUNDING
University of Oslo (to H.N. and B.T., group leader); Functional Genomics program of the Research Council of Norway (to H.N.); My First AIRC (Italian Association for Cancer Research) [MFAG11509 to N.V., recipient]; Swedish Research Council (to J.H. and T.R.B.). K.D.A. and H.K. were recipients of travel grants from the Nordic C. elegans Researcher Network of Shared Technology Platforms. Funding for open access charge: University of Oslo.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors also acknowledge the technical assistance of Christian Koehler for peptide labeling, Magnus Ø. Arntzen for data analysis, Joanna Proszek for help with NTH-1 protein purification and Johan Dethlefsen for COPAS technical support. Klaus Richter, Joohong Ahn for antibodies, and Wim Vermeulen and Hannes Lans for constructs. The Affymetrix service was provided by the NTNU Genomics Resource Center (Trondheim, Norway). The authors would like to acknowledge and thank Miral Dizdaroglu (NIST) for sharing his scientific expertise regarding the DNA-damage mass spectrometry experiments. Members of the NordForsk sponsored C. elegans Researcher Networks for helpful discussion.
REFERENCES
- 1.Harman D. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 2.Kamileri I, Karakasilioti I, Garinis GA. Nucleotide excision repair: new tricks with old bricks. Trends Genet. 2012;28:566–573. doi: 10.1016/j.tig.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 4.van der Pluijm I, Garinis GA, Brandt RM, Gorgels TG, Wijnhoven SW, Diderich KE, de Wit J, Mitchell JR, van Oostrom C, Beems R, et al. Impaired genome maintenance suppresses the growth hormone–insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 2007;5:e2. doi: 10.1371/journal.pbio.0050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garinis GA, van der Horst GT, Vijg J, Hoeijmakers JH. DNA damage and ageing: new-age ideas for an age-old problem. Nat. Cell Biol. 2008;10:1241–1247. doi: 10.1038/ncb1108-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 7.Friedman DB, Johnson TE. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics. 1988;118:75–86. doi: 10.1093/genetics/118.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 9.Garinis GA, Uittenboogaard LM, Stachelscheid H, Fousteri M, van Ijcken W, Breit TM, van Steeg H, Mullenders LH, van der Horst GT, Bruning JC, et al. Persistent transcription-blocking DNA lesions trigger somatic growth attenuation associated with longevity. Nat. Cell Biol. 2009;11:604–615. doi: 10.1038/ncb1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Clauson CL, Robbins PD, Niedernhofer LJ, Wang Y. The oxidative DNA lesions 8,5'-cyclopurines accumulate with aging in a tissue-specific manner. Aging cell. 2012;11:714–716. doi: 10.1111/j.1474-9726.2012.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pascucci B, D'Errico M, Parlanti E, Giovannini S, Dogliotti E. Role of nucleotide excision repair proteins in oxidative DNA damage repair: an updating. Biochemistry. Biokhimiia. 2011;76:4–15. doi: 10.1134/s0006297911010032. [DOI] [PubMed] [Google Scholar]
- 12.Kuraoka I, Bender C, Romieu A, Cadet J, Wood RD, Lindahl T. Removal of oxygen free-radical-induced 5',8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl Acad. Sci. USA. 2000;97:3832–3837. doi: 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antebi A. Genetics of aging in Caenorhabditis elegans. PLoS Genet. 2007;3:1565–1571. doi: 10.1371/journal.pgen.0030129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morinaga H, Yonekura S, Nakamura N, Sugiyama H, Yonei S, Zhang-Akiyama QM. Purification and characterization of Caenorhabditis elegans NTH, a homolog of human endonuclease III: essential role of N-terminal region. DNA Repair. 2009;8:844–851. doi: 10.1016/j.dnarep.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 15.Lans H, Vermeulen W. Nucleotide excision repair in Caenorhabditis elegans. Mol. Biol. Int. 2011;2011:542–795. doi: 10.4061/2011/542795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lans H, Marteijn JA, Schumacher B, Hoeijmakers JH, Jansen G, Vermeulen W. Involvement of global genome repair, transcription coupled repair, and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet. 2010;6:e1000941. doi: 10.1371/journal.pgen.1000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naegeli H, Sugasawa K. The xeroderma pigmentosum pathway: decision tree analysis of DNA quality. DNA Repair. 2011;10:673–683. doi: 10.1016/j.dnarep.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 18.Sugasawa K, Akagi J, Nishi R, Iwai S, Hanaoka F. Two-step recognition of DNA damage for mammalian nucleotide excision repair: directional binding of the XPC complex and DNA strand scanning. Mol. Cell. 2009;36:642–653. doi: 10.1016/j.molcel.2009.09.035. [DOI] [PubMed] [Google Scholar]
- 19.Fensgard O, Kassahun H, Bombik I, Rognes T, Lindvall JM, Nilsen H. A Two-tiered compensatory response to loss of DNA repair modulates aging and stress response pathways. Aging. 2010;2:26. doi: 10.18632/aging.100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Link CD, Cypser JR, Johnson CJ, Johnson TE. Direct observation of stress response in Caenorhabditis elegans using a reporter transgene. Cell Stress Chaperon. 1999;4:235–242. doi: 10.1379/1466-1268(1999)004<0235:doosri>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 23.Hill AA, Hunter CP, Tsung BT, Tucker-Kellogg G, Brown EL. Genomic analysis of gene expression in C. elegans. Science. 2000;290:809–812. doi: 10.1126/science.290.5492.809. [DOI] [PubMed] [Google Scholar]
- 24.Arczewska KD, Baumeier C, Kassahun H, Sengupta T, Bjoras M, Kusmierek JT, Nilsen H. Caenorhabditis elegans NDX-4 is a MutT-type enzyme that contributes to genomic stability. DNA Repair. 2011;10:176–187. doi: 10.1016/j.dnarep.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Tomazella GG, Kassahun H, Nilsen H, Thiede B. Quantitative proteome analysis reveals RNA processing factors as modulators of ionizing radiation-induced apoptosis in the C. elegans germline. J. Proteome Res. 2012;11:4277–4288. doi: 10.1021/pr300386z. [DOI] [PubMed] [Google Scholar]
- 26.Koehler CJ, Arntzen MO, Strozynski M, Treumann A, Thiede B. Isobaric peptide termini labeling utilizing site-specific N-terminal succinylation. Anal. Chem. 2011;83:4775–4781. doi: 10.1021/ac200229w. [DOI] [PubMed] [Google Scholar]
- 27.Gaiser AM, Brandt F, Richter K. The non-canonical Hop protein from Caenorhabditis elegans exerts essential functions and forms binary complexes with either Hsc70 or Hsp90. J. Mol. Biol. 2009;391:621–634. doi: 10.1016/j.jmb.2009.06.051. [DOI] [PubMed] [Google Scholar]
- 28.Son le T, Ko KM, Cho JH, Singaravelu G, Chatterjee I, Choi TW, Song HO, Yu JR, Park BJ, Lee SK, et al. DHS-21, a dicarbonyl/L-xylulose reductase (DCXR) ortholog, regulates longevity and reproduction in Caenorhabditis elegans. FEBS Lett. 2011;585:1310–1316. doi: 10.1016/j.febslet.2011.03.062. [DOI] [PubMed] [Google Scholar]
- 29.Birincioglu M, Jaruga P, Chowdhury G, Rodriguez H, Dizdaroglu M, Gates KS. DNA base damage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine) J. Am. Chem. Soc. 2003;125:11607–11615. doi: 10.1021/ja0352146. [DOI] [PubMed] [Google Scholar]
- 30.Jaruga P, Xiao Y, Nelson BC, Dizdaroglu M. Measurement of (5'R)- and (5'S)-8,5'-cyclo-2'-deoxyadenosines in DNA in vivo by liquid chromatography/isotope-dilution tandem mass spectrometry. Biochem. Biophys. Res. Commun. 2009;386:656–660. doi: 10.1016/j.bbrc.2009.06.107. [DOI] [PubMed] [Google Scholar]
- 31.Dizdaroglu M. The use of capillary gas chromatography-mass spectrometry for identification of radiation-induced DNA base damage and DNA base-amino acid cross-links. J. Chromatogr. 1984;295:103–121. doi: 10.1016/s0021-9673(01)87602-0. [DOI] [PubMed] [Google Scholar]
- 32.Dizdaroglu M. Application of capillary gas chromatography-mass spectrometry to chemical characterization of radiation-induced base damage of DNA: implications for assessing DNA repair processes. Anal. Biochem. 1985;144:593–603. doi: 10.1016/0003-2697(85)90158-7. [DOI] [PubMed] [Google Scholar]
- 33.Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free radical-induced damage to DNA: mechanisms and measurement. Free Radical Bio. Med. 2002;32:1102–1115. doi: 10.1016/s0891-5849(02)00826-2. 34. [DOI] [PubMed] [Google Scholar]
- 34.Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arntzen MO, Koehler CJ, Barsnes H, Berven FS, Treumann A, Thiede B. IsobariQ: software for isobaric quantitative proteomics using IPTL, iTRAQ, and TMT. J. Proteome Res. 2011;10:913–920. doi: 10.1021/pr1009977. [DOI] [PubMed] [Google Scholar]
- 36.Boyd WA, Crocker TL, Rodriguez AM, Leung MC, Lehmann DW, Freedman JH, Van Houten B, Meyer JN. Nucleotide excision repair genes are expressed at low levels and are not detectably inducible in Caenorhabditis elegans somatic tissues, but their function is required for normal adult life after UVC exposure. Mutat. Res. 2010;683:57–67. doi: 10.1016/j.mrfmmm.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fensgard O, Kassahun H, Bombik I, Rognes T, Lindvall JM, Nilsen H. A two-tiered compensatory response to loss of DNA repair modulates aging and stress response pathways. Aging. 2010;2:133–159. doi: 10.18632/aging.100127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirienko NV, McEnerney JD, Fay DS. Coordinated regulation of intestinal functions in C. elegans by LIN-35/Rb and SLR-2. PLoS Genet. 2008;4:e1000059. doi: 10.1371/journal.pgen.1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, Blackwell TK. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell. 2009;8:524–541. doi: 10.1111/j.1474-9726.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, Robida-Stubbs S, Tullet JM, Rual JF, Vidal M, Blackwell TK. RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet. 2010;6:e1001048. doi: 10.1371/journal.pgen.1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, Janette J, Chen C, Alves P, Preston E, et al. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res. 2011;21:245–254. doi: 10.1101/gr.114587.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009;5:1512–1526. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maier T, Guell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 44.Alexeyenko A, Sonnhammer EL. Global networks of functional coupling in eukaryotes from comprehensive data integration. Genome Res. 2009;19:1107–1116. doi: 10.1101/gr.087528.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kahn NW, Rea SL, Moyle S, Kell A, Johnson TE. Proteasomal dysfunction activates the transcription factor SKN-1 and produces a selective oxidative-stress response in Caenorhabditis elegans. Biochem. J. 2008;409:205–213. doi: 10.1042/BJ20070521. [DOI] [PubMed] [Google Scholar]
- 46.Li X, Matilainen O, Jin C, Glover-Cutter KM, Holmberg CI, Blackwell TK. Specific SKN-1/Nrf stress responses to perturbations in translation elongation and proteasome activity. PLoS Genet. 2011;7:e1002119. doi: 10.1371/journal.pgen.1002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vilchez D, Boyer L, Morantte I, Lutz M, Merkwirth C, Joyce D, Spencer B, Page L, Masliah E, Berggren WT, et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature. 2012;489:304–308. doi: 10.1038/nature11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vilchez D, Morantte I, Liu Z, Douglas PM, Merkwirth C, Rodrigues AP, Manning G, Dillin A. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature. 2012;489:263–268. doi: 10.1038/nature11315. [DOI] [PubMed] [Google Scholar]
- 49.Norman KR, Moerman DG. Alpha spectrin is essential for morphogenesis and body wall muscle formation in Caenorhabditis elegans. J. Cell Biol. 2002;157:665–677. doi: 10.1083/jcb.200111051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Praitis V, Ciccone E, Austin J. SMA-1 spectrin has essential roles in epithelial cell sheet morphogenesis in C. elegans. Dev. Biol. 2005;283:157–170. doi: 10.1016/j.ydbio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 51.Brooks PJ. The case for 8,5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience. 2007;145:1407–1417. doi: 10.1016/j.neuroscience.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.An JH, Blackwell TK. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Gene. Dev. 2003;17:1882–1893. doi: 10.1101/gad.1107803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirienko NV, Fay DS. SLR-2 and JMJC-1 regulate an evolutionarily conserved stress-response network. EMBO J. 2010;29:727–739. doi: 10.1038/emboj.2009.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: an HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 55.Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 56.Scheibye-Knudsen M, Ramamoorthy M, Sykora P, Maynard S, Lin PC, Minor RK, Wilson DMIII, Cooper M, Spencer R, de Cabo R, et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 2012;209:855–869. doi: 10.1084/jem.20111721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pascucci B, Lemma T, Iorio E, Giovannini S, Vaz B, Iavarone I, Calcagnile A, Narciso L, Degan P, Podo F, et al. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell. 2012;11:520–529. doi: 10.1111/j.1474-9726.2012.00815.x. [DOI] [PubMed] [Google Scholar]
- 58.Arbault S, Sojic N, Bruce D, Amatore C, Sarasin A, Vuillaume M. Oxidative stress in cancer prone xeroderma pigmentosum fibroblasts. Real-time and single cell monitoring of superoxide and nitric oxide production with microelectrodes. Carcinogenesis. 2004;25:509–515. doi: 10.1093/carcin/bgh046. [DOI] [PubMed] [Google Scholar]
- 59.Bacsi A, Chodaczek G, Hazra TK, Konkel D, Boldogh I. Increased ROS generation in subsets of OGG1 knockout fibroblast cells. Mech. Ageing Dev. 2007;128:637–649. doi: 10.1016/j.mad.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Gene. Dev. 2009;23:496–511. doi: 10.1101/gad.1775409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.D'Errico M, Parlanti E, Teson M, de Jesus BM, Degan P, Calcagnile A, Jaruga P, Bjoras M, Crescenzi M, Pedrini AM, et al. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006;25:4305–4315. doi: 10.1038/sj.emboj.7601277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.D'Errico M, Parlanti E, Teson M, Degan P, Lemma T, Calcagnile A, Iavarone I, Jaruga P, Ropolo M, Pedrini AM, et al. The role of CSA in the response to oxidative DNA damage in human cells. Oncogene. 2007;26:4336–4343. doi: 10.1038/sj.onc.1210232. [DOI] [PubMed] [Google Scholar]
- 63.Sontz PA, Mui TP, Fuss JO, Tainer JA, Barton JK. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc. Natl Acad. Sci. USA. 2012;109:1856–1861. doi: 10.1073/pnas.1120063109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fuss JO, Tainer JA. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair. 2011;10:697–713. doi: 10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- 66.Candiano G, Bruschi M, Musante L, Santucci L, Ghiggeri GM, Carnemolla B, Orecchia P, Zardi L, Righetti PG. Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis. 2004;25:1327–1333. doi: 10.1002/elps.200305844. [DOI] [PubMed] [Google Scholar]
- 67.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ventura N, Rea SL, Schiavi A, Torgovnick A, Testi R, Johnson TE. p53/CEP-1 increases or decreases lifespan, depending on level of mitochondrial bioenergetic stress. Aging Cell. 2009;8:380–393. doi: 10.1111/j.1474-9726.2009.00482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. R Core Team (2012). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, http://www.R-project.org/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.