

Abstract
Extracellular signal-regulated kinase 5 (ERK5), also known as big mitogen-activated protein kinase (MAPK) 1, is implicated in a wide range of biologic processes, which include proliferation or vascularization. Here, we show that ERK5 is degraded through the ubiquitin-proteasome system, in a process mediated by the tumor suppressor von Hippel-Lindau (VHL) gene, through a prolyl hydroxylation-dependent mechanism. Our conclusions derive from transient transfection assays in Cos7 cells, as well as the study of endogenous ERK5 in different experimental systems such as MCF7, HMEC, or Caki-2 cell lines. In fact, the specific knockdown of ERK5 in pVHL-negative cell lines promotes a decrease in proliferation and migration, supporting the role of this MAPK in cellular transformation. Furthermore, in a short series of fresh samples from human clear cell renal cell carcinoma, high levels of ERK5 correlate with more aggressive and metastatic stages of the disease. Therefore, our results provide new biochemical data suggesting that ERK5 is a novel target of the tumor suppressor VHL, opening a new field of research on the role of ERK5 in renal carcinomas.
Introduction
Extracellular signal-regulated kinase 5 (ERK5), also known as big mitogen-activated protein kinase (MAPK) 1, is a member of the MAPK family that shows greatest similarity to the ERK1/2 family members, sharing 66% sequence identity in the amino-terminal half, as well as in the activation loop motif (Thr-Glu-Tyr), while the carboxy-terminal half of ERK5 is unique [1]. ERK5 is activated in response to cell stress and growth factors [2,3] through its selective phosphorylation by mitogen-activated protein kinase kinase 5 (MEK5) [4]. In contrast to the detailed knowledge about the regulation of its activity, the molecular mechanisms controlling ERK5 protein expression levels remain poorly understood. A recent report suggested a role for c-Abl in the regulation of ERK5 half-life, but the mechanism is still unclear [5].
ERK5 participates in several processes including proliferation, angiogenesis, and vasculature maintenance [6,7]. ERK5 is known to mediate the effects of different oncogenes [8,9], and its signaling has been found altered in several human tumors [10–12]. In particular, the role of ERK5 in angiogenesis and endothelial function has been clearly demonstrated in several experimental systems [13,14]. In this regard, several studies have shown that hypoxia-inducible factor 1, α subunit (HIF-1α), a critical mediator in the cellular response to hypoxia and angiogenesis, is regulated by several MAPKs including ERK5 [15–17]. One of the proposed mechanisms involves ubiquitindependent degradation of HIF-1α mediated by ERK5 [15]. Interestingly, gene profiling studies demonstrated that there is a large overlap between the gene expression patterns regulated by ERK5 and HIF-1α, with 82% of the genes specifically regulated by ERK5 being modulated in response to hypoxia through HIF-1α [18]. Under normoxia, HIF-1α is efficiently hydroxylated at two proline residues by a family of dioxygenases [EGL nine homologs (EGLNs), also known as prolyl hydroxylase domain proteins (PHDs)] that require oxygen as co-substrate. This posttranslational modification labels HIF-1α for proteasomal degradation, as the proline-hydroxylated form is recognized by an E3 ubiquitin ligase complex that contains the von Hippel-Lindau (pVHL) tumor suppressor protein. Thus, under normal oxygen tension, HIF-1α half-life is extremely short and normoxic protein levels are very low [19]. Importantly, VHL is a key tumor suppressor in clear cell renal cell carcinoma (CCRCC), where up to 75% to 80% of the cases present a loss of function of the VHL [20].
Our results demonstrate that ERK5 is a novel target for the pVHL tumor suppressor that is labeled for ubiquitin-proteasome system (UPS)-mediated degradation upon proline hydroxylation. Moreover, there was a strong correlation between ERK5 expression and poor prognosis in human samples from CCRCC, suggesting that ERK5 deregulation could contribute to tumor progression and may represent a novel target for therapeutic intervention using drugs that block ERK5 activity.
Materials and Methods
Cell Lines and Plasmids
Cells were maintained in 5% CO2 and 37°C. All culture reagents were provided by Lonza (Madrid, Spain). Cos7 cells were purchased from ATCC (LGC Promochem, Barcelona, Spain), and cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS and 1% glutamine plus antibiotics. 786-0 (ATCC), 769-P cells (ATCC), and Caki-2 (kindly provided by Dr A. Meseguer, Centre d'Investigació en Bioquímica i Biologia Molecular, Barcelona, Spain) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 1% glutamine plus antibiotics, and 1% nonessential amino acids (Sigma-Aldrich, Madrid, Spain). MCF7 cells have been previously described [21]. HMEC cells were kindly provided by Dr L. Alvarez-Vallina (Hospital Universitario Puerta de Hierro, Madrid, Spain) and cultured in 95% EBM-2 plus bovine brain extract (BBE), epidermal growth factor, hydrocortisone, GA-1000 antibiotics, and 5% FBS. Plasmids encoding for green fluorescent protein (GFP), haemagglutinin (HA)-ERK5 wild type (WT), and MEK5 hyperactive (DD) in pCEFL were kindly provided by Dr S. Gutkind [Oral and Pharyngeal Cancer Branch, National Institutes of Health (NIH), Bethesda, MD]. WT HA-ERK5 and mutants forms AEF and Δ713 in pCDNA3 were generous gifts from Dr M. Buschbeck (Institut de Medicina Predictiva i Personalitzada del Cancer, Badalona, Spain). Flag-tagged pVHL was obtained by conventional polymerase chain reaction (PCR) procedures using as template a plasmid coding HA-pVHL kindly provided by Dr M. Ortiz de Landazuri (Hospital Universitario de La Princesa, Madrid, Spain). Briefly, the following primers were used: forward, 5′-ACAGGATCCATGGACTACAAGGACGACGATGAC-AAGCCCCGGAGGGCGGAGAACTGG-3′, which include a BamHI site plus 3X Flag-tagged epitope between codons 1 and 2, and reverse, 5′-CACAGAATTCTCAATCTCCCATCCGTTGATGTGC-3′ including an EcoRI site. PCR conditions were 95°C for 2 minutes for the first cycle and then 35 cycles of 95°C for 30 seconds, 60°C for 1 minute, and 72°C for 1 minute with a final extension of 72°C for 5 minutes. The PCR products were cloned in pCDNA3.1 (Invitrogen, Barcelona, Spain) vectors using the BamHI/EcoRI sites. DNA was confirmed by automatic sequencing. HA-pVHL WT and C162F mutant form in pRc/CMV vector were kindly provided by Dr W. Kaelin through Addgene (Plasmid Nos 19999 and 22042; Cambridge, MA). Plasmids coding for Flag-tagged PHD-1 and PHD-3 were kindly provided by Dr F. S. Lee (School of Medicine, University of Pennsylvania, Philadelphia, PA).
Chemicals and Antibodies
Antibodies against VHL, ubiquitin, and hydroxylated HIF were purchased from Cell Signaling Technology (Izasa, Barcelona, Spain). Antibodies against ERK5 were produced in our laboratory [21] or from Cell Signaling Technology. HA antibody was purchased from Covance (Princeton, NJ). Antibodies against ERK2 and tubulin were from Santa Cruz Biotechnology (Quimigen, Madrid, Spain). Antibody against Flag, cycloheximide, dimethyloxalylglycine (DMOG), and 4′,6-diamidino-2-phenylindole (DAPI) were obtained from Sigma-Aldrich. MG-132 was purchased from Calbiochem (Bionova, Madrid, Spain).
Transfections
Cells were transiently transfected by using Lipofectamine (Invitrogen) following the manufacturer's instruction. The total amount of DNA was normalized using an empty vector. Transfected cells were used 36 to 48 hours after transfection for the different assays.
Western Blot Analysis, Immunoprecipitation, and Co-Immunoprecipitation Assays
Cells were collected in lysis buffer [100 mM Hepes (pH7.5), 50 mM NaCl, 0.1% Triton X-100, 5 mM EDTA, and 0.125 M EGTA]. Protease and phosphatase inhibitors [0.2 µg/ml leupeptin, 2 µg/ml, aprotinin, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 0.1 mM Na3VO4] were added before lysis. Indicated amounts of protein were loaded onto 6% to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride (PVDF) filters, and blotted against different proteins using specific antibodies. In the case of human samples, tissues were disaggregated by using the POLYTRON Dispersing System PT 2100 (Kinematica AG, Lucerne, Switzerland) in lysis buffer and processed as in the rest of the cases. Protein quantification was performed using the BCA Protein Assay Kit (Pierce, Madrid, Spain) following the manufacturer's instructions. In the immunoprecipitation assays, extracts were precleared and soluble fractions were incubated with the indicated antibody. After 2 hours, extracts were incubated for 45 minutes in the presence of protein G (Gamma bind Sepharose; Pharmacia Biotech, Uppsala, Sweden) and then washed three times in the same lysis buffer. Then, immunocomplexes were resuspended in loading buffer and loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels. For the co-immunoprecipitation assays, 293T cells were transfected with 3 µg of indicated plasmid by using Lipofectamine and, 48 hours later, were lysated in HNTG buffer [22] and processed as in immunoprecipitation assays. Antibody detection was achieved by enhanced chemiluminescence (Amersham, GE Healthcare, Barcelona, Spain). Results show a representative blot of three with nearly identical results. Images were quantified by using ImageJ software (NIH).
Immunocytochemistry
Samples were processed as previously described [23]. In the case of exogenous protein, cells were grown onto glass coverslips and then transfected as described above. Samples were then incubated with the indicated antibody overnight and, after extensive wash, incubated 60 minutes with Alexa Fluor 488- or Alexa Fluor 546- conjugated anti-rabbit or anti-mouse antibodies (Invitrogen Molecular Probes). Then, samples were mounted with Fluorosave (Dako, Barcelona, Spain). Positive immunofluorescence was detected using a Zeiss LSM-710 confocal microscope. Images were acquired and processed using Zen 2009 Light Edition program.
Patient's Samples and Analysis
Fresh samples of 19 cases were obtained from patients diagnosed and surgically treated for CCRCC in the Urology Department of the University Complex of Albacete, under the supervision of the local ethical committee and the pathologist with the purpose of not interfering in the histologic evaluation. All cases were reviewed and diagnosed according to the criteria of the World Health Organization classification. Bivariate analysis was performed with the Pearson chi-squared test to evaluate the correlation between tumor stage and Fuhrman grade with the expression level of ERK5. Stage variable was recorded at low risk of disease progression (stages I and II) and high risk (stages III–IV) by using PASW Statistics 18 v.18.0.0 program.
RNA Isolation, Reverse Transcription, and Real-Time Quantitative PCR
Total RNA was obtained, and reverse transcription (RT) performed as previously described [23]. Changes in the mRNA expression of ERK5 and VHL were examined by real-time quantitative PCR using an ABI PRISM 7500 FAST Sequence Detection System (Applied Biosystems, Madrid, Spain). cDNA was amplified using SYBR1 Green PCR Master Mix (Applied Biosystems) in the presence of specific oligonucleotides. The PCR conditions and quantification were performed as previously described [23]. Primers for all target sequences were designed using the computer Primer Express software program especially provided with the 7000 Sequence Detection System (Applied Biosystems).
Chosen PCR primers were given as follows:
ERK5: sense, 5′-GGCCCCTGAAAGAATAAACCC-3′; antisense, 5′-CGAAGGATGGCCAACTCAATC-3′;
VHL: sense, 5′-GACCTGGAGCGGCTGACA-3′; antisense, 5′-TACCATCAAAAGCTGAGATGAAACA-3′;
GAPDH: sense, 5′-TCGTGGAAGGACTCATGACCA-3′; antisense, 5′-CAGTCTTCTGGGTGGCAGTGA-3′.
Interference Assays
SiRNA for VHL was purchased from Dharmacon (Thermo Fisher Scientific, Inc, Waltham, MA; ON-TARGET plus SMART pool Human VHL, Catalog No. L-003936-00 and ON-TARGETplus CONTROL pool, Catalog No. D-001810-10-05) and used following the manufacturer's recommendations. For siRNA assays, cells were transfected by using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions.
Stable knockdown of endogenous ERK5 in 769-P cells was performed by using lentiviral vectors containing shRNA for ERK5 from Sigma-Aldrich (Catalog No. NM_139034). Lentivirus production and infections were performed as previously described [23]. 769-P cells were selected with puromycin (3 µg/ml) and best performing shRNA was selected.
Cell Proliferation Measurements
Subconfluent monolayer cultures were trypsinized, and cells were plated in 24-well plates at a density of 10,000 cells per well. Cell proliferation was analyzed at 1, 2, 3, 4, and 5 days by an MTT-based assay. Briefly, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at 0.5 mg/ml was added to the medium in each well and plates were returned to the incubator for 1 hour. The medium-MTT was then removed, 500 µl of DMSO was added to each well, and the plate was kept in agitation for 5 minutes in the dark to dissolve the MTT-formazan crystals. The absorbance of the samples was then recorded at 570 nm. Four wells were analyzed for each condition, and wells containing medium plus MTT but no cells were used as blanks.
Migration Assays
To perform wound healing assays, cells were grown to confluence (>90%) in six-well dishes. A small area was then disrupted by scratching the monolayer with a 1000-µl plastic pipette tip. Cells were inspected microscopically every 12 hours. The remaining wound area was calculated using ImageJ software (NIH), and the migration distance of the cells was estimated on the basis of that calculation.
Data Analysis
Results are represented as means ± SD of at least three independent experiments. Statistical analysis was performed using the GraphPad Prism 5.00 software. Significance was determined using a t test. The statistical significance of differences was indicated in the figures by asterisks as follows: *P < .05, **P < .01, and ***P < .001.
Results
ERK5 Is Degraded through the UPS
To study the mechanism controlling ERK5 protein expression level, we transiently transfected Cos7 cells with an HA-tagged version of ERK5 and determined protein levels at different time points after inhibition of protein synthesis with cycloheximide. As shown in Figure 1A, the half-life of exogenous HA-ERK5 was much shorter than that of HA-ERK1/2. To investigate the participation of the proteasome in the degradation of these proteins, we used the well-established inhibitor MG132 [24]. This experiment revealed that ERK5, but not ERK1 or ERK2, accumulated upon proteasomal blockade (Figure 1B). Furthermore, similar result was obtained when endogenous ERK5 was analyzed in response to MG132 (Figure 1C).
Figure 1.
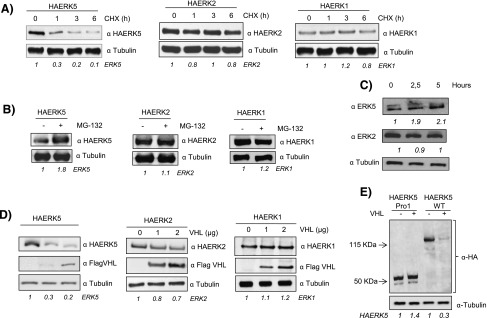
HA-ERK5 is degraded through the proteasome. (A) Cos7 cells were transfected with 0.5 µg of HA-ERK5, HA-ERK2, and HA-ERK1 and, 36 hours later, treated with 100 µM cycloheximide for the indicated times. Then, 30 µg of total cell lysates (TCLs) were blotted against indicated antibodies. (B) Cos7 cells were transfected as in A and treated with 20 µM MG132 for 5 hours. Then, 30 µg of TCLs were blotted against HA and tubulin. (C) Cos7 cells were treated with 20 µM MG132 for indicated times. Then, 60 µg of TCLs were blotted against ERK5, ERK2, and tubulin. (D) Cos7 cells were transfected with 0.5 µg of HA-ERK5, HA-ERK2, and HA-ERK1 plus increasing amounts of Flag-VHL. Thirty-six hours later, 30 µg of TCLs were blotted against the indicated antibodies. (E) Western blot of Cos7 cells transfected with 0.5 µg of HA-ERK5 Pro1 or HA-ERK5 WT in the presence or absence of 2 µg of Flag-VHL. Lysates were blotted against HA and tubulin as loading control. Fold variation of these experiments for each MAPK is shown at the bottom of each panel.
In the proteasome-mediated degradation, proteins are labeled for degradation by covalent binding to the protein ubiquitin in a reaction that requires an E3 complex containing a specific substrate recognition subunit. In the case of HIF-1α, the specificity of the E3 ligase complex is conferred by the protein pVHL [25]. Thus, given the functional similitude between ERK5 and HIF-1α, we studied the role of pVHL as a putative E3 ubiquitin ligase for ERK5. To this end, HA-tagged versions of ERK5, ERK2, and ERK1 were transiently co-transfected with increasing amounts of a plasmid coding for Flag-tagged pVHL. As shown in Figure 1D, overexpression of pVHL results in a marked reduction of HA-ERK5 levels, whereas HA-ERK2 and HA-ERK1 remained largely unaffected. A mutant lacking C-terminal region of ERK5 (HA-ERK5 Pro1), which renders a protein highly similar to ERK1/2 [4,26], was not affected by the overexpression of pVHL (Figure 1E).
To further confirm the role of VHL as a putative E3 ubiquitin ligase for ERK5, HA-ERK5 and Flag-VHL were co-transfected in Cos7 cells and their ubiquitination pattern was evaluated in the presence or absence of MG132. As expected (Figure 2A), overexpression of pVHL resulted in the accumulation of ubiquitinated forms of HA-ERK5. Indeed, the use of a mutant form of pVHL as C162F with impaired binding to Cul2 and elongins B and C [27] did not show a detectable effect onto HA-ERK5 compared to WT (Figure W1), supporting the role of pVHL as an E3 ubiquitin ligase for ERK5. Moreover, we observed physical interaction between HA-ERK5 and Flag-pVHL (Figure 2, B and C) as well as co-localization (Figure 2D). We next asked if the subcellular localization could influence the activity of pVHL on HA-ERK5. To this end, we transfected Cos7 cells with a truncated form of ERK5 (Δ713) that preferentially localizes in the nucleus [28] and found that pVHL promoted ERK5 degradation regardless of its subcellular localization (Figure 2E).
Figure 2.
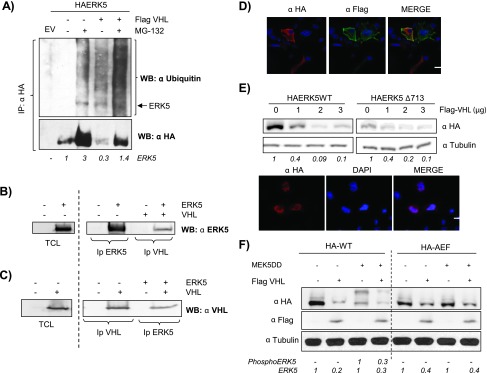
pVHL promotes ERK5 degradation. (A) Cos7 cells were transfected with 0.5 µg of HA-ERK5 and with 3 µg of Flag-VHL. Thirty-six hours after transfection, cells were incubated in the presence or absence of 20 µM MG-132 for 5 hours. Cells were immunoprecipitated against HA and blotted against ubiquitin. Lower panel showed reblotting of the membrane against HA. HA-ERK5 fold variation observed in this experiment is shown at the bottom. (B) 293T cells were transfected with 5 µg of HA-ERK5 and 5 µg of Flag-VHL. Samples were immunoprecipitated and immunoblotted with indicated antibodies. As positive controls, TCLs overexpressing HA-ERK5 were blotted against HA. (C) Same as B. As positive controls, TCLs overexpressing Flag-VHL were blotted against Flag. (D) Cos7 cells were transfected with 0.25 µg of HA-ERK5 and 0.25 µg of Flag-VHL, and subcellular distributions of both proteins were evaluated by immunofluorescence. Image shows a representative field of five. The scale bar represents 10 µm. (E) Upper panel: Cos7 cells were transfected with 0.5 µg of HA-ERK5 WT or HA-ERK5Δ713 with increasing amounts of Flag-VHL. Thirty-six hours later, 30 µg of TCLs were blotted against HA or tubulin. Fold variation of this experiment is shown at the bottom. Lower panel: Cos7 cells were transfected with 0.25 µg of HA-ERK5Δ713 and processed as in D. (F) Cos7 cells were transfected with 0.5 µg of HA-ERK5 or HA-ERK5-AEF, 1.5 µg of MEK5DD, and 3 µg of Flag-VHL at the indicated combinations. TCLs were processed as in E. Fold variation for both proteins in this experiment is shown at the bottom.
Next, we evaluated if activation of ERK5 could be a determinant in the effect of pVHL onto ERK5. Cos7 cells were co-transfected with HA-ERK5 WT or a mutant resistant to activation (HA-AEF-ERK5) in the presence/absence of Flag-pVHL and a constitutively active form of MEK5 (MEK5-DD). As shown in Figure 2F, both the basal and activated forms of HA-ERK5 (achieved by the mobility shift) were affected by the presence of pVHL. Moreover, although to a lower extent, pVHL was able to mediate the degradation of the nonactivable form of ERK5 (Figure 2F), which showed a similar binding to pVHL and subcellular distribution than the WT (Figure W2).
In summary, our results indicate that pVHL binds to ERK5, leading to its ubiquitination and proteasomal degradation regardless of its localization and activation status.
VHL Mediates ERK5 Degradation through Prolyl Hydroxylation-Dependent Mechanism
pVHL binding to HIF-1α is critically dependent on the hydroxylation of specific proline residues within HIF-1α proteins. This posttranslational modification is catalyzed by a family of 2-oxoglutarate-dependent dioxygenases termed EGLNs or PHDs [29,30]. Therefore, we next sought to investigate if a similar mechanism was applicable to ERK5. As a first approach, Cos7 cells were transiently transfected with HA-ERK5 and then incubated in the presence/absence of DMOG, a specific inhibitor of prolyl hydroxylation, and found a marked increase in the expression levels of HA-ERK5 (Figure 3A). In contrast, exogenously expressed HA-ERK2 was not affected by DMOG treatment (Figure 3A), suggesting that the effect was specific for ERK5. Importantly, we also observed stabilization of endogenous ERK5, but not ERK2, in MCF7 and HMEC cells exposed to DMOG(Figure 3B), demonstrating that endogenous ERK5 could also be regulated through a prolyl hydroxylation mechanism. In agreement, overexpression of PHD-1 and PHD-3 promoted a marked reduction in HA-ERK5 levels with almost no effect on HA-ERK2 (Figure 3C). Next, we sought to investigate whether ERK5 was subjected to proline hydroxylation. To this end, we probed HA-ERK5 with antibodies raised against the hydroxyproline-containing epitopes within HIF-1α and reasoned that they might detect other hydroxylated proteins when overexpressed. As shown in Figure 3D, a specific band was observed after immunoprecipitation of HA-ERK5 from MG132-treated samples. Furthermore, the band intensity was decreased in samples exposed to DMOG and increased upon overexpression of PHD-3 (Figure 3D).
Figure 3.
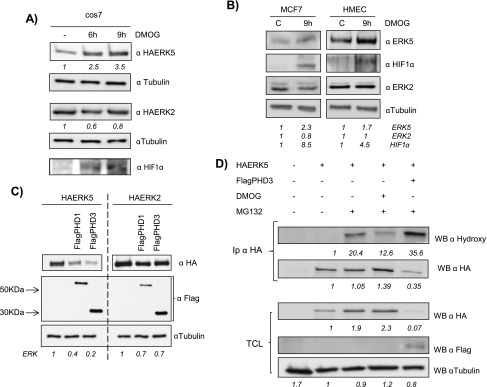
ERK5 levels are regulated through a prolyl hydroxylation mechanism. (A) Cos7 cells were transfected as in Figure 1A. Thirty-six hours later, cells were treated with 1.5 mM DMOG at indicated times. TCLs were blotted against HA, HIF-1α, and tubulin. (B) Subconfluent cultures of MCF7 and HMEC cell lines were treated with 1.5 mM DMOG for 9 hours and endogenous levels of ERK5 (60 µg), ERK2 (30 µg), HIF-1α (60 µg), and tubulin (10 µg) were detected by immunoblot analysis using TCL. (C) Cos7 cells were transfected with 0.5 µg of HA-ERK5 or HA-ERK2 in the presence/absence of 2 µg of FlagPHD-1 or FlagPHD-3 and processed as in Figure 1C. (D) Cos7 cells were transfected with 0.5 µg of HA-ERK5 alone or with 2 µg of FlagPHD-3. Thirty-six hours later, cells, except control, were treated with 20 µM MG132 in the presence/absence of 1.5 mM DMOG for 12 hours. Then, extracts were collected and immunoprecipitated against HA and blotted with the indicated antibody and reblotted against HA. Thirty micrograms of TCL were blotted against HA, Flag, and tubulin. Fold variations for HA-tagged proteins or endogenous proteins in each experiment are indicated at the bottom of the panels.
To further explore the role of pVHL on ERK5 stability, we used a genetic approach based on RNAi. To this end, we chose two CCRCC-derived cell lines, Caki-2 and 769-P, showing normal or defective pVHL activity, respectively [31]. Caki-2 cells showed a lower level of ERK5 protein than 769-P cells (Figure 4A), but no differences were observed in mRNA levels (Figure 4B). To demonstrate the role of the different VHL status, both cell lines were transfected with siRNA against VHL or RNAi control. This treatment resulted in a marked reduction of VHL levels [>90%, as assessed by quantitative RT-PCR (qRT-PCR); Figure 4B] that correlated with an increase in ERK5 protein expression levels in Caki-2, while no effect was observed in the 769-P cell line (Figure 4, B and C). Furthermore, pVHL depletion affected ERK5 expression posttranscriptionally as mRNA levels were not affected by the interference of VHL (Figure 4B). Finally, DMOG treatment resulted in a marked increase in ERK5 protein in the VHL functional cell line—Caki-2—whereas it had no effect on 769-P ERK5 levels (Figure 4D). Altogether, these experiments strongly support the regulation of ERK5 protein levels by its interaction with VHL in a hydroxyproline-dependent manner.
Figure 4.
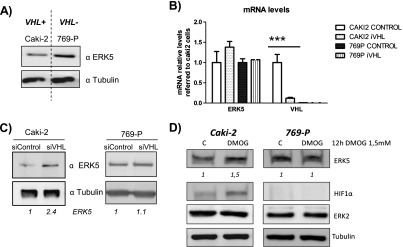
VHL mediates ERK5 expression level in renal carcinoma-derived cell lines. (A) Caki-2 and 769-P cell lines were tested for ERK5 (60 µg) and tubulin (10 µg) expression by Western blot by using lysates from subconfluent cultures. (B) Levels of RNA ERK5 were analyzed by qRT-PCR in Caki-2 and 769-P cells 48 hours after transfection of control or VHL siRNA cells. (C) Caki-2 and 769-P were transfected as in B, and 60 hours later, ERK5 protein levels were analyzed by using 60 µg of cell lysates. Tubulin (10 µg) was used as loading control. (D) Subconfluent cultures of Caki-2 and 769-P cell lines were treated with 1.5 mM DMOG for 9 hours. Then, TCLs were collected and 60 µg were blotted against ERK5 and 10 µg against tubulin. Fold variation of endogenous protein in each experiment is indicated at the bottom of the panels.
ERK5 Is Implicated in Renal Cell Carcinoma
In light of our findings, we decided to investigate the role of ERK5 in CCRCC, a type of tumor in which loss of pVHL function is a hallmark [28]. To this end, we knocked down ERK5 expression in 769-P cells by infection with lentiviral particles encoding for shRNA against ERK5. The treatment resulted in effective knockdown of ERK5 at the mRNA and protein levels in selected pools (Figure 5A). Interestingly, low levels of ERK5 correlated with impaired cell growth under complete and low serum conditions (Figure 5B and data not shown) and in soft agar assays (Figure W3). In addition, ERK5 knockdown resulted in delayed migration in wound healing assays (Figure 5C), supporting a role for this MAPK in the growth and migration of 769-P cells. Similar results were obtained in other experimental models lacking VHL function, such as 786-O, underlying the importance of ERK5 for these processes in CCRCC cells (Figure W4).
Figure 5.
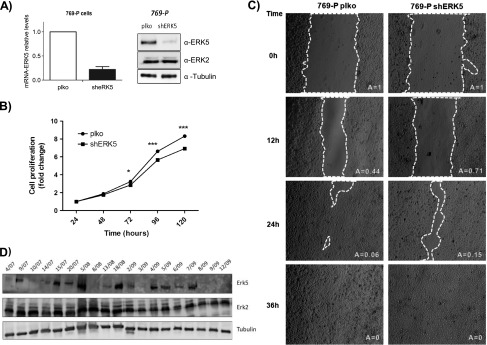
ERK5 is altered in renal cell carcinoma. (A) 769-P cells were infected with retroviral vector control (pLKO) or carrying shRNA against ERK5 (shERK5). Selected pools were evaluated by qRT-PCR (left panel) and Western blot analysis (right panel) by using the indicated antibodies. Image shows a representative experiment of three. (B) The results of proliferation assays are presented as the means ± SD. Values of OD at 570 nm at 24 hours were referred as 1. (C) Wound healing assays were performed in 769-P cells infected with empty vector (PLKO) or coding for shRNA against ERK5 (shERK5), and migration was evaluated at indicated time points. Images show a representative experiment of three independent experiments performed in duplicated cultures. (D) Western blot analysis against ERK5 (120 µg), ERK1/2 (60 µg), and tubulin (10 µg) in different tumors diagnosed as CCRCC.
Given the functional role of EKR5 in CCRCC cells, next we decided to study its role on CCRCC tumor progression. Although we could not test the effect of ERK5 interference in a xenograft model of 769-P cells due to the low tumorigenicity of the cell lines (Figure W5), we explored ERK5 expression levels in primary samples from 19 patients diagnosed with CCRCC (Figure W6). Clinicopathologic data from these patients are shown in Table 1. The mean age was 62.60 years (SD, 11.50; range, 39.08–79.08 years; Table 1). A marked positivity for ERK5 was observed in 9 cases (47.3%), while the remaining 10 cases showed middle to low positivity (15.7%) or not detectable ERK5 protein (36.8%; Table 1 and Figure 5D). ERK2 levels were also analyzed, showing a marked positivity in all the cases with almost no differences among them. A statistically significant (P < .001) correlation between ERK5 positivity and tumor stage at high risk (stages III and IV, in which four of them died or needed chemotherapy during the follow-up; Table 1) was found. Furthermore, tumors with high levels of ERK5 showed a tendency for metastases at the moment of diagnosis (5 of 9), while tumors negative or with low positivity for ERK5 did not show any metastases at the moment of diagnosis (10 of 10). This set of experiments suggests that ERK5 could be a novel biomarker in CCRCC.
Table 1.
Clinicopathologic Data of Patients Studied.
| Case | Age (Years) | Sex | Fuhrman | pT | pN | pM | TNM Stage | ERK5 | Follow-Up (Months) | Others |
| 04/07 | 48.48 | ♂ | 3 | T1b | Nx | Mx | I | - | 42.97 | W/O treatment |
| 09/07 | 50.16 | ♂ | 2 | T1a | Nx | M1 | IV | + | 39.85 | W/O treatment |
| 10/07 | 65.56 | ♂ | 2 | T1b | Nx | Mx | I | - | 39.75 | W/O treatment |
| 14/07 | 76.71 | ♂ | 2 | T2a | N0 | Mx | II | +/- | 36.37 | W/O treatment |
| 15/07 | 63.27 | ♂ | 4 | T3b | N1 | M1 | IV | + | 21.75 | Exitus letalis |
| 20/07 | 60.00 | ♂ | 2 | T3a | Nx | Mx | III | + | 33.38 | W/O treatment |
| 05/08 | 73.83 | ♀ | 4 | T3a | Nx | Mx | III | + | 29.86 | W/O treatment |
| 08/08 | 60.55 | ♂ | 1 | T1a | Nx | Mx | I | - | 27.79 | W/O treatment |
| 13/08 | 74.80 | ♀ | 2 | T2a | N0 | Mx | II | +/- | 21.82 | W/O treatment |
| 18/08 | 64.15 | ♂ | 2 | T4 | Nx | M1 | IV | + | 28.94 | Chemotherapy |
| 02/09 | 79.88 | ♂ | 3 | T2a | N0 | Mx | II | - | 26.68 | Lost follow-up |
| 03/09 | 42.96 | ♀ | 2 | T2a | N0 | Mx | II | - | 17.05 | W/O treatment |
| 04/09 | 57.11 | ♂ | 2 | T3a | Nx | Mx | III | + | 17.28 | W/O treatment |
| 05/09 | 70.81 | ♀ | 3 | T2b | Nx | Mx | III | + | 18.37 | W/O treatment |
| 06/09 | 54.95 | ♂ | 4 | T4 | N1 | M1 | IV | + | 5.02 | Exitus letalis |
| 07/09 | 66.86 | ♀ | 4 | T1b | Nx | M1 | IV | + | 13.77 | Chemotherapy |
| 08/09 | 39.08 | ♂ | 2 | T1a | Nx | Mx | I | - | 13.86 | W/O treatment |
| 09/08 | 68.38 | ♀ | 1 | T1b | Nx | Mx | I | - | 14.00 | W/O treatment |
| 12/09 | 71.79 | ♀ | 1 | T3a | Nx | Mx | II | - | 10.18 | W/O treatment |
The above table summarizes demographic and pathologic data (sex, age, Fuhrman grade, and TNM stage) of the patients studied and their follow-up (end of the study; TNM means tumor-node-metastasis, W/O means without, + means strong positivity, +/- means low positivity, and - means negative).
Discussion
The first conclusion from the present study is that ERK5's expression is tightly regulated, at the protein level, through the UPS. It has been reported that ERK1/2 could be also ubiquitinated in stress conditions, through the PHD domain of mitogen-activated protein kinase kinase kinase 1 (MEKK1) [32], but no effect has been proposed in nonstress conditions. The regulation of ERK5 by the proteasome fits with its proposed role in cellular processes, such as REDOX or hypoxia [2,33], that require a rapid response. Indeed, we have observed an increase in ERK5 protein levels when cell lines as MCF7 or Caki-2 were exposed to hypoxic conditions (Figure W7). Interestingly, ERK5 transcriptional activity has been shown to be affected by the SUMOylation machinery, through its mitogen-activated protein kinase kinase (MAPKK) [34]. However, our data demonstrate a lack of involvement of MEK5 activity in the ubiquitination of ERK5. Therefore, ERK5 biologic levels and functions are probably regulated through complex mechanisms involving SUMOylation [35], ubiquitination (this report), and other processes such as autophosphorylation [36,37].
Our second conclusion is that pVHL is an ERK5 ubiquitin ligase and that ERK5 needs to be proline hydroxylated to be targeted for proteasomal degradation. This represents a novel and important finding in the ERK5 field and suggests that ERK5 is a novel member of the growing list of EGLN/PHD-regulated proteins [38–43]. It is noteworthy that ERK5 does not have an LXXLAP hydroxylation motif described for HIF-1α [30,44]. However, it has been reported that the main sequence determinant for PHD activity is the presence of the proline hydroxy acceptor [45] and it has also been shown that positions -1, -2, and -5 relative to proline hydroxy acceptor can accept a large variety of substitutions [46,47]. In agreement, other well-characterized substrates of PHDs, such as ATF4, do not have an LXXLAP motif [40]. In this regard, the data obtained with the ERK5-Pro1 mutant form lacking the C-terminal region that includes the two specific proline-rich domains of ERK5, residues 434 to 485 and 578 to 701 with more than 60 proline residues, support the idea that proline(s) affected by PHD could lie in these regions.
Interestingly, previous observation showed that 82% of genes that seem to be specifically regulated by ERK5 under normoxic conditions are also targets of HIF-1α in hypoxia [18]. Therefore, the control exerted by pVHL onto ERK5 and HIF-1α at the same time could ensure that the shared target genes receive a coherent set of input signals and will allow the expression of target genes for HIF-1α not only in hypoxic conditions. To this end, one possibility could be a different sensitivity for this pVHL-prolyl hydroxylase-dependent mechanism. In this model, HIF-1α is extremely sensitive to this mechanism, while ERK5 could be less sensitive. In addition, our data strongly support a model in which VHL regulates ERK5 expression by affecting its degradation rather than at the RNA level, as has been reported for other proteins as insulin-like growth factor 1 receptor (IGF1R) [48], in agreement with the mechanism described for HIF-1α. Nonetheless, other possibilities in addition to VHL, such as c-Abl [5], should be considered to fully understand the molecular basis of ERK5 expression levels and function, especially in a tumoral context, where deregulation of tyrosine phosphorylation, protein degradation, and many other processes are well established. Therefore, further studies are necessary to fully clarify the molecular mechanism that controls ERK5 in CCRCC.
Third, the role of VHL as a tumor suppressor gene is in nice agreement with its inhibitory effect onto ERK5, a signaling molecule activated by oncogenes and cell proliferation and that contributes to cancer [49]. Therefore, ERK5 could be considered as a novel target of the tumor suppressor pVHL, as has recently been proposed for phosphorylated JAK2 [50], although the latter does not require proline hydroxylation.
Finally, our findings demonstrate that high levels of ERK5 correlate with stages associated to a worse prognosis in CCRCC [51], suggesting that ERK5 could be considered a novel biomarker and a potential therapeutic target. In fact, our data provide a possible novel explanation for the characteristic vasculature of CCRCC [52]. For example, ERK5 exerts an inhibitory effect on thrombospondin-1 [53,54], known to mediate angiogenesis, proliferation, and tumor aggressiveness in CCRCC [55]. In our experimental system or in CCRCC samples, the lack of ERK5 results in decreased cell motility in vitro and seems to correlate with a low metastatic potential. Interestingly, in breast cancer, expression of ERK5 correlated with a worse prognosis [11]. Therefore, it is possible that ERK5 targeting may be therapeutically useful in CCRCC and probably in several solid tumors. However, in lung cancer, a recent report indicated that loss of ERK5 function may be linked to aggressiveness [56] and that ERK5 is also known to mediate the effect of antiangiogenic factors such as pigment epithelium-derived factor (PEDF) [57]. Therefore, studies with ERK5-specific inhibitors, such as XMD8-92 [58], and with other drugs that interfere with ERK5 activity, such as TG02 that is currently in a phase I clinical trial, will help to elucidate the value of ERK5 targeting in cancer.
In summary, this report presents a novel mechanism for the control of ERK5 protein level through the ubiquitin-proteasome machinery, in which pVHL acts as the E3 ubiquitin ligase through a prolyl hydroxylation-dependent mechanism. This new mechanism for controlling ERK5 expression could have potential implications in tumors, as CCRCC, in which VHL inactivation is a critical step.
Supplementary Material
Acknowledgments
We appreciate the technical help of Elena García. We also appreciate the comments and suggestions of J. Aragonés and I. Sánchez-Pérez.
Abbreviations
- ERK5/ERK5
extracellular signal-regulated kinase 5 (protein/gene)
- pVHL/VHL
von Hippel-Lindau protein/gene
- CCRCC
clear cell renal cell carcinoma
- HIF1α/HIF1α
hypoxia-inducible factor 1α (protein/gene)
- UPS
ubiquitin-proteasome system
- DMOG
dimethyloxalylglycine
- PHDs
prolyl hydroxylase domain proteins
Footnotes
This work was supported by grants from Fundación Leticia Castillejo Castillo and Ministerio de Economía y Competitividad (SAF2009-07329 and SAF2012-30862) and grant JCCM PPII10-0141-040 to R.S.P., grant FIS PI080432 and a grant from Fundación Para la Investigación en Urología to A.S.S.-S., grant FISCAM PI2007-38 to J.M.G.-B., and grant FIS PS09/00868 to A.E.-O. R.S.P. and A.P. Research Institutes and the work carried out in their laboratories receive support from the European Community through the Regional Development Funding Program (FEDER). The authors declare that there are no competing financial interests in relation to the work described.
This article refers to supplementary materials, which are designated by Figures W1 to W7 and are available online at www.neoplasia.com.
References
- 1.Nishimoto S, Nishida E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 2006;7:782–786. doi: 10.1038/sj.embor.7400755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abe J, Kusuhara M, Ulevitch RJ, Berk BC, Lee JD. Bigmitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J Biol Chem. 1996;271:16586–16590. doi: 10.1074/jbc.271.28.16586. [DOI] [PubMed] [Google Scholar]
- 3.Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998;395:713–716. doi: 10.1038/27234. [DOI] [PubMed] [Google Scholar]
- 4.Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- 5.Buschbeck M, Hofbauer S, Di CL, Keri G, Ullrich A. Abl-kinase-sensitive levels of ERK5 and its intrinsic basal activity contribute to leukaemia cell survival. EMBO Rep. 2005;6:63–69. doi: 10.1038/sj.embor.7400316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts OL, Holmes K, Muller J, Cross DA, Cross MJ. ERK5 and the regulation of endothelial cell function. Biochem Soc Trans. 2009;37:1254–1259. doi: 10.1042/BST0371254. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006;18:753–760. doi: 10.1016/j.cellsig.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Chiariello M, Marinissen MJ, Gutkind JS. Multiple mitogen-activated protein kinase signaling pathways connect the cot oncoprotein to the c-jun promoter and to cellular transformation. Mol Cell Biol. 2000;20:1747–1758. doi: 10.1128/mcb.20.5.1747-1758.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.English JM, Pearson G, Hockenberry T, Shivakumar L, White MA, Cobb MH. Contribution of the ERK5/MEK5 pathway to Ras/Raf signaling and growth control. J Biol Chem. 1999;274:31588–31592. doi: 10.1074/jbc.274.44.31588. [DOI] [PubMed] [Google Scholar]
- 10.Mehta PB, Jenkins BL, McCarthy L, Thilak L, Robson CN, Neal DE, Leung HY. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene. 2003;22:1381–1389. doi: 10.1038/sj.onc.1206154. [DOI] [PubMed] [Google Scholar]
- 11.Montero JC, Ocana A, Abad M, Ortiz-Ruiz MJ, Pandiella A, Esparis-Ogando A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS One. 2009;4:e5565. doi: 10.1371/journal.pone.0005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sticht C, Freier K, Knopfle K, Flechtenmacher C, Pungs S, Hofele C, Hahn M, Joos S, Lichter P. Activation of MAP kinase signaling through ERK5 but not ERK1 expression is associated with lymph node metastases in oral squamous cell carcinoma (OSCC) Neoplasia. 2008;10:462–470. doi: 10.1593/neo.08164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi M, Tapping RI, Chao TH, Lo JF, King CC, Yang Y, Lee JD. BMK1 mediates growth factor-induced cell proliferation through direct cellular activation of serum and glucocorticoid-inducible kinase. J Biol Chem. 2001;276:8631–8634. doi: 10.1074/jbc.C000838200. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pi X, Garin G, Xie L, Zheng Q, Wei H, Abe J, Yan C, Berk BC. BMK1/ERK5 is a novel regulator of angiogenesis by destabilizing hypoxia inducible factor 1α. Circ Res. 2005;96:1145–1151. doi: 10.1161/01.RES.0000168802.43528.e1. [DOI] [PubMed] [Google Scholar]
- 16.Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, Gutkind JS. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1α. Cancer Res. 2000;60:4873–4880. [PubMed] [Google Scholar]
- 17.Sutton KM, Hayat S, Chau NM, Cook S, Pouyssegur J, Ahmed A, Perusinghe N, Le FR, Yang J, Ashcroft M. Selective inhibition of MEK1/2 reveals a differential requirement for ERK1/2 signalling in the regulation of HIF-1 in response to hypoxia and IGF-1. Oncogene. 2007;26:3920–3929. doi: 10.1038/sj.onc.1210168. [DOI] [PubMed] [Google Scholar]
- 18.Schweppe RE, Cheung TH, Ahn NG. Global gene expression analysis of ERK5 and ERK1/2 signaling reveals a role for HIF-1 in ERK5-mediated responses. J Biol Chem. 2006;281:20993–21003. doi: 10.1074/jbc.M604208200. [DOI] [PubMed] [Google Scholar]
- 19.Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE. 2007 doi: 10.1126/stke.4072007cm8. cm8. [DOI] [PubMed] [Google Scholar]
- 20.Kaelin WG., Jr The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma. Clin Cancer Res. 2007;13:680s–684s. doi: 10.1158/1078-0432.CCR-06-1865. [DOI] [PubMed] [Google Scholar]
- 21.Borges J, Pandiella A, Esparis-Ogando A. Erk5 nuclear location is independent on dual phosphorylation, and favours resistance to TRAIL-induced apoptosis. Cell Signal. 2007;19:1473–1487. doi: 10.1016/j.cellsig.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 22.Sanchez-Prieto R, Rojas JM, Taya Y, Gutkind JS. A role for the p38 mitogen-activated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000;60:2464–2472. [PubMed] [Google Scholar]
- 23.de la Cruz-Morcillo MA, Valero ML, Callejas-Valera JL, Arias-Gonzalez L, Melgar-Rojas P, Galan-Moya EM, Garcia-Gil E, Garcia-Cano J, Sanchez-Prieto R. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: implication in resistance. Oncogene. 2012;31:1073–1085. doi: 10.1038/onc.2011.321. [DOI] [PubMed] [Google Scholar]
- 24.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell. 1994;78:773–785. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 25.Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1α ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci USA. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JD, Ulevitch RJ, Han J. Primary structure of BMK1: a new mammalian map kinase. Biochem Biophys Res Commun. 1995;213:715–724. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- 27.Lonergan KM, Iliopoulos O, Ohh M, Kamura T, Conaway RC, Conaway JW, Kaelin WG., Jr Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol. 1998;18:732–741. doi: 10.1128/mcb.18.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buschbeck M, Ullrich A. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J Biol Chem. 2005;280:2659–2667. doi: 10.1074/jbc.M412599200. [DOI] [PubMed] [Google Scholar]
- 29.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 30.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 31.Shinojima T, Oya M, Takayanagi A, Mizuno R, Shimizu N, Murai M. Renal cancer cells lacking hypoxia inducible factor (HIF)-1α expression maintain vascular endothelial growth factor expression through HIF-2α. Carcinogenesis. 2007;28:529–536. doi: 10.1093/carcin/bgl143. [DOI] [PubMed] [Google Scholar]
- 32.Lu Z, Xu S, Joazeiro C, Cobb MH, Hunter T. The PHD domain of MEKK1 acts as an E3 ubiquitin ligase and mediates ubiquitination and degradation of ERK1/2. Mol Cell. 2002;9:945–956. doi: 10.1016/s1097-2765(02)00519-1. [DOI] [PubMed] [Google Scholar]
- 33.Sohn SJ, Sarvis BK, Cado D, Winoto A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J Biol Chem. 2002;277:43344–43351. doi: 10.1074/jbc.M207573200. [DOI] [PubMed] [Google Scholar]
- 34.Shishido T, Woo CH, Ding B, McClain C, Molina CA, Yan C, Yang J, Abe J. Effects of MEK5/ERK5 association on small ubiquitin-related modification of ERK5: implications for diabetic ventricular dysfunction after myocardial infarction. Circ Res. 2008;102:1416–1425. doi: 10.1161/CIRCRESAHA.107.168138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo CH, Shishido T, McClain C, Lim JH, Li JD, Yang J, Yan C, Abe J. Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ Res. 2008;102:538–545. doi: 10.1161/CIRCRESAHA.107.156877. [DOI] [PubMed] [Google Scholar]
- 36.Diaz-Rodriguez E, Pandiella A. Multisite phosphorylation of Erk5 in mitosis. J Cell Sci. 2010;123:3146–3156. doi: 10.1242/jcs.070516. [DOI] [PubMed] [Google Scholar]
- 37.Inesta-Vaquera FA, Campbell DG, Tournier C, Gomez N, Lizcano JM, Cuenda A. Alternative ERK5 regulation by phosphorylation during the cell cycle. Cell Signal. 2010;22:1829–1837. doi: 10.1016/j.cellsig.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 38.Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, et al. Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NFκB activity. Proc Natl Acad Sci USA. 2006;103:18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu J, Menzies K, Freeman RS, Taubman MB. EGLN3 prolyl hydroxylase regulates skeletal muscle differentiation and myogenin protein stability. J Biol Chem. 2007;282:12410–12418. doi: 10.1074/jbc.M608748200. [DOI] [PubMed] [Google Scholar]
- 40.Koditz J, Nesper J, Wottawa M, Stiehl DP, Camenisch G, Franke C, Myllyharju J, Wenger RH, Katschinski DM. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110:3610–3617. doi: 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- 41.Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG, Jr, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8:155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 42.Luo W, Hu H, Chang R, Zhong J, Knabel M, O'Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mikhaylova O, Ignacak ML, Barankiewicz TJ, Harbaugh SV, Yi Y, Maxwell PH, Schneider M, Van GK, Carmeliet P, Revelo MP, et al. The von Hippel-Lindau tumor suppressor protein and Egl-9-type proline hydroxylases regulate the large subunit of RNA polymerase II in response to oxidative stress. Mol Cell Biol. 2008;28:2701–2717. doi: 10.1128/MCB.01231-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang J, Zhao Q, Mooney SM, Lee FS. Sequence determinants in hypoxia-inducible factor-1α for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem. 2002;277:39792–39800. doi: 10.1074/jbc.M206955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landazuri MO, Vara-Vega A, Viton M, Cuevas Y, Del PL. Analysis of HIF-prolyl hydroxylases binding to substrates. Biochem Biophys Res Commun. 2006;351:313–320. doi: 10.1016/j.bbrc.2006.09.170. [DOI] [PubMed] [Google Scholar]
- 47.Li D, Hirsila M, Koivunen P, Brenner MC, Xu L, Yang C, Kivirikko KI, Myllyharju J. Many amino acid substitutions in a hypoxia-inducible transcription factor (HIF)-1α-like peptide cause only minor changes in its hydroxylation by the HIF prolyl 4-hydroxylases: substitution of 3,4-dehydroproline or azetidine-2-carboxylic acid for the proline leads to a high rate of uncoupled 2-oxoglutarate decarboxylation. J Biol Chem. 2004;279:55051–55059. doi: 10.1074/jbc.M410287200. [DOI] [PubMed] [Google Scholar]
- 48.Yuen JS, Cockman ME, Sullivan M, Protheroe A, Turner GD, Roberts IS, Pugh CW, Werner H, Macaulay VM. The VHL tumor suppressor inhibits expression of the IGF1R and its loss induces IGF1R upregulation in human clear cell renal carcinoma. Oncogene. 2007;26:6499–6508. doi: 10.1038/sj.onc.1210474. [DOI] [PubMed] [Google Scholar]
- 49.Drew BA, Burow ME, Beckman BS. MEK5/ERK5 pathway: the first fifteen years. Biochim Biophys Acta. 2011;1825:37–48. doi: 10.1016/j.bbcan.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Russell RC, Sufan RI, Zhou B, Heir P, Bunda S, Sybingco SS, Greer SN, Roche O, Heathcote SA, Chow VW, et al. Loss of JAK2 regulation via a heterodimeric VHL-SOCS1 E3 ubiquitin ligase underlies Chuvash polycythemia. Nat Med. 2011;17:845–853. doi: 10.1038/nm.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klatte T, Lam JS, Shuch B, Belldegrun AS, Pantuck AJ. Surveillance for renal cell carcinoma: why and how?When and how often? Urol Oncol. 2008;26:550–554. doi: 10.1016/j.urolonc.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 52.Qian CN, Huang D, Wondergem B, Teh BT. Complexity of tumor vasculature in clear cell renal cell carcinoma. Cancer. 2009;115:2282–2289. doi: 10.1002/cncr.24238. [DOI] [PubMed] [Google Scholar]
- 53.Doebele RC, Schulze-Hoepfner FT, Hong J, Chlenski A, Zeitlin BD, Goel K, Gomes S, Liu Y, Abe MK, Nor JE, et al. A novel interplay between Epac/Rap1 and mitogen-activated protein kinase kinase 5/extracellular signal-regulated kinase 5 (MEK5/ERK5) regulates thrombospondin to control angiogenesis. Blood. 2009;114:4592–4600. doi: 10.1182/blood-2009-04-217042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan BK, Adya R, Chen J, Farhatullah S, Heutling D, Mitchell D, Lehnert H, Randeva HS. Metformin decreases angiogenesis via NF-κB and Erk1/2/Erk5 pathways by increasing the antiangiogenic thrombospondin-1. Cardiovasc Res. 2009;83:566–574. doi: 10.1093/cvr/cvp131. [DOI] [PubMed] [Google Scholar]
- 55.Zubac DP, Bostad L, Kihl B, Seidal T, Wentzel-Larsen T, Haukaas SA. The expression of thrombospondin-1 and p53 in clear cell renal cell carcinoma: its relationship to angiogenesis, cell proliferation and cancer specific survival. J Urol. 2009;182:2144–2149. doi: 10.1016/j.juro.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 56.Chen R, Yang Q, Lee JD. BMK1 kinase suppresses epithelialmesenchymal transition through the Akt/GSK3β signaling pathway. Cancer Res. 2012;72:1579–1587. doi: 10.1158/0008-5472.CAN-11-2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biyashev D, Veliceasa D, Kwiatek A, Sutanto MM, Cohen RN, Volpert OV. Natural angiogenesis inhibitor signals through Erk5 activation of peroxisome proliferator-activated receptor γ (PPARγ) J Biol Chem. 2010;285:13517–13524. doi: 10.1074/jbc.M110.117374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, Yates JR, III, Gray NS, Lee JD. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell. 2010;18:258–267. doi: 10.1016/j.ccr.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Supplemental Reference
- 1.Guerrero C, Martín-Encabo S, Fernández-Medarde A, Santos E. C3G-mediated suppression of oncogene-induced focus formation in fibroblasts involves inhibition of ERK activation, cyclin A expression and alterations of anchorage-independent growth. Oncogene. 2004;23:4885–4893. doi: 10.1038/sj.onc.1207622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.