

Summary
Recurrent exertional rhabdomyolysis is a heritable disorder that results in painful skeletal muscle cramping with exercise in up to 10% of all Thoroughbred racehorses. Here we report a genome-wide association study with 48,282 SNPs analyzed among 48 case and 37 control Thoroughbreds. The most significant SNPs spanned approximately 13 Mb on ECA16, and the p-value of the most significant SNP after correcting for population structure was 8.0 × 10−6. This region on ECA16 was further evaluated by genotyping 247 SNPs in both the initial population and a second population of 34 case and 98 control Thoroughbreds. Several SNPs across the 13 Mb region on ECA16 showed significance when each population was analyzed separately; however, the exact positions of the most significant SNPs within this region on ECA16 varied between populations. This variability in location may be attributed to lack of power due to insufficient sample sizes within each population individually, or to the relative distribution of long conserved haplotypes, which are characteristic of the Thoroughbred breed. Future genome-wide association studies with additional horses would likely improve the power to resolve casual loci located on ECA16 and increase the likelihood of detecting any additional loci on other chromosomes contributing to disease susceptibility.
Keywords: Equine SNP chip, Whole genome association study, GWAS, RER, Tying up, Skeletal muscle
Introduction
Equine exertional rhabdomyolysis (ER), often referred to as “tying-up”, is a clinical syndrome characterized by painful muscle contractures with exercise and skeletal muscle fiber necrosis (MacLeay et al. 1999b). Thoroughbred horses are particularly susceptible to ER, with 5 to 10 % of all Thoroughbreds developing ER during a racing season (MacLeay et al. 1999a; McGowan et al. 2002), and many having more than four episodes during a season. Most horses with repeated episodes of ER are believed to have a specific disease, termed recurrent exertional rhabdomyolysis (RER) (Lentz et al. 1999). A diagnosis of RER is made on the basis of history and recurring signs of muscle stiffness in association with high serum levels of creatine kinase (CK) and aspartate aminotransferase (AST) subsequent to muscle cell necrosis (MacLeay et al. 1999b; Valberg et al. 1999). A diagnosis of RER is also supported by skeletal muscle biopsy, where histological features include an increased number of centrally located nuclei in mature muscle fibers and the absence of abnormal polysaccharide and excessive glycogen, which are characteristic of polysaccharide storage myopathy (PSSM) (McCue et al. 2006). Previous work evaluating the in vitro sensitivity of surgically-excised external intercostal muscle bundles found an increased sensitivity to contractures induced by halothane and caffeine within muscle from RER horses. It was therefore suggested that RER may be attributed to abnormal regulation of muscle contraction (Lentz et al. 1999, 2002).
There is strong evidence that RER susceptibility has an underlying genetic basis; however, the mode of inheritance has not been conclusively defined. A previous analysis of pedigrees of RER affected and clinically normal Thoroughbreds found support for dominant inheritance with variable expression originating from a founder stallion (MacLeay et al. 1999b). Further, a multi-year controlled breeding trial suggested Mendelian segregation of RER when individuals were phenotyped by the in vitro halothane-caffeine contracture test (Dranchak et al. 2005). Another study estimated the heritability of tying-up in Thoroughbreds to be approximately 0.42 (Oki et al. 2005). Variability in heritability estimates may reflect differences in RER diagnostic criteria or the contribution of non-genetic factors such as temperament, sex, age, diet, exercise routine, and lameness (MacLeay et al. 1999a; McGowan et al. 2002; Wilsher et al. 2006). Young, nervous female Thoroughbreds on the racetrack are particularly at risk. Similar risk factors for ER have also been reported in the Standardbred racehorse (Isgren et al. 2010).
The purpose of this study was to perform a genome-wide association study (GWAS) to identify a genomic region(s) associated with RER in a cohort of North American Thoroughbreds and to confirm the resulting region of significance in a second, independent population of North American Thoroughbred RER cases and controls.
Materials and methods
Selection of cases and controls
A total of 217 Thoroughbreds bred and raced in North America, consisting of 27 male cases, 55 female cases, 59 male controls, and 76 female controls, were included in this study. The RER cases included horses acquired from previous studies (Dranchak et al. 2005; MacLeay et al. 1999b), the submission of blood samples from trainers, and biopsy submissions to the Neuromuscular Disease Laboratory at the University of Minnesota. Each RER case was evaluated on the severity of clinical signs, number and frequency of rhabdomyolysis episodes, muscle biopsy findings, elevations in CK and AST activities, and absence of other known muscle diseases (Supplementary Methods).
Thoroughbred control horses were selected from previous studies and by collaborating veterinarians to have completed at least one racing season or one year of intense training with no observed episodes of ER. Records were obtained whenever possible to determine if horses were trained or raced on the racetrack (Supplementary Methods).
Population one
Population one consisted of 33 female cases, 15 male cases, 18 female controls, and 19 male controls used in a whole-genome association study (GWAS). Horses in this population were selected based on severity of clinical signs and pedigree relationships. RER cases in population one had severity scores of 2-3, on a scale of 0-3 (Supplementary Methods). Pedigree relationships were used to limit horses in population one to individuals that traced back to a common Thoroughbred stallion, in an attempt to maximize the length of linkage disequilibrium (LD) and minimize heterogeneity at putative RER loci (Supplementary Table 1).
Population Two
Population two consisted of 22 female cases, 12 male cases, 58 female controls, and 40 male controls, utilized for validation of GWAS results. RER cases in population two were selected with the same phenotypic criteria as population one; however, relationship standards were much less stringent (Supplementary Methods & Supplementary Table 2).
Genotyping platforms
Whole genome SNP data was generated with the Illumina iSelect Equine SNP50 bead chip, which consists of 54,602 SNPs distributed across all equine autosomes and the X chromosome. After data quality control, 48,282 SNPs remained in the GWAS (Supplementary Methods).
GWAS results were validated in a second cohort of RER cases and controls genotyped using the Sequenom platform (van den Boom & Ehrich 2007). A total of 411 ECA16 SNPs were selected from the Broad Institute EquCab2.0 SNP discovery project (http://www.broadinstitute.org/mammals/horse/snp) based on genomic position and suitability in multiplex assays designed with SpectroDESIGNER software. After assay design and data quality control, 247 SNPs remained in the data analysis (Supplementary Methods).
Identification of cryptic relatedness and population structure
Cryptic relatedness and population structure in population one was determined using the proportion of the genome identical-by-descent (IBD) between each pair of individuals and principal components analysis as described in Supplementary Methods. The mean pair-wise IBD value among the 85 Thoroughbreds in population one was 0.026, and members of the control cohort were more closely related to each other than members of the case cohort (p-value = 0.00276). The first principal component accounted for significant differences between the case and control cohorts; therefore, genome-wide association tests were performed using a mixed model with the IBD relationship matrix and the first principal component as a covariate to correct for relatedness and population stratification.
Genome-wide association tests
All association tests were performed with the snpMatrix package in R (Clayton & Leung 2007). The first genome-wide association test was a logistic regression modeling for additive allelic effect using gender and racing history as covariate because RER is most clearly expressed in females and horses that are nervous on the racetrack (Isgren et al. 2010; MacLeay et al. 1999a; McGowan et al. 2002; Wilsher et al. 2006). Logistic regression tests modeling for additive allelic effect were run with sex as a covariate, and with both sex and racing history as covariates. A total of 10,000 case/control label-swapping permutations were performed to correct for multiple testing, and after permutation testing, a max(T) corrected p-value (EMP2) < 0.05 was considered significant at the genome-wide level (Purcell et al. 2007).
The second genome-wide association test accounted for population stratification with a combination of principal component and mixed model analyses. Relationship matrices used in the mixed model were calculated from estimations of IBD between every pair-wise combination of individuals, and the first principal component was used as a covariate in an additive logistic regression test along with sex and racing history. The mixed model calculation was modified from PedigreeMM (Vazquez et al. 2010) in R (R Development Core Team 2011).
Fine-mapping association tests
Association tests for the 247 fine-mapping SNPs were performed in population one by itself (n=85), population two by itself (n=132), and both populations together (n=217). The data sets for population one and both populations together were analyzed in additive logistic regression tests with sex as a covariate and with sex and racing history as covariates. Racing history was not needed as a covariate when population two was analyzed by itself because all of the controls in population two had been raced on the track. Fine-mapping SNPs were assessed for all 217 horses in a mixed model using a relationship matrix computed from pedigree information. Bonferroni correction was applied to all association tests with the 247 fine-mapping SNPs. All results of the association tests were plotted in graphs with R (R Development Core Team 2011).
Evaluation of ECA16 haplotype blocks
A case/control association test with 10,000 permutations was performed in Haploview (Barrett et al. 2005) for the haplotypes of the 247 fine-mapping SNPs, as predicted by the confidence intervals algorithm (Gabriel et al. 2002) in population one, population two, and both populations together. The boundaries of the most significant haplotypes were used to extract data from the fastPHASE results (Scheet & Stephens 2006), and these haplotypes were then evaluated in logistic regression tests with sex as a covariate and with both sex and racing history as covariates for population one, population two, and both populations together using R statistical software (R Development Core Team 2011).
ECA16 gene predictions
The web interface MartView was used to query the BioMart Central Portal (Haider et al. 2009) for all Ensembl equine protein-coding gene predictions within the range of 25 - 45 Mb on ECA16.
Results
The GWAS with population one included 85 North American Thoroughbreds, consisting of 15 male RER cases, 33 female RER cases, 19 male controls, and 18 female controls. After quality control, a total of 48,282 SNPs positioned across all autosomes and the X chromosome were included in the analysis.
Results for a logistic regression modeling for additive allelic effect with sex and racing history as covariates are shown in Figure 1. An association was identified on equine chromosome 16 (ECA16) with little evidence for other genomic regions of interest. The most significant SNP on ECA16 was located at base pair position 41996587, with a p-value of 1.3 × 10−5 (EMP2 = 0.14) in the additive logistic regression test with only sex as a covariate, and a p-value of 3.0 × 10−6 (EMP2 = 0.04) in the same test with both sex and racing history as covariates. While chr16.41996587 was the only significant SNP when sex and racing history were used as covariates, the SNPs located at chr16.29349242 and chr16.29314271 also showed suggestive significance (respective p-values = 3.7 × 10−5, EMP2 = 0.39 and 3.9 × 10−5, EMP2 = 0.40) in an additive logistic regression test with only sex as covariate (Table 1). Therefore, these GWAS association tests identified a significant association at approximately 42.0 Mb on ECA16 and a less significant association at approximately 29.3 Mb on ECA16.
Figure 1. Manhattan plots for GWAS results.
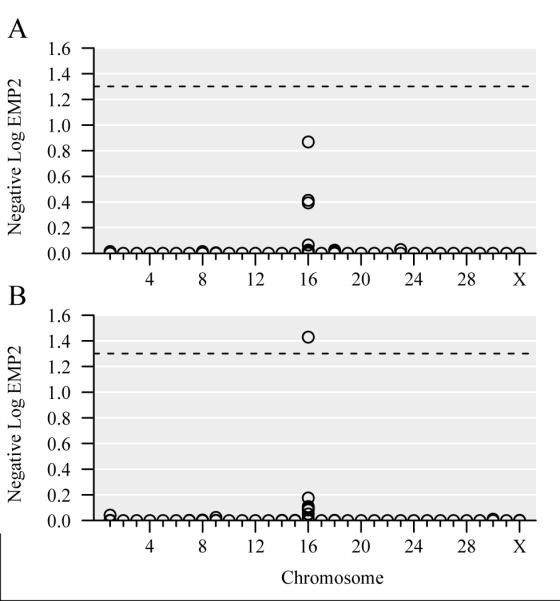
Both panels display GWAS results from additive logistic regression tests with 10,000 permutations. Sex was used as a covariate in panel A, and sex and racing history were used as covariates in panel B. Each SNP is denoted by a circular point on the plot, and SNPs with the same p-value overlap. The horizontal axis indicates chromosomes 1-31 and X, and the vertical axis shows negative log EMP2 values. A dashed line is drawn where p-value=0.05 (negative log EMP2 = 1.3).
Table 1.
The p-values of the most significant SNPs in the GWAS.
| SNP Position1 | Covariates2 | MAF3 | p-value4 | EMP25 | PC1 and mixed model p-value6 |
|---|---|---|---|---|---|
| chr16.41996587 | Sex and Raced | 0.30 | 3.0 × 10−6 (5.52) | 0.04 (1.43) | 8.0 × 10−6 (5.10) |
| chr16.41996587 | Sex | 0.30 | 1.3 × 10−5 (4.89) | 0.14 (0.87) | 4.2 × 10−5 (4.38) |
| chr16.29349242 | Sex | 0.42 | 3.7 × 10−5 (4.43) | 0.39 (0.41) | 5.8 × 10−5 (4.24) |
| chr16.29314271 | Sex | 0.40 | 3.9 × 10−5 (4.41) | 0.40 (0.39) | 1.4 × 10−4 (3.84) |
Base pair position of the SNP on chromosome 16.
The possible covariates of additive logistic regression tests included gender and whether or not the horse was raced on the track.
Minor allele frequency of the SNP.
The resulting p-value (negative log p-value is adjacently listed in parentheses).
The EMP2 value after 10,000 case/control label-swapping permutations (negative log EMP2 is adjacently listed in parentheses).
The p-value after correcting for population stratification with the first principal component as a covariate in a mixed model with the IBD relationship matrix (negative log p-value is adjacently listed in parentheses).
The comparatively high levels of relatedness among the GWAS control cohort compared to the case cohort created a degree of population stratification which could result in spurious associations. Therefore, a second genome-wide association analysis was performed utilizing a combination of principal component and mixed model analyses to confirm the association on ECA16, and rule out potential confounding due to population stratification. After correcting for population stratification, the resulting p-values revealed that the chr16.41996587 SNP was once again the most significant SNP (p-value = 8.0 × 10−6), albeit at a level of significance slightly less than that of the raw p-value previously described (raw p-value = 3.0 × 10−6) (Table 1). These analyses demonstrate that the significance on ECA16 is unlikely to be a spurious association driven by underlying population stratification.
After quality control, a total of 247 SNPs, spanning 18.15 Mb on ECA16 (chr16.25727220 to chr16.43876419), were selected to confirm the GWAS results and attempt to fine-map the region of significance on ECA16. The average distance between SNPs decreased from approximately 47 kb to 29 kb after the addition of the 247 fine-mapping SNPs in this region on ECA16. These SNPs were genotyped across the GWAS population (n = 85 horses), as well as a second population of 132 Thoroughbred horses consisting of 12 male cases, 22 female cases, 40 male controls, and 58 female controls. After Bonferroni correction, a total of 23 fine-mapping SNPs spanning chr16.28806500 to chr16.41997223 showed significance (p<0.05) in either population one, population two, or both populations together (Figures 2 and 3). The SNPs with the strongest associations are shown in Table 2. Five SNPs were significant in population one when only sex was used as a covariate, and nine SNPs were significant when sex and racing history were used as covariates. Two SNPs were significant when population two was analyzed by itself, and 16 SNPs were significant when all horses were analyzed together. The minor allele frequencies (MAF) of the most significant ECA16 SNPs are listed in Supplementary Table 3.
Figure 2. Manhattan plots for ECA16 fine-mapping SNPs with sex as a covariate.

All panels show results for 247 SNPs after additive logistic regression tests with sex a covariate and Bonferroni correction. Circular points represent SNPs, ECA16 Mb positions are on the horizontal axis, negative log p-values are on the vertical axis, and the dashed line represents p-value=0.05 (negative log p-value=1.3). Panel A shows population one (n = 85 horses), panel B shows population two (n = 132 horses), panel C shows both populations (n = 217 horses), and panel D shows both populations in a mixed model using a relationship matrix derived from pedigrees (n = 217 horses).
Figure 3. Manhattan plots for ECA16 fine-mapping SNPs with sex and racing history as covariates.

All panels show results for 247 SNPs after additive logistic regression tests with sex and racing history as covariates and Bonferroni correction. Circular points represent SNPs, ECA16 Mb positions are on the horizontal axis, negative log p-values are on the vertical axis, and the dashed line represents p-value=0.05 (negative log p-value=1.3). All of the controls in population two were raced, so race was not needed as a covariate when population two was analyzed alone. Panel A shows population one (n = 85 horses), panel B shows both populations (n = 217 horses), and panel C shows both populations in a mixed model using a relationship matrix derived from pedigrees (n = 217 horses).
Table 2.
The p-values of the most significant ECA16 fine-mapping SNPs after additive logistic regression and Bonferroni correction.
| Sex as a Covariate2 | Sex and Raced as Covariates3 | ||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| SNP Position1 | Pop 1 | Pop 2 | All | All, Mixed Model |
Pop 1 | All | All, Mixed Model |
| chr16.28806500 | 1.0 (0) | 0.042 (1.38) * | 0.005 (2.28) * | 0.005 (2.28) * | 1.0 (0) | 0.007 (2.13) * | 0.007 (2.16) * |
| chr16.28961378 | 0.044 (1.35) * | 1.0 (0) | 0.012 (1.92) * | 0.019 (1.72) * | 0.071 (1.15) | 0.011 (1.95) * | 0.016 (1.79) * |
| chr16.29349242 | 0.009 (2.07) * | 1.0 (0) | 0.024 (1.63) * | 0.022 (1.65) * | 0.024 (1.61) * | 0.012 (1.93) * | 0.011 (1.97) * |
| chr16.29879300 | 0.065 (1.19) | 1.0 (0) | 0.022 (1.65) * | 0.048 (1.32) * | 0.063 (1.20) | 0.029 (1.53) * | 0.050 (1.30) * |
| chr16.29937753 | 0.072 (1.14) | 1.0 (0) | 0.019 (1.73) * | 0.039 (1.41) * | 0.074 (1.13) | 0.023 (1.64) * | 0.039 (1.41) * |
| chr16.30549587 | 1.0 (0) | 0.037 (1.43) * | 0.043 (1.37) * | 0.031 (1.51) * | 1.0 (0) | 0.030 (1.52) * | 0.023 (1.64) * |
| chr16.31717513 | 1.0 (0) | 0.201 (0.70) | 0.009 (2.04) * | 0.008 (2.12) * | 1.0 (0) | 0.008 (2.09) * | 0.007 (2.13) * |
| chr16.31837350 | 0.127 (0.90) | 1.0 (0) | 0.019 (1.73) * | 0.019 (1.72) * | 0.156 (0.81) | 0.020 (1.69) * | 0.020 (1.71) * |
| chr16.31837487 | 0.127 (0.90) | 1.0 (0) | 0.019 (1.73) * | 0.019 (1.72) * | 0.156 (0.81) | 0.020 (1.69) * | 0.020 (1.71) * |
| chr16.31943558 | 0.537 (0.27) | 0.914 (0.04) | 0.003 (2.57) * | 0.004 (2.39) * | 1.0 (0) | 0.002 (2.62) * | 0.004 (2.45) * |
| chr16.32011126 | 0.537 (0.27) | 0.914 (0.04) | 0.003 (2.57) * | 0.004 (2.39) * | 1.0 (0) | 0.002 (2.62) * | 0.004 (2.45) * |
| chr16.32340326 | 0.188 (0.73) | 0.962 (0.02) | 0.001 (3.00) * | 0.001 (3.00) * | 1.0 (0) | 0.0003 (3.46) * | 0.0003 (3.51) * |
| chr16.32365482 | 0.246 (0.61) | 1.0 (0) | 0.043 (1.36) * | 0.032 (1.50) * | 1.0 (0) | 0.024 (1.62) * | 0.019 (1.73) * |
| chr16.32428436 | 0.534 (0.27) | 0.647 (0.19) | 0.001 (3.27) * | 0.001 (3.02) * | 1.0 (0) | 0.0002 (3.65) * | 0.0004 (3.44) * |
| chr16.33060460 | 0.587 (0.23) | 1.0 (0) | 1.0 (0) | 1.0 (0) | 0.026 (1.59) * | 1.0 (0) | 1.0 (0) |
| chr16.33077036 | 0.587 (0.23) | 1.0 (0) | 1.0 (0) | 1.0 (0) | 0.026 (1.59) * | 1.0 (0) | 1.0 (0) |
| chr16.33085358 | 0.587 (0.23) | 1.0 (0) | 1.0 (0) | 1.0 (0) | 0.026 (1.59) * | 1.0 (0) | 1.0 (0) |
| chr16.33094013 | 0.583 (0.23) | 1.0 (0) | 1.0 (0) | 1.0 (0) | 0.025 (1.61) * | 1.0 (0) | 1.0 (0) |
| chr16.33132124 | 0.587 (0.23) | 1.0 (0) | 1.0 (0) | 1.0 (0) | 0.026 (1.59) * | 1.0 (0) | 1.0 (0) |
| chr16.36163336 | 1.0 (0) | 0.063 (1.20) | 0.023 (1.64) * | 0.036 (1.44) * | 1.0 (0) | 0.034 (1.47) * | 0.046 (1.34) * |
| chr16.41773281 | 0.010 (1.99) * | 1.0 (0) | 0.035 (1.46) * | 0.033 (1.48) * | 0.003 (2.47) * | 0.039 (1.41) * | 0.034 (1.46) * |
| chr16.41818090 | 0.010 (1.99) * | 1.0 (0) | 0.157 (0.81) | 0.163 (0.79) | 0.003 (2.47) * | 0.175 (0.76) | 0.171 (0.77) |
| chr16.41997223 | 0.005 (2.29) * | 1.0 (0) | 0.055 (1.26) | 0.054 (1.27) | 0.001 (2.99) * | 0.064 (1.19) | 0.059 (1.23) |
Position of the SNP on chromosome 16.
The p-values after additive logistic regression with sex as a covariate and Bonferroni correction for horses in population 1, population 2, both populations together, and both populations together with a mixed model using the relationship matrix derived from pedigrees (negative log p-values are adjacently listed in parentheses).
The p-values after additive logistic regression with sex and racing history as covariates and Bonferroni correction for horses in population 1, both populations together, and both populations together with a mixed model using the relationship matrix derived from pedigrees (negative log p-values are adjacently listed in parentheses).
p-value < 0.05
The mixed model using the pedigree-derived relationship matrix was applied to tests that included the fine-mapping data for all horses, and the resulting p-values were similar but slightly less significant than the raw p-values (Table 2). Although the most significant SNPs were not found at the exact same positions in each population, all associations were identified within approximately 13 Mb on ECA16.
Haplotype blocks were predicted from the 247 fine-mapping SNPs for population one, population two, and both populations together (Supplementary Tables 4A, 5A, and 6A). The average haplotype block was comprised of approximately six SNPs spanning 250 kb, the smallest haplotype block consisted of two SNPs spanning 0.87 kb, and the largest haplotype block consisted of nine SNPs spanning approximately one megabase (Mb).
Haplotypes were evaluated with Pearson’s chi-square tests and 10,000 permutations in population one, population two, and both populations together (Supplementary Methods, Supplementary Tables 4B, 5B, and 6B). The most significant haplotype blocks on ECA16 (Supplementary Table 7) were then analyzed in logistic regression tests with covariates, and the resulting raw p-values are shown in Table 3. Overall, the significant haplotypes contained individual SNPs that were previously found to have the strongest associations with RER. One notable exception was the significance of the 36.97-37.54 Mb haplotype (raw p-value = 1.1 × 10−5) from the analysis of population one with sex and racing history as covariates, and the absence of this significance when the 36.97-37.54 Mb region was analyzed with single SNPs alone.
Table 3.
The raw p-values of the most significant haplotypes formed from 247 fine-mapping SNPs spanning chr16:25727220-43876419.
| Sex as a Covariate3 | Sex and Raced as Covariates4 | |||||
|---|---|---|---|---|---|---|
| Haplotype Position1 | SNPs2 | Pop 1 | Pop 2 | All | Pop 1 | All |
| chr16.28806500-28896478 | 4 | 8.8 × 10−3 (2.05) | 2.7 × 10−4 (3.56) | 1.1 × 10−4 (3.95) | 8.5 × 10−3 (2.07) | 1.4 × 10−4 (3.86) |
| chr16.29228158-29334863 | 6 | 1.8 × 10−4 (3.75) | 5.5 × 10−2 (1.26) | 1.4 × 10−4 (3.86) | 4.6 × 10−4 (3.34) | 9.4 × 10−5 (4.03) |
| chr16.29854244-29937753 | 4 | 2.2 × 10−3 (2.66) | 1.3 × 10−1 (0.89) | 3.7 × 10−3 (2.44) | 1.2 × 10−3 (2.93) | 4.6 × 10−3 (2.34) |
| chr16.30375999-30637486 | 5 | 8.7 × 10−3 (2.06) | 2.2 × 10−3 (2.65) | 8.9 × 10−5 (4.05) | 2.0 × 10−3 (2.71) | 8.5 × 10−5 (4.07) |
| chr16.30489154-30817982 | 12 | 1.3 × 10−2 (1.88) | 5.9 × 10−4 (3.23) | 3.4 × 10−5 (4.47) | 5.0 × 10−3 (2.31) | 3.8 × 10−5 (4.42) |
| chr16.31808864-31859044 | 4 | 6.9 × 10−4 (3.16) | 2.9 × 10−2 (1.53) | 1.8 × 10−4 (3.74) | 9.6 × 10−4 (3.02) | 1.8 × 10−4 (3.75) |
| chr16.31924094-32234376* | 8 | 4.4 × 10−3 (2.36) | 7.3 × 10−2 (1.14) | 2.8 × 10−4 (3.55) | 1.9 × 10−3 (2.73) | 2.0 × 10−4 (3.69) |
| chr16.32255697-32407166 | 9 | 2.6 × 10−3 (2.59) | 3.7 × 10−2 (1.43) | 6.7 × 10−5 (4.18) | 1.2 × 10−2 (1.91) | 2.4 × 10−5 (4.62) |
| chr16.32381149-32550636 | 7 | 1.2 × 10−2 (1.93) | 2.6 × 10−2 (1.59) | 3.4 × 10−5 (4.47) | 2.4 × 10−2 (1.62) | 2.1 × 10−5 (4.68) |
| chr16.32880400-32980666 | 5 | 4.5 × 10−3 (2.35) | 0.25 (0.60) | 3.5 × 10−3 (2.46) | 1.2 × 10−3 (2.93) | 3.4 × 10−3 (2.47) |
| chr16.36970325-37536110 | 7 | 4.2 × 10−3 (2.38) | 3.0 × 10−1 (0.52) | 4.4 × 10−2 (1.36) | 1.1 × 10−5 (4.95) | 3.3 × 10−2 (1.49) |
| chr16.41947080-42152983 | 6 | 2.1 × 10−4 (3.68) | 6.6 × 10−3 (2.18) | 2.2 × 10−4 (3.66) | 3.5 × 10−5 (4.46) | 3.3 × 10−4 (3.48) |
| chr16.41947080-42340193 | 8 | 5.4 × 10−3 (2.27) | 2.4 × 10−2 (1.62) | 7.9 × 10−4 (3.10) | 5.8 × 10−4 (3.24) | 1.3 × 10−3 (2.87) |
Boundaries of the haplotype on chromosome 16.
Number of SNPs within the haplotype.
The p-values (negative log p-values are adjacently listed in parentheses) after additive logistic regression with gender as a covariate for population 1, population 2, and both populations together.
The p-values (negative log p-values are adjacently listed in parentheses) after additive logistic regression with gender and racing history as covariates for population 1 and both populations together.
Position chr16.31924094-32234376 has the same significance as chr16.31943558-32234376
Discussion
The GWAS of 48 RER case and 37 control Thoroughbreds from North America revealed an association spanning approximately 29-42 Mb on ECA16. Additive logistic regression tests were performed with sex as a covariate and with both sex and racing history as covariates because the complexity and relatively unknown pathophysiology of RER did not allow the best method to be chosen a priori. Analyses with the covariates of sex and racing history yielded more significant p-values than with sex as the only covariate, but the results of both methods were comparable.
The region of interest on ECA16 was further evaluated by genotyping 12 SNPs from the initial GWAS and 235 additional SNPs in both the initial population and a second population of 34 cases and 98 controls. Several SNPs and haplotypes showed significance when each population was analyzed separately; however, the positions of the most significant ECA16 SNPs varied between populations. The levels of significance increased considerably when both populations were evaluated together. It is possible that each population did not have enough power to identify the actual position of significance on its own, and this limitation was overcome by analyzing both populations together.
It is also possible that the most significant SNPs were located near breakpoints in LD, so that the position of the strongest association within the 13 Mb interval on ECA16 was determined by the informative nature of the marker more than its actual proximity to a casual mutation. Thoroughbreds have the highest levels of LD of any horse breed studied to date (Wade et al. 2009), and while this may allow for whole-genome associations to be found with low marker coverage, the long, conserved haplotypes characteristic of the Thoroughbred breed pose challenges in pinpointing causative mutations. Selecting a smaller region on ECA16 to investigate based on the location of potential candidate genes is not feasible, as Ensembl predicts a total of 276 protein-coding genes within the 25 – 45 Mb interval on ECA16 (Haider et al. 2009).
The results of this study did not confirm the recently published loci on ECA12 and ECA20 identified as candidate regions for “tying-up syndrome” in a population of Japanese Thoroughbred horses (Tozaki et al. 2010). Disparities between the results of these two studies may be due, in part, to the presence of different RER susceptibility loci in different Thoroughbred populations, or significantly different phenotyping and analysis methods between the studies. The horses in our study were carefully phenotyped to ensure as much as possible that cases had the same muscular disorder. There are numerous causes for ER in horses, including sporadic causes that have a nutritional or environmental basis, medical causes (MacLeay et al. 1999b), or genetic causes such as PSSM (McCue et al. 2006, 2008) and malignant hyperthermia (MH) (Aleman et al. 2009).
In addition, the Tozaki et al study employed microsatellite markers and two different analysis methods; a multipoint Markov chain Monte Carlo linkage analysis with 117 markers in 5 half-sib families and a 986 marker GWAS with pooled DNA samples from cases and controls (Tozaki et al. 2010). The GWAS reported here likely benefitted from a denser, more evenly spaced set of markers and statistical control for population structure. Furthermore, the gender and racing status of every horse was taken into account in our RER association tests by including sex and racing history as covariates in a logistic regression test modeling for additive allelic effect. This was important because gender, exposure to the racetrack, age, temperament, diet and exercise routines have been reported to affect the expression of clinical signs of RER and, therefore, may confound genetic association or linkage studies (MacLeay et al. 1999a; MacLeay et al. 2000; McGowan et al. 2002). While it was not possible to account for all of these factors, gender and racing history were included in this study because being female and racing on the track are considered the most significant risk factors in the development of RER (MacLeay et al. 1999a; McGowan et al. 2002; Wilsher et al. 2006).
In conclusion, we identified an association on ECA16 in a genome-wide search for RER causal loci in Thoroughbreds from North America, and SNPs within this 13 Mb region on ECA16 were also significant in a second population of North American Thoroughbreds. To account for the possibility that different RER susceptibility loci exist within different Thoroughbred lineages, the RER cases in this study all traced back to a common ancestral stallion. Although we found support for only the ECA16 locus in this study, the genetic model and pattern of inheritance for RER has not yet been conclusively defined, and it is unlikely that a single causative mutation is responsible for RER susceptibility. Rather, RER is likely be a complex genetic disease resulting from one or more genes of major effect, as well as modifying genes, gender, and environmental factors. A future GWAS with a larger number of RER cases and controls could narrow the region on ECA16 to a smaller interval that is likely to contain a causal RER mutation, as well as increase the power to identify additional risk loci across the genome.
Supplementary Material
Acknowledgements
Our appreciation goes out to Michelle Lucio for organizing and aliquoting the DNA samples and Robert Schaefer for formatting the data files. This project was funded by Minnesota Agricultural Experiment Station, Morris Animal Foundation D07EQ-500, The University of Minnesota Equine Center, National Research Initiative Competitive Grant no. 2008-35205-18766 from the USDA National Institute of Food and Agriculture, NIH NIAMS 1K08AR055713-01A2, and NIH NIAMS T32-AR0076I2.
References
- Aleman M, Nieto JE, Magdesian KG. Malignant hyperthermia associated with ryanodine receptor 1 (C7360G) mutation in Quarter Horses. Journal of Veterinary Internal Medicine. 2009;23:329–34. doi: 10.1111/j.1939-1676.2009.0274.x. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Clayton D, Leung HT. An R package for analysis of whole-genome association studies. Human Heredity. 2007;64:45–51. doi: 10.1159/000101422. [DOI] [PubMed] [Google Scholar]
- Dranchak PK, Valberg SJ, Onan GW, Gallant EM, MacLeay JM, McKenzie EC, de la Corte FD, Ekenstedt K, Mickelson JR. Inheritance of recurrent exertional rhabdomyolysis in Thoroughbreds. Journal of the American Veterinary Medical Association. 2005;227:762–7. doi: 10.2460/javma.2005.227.762. [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly ML, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Haider S, Ballester B, Smedley D, Zhang J, Rice P, Kasprzyk A. BioMart Central Portal--unified access to biological data. Nucleic Acids Research. 2009;37:W23–7. doi: 10.1093/nar/gkp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isgren CM, Upjohn MM, Fernandez-Fuente M, Massey C, Pollott G, Verheyen KL, Piercy RJ. Epidemiology of exertional rhabdomyolysis susceptibility in Standardbred horses reveals associated risk factors and underlying enhanced performance. PLoS ONE. 2010;5:e11594. doi: 10.1371/journal.pone.0011594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz LR, Valberg SJ, Balog EM, Mickelson JR, Gallant EM. Abnormal regulation of muscle contraction in horses with recurrent exertional rhabdomyolysis. American Journal of Veterinary Research. 1999;60:992–9. [PubMed] [Google Scholar]
- Lentz LR, Valberg SJ, Herold LV, Onan GW, Mickelson JR, Gallant EM. Myoplasmic calcium regulation in myotubes from horses with recurrent exertional rhabdomyolysis. American Journal of Veterinary Research. 2002;63:1724–31. doi: 10.2460/ajvr.2002.63.1724. [DOI] [PubMed] [Google Scholar]
- MacLeay JM, Sorum SA, Valberg SJ, Marsh WE, Sorum MD. Epidemiologic analysis of factors influencing exertional rhabdomyolysis in Thoroughbreds. American Journal of Veterinary Research. 1999a;60:1562–6. [PubMed] [Google Scholar]
- MacLeay JM, Valberg SJ, Pagan JD, Xue JL, de la Corte FD, Roberts J. Effect of ration and exercise on plasma creatine kinase activity and lactate concentration in Thoroughbred horses with recurrent exertional rhabdomyolysis. American Journal of Veterinary Research. 2000;61:1390–5. doi: 10.2460/ajvr.2000.61.1390. [DOI] [PubMed] [Google Scholar]
- MacLeay JM, Valberg SJ, Sorum SA, Sorum MD, Kassube T, Santschi EM, Mickelson JR, Geyer CJ. Heritability of recurrent exertional rhabdomyolysis in Thoroughbred racehorses. American Journal of Veterinary Research. 1999b;60:250–6. [PubMed] [Google Scholar]
- McCue ME, Ribeiro WP, Valberg SJ. Prevalence of polysaccharide storage myopathy in horses with neuromuscular disorders. Equine Veterinary Journal Supplement. 2006;36:340–4. doi: 10.1111/j.2042-3306.2006.tb05565.x. [DOI] [PubMed] [Google Scholar]
- McCue ME, Valberg SJ, Miller MB, Wade C, DiMauro S, Akman HO, Mickelson JR. Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis. Genomics. 2008;91:458–66. doi: 10.1016/j.ygeno.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan CM, Fordham T, Christley RM. Incidence and risk factors for exertional rhabdomyolysis in Thoroughbred racehorses in the United Kingdom. Veterinary Record. 2002;151:623–6. doi: 10.1136/vr.151.21.623. [DOI] [PubMed] [Google Scholar]
- Oki H, Miyake T, Hasegawa T, Sasaki Y. Estimation of heritability for Tying-up syndrome in the Thoroughbred racehorse by Gibbs sampling. Journal of Animal Breeding and Genetics. 2005;122:289–93. doi: 10.1111/j.1439-0388.2005.00539.x. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team R: a language and environment for statistical computing. 2011 http://www.R-project.org.
- Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. American Journal of Human Genetics. 2006;78:629–44. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozaki T, Hirota K, Sugita S, Ishida N, Miyake T, Oki H, Hasegawa T. A genome-wide scan for tying-up syndrome in Japanese Thoroughbreds. Animal Genetics Supplement. 2010;41:80–6. doi: 10.1111/j.1365-2052.2010.02112.x. [DOI] [PubMed] [Google Scholar]
- Valberg SJ, Mickelson JR, Gallant EM, MacLeay JM, Lentz L, de la Corte F. Exertional rhabdomyolysis in quarter horses and Thoroughbreds: one syndrome, multiple aetiologies. Equine Veterinary Journal Supplement. 1999;30:533–8. doi: 10.1111/j.2042-3306.1999.tb05279.x. [DOI] [PubMed] [Google Scholar]
- van den Boom D, Ehrich M. Discovery and identification of sequence polymorphisms and mutations with MALDI-TOF MS. Methods in Molecular Biology. 2007;366:287–306. doi: 10.1007/978-1-59745-030-0_16. [DOI] [PubMed] [Google Scholar]
- Vazquez AI, Bates DM, Rosa GJ, Gianola D, Weigel KA. Technical note: an R package for fitting generalized linear mixed models in animal breeding. Journal of Animal Science. 2010;88:497–504. doi: 10.2527/jas.2009-1952. [DOI] [PubMed] [Google Scholar]
- Wade CM, Giulotto E, Sigurdsson S, Zoli M, Gnerre S, Imsland F, Lear TL, Adelson DL, Bailey E, Bellone RR, Blocker H, Distl O, Edgar RC, Garber M, Leeb T, Mauceli E, MacLeod JN, Penedo MC, Raison JM, Sharpe T, Vogel J, Andersson L, Antczak DF, Biagi T, Binns MM, Chowdhary BP, Coleman SJ, Della VG, Fryc S, Guerin G, Hasegawa T, Hill EW, Jurka J, Kiialainen A, Lindgren G, Liu J, Magnani E, Mickelson JR, Murray J, Nergadze SG, Onofrio R, Pedroni S, Piras MF, Raudsepp T, Rocchi M, Roed KH, Ryder OA, Searle S, Skow L, Swinburne JE, Syvanen AC, Tozaki T, Valberg SJ, Vaudin M, White JR, Zody MC, Lander ES, Lindblad-Toh K. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science. 2009;326:865–7. doi: 10.1126/science.1178158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilsher S, Allen WR, Wood JL. Factors associated with failure of Thoroughbred horses to train and race. Equine Veterinary Journal. 2006;38:113–8. doi: 10.2746/042516406776563305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.