

Abstract
BACKGROUND
Neuroendocrine differentiation (NED) is one of the mechanisms underlying development of castration-resistant prostate cancer. In this study, we investigated IL-6-induced NED in two LNCaP sublines.
METHODS
LNCaP-S17, an LNCaP subline that secretes IL-6, and LNCaP-C3, a control subline that does not express IL-6, were analyzed for IL-6-induced NED, activation of JAK2 and STAT3 pathways, and expression of IL-6/IL-6R signaling proteins and downstream target genes.
RESULTS
IL-6 did not induce NED in LNCaP-S17 cells, even though IL-6 induced NED in LNCaP-C3 cells. IL-6 activated JAK2 and STAT3 pathways in LNCaP-C3 cells but not in LNCaP-S17 cells. IL-6 did not activate ERK1/2, AKT, or NF-κB pathways in either cell line. Both LNCaP-C3 and LNCaP-S17 cell lines expressed IL-6R, gp130, and TYK2 at almost the same levels and did not express JAK1 or JAK3. The basal level of JAK2 expression was slightly higher in LNCaP-C3 cells than in LNCaP-S17 cells. Two suppressors of cytokine signaling, SOCS7 and CIS, were expressed constitutively at higher levels in LNCaP-S17 cells than in LNCaP-C3 cells, while SOCS1 to SOCS6 were expressed at approximately the same levels. Using siRNA to knockdown SOCS7 and CIS expression in LNCaP-S17 cells led to increased phosphorylation of STAT3 upon IL-6 stimulation.
CONCLUSIONS
LNCaP-S17 cells are resistant to exogenous IL-6-induced NED due to increased levels of CIS/SOCS7 that block activation of JAK2-STAT3 pathways.
Keywords: IL-6, neuroendocrine, prostate cancer
INTRODUCTION
Prostate cancer is the most common malignant disease in men in the developed countries. Initially, most prostate cancers respond to androgen ablative (or castration) therapy. However, even under the castrate levels of testosterone, prostate cancer eventually re-grows and becomes known as castration-resistant prostate cancer (CRPC), a lethal form of cancer [1,2]. The molecular mechanisms underlying the progression from a hormone sensitive cancer to CRPC include alterations of androgen receptor (AR) signaling pathways, alterations of apoptosis-related genes such as up-regulation of bcl-2 [3] and clusterin [4,5], and neuroendocrine differentiation (NED) of prostate cancer [6]. AR gene amplification [7,8], increase in AR expression [9], and AR gene mutations [10,11] are important factors in development of CRPC. AR can also be activated by intraprostatic tissue androgens (so called intracrine androgens) [12,13]. Ligand-independent AR variants derived from splicing of cryptic exons have been associtated with CRPC [14]. Some growth factors can activate AR signaling pathways in the absence of androgen [15]. Activation of HER-2/neu [16] and Ras/mitogen-activated protein kinase (MAPK) pathways can activate AR signaling pathways in androgen-independent manner [17]. Transcriptional coactivators are also involved in the ligand-independent activation of AR signaling pathways [18,19].
NED of prostate cancer has not been studied as extensively as AR signaling. Neuroendocrine (NE) cells, part of the amine precursor uptake and decarboxylation system, are specialized endocrine cells of the nervous system that produce neurohormones. In the prostate, NE cells are present in the developing and mature gland, forming the prostatic epithelium with the epithelial and basal cells [20]. NE cells are characterized morphologically by the presence of cytoplasmic dense core granules and irregular dendrite-like processes extending underneath and between adjacent epithelial cells [21]. The prostatic NE cells express neurotensin (NTS), synaptotagmin I (SYT1), gastrin-releasing peptide (GRP), neuron-specific enolase (NSE), midkine (MDK), and other neurohormones [6,22]. NE cells are generally non-mitotic/growth-arrested [23,24], yet proliferating carcinoma cells have been identified in close proximity to them [24,25], indicating a tumor-promoting role of NE cells through secretion of neurohormones [26–29]. NE cells are found in primary and metastatic prostate cancers and may have arisen from NED [30–32]. Focal NED is a common feature of prostate cancer, occurring in about 30% to 100% of tumors studied [21,33,34]. Recent studies suggest that NED is enhanced after long-term androgen ablative therapy and may promote the development of CRPC [35–38]. NED is typically exemplified by a human prostate cancer cell line, LNCaP, under conditions of androgen deprivation, increased intracellular cAMP, activated MAPK pathway, and IL-6 treatment [23,39–49]. IL-6-induced NED is clinically relevant because elevated serum IL-6 levels are associated with CRPC [50–54].
Studies in IL-6 and prostate cancer have revealed complex effects of IL-6 on proliferation of prostate cancer cells (see a recent review by Culig and Puhr [55]). Giri et al. reported that exogenous IL-6 treatment increased cell proliferation in LNCaP cells [56]. In contrast, several groups of investigators independently found that exogenous IL-6 caused growth arrest in LNCaP cells, which was associated with NED [44,47,57–59]. The causes of the discrepancy are not known. Nevertheless, the molecular mechanisms of IL-6’s actions have been identified. IL-6 cytokine first binds to a nonsignaling receptor, IL-6R, and then forms a hexameric complex with gp130 [60]. Gp130 is a signal-transducing receptor shared among the IL-6 family of cytokines including IL-6, IL-11, leukemia inhibitory factor, ciliary neurotrophic factor, oncostatin-M, and others [61,62]. IL-6 leads to dimerization of gp130, resulting in phosphorylation and activation of Janus kinases (JAK1, JAK2, JAK3, and TYK2). Subsequently, the cytoplasmic tail of gp130 is phosphorylated, opening docking sites for STATs (STAT1 and STAT3). STATs also become phosphorylated, form dimers and enter the nucleus, where they regulate target gene expression [63,64].
LNCaP cell line does not express IL-6 but does express its receptors IL-6R and gp130 [65,66]. Previously, we established an LNCaP subline stably expressing IL-6 by introduction of a full-length IL-6 cDNA into LNCaP cells [66]. We reported that IL-6-expressing LNCaP cells (i.e., LNCaP cells with autocrine IL-6 expression) acquire the capacity to grow in the absence of androgen in vitro and in vivo [67]. In the current study, we found that IL-6-expressing LNCaP cells were resistant to exogenous IL-6-induced NED because the cells constitutively expressed high levels of negative feedback inhibitors, suppressor of cytokine signaling 7 (SOCS7) and cytokine-inducible SH2 protein (CIS). The results provide new insights into the molecular mechanisms of NED in prostate cancer.
MATERIALS AND METHODS
Cell Culture and In Vitro Cell Growth
IL-6-expressing LNCaP subline (LNCaP-S17, expressing IL-6 at 2,743 pg/ml/million cells) and control subline LNCaP-C3 (transfected with empty vector, not expressing IL-6) were described previously [66]. The cell lines were cultured in T-medium (Invitrogen) with 5% FBS in a 37°C, 5% CO2 humidified incubator. To analyze cell growth under exogenous IL-6 treatment, LNCaP-C3 and LNCaP-S17 cells were cultured in serum-free medium with 50 ng/ml IL-6 for 4 days; then the cell number was determined using the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma) according to the manufacturer’s instructions [67]. To confirm if IL-6’s effects were indeed mediated through IL-6 receptor, humanized anti-human IL-6R monoclonal antibodies (MRA, a gift from Chugai Pharmaceutical Co., Ltd, Tokyo, Japan) were added to block IL-6’s action.
Induction of NED and Reverse Transcription-Quantitative Real-time PCR
LNCaP-C3 and LNCaP-S17 cells were cultured separately in serum-free medium with 50 ng/ml IL-6 for 4 days to induce NED. For co-culture of LNCaP-C3 and LNCaP-S17 cells, 100-mm dishes were separated into two halves by a dam made with a 1-cm strip of sterilized paper pad; the two cell lines were plated in each half of the dish side-by-side; after the cells were attached overnight, the dam was removed; serum-free medium was added to allow free exchange of secreted factors between LNCaP-C3 and LNCaP-S17 cells for 4 days. The cells were photographed using a digital camera. RNA was isolated from the cells using an RNeasy kit (QIAGEN); reverse transcription was performed using Superscript First-Strand Synthesis System (Invitrogen); real-time PCR was performed on the iQ5 PCR instrument with SYBR Green reagents (Bio-Rad). PCR primer sequences are shown in Table I. The mRNA levels were first normalized by that of GAPDH and then the relative mRNA fold change was calculated as 2ΔΔCt compared to the basal levels (either without IL-6 treatment or at Day 0).
TABLE I.
Primers used in real-time PCR analysis
| Gene name | Sequence (5′ to 3′) | |
|---|---|---|
| NTS | sense | GAGGAGCTTGTTGCAAGAAGGA |
| anti-sense | GCCCTGCTGTGACAGATTTTGT | |
| SYT1 | sense | AACATGGGGTTGGCTGTTT |
| anti-sense | CGGCAGACGGTTATTTTCCT | |
| GRP | sense | AGTCTCTGCTCTTCCAGCC |
| anti-sense | CCGATGGACAACCAATCTAAG | |
| NSE | sense | TGTCTGCTGCTCAAGGTCAA |
| anti-sense | CGATGACTCACCATGACCC | |
| MDK | sense | CGACTGCAAGTACAAGTTTGAGAAC |
| anti-sense | CTTGGTGACGCGGATGGT | |
| GAPDH | sense | TAAAAGCAGCCCTGGTGACC |
| anti-sense | CCACATCGCTCAGACACCAT | |
Western Blot Analysis and Immunoprecipitation
The cells were washed with ice-cold phosphate-buffered saline and collected. Whole cell lysates were prepared with RIPA buffer containing 1% Nonidet P-40, 5% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride, 100 mM sodium orthovanadate, and protease inhibitors mixture (Sigma). The protein concentrations were determined using the Bradford protein assay (Bio-Rad). Equal amounts of protein extracts were subjected to 10% SDS-polyacrylamide gel electrophoresis and transferred to a PVDF membrane by electroblotting (Bio-Rad). The membranes were blocked with 5% nonfat dry milk in 1 x TBST (25mM Tris-HCl, 125 mM NaCl, 0.1% Tween 20) for 2 h, probed with primary antibodies overnight at 4 °C, and then with fluorphores-conjugated secondary antibodies (LI-COR) for 1 h. The results were visualized using an Odyssey Infrared Imaging System (LI-COR). For internal control, the blots were stripped and re-probed with anti-GAPDH or anti-β-tubulin antibodies. For immunoprecipitation of p-JAK2, 1 μg of anti-p-JAK2 antibodies were incubated with equal amounts of protein extracts for 30 min, followed by incubation with 10 μl of protein A- Sepharose beads overnight at 4 °C; after washing 4 times, the precipitated protein was analyzed using the procedures described above. For siRNA knockdown assays, LNCaP-S17 cells were plated at 70% confluence in 60-mm dishes; 100 nM of SOCS7 siRNA (siGENOME SMARTpool D-027197-04), CIS siRNA (siGENOME SMARTpool D-017381-04), or non-targeting scrambled control siRNA were transfected into the cells using DharmaFECT®2 reagents according to the manufacturer’s instructions (all reagents including siRNAs were obtained from Thermo Fisher Scientific Dharmacon Products, Lafayette, CO); 4 days later, the cells were treated with 50 ng/ml of IL-6 in serum-free medium for 5 min; and the whole cell lysates were analyzed for expression of p-STAT3, SOCS7, and CIS, respectively, by Western blot. The primary antibodies were purchased from the following commercial sources: antibodies against STAT3, p-STAT3 (Y705), JAK1, JAK2, JAK3, TYK2, p-JAK2, AKT, p-AKT, ERK1/2, IκBα, and p-IκBα were obtained from Cell Signaling Technology; antibodies against IL-6R, gp130, GAPDH, and β-tubulin were obtained from R&D Systems; and antibodies against p-ERK1/2, CIS, and SOCS1 to SOCS7 were obtained from Santa Cruz Biotechnology.
Statistical Analysis
All experiments were repeated three times. Photomicrographs and Western blot results are presented from a representative experiment. Student’s t test (two-tailed) was used to determine the significance between the control and treatment groups of LNCaP-C3 and LNCaP-S17 cells in the cell growth analysis, and P < 0.05 was considered statistically significant.
RESULTS
LNCaP-S17 Cells Were Resistant to IL-6-induced NED
We previously showed that LNCaP-S17 cells could grow in the absence of androgen [67]. To test if the cells were still able to undergo NED, we treated LNCaP-C3 and LNCaP-S17 cells with exogenous IL-6 for 4 days. We found that LNCaP-C3 cells showed irregular dendrite-like processes typical of NE cells (Fig. 1B, compared to Fig. 1A). In contrast, the LNCaP-S17 cells did not show any obvious change in cell morphology under the same exogenous IL-6 treatment (Fig. 1E, compared to Fig. 1D). To confirm that LNCaP-S17 cells secreted IL-6, we co-cultured LNCaP-C3 and LNCaP-S17 cells in a system such that IL-6 secreted by LNCaP-S17 cells could move freely to LNCaP-C3 cells but the two cell lines did not mix together. Indeed, we found that the co-cultured LNCaP-C3 cells extended dendrite-like processes (Fig. 1C), whereas LNCaP-S17 cells did not show any processes (Fig. 1F). Because NE cells are generally non-mitotic/growth-arrested [23,24], we examined if IL-6 treatment induced growth arrest in the two cell lines. We found that IL-6 induced approximately 50% reduction in the number of LNCaP-C3 cells (Fig. 2A, comparing group 2 versus group 1; p = 0.007), whereas the number of LNCaP-S17 cells was similar to the untreated control group (Fig. 2A, comparing group 2 versus group 1). The cell growth arrest observed in LNCaP-C3 cells was specifically induced by IL-6, as the anti-IL-6R antibody MRA completely blocked IL-6’s function and rescued cell growth in LNCaP-C3 cells (Fig. 2A, comparing group 4 versus group 2). To further confirm that exogenous IL-6 induced NED in LNCaP-C3 cells but not in LNCaP-S17 cells, we examined five markers of NED. As shown in Fig. 2B, exogenous IL-6 dramatically induced mRNA expression of NTS, SYT1, GRP, NSE, and MDK in LNCaP-C3 cells, particularly NTS mRNA that was increased by approximately 68 fold. In contrast, exogenous IL-6 minimally induced the expression of these markers in LNCaP-S17 cells, e.g., only 2.6 fold increase in NTS mRNA (Fig. 2B). Similarly, when the two cell lines were co-cultured for 4 days, IL-6 secreted by LNCaP-S17 cells considerably induced NTS and SYT1 mRNA expression in LNCaP-C3 cells but only minimally in LNCaP-S17 cells (Fig. 2C). In addition, we found that induction of NTS and NSE expression occurred mainly on the 3rd and 4th day of exogenous IL-6 treatment (Fig. 1D).
Fig. 1. IL-6 induced formation of dentrite-like processes in LNCaP-C3 but not in LNCaP-S17 cells.
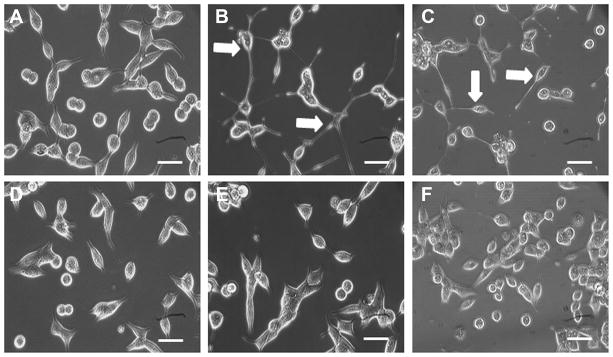
A. LNCaP-C3 cells without treatment (Day 0). B. LNCaP-C3 cells treated with 50 ng/ml IL-6 for 4 days in serum-free culture medium. C. LNCaP-C3 cells co-cultured with LNCaP-S17 cells for 4 days. D. LNCaP-S17 cells without treatment (Day 0). E. LNCaP-S17 cells treated with 50 ng/ml IL-6 for 4 days in serum-free culture medium. F. LNCaP-S17 cells co-cultured with LNCaP-C3 cells for 4 days. Arrows indicate LNCaP-C3 cells with dentrite-like processes. Scale bars, 10 μm.
Fig. 2. IL-6 induced growth arrest and expression of NED markers in LNCaP-C3 but not in LNCaP-S17 cells.

A. LNCaP-C3 and LNCaP-S17 cells were cultured in 12-well plates with serum-free medium, with or without 50 ng/ml IL-6 and/or 100 μg/ml MRA for 4 days. The cell growth was determined by MTT assay and normalized to the control group. Asterisk indicates p = 0.007, comparing between group 1 and group 2 of LNCaP-C3 cells. B. LNCaP-C3 and LNCaP-S17 cells were treated with or without 50 ng/ml IL-6 in serum-free medium for 4 days. The mRNA levels were determined with quantitative real-time PCR. C. LNCaP-C3 and LNCaP-S17 cells were co-cultured in a custom-made system for 4 days. The two cell lines were separately harvested and mRNA levels were determined with quantitative real-time PCR. D. LNCaP-C3 (C3) and LNCaP-S17 (S17) cells were treated without or with 50 ng/ml IL-6 for the indicated time. The mRNA levels were determined with quantitative real-time PCR. All experiments were performed in triplicate and the results represented the means and standard deviations (error bars) of three independent experiments.
Activation of STAT3 Pathway Was Inhibited in LNCaP-S17 Cells
As we had noticed the differences in IL-6-induced NED between LNCaP-C3 and LNCaP-S17 cells, we investigated the underlying molecular mechanisms. Because IL-6 has been shown to activate JAK-STAT3 and/or PI3K-Etk-STAT3 pathways for induction of NED [44,47,57–59], we examined phosphorylation of STAT3, ERK1/2, AKT, and IκBα. As shown in Fig. 3A, exogenous IL-6 induced remarkable phosphorylation of STAT3 as early as 5 min upon IL-6 treatment in LNCaP-C3 cells, whereas there was barely any induction of p-STAT3 in LNCaP-S17 cells. The basal levels of p-AKT were very high in both cell lines due to lack of PTEN expression in LNCaP cells, thus there was no obvious induction of p-AKT in either cell line. The basal levels of p-ERK1/2 and p-IκBα were higher in LNCaP-C3 cells than in LNCaP-S17 cells, but no apparent induction was found in either cell line (Fig. 3A). To confirm that LNCaP-S17 cells secreted IL-6 as previously measured [66], we collected the conditioned medium from the cultured LNCaP-S17 cells. Addition of the conditioned medium induced p-STAT3 in LNCaP-C3 cells but barely in LNCaP-S17 cells (Fig. 3B).
Fig. 3. IL-6 induced phosphorylation of STAT3 in LNCaP-C3 but not in LNCaP-S17 cells.

A. LNCaP-C3 and LNCaP-S17 cells were treated without or with 50 ng/ml IL-6 in serum-free medium for the indicated time. B. LNCaP-S17 cells were cultured in serum-free medium for 24 h. The medium was harvested and centrifuged at 4000 rpm for 10 min and the supernatant was collected as LNCaP-S17 conditioned medium (S17 CM), which was used to treat the cells for the indicated time. Western blot analysis was performed as described under “Materials and Methods”.
To demonstrate that induction of p-STAT3 in LNCaP-C3 cells was specifically through IL-6/IL-6R signaling pathway, we examined the dose-dependence and effects of specific inhibitors. We found that IL-6 induced p-STAT3 at a dosage as low as 1 ng/ml, but the induction was maximal at a dosage of 50 ng/ml (Fig. 4A). The anti-IL-6R antibody MRA dose-dependently inhibited IL-6-induced p-STAT3 (Fig. 4B). As expected, a single dose of 100 μg/ml MRA blocked IL-6’s action over the 4-day treatment period (Fig. 4C).
Fig. 4. IL-6 induced phosphorylation of STAT3 in LNCaP-C3 cells via IL-6R.

A. LNCaP-C3 cells were treated without or with 1, 5, 10, and 50 ng/ml IL-6 for 5 min. B. LNCaP-C3 cells were treated without or with 50 ng/ml IL-6 in serum-free medium or in combination with different dosages of anti-IL-6R mAb (MRA) for 5 min. C. LNCaP-C3 cells were treated without or with 50 ng/ml IL-6 in serum-free medium or in combination with 100 μg/ml MRA for the indicated days. Western blot analysis was performed as described under “Materials and Methods”.
LNCaP-S17 Cells Constitutively Expressed High Levels of SOCS7 and CIS
To reveal the mechanisms that caused the differences in STAT3 activation between LNCaP-C3 and LNCaP-S17 cells, we systematically examined the IL-6/IL-6R signaling pathways. First, we determined the protein levels of IL-6 receptors. As shown in Fig. 5A, the 80-kDa and 55-kDa forms of IL-6R as well as gp130 were expressed at similar levels in LNCaP-C3 and LNCaP-S17 cells. Downstream to the IL-6 receptors are the JAK kinase family members. We found that JAK1 and JAK3 were not expressed in either cell line. TYK2 was expressed at the same levels in both cell lines. JAK2 expression was slightly higher in LNCaP-C3 cells than in LNCaP-S17 cells (Fig. 5A). Upon exogenous IL-6 treatment, phosphorylation of JAK2 was clearly induced in LNCaP-C3 cells but not in LNCaP-S17 cells (Fig. 5B). To further confirm our findings, we did immunoprecipitation to enrich p-JAK2. Indeed, we found that exogenous IL-6 induced noticeable p-JAK2 accompanied by remarkable degradation of JAK2 in LNCaP-C3 cells but not in LNCaP-S17 cells (Fig. 5C). To identify what factors were inhibiting JAK2 phosphorylation, we examined the well-known negative feedback regulators CIS/SOCS family. This family of inhibitors has 8 members including CIS and SOCS1 to SOCS7. As shown in Fig. 6A, the basal levels of SOCS1 to SOCS6 were almost the same between LNCaP-C3 and LNCaP-S17 cells. There was not any obvious difference upon IL-6 treatment. However, the basal levels of SOCS7 and CIS were clearly higher in LNCaP-S17 cells than in LNCaP-C3 cells (Fig. 6A). IL-6 treatment within the short time did not noticeably change the levels of SOCS7 and CIS protein in either cell line (Fig. 6A). Because SOCS3 is a known regulator of p-STAT3 in other cell systems and our short time IL-6 treatment did not change SOCS3 expression, we then treated LNCaP-C3 cells with IL-6 for up to 4 hours. We found that IL-6 induced phosphorylation of STAT3 from 30 min to 4 hours, but SOCS3 protein levels were not increased (Fig. 6B). To confirm if the increased levels of SOCS7 and CIS were responsible for IL-6 resistance in LNCaP-S17 cells, we used specific siRNAs to knockdown SOCS7 and CIS expression in LNCaP-S17 cells and then examined IL-6-induced phosphorylation of STAT3. As shown in Figure 6C, SOCS7 siRNA down-regulated SOCS7 expression and increased phosphorylation of STAT3 compared to the control siRNA and control groups. CIS siRNA also slightly down-regulated CIS expression and increased phosphorylation of STAT3 (Fig. 6D). Taken together, these results suggest that the increased basal levels of CIS/SOCS7 in LNCaP-S17 cells inhibit phosphorylation of JAK2 and subsequently STAT3 (Fig. 6E), leading to reduced response in IL-6-induced NED.
Fig. 5. IL-6 induced phosphorylation of JAK2 in LNCaP-C3 but not in LNCaP-S17 cells.

A. Expression of IL-6R, gp130, and JAK family in LNCaP-C3 (C3) and LNCaP-S17 (S17) cells in regular culture medium without any treatment. B. Phosphorylation of JAK2 in LNCaP-C3 (C3) and LNCaP-S17 (S17) cells treated without or with 50 ng/ml IL-6 in serum-free medium for the indicated time. C. LNCaP-C3 (C3) and LNCaP-S17 (S17) cells were treated without or with 50 ng/ml IL-6 in serum-free medium for 5 min. Equal amounts of total proteins were precipitated with anti-p-JAK2 antibodies and protein A-sepharose beads. Western blot analysis was performed as described under “Materials and Methods”.
Fig. 6. LNCaP-S17 cells expressed higher basal levels of SOCS7 and CIS than LNCaP-C3 cells.

A and B. LNCaP-C3 and LNCaP-S17 cells were treated without or with 50 ng/ml IL-6 in serum-free medium for the indicated time. Protein expression of CIS/SOCS family members, p-STAT3, and STAT3 was determined by Western blot analysis. C and D. LNCaP-S17 cells were not transfected (control group) or transfected with 100 nM of non-targeting scrambled control siRNA, SOCS7 siRNA, or CIS siRNA, respectively; 4 days later, the cells were treated with 50 ng/ml of IL-6 in serum-free medium for 5 min; and p-STAT3, SOCS7, and CIS expression was analyzed by Western blot. E. Proposed working model. IL-6 acts via IL-6R/gp130 to activate JAK2/STAT3 pathway to induce expression of CIS/SOCS7; long-term IL-6 stimuli lead to increased basal levels of CIS and SOCS7, which act in a negative feedback loop to inhibit activation of JAK2/STAT3.
DISCUSSION
NED is an important mechanism in the development of the lethal CRPC [6]. IL-6 is a critical cytokine for induction of NED because elevated serum IL-6 levels are associated with CRPC [50–54]. Several studies have independently demonstrated that exogenous IL-6 induces NED in LNCaP cells [40,44,47,49]. Our results showed that exogenous IL-6 treatment induced NED in LNCaP-C3 cells as evidenced by the morphologic changes (i.e., dendrite-like processes), growth arrest, and expression of NED markers (i.e., NTS, SYT1, GRP, NSE, and MDK). These results are consistent with the literature reports [40,44,47,49]. To our surprise, IL-6 did not induce NED in LNCaP-S17 cells. Both LNCaP-C3 and LNCaP-S17 cell lines are derived from the parental LNCaP cells, in which LNCaP-C3 is a control cell line transfected with empty vector, and LNCaP-S17 is transfected with full-length IL-6 cDNA [66]. Thus, the difference is that LNCaP-C3 cells do not express IL-6 but LNCaP-S17 cells secrete IL-6 in an autocrine manner.
To reveal the molecular basis underlying the differences in IL-6-induced NED between LNCaP-C3 and LNCaP-S17 cell lines, we first examined activation of STAT3 by IL-6 because it has been well established that IL-6 activates JAK-STAT3 and/or PI3K-Etk-STAT3 pathways to induce NED in LNCaP cells [44,47,57–59]. We identified that a remarkable difference between LNCaP-C3 and LNCaP-S17 cell lines is in their response to exogenous IL-6 in terms of phosphorylation of STAT3. IL-6 induced p-STAT3 in a dose-dependent manner for a period of 4 days after a single dose treatment in LNCaP-C3 cells, which was dependent on IL-6R as anti-IL-6R antibodies could abolish the induction completely. In contrast, IL-6 failed to induce any noticeable p-STAT3 in LNCaP-S17 cells. IL-6 did not activate ERK1/2, AKT or NF-κB pathways in either LNCaP-C3 or LNCaP-S17 cell line, suggesting that IL-6 specifically activates the STAT3 pathway to induce NED.
It is known that IL-6 acts through IL-6R and gp130 [60]. Gp130 then activates JAKs, followed by activation of STAT3 [63,64]. Our results suggest that LNCaP-C3 and LNCaP-S17 cell lines express similar levels of IL-6R and gp130. In addition, JAK family member TYK2 is expressed at the same levels in the two cell lines while JAK1 and JAK3 are not expressed in either cell line. Thus, IL-6R, gp130, JAK1, JAK3, and TYK2 do not contribute to the differential responses observed in the two cell lines. The basal level of JAK2 expression was slightly higher in LNCaP-C3 cell line than in LNCaP-S17 cell line. IL-6 induced phosphorylation of JAK2 in LNCaP-C3 cell line but not in LNCaP-S17 cell line. This cannot be completely attributed to the increased basal level of JAK2 in LNCaP-C3 cell line because LNCaP-S17 cell line also expresses a decent level of JAK2. Thus, it is more likely that activation of JAK2 is inhibited by some factors. Given that there are 8 well-known negative regulators of cytokine signaling including CIS and SOCS1-7 [68], we examined the expression of all of them in LNCaP-C3 and LNCaP-S17 cell lines. Our results suggest that SOCS1-6 are equally expressed in the two cell lines, thus SOCS1-6 do not contribute to the differential inhibition of JAK2/STAT3 signaling. However, SOCS7 and CIS levels were constitutively higher in LNCaP-S17 cell line than in LNCaP-C3 cell line, suggesting that SOCS7 and CIS are responsible for the observed differences in IL-6-induced NED between the two cell lines. It has been shown that CIS and SOCS proteins can directly bind to JAKs and reduce their tyrosine-kinase activities and activation of STATs [69]. IL-6 can induce expression of CIS and SOCS7 [70,71], thus CIS and SOCS7 act in a negative feedback loop to inhibit IL-6-induced activation of JAK2/STAT3 pathway. Furthermore, down-regulation of SOCS7 and CIS expression by siRNA transfection increased IL-6-induced phosphorylation of STAT3, which confirms that SOCS7 and CIS are responsible for the IL-6 resistance in LNCaP-S17 cells.
The results presented here significantly extended our previous studies [66,67,72]. We demonstrated that LNCaP-S17 cells constitutively express high levels of CIS and SOCS7, which in turn inhibit activation of JAK2 and STAT3, leading to a failure to respond to IL-6-induced NED. We speculate that the high levels of CIS and SOCS7 expression are caused by the chronic stimuli of IL-6 secreted by LNCaP-S17 cells because CIS and SOCS7 are downstream target genes induced by IL-6. We and other investigators have shown that short-term exogenous IL-6 treatment induces NED, whereas long-term exogenous IL-6 stimuli not only cause LNCaP cells to express endogenous IL-6 but also lead to increased cell proliferation [72,73]. The mechanisms are reportedly due to a decrease in IL-6 binding capacity and/or an increase in androgen binding capacity and AR signaling after long-term IL-6 treatment [72,73]. The present study provides a new mechanism that CIS and SOCS7 levels are increased in LNCaP cells exposed to long-term autocrine IL-6. Thus, the increased levels of CIS and SOCS7 expression desensitize LNCaP cells’ response to IL-6-induced NED.
In conclusion, we demonstrated in this study that LNCaP cells with autocrine IL-6 expression are resistant to exogenous IL-6-induced NED due to increased levels of CIS/SOCS7 that block activation of JAK2-STAT3 pathways.
Acknowledgments
We thank Dr. Prescott L. Deininger (Director of Tulane Cancer Center, PI of the COBRE grant 2P20RR020152-06) and Tulane Cancer Center Core Facilities for research support. We thank Chugai Pharmaceutical Co., Ltd, Tokyo, Japan, for the gift of MRA.
Grant sponsors: National Institutes of Health’s Centers of Biomedical Research Excellence (COBRE) grant (2P20RR020152-06), Department of Defense grants (W81XWH-05-1-0567 and W81XWH-10-1-0937), the developmental funds of the Tulane Cancer Center, Louisiana Cancer Research Consortium, and Tulane Framework for Global Health Seed Grant.
Abbreviations
- AR
androgen receptor
- CIS
cytokine-inducible SH2 protein
- CRPC
castration-resistant prostate cancer
- GRP
gastrin-releasing peptide
- MDK
midkine
- MRA
humanized anti-human IL-6R monoclonal antibodies
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NE
neuroendocrine
- NED
neuroendocrine differentiation
- NSE
neuron-specific enolase
- NTS
neurotensin
- siRNA
short interfering RNA
- SOCS
suppressor of cytokine signaling
- SYT1
synaptotagmin I
References
- 1.Chodak GW, Vogelzang NJ, Caplan RJ, Soloway M, Smith JA. Independent prognostic factors in patients with metastatic (stage D2) prostate cancer. The Zoladex Study Group. Jama. 1991;265(5):618–621. [PubMed] [Google Scholar]
- 2.Roudier MP, True LD, Higano CS, Vesselle H, Ellis W, Lange P, Vessella RL. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Hum Pathol. 2003;34(7):646–653. doi: 10.1016/s0046-8177(03)00190-4. [DOI] [PubMed] [Google Scholar]
- 3.McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, Hsieh JT, Tu SM, Campbell ML. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992;52(24):6940–6944. [PubMed] [Google Scholar]
- 4.July LV, Akbari M, Zellweger T, Jones EC, Goldenberg SL, Gleave ME. Clusterin expression is significantly enhanced in prostate cancer cells following androgen withdrawal therapy. Prostate. 2002;50(3):179–188. doi: 10.1002/pros.10047. [DOI] [PubMed] [Google Scholar]
- 5.Gleave M, Jansen B. Clusterin and IGFBPs as antisense targets in prostate cancer. Ann N Y Acad Sci. 2003;1002:95–104. doi: 10.1196/annals.1281.020. [DOI] [PubMed] [Google Scholar]
- 6.Abrahamsson PA. Neuroendocrine cells in tumour growth of the prostate. Endocr Relat Cancer. 1999;6(4):503–519. doi: 10.1677/erc.0.0060503. [DOI] [PubMed] [Google Scholar]
- 7.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9(4):401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 8.Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A, Visakorpi T, Kallioniemi OP. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57(2):314–319. [PubMed] [Google Scholar]
- 9.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 10.Newmark JR, Hardy DO, Tonb DC, Carter BS, Epstein JI, Isaacs WB, Brown TR, Barrack ER. Androgen receptor gene mutations in human prostate cancer. Proc Natl Acad Sci U S A. 1992;89(14):6319–6323. doi: 10.1073/pnas.89.14.6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332(21):1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 12.Mohler JL, Gregory CW, Ford OH, 3rd, Kim D, Weaver CM, Petrusz P, Wilson EM, French FS. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10(2):440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 13.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66(5):2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 14.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54(20):5474–5478. [PubMed] [Google Scholar]
- 16.Mellon K, Thompson S, Charlton RG, Marsh C, Robinson M, Lane DP, Harris AL, Horne CH, Neal DE. p53, c-erbB-2 and the epidermal growth factor receptor in the benign and malignant prostate. J Urol. 1992;147(2):496–499. doi: 10.1016/s0022-5347(17)37287-7. [DOI] [PubMed] [Google Scholar]
- 17.Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nat Med. 1999;5(3):280–285. doi: 10.1038/6495. [DOI] [PubMed] [Google Scholar]
- 18.Unni E, Sun S, Nan B, McPhaul MJ, Cheskis B, Mancini MA, Marcelli M. Changes in androgen receptor nongenotropic signaling correlate with transition of LNCaP cells to androgen independence. Cancer Res. 2004;64(19):7156–7168. doi: 10.1158/0008-5472.CAN-04-1121. [DOI] [PubMed] [Google Scholar]
- 19.Debes JD, Schmidt LJ, Huang H, Tindall DJ. p300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res. 2002;62(20):5632–5636. [PubMed] [Google Scholar]
- 20.Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma. Prostate. 1999;39(2):135–148. doi: 10.1002/(sici)1097-0045(19990501)39:2<135::aid-pros9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 21.di Sant’ Agnese PA. Neuroendocrine differentiation in carcinoma of the prostate. Diagnostic, prognostic, and therapeutic implications. Cancer. 1992;70(1 Suppl):254–268. doi: 10.1002/1097-0142(19920701)70:1+<254::aid-cncr2820701312>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 22.Vashchenko N, Abrahamsson PA. Neuroendocrine differentiation in prostate cancer: implications for new treatment modalities. Eur Urol. 2005;47(2):147–155. doi: 10.1016/j.eururo.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Bang YJ, Pirnia F, Fang WG, Kang WK, Sartor O, Whitesell L, Ha MJ, Tsokos M, Sheahan MD, Nguyen P, et al. Terminal neuroendocrine differentiation of human prostate carcinoma cells in response to increased intracellular cyclic AMP. Proc Natl Acad Sci U S A. 1994;91(12):5330–5334. doi: 10.1073/pnas.91.12.5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonkhoff H, Stein U, Remberger K. Endocrine-paracrine cell types in the prostate and prostatic adenocarcinoma are postmitotic cells. Hum Pathol. 1995;26(2):167–170. doi: 10.1016/0046-8177(95)90033-0. [DOI] [PubMed] [Google Scholar]
- 25.Bonkhoff H, Wernert N, Dhom G, Remberger K. Relation of endocrine-paracrine cells to cell proliferation in normal, hyperplastic, and neoplastic human prostate. Prostate. 1991;19(2):91–98. doi: 10.1002/pros.2990190202. [DOI] [PubMed] [Google Scholar]
- 26.Dizeyi N, Bjartell A, Nilsson E, Hansson J, Gadaleanu V, Cross N, Abrahamsson PA. Expression of serotonin receptors and role of serotonin in human prostate cancer tissue and cell lines. Prostate. 2004;59(3):328–336. doi: 10.1002/pros.10374. [DOI] [PubMed] [Google Scholar]
- 27.Levine L, Lucci JA, 3rd, Pazdrak B, Cheng JZ, Guo YS, Townsend CM, Jr, Hellmich MR. Bombesin stimulates nuclear factor kappa B activation and expression of proangiogenic factors in prostate cancer cells. Cancer Res. 2003;63(13):3495–3502. [PubMed] [Google Scholar]
- 28.Iwamura M, di Sant’Agnese PA, Wu G, Benning CM, Cockett AT, Deftos LJ, Abrahamsson PA. Immunohistochemical localization of parathyroid hormone-related protein in human prostate cancer. Cancer Res. 1993;53(8):1724–1726. [PubMed] [Google Scholar]
- 29.Jin RJ, Wang Y, Masumori N, Ishii K, Tsukamoto T, Shappell SB, Hayward SW, Kasper S, Matusik RJ. NE-10 neuroendocrine cancer promotes the LNCaP xenograft growth in castrated mice. Cancer Res. 2004;64(15):5489–5495. doi: 10.1158/0008-5472.CAN-03-3117. [DOI] [PubMed] [Google Scholar]
- 30.Di Sant’Agnese PA, Cockett AT. The prostatic endocrine-paracrine (neuroendocrine) regulatory system and neuroendocrine differentiation in prostatic carcinoma: a review and future directions in basic research. J Urol. 1994;152(5 Pt 2):1927–1931. doi: 10.1016/s0022-5347(17)32417-5. [DOI] [PubMed] [Google Scholar]
- 31.di Sant’Agnese PA, de Mesy Jensen KL. Neuroendocrine differentiation in prostatic carcinoma. Hum Pathol. 1987;18(8):849–856. doi: 10.1016/s0046-8177(87)80060-6. [DOI] [PubMed] [Google Scholar]
- 32.Bonkhoff H, Stein U, Remberger K. Multidirectional differentiation in the normal, hyperplastic, and neoplastic human prostate: simultaneous demonstration of cell-specific epithelial markers. Hum Pathol. 1994;25(1):42–46. doi: 10.1016/0046-8177(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 33.Abrahamsson PA, di Sant’Agnese PA. Neuroendocrine cells in the human prostate gland. J Androl. 1993;14(5):307–309. [PubMed] [Google Scholar]
- 34.Angelsen A, Syversen U, Haugen OA, Stridsberg M, Mjolnerod OK, Waldum HL. Neuroendocrine differentiation in carcinomas of the prostate: do neuroendocrine serum markers reflect immunohistochemical findings? Prostate. 1997;30(1):1–6. doi: 10.1002/(sici)1097-0045(19970101)30:1<1::aid-pros1>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 35.Ismail AH, Landry F, Aprikian AG, Chevalier S. Androgen ablation promotes neuroendocrine cell differentiation in dog and human prostate. Prostate. 2002;51(2):117–125. doi: 10.1002/pros.10066. [DOI] [PubMed] [Google Scholar]
- 36.Sciarra A, Di Silverio F. Effect of nonsteroidal antiandrogen monotherapy versus castration therapy on neuroendocrine differentiation in prostate carcinoma. Urology. 2004;63(3):523–527. doi: 10.1016/j.urology.2003.10.043. [DOI] [PubMed] [Google Scholar]
- 37.Hirano D, Okada Y, Minei S, Takimoto Y, Nemoto N. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur Urol. 2004;45(5):586–592. doi: 10.1016/j.eururo.2003.11.032. discussion 592. [DOI] [PubMed] [Google Scholar]
- 38.Huss WJ, Gregory CW, Smith GJ. Neuroendocrine cell differentiation in the CWR22 human prostate cancer xenograft: association with tumor cell proliferation prior to recurrence. Prostate. 2004;60(2):91–97. doi: 10.1002/pros.20032. [DOI] [PubMed] [Google Scholar]
- 39.Cox ME, Deeble PD, Lakhani S, Parsons SJ. Acquisition of neuroendocrine characteristics by prostate tumor cells is reversible: implications for prostate cancer progression. Cancer Res. 1999;59(15):3821–3830. [PubMed] [Google Scholar]
- 40.Deeble PD, Murphy DJ, Parsons SJ, Cox ME. Interleukin-6- and cyclic AMP-mediated signaling potentiates neuroendocrine differentiation of LNCaP prostate tumor cells. Mol Cell Biol. 2001;21(24):8471–8482. doi: 10.1128/MCB.21.24.8471-8482.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim J, Adam RM, Freeman MR. Activation of the Erk mitogen-activated protein kinase pathway stimulates neuroendocrine differentiation in LNCaP cells independently of cell cycle withdrawal and STAT3 phosphorylation. Cancer Res. 2002;62(5):1549–1554. [PubMed] [Google Scholar]
- 42.Cox ME, Deeble PD, Bissonette EA, Parsons SJ. Activated 3′,5′-cyclic AMP-dependent protein kinase is sufficient to induce neuroendocrine-like differentiation of the LNCaP prostate tumor cell line. J Biol Chem. 2000;275(18):13812–13818. doi: 10.1074/jbc.275.18.13812. [DOI] [PubMed] [Google Scholar]
- 43.Juarranz MG, Bolanos O, Gutierrez-Canas I, Lerner EA, Robberecht P, Carmena MJ, Prieto JC, Rodriguez-Henche N. Neuroendocrine differentiation of the LNCaP prostate cancer cell line maintains the expression and function of VIP and PACAP receptors. Cell Signal. 2001;13(12):887–894. doi: 10.1016/s0898-6568(01)00199-1. [DOI] [PubMed] [Google Scholar]
- 44.Spiotto MT, Chung TD. STAT3 mediates IL-6-induced neuroendocrine differentiation in prostate cancer cells. Prostate. 2000;42(3):186–195. doi: 10.1002/(sici)1097-0045(20000215)42:3<186::aid-pros4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 45.Shi XB, Ma AH, Tepper CG, Xia L, Gregg JP, Gandour-Edwards R, Mack PC, Kung HJ, deVere White RW. Molecular alterations associated with LNCaP cell progression to androgen independence. Prostate. 2004;60(3):257–271. doi: 10.1002/pros.20039. [DOI] [PubMed] [Google Scholar]
- 46.Burchardt T, Burchardt M, Chen MW, Cao Y, de la Taille A, Shabsigh A, Hayek O, Dorai T, Buttyan R. Transdifferentiation of prostate cancer cells to a neuroendocrine cell phenotype in vitro and in vivo. J Urol. 1999;162(5):1800–1805. [PubMed] [Google Scholar]
- 47.Qiu Y, Robinson D, Pretlow TG, Kung HJ. Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3′-kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proc Natl Acad Sci U S A. 1998;95(7):3644–3649. doi: 10.1073/pnas.95.7.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adam RM, Kim J, Lin J, Orsola A, Zhuang L, Rice DC, Freeman MR. Heparin-binding epidermal growth factor-like growth factor stimulates androgen-independent prostate tumor growth and antagonizes androgen receptor function. Endocrinology. 2002;143(12):4599–4608. doi: 10.1210/en.2002-220561. [DOI] [PubMed] [Google Scholar]
- 49.Wang Q, Horiatis D, Pinski J. Interleukin-6 inhibits the growth of prostate cancer xenografts in mice by the process of neuroendocrine differentiation. Int J Cancer. 2004;111(4):508–513. doi: 10.1002/ijc.20286. [DOI] [PubMed] [Google Scholar]
- 50.Michalaki V, Syrigos K, Charles P, Waxman J. Serum levels of IL-6 and TNF-alpha correlate with clinicopathological features and patient survival in patients with prostate cancer. Br J Cancer. 2004;90(12):2312–2316. doi: 10.1038/sj.bjc.6601814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corcoran NM, Costello AJ. Interleukin-6: minor player or starring role in the development of hormone-refractory prostate cancer? BJU Int. 2003;91(6):545–553. doi: 10.1046/j.1464-410x.2003.04025.x. [DOI] [PubMed] [Google Scholar]
- 52.Adler HL, McCurdy MA, Kattan MW, Timme TL, Scardino PT, Thompson TC. Elevated levels of circulating interleukin-6 and transforming growth factor-beta1 in patients with metastatic prostatic carcinoma. J Urol. 1999;161(1):182–187. [PubMed] [Google Scholar]
- 53.Drachenberg DE, Elgamal AA, Rowbotham R, Peterson M, Murphy GP. Circulating levels of interleukin-6 in patients with hormone refractory prostate cancer. Prostate. 1999;41(2):127–133. doi: 10.1002/(sici)1097-0045(19991001)41:2<127::aid-pros7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 54.Nakashima J, Tachibana M, Horiguchi Y, Oya M, Ohigashi T, Asakura H, Murai M. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin Cancer Res. 2000;6(7):2702–2706. [PubMed] [Google Scholar]
- 55.Culig Z, Puhr M. Interleukin-6: A multifunctional targetable cytokine in human prostate cancer. Mol Cell Endocrinol. 2011 doi: 10.1016/j.mce.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giri D, Ozen M, Ittmann M. Interleukin-6 is an autocrine growth factor in human prostate cancer. Am J Pathol. 2001;159(6):2159–2165. doi: 10.1016/S0002-9440(10)63067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsai YT, Su YH, Fang SS, Huang TN, Qiu Y, Jou YS, Shih HM, Kung HJ, Chen RH. Etk, a Btk family tyrosine kinase, mediates cellular transformation by linking Src to STAT3 activation. Mol Cell Biol. 2000;20(6):2043–2054. doi: 10.1128/mcb.20.6.2043-2054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grossmann ME, Huang H, Tindall DJ. Androgen receptor signaling in androgen-refractory prostate cancer. J Natl Cancer Inst. 2001;93(22):1687–1697. doi: 10.1093/jnci/93.22.1687. [DOI] [PubMed] [Google Scholar]
- 59.Kim J, Adam RM, Solomon KR, Freeman MR. Involvement of cholesterol-rich lipid rafts in interleukin-6-induced neuroendocrine differentiation of LNCaP prostate cancer cells. Endocrinology. 2004;145(2):613–619. doi: 10.1210/en.2003-0772. [DOI] [PubMed] [Google Scholar]
- 60.Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300(5628):2101–2104. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- 61.Murakami M, Hibi M, Nakagawa N, Nakagawa T, Yasukawa K, Yamanishi K, Taga T, Kishimoto T. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science. 1993;260(5115):1808–1810. doi: 10.1126/science.8511589. [DOI] [PubMed] [Google Scholar]
- 62.Benigni F, Fantuzzi G, Sacco S, Sironi M, Pozzi P, Dinarello CA, Sipe JD, Poli V, Cappelletti M, Paonessa G, Pennica D, Panayotatos N, Ghezzi P. Six different cytokines that share GP130 as a receptor subunit, induce serum amyloid A and potentiate the induction of interleukin-6 and the activation of the hypothalamus-pituitary-adrenal axis by interleukin-1. Blood. 1996;87(5):1851–1854. [PubMed] [Google Scholar]
- 63.Ihle JN. Cytokine receptor signalling. Nature. 1995;377(6550):591–594. doi: 10.1038/377591a0. [DOI] [PubMed] [Google Scholar]
- 64.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334 ( Pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chung TD, Yu JJ, Spiotto MT, Bartkowski M, Simons JW. Characterization of the role of IL-6 in the progression of prostate cancer. Prostate. 1999;38(3):199–207. doi: 10.1002/(sici)1097-0045(19990215)38:3<199::aid-pros4>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 66.Lou W, Ni Z, Dyer K, Tweardy DJ, Gao AC. Interleukin-6 induces prostate cancer cell growth accompanied by activation of stat3 signaling pathway. Prostate. 2000;42(3):239–242. doi: 10.1002/(sici)1097-0045(20000215)42:3<239::aid-pros10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 67.Lee SO, Lou W, Hou M, de Miguel F, Gerber L, Gao AC. Interleukin-6 promotes androgen-independent growth in LNCaP human prostate cancer cells. Clin Cancer Res. 2003;9(1):370–376. [PubMed] [Google Scholar]
- 68.Nicholson SE, Hilton DJ. The SOCS proteins: a new family of negative regulators of signal transduction. J Leukoc Biol. 1998;63(6):665–668. doi: 10.1002/jlb.63.6.665. [DOI] [PubMed] [Google Scholar]
- 69.Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, Fujita T, Kanakura Y, Komiya S, Yoshimura A. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387(6636):921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 70.Campbell JS, Prichard L, Schaper F, Schmitz J, Stephenson-Famy A, Rosenfeld ME, Argast GM, Heinrich PC, Fausto N. Expression of suppressors of cytokine signaling during liver regeneration. J Clin Invest. 2001;107(10):1285–1292. doi: 10.1172/JCI11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dogusan Z, Hooghe-Peters EL, Berus D, Velkeniers B, Hooghe R. Expression of SOCS genes in normal and leukemic human leukocytes stimulated by prolactin, growth hormone and cytokines. J Neuroimmunol. 2000;109(1):34–39. doi: 10.1016/s0165-5728(00)00300-3. [DOI] [PubMed] [Google Scholar]
- 72.Lee SO, Chun JY, Nadiminty N, Lou W, Gao AC. Interleukin-6 undergoes transition from growth inhibitor associated with neuroendocrine differentiation to stimulator accompanied by androgen receptor activation during LNCaP prostate cancer cell progression. Prostate. 2007;67(7):764–773. doi: 10.1002/pros.20553. [DOI] [PubMed] [Google Scholar]
- 73.Hobisch A, Ramoner R, Fuchs D, Godoy-Tundidor S, Bartsch G, Klocker H, Culig Z. Prostate cancer cells (LNCaP) generated after long-term interleukin 6 (IL-6) treatment express IL-6 and acquire an IL-6 partially resistant phenotype. Clin Cancer Res. 2001;7(9):2941–2948. [PubMed] [Google Scholar]