

Abstract
We recently reported that cranial bones of Fgfr2C342Y/+ craniosynostotic mice are diminished in density when compared to those of wild type mice, and that cranial bone cells isolated from the mutant mice exhibit inhibited late stage osteoblast differentiation. To provide further support for the idea that craniosynostosis-associated Fgfr mutations lead to cell autonomous defects in osteoblast differentiation and mineralized tissue formation, here we tested bone marrow stromal cells isolated from Fgfr2C342Y/+ mice for their ability to differentiate into osteoblasts. Additionally, to determine if the low bone mass phenotype of Crouzon syndrome includes the appendicular skeleton, long bones were assessed by micro CT. Fgfr2C342Y/+ cells showed increased osteoblastic gene expression during early osteoblastic differentiation but decreased expression of alkaline phosphatase mRNA and enzyme activity, and decreased mineralization during later stages of differentiation, when cultured under 2D in vitro conditions. Cells isolated from Fgfr2C342Y/+ mice also formed less bone when allowed to differentiate in a 3D matrix in vivo. Cortical bone parameters were diminished in long bones of Fgfr2C342Y/+ mice. These results demonstrate that marrow stromal cells of Fgfr2C342Y/+ mice have an autonomous defect in osteoblast differentiation and bone mineralization, and that the Fgfr2C342Y mutation influences both the axial and appendicular skeletons.
1. Introduction
Craniosynostosis is a debilitating pediatric condition characterized by the premature fusion of cranial bones. This fusion leads to high intracranial pressure and abnormal skull and facial shapes presumably resulting from limited growth at fused craniofacial sutures with compensating overgrowth at nonfused cranial sutures [1–5]. Untreated craniosynostosis can lead to blindness, seizures, and death [6–10]. Current treatment options for craniosynostosis and its associated craniofacial abnormalities are limited to surgery with genetic counseling, orthodontic, medical, and social support [11]. Notably, even with an appropriately early and accurate diagnosis, craniosynostosis can carry high morbidity, with some patients requiring multiple surgeries throughout infancy and childhood for treatment of recurring craniosynostosis and normalization of skull and facial shapes [12].
It has been known for over a decade that craniosynostosis occurs in association with mutations in the genes for fibroblast growth factor receptors (Fgfr's). Mutations in Fgfr2 cause Apert, Crouzon, Jackson-Weiss, and Pfeiffer craniosynostosis syndromes, while mutations in Fgfr1 cause Pfeiffer syndrome and mutations in Fgfr3 cause Muenke craniosynostosis syndrome and Crouzon syndrome with acanthosis nigricans [13–17]. Numerous prior studies have shown that these craniosynostosis-associated Fgfr mutations lead to a gain of function in terms of Fgfr signaling [18–22]. Yet, despite our extensive knowledge in the genetics underlying syndromic craniosynostosis, the pathogenesis by which mutations in Fgfr's lead to craniosynostosis has yet to be fully elucidated. Because Fgf/Fgfr signaling is an important form of intercellular communication, it has been proposed that craniosynostosis-associated mutations in Fgf receptors lead to craniosynostosis by causing inappropriate signaling to cranial cells from neighboring tissues such as the dura mater [23–25]. In contrast, it has also been proposed that craniosynostosis-associated mutations in Fgf receptors lead to intrinsic defects in the behavior of cranial bone cells and tissues [26–28]. Importantly, while the biologic pathogenesis of craniosynostosis remains unknown, the only treatment for craniosynostosis will remain that of surgical intervention.
To advance our understanding of mechanisms that lead to craniosynostosis, we are investigating the Fgfr2C342Y/+ mouse model of Crouzon syndrome. Common features of Crouzon syndrome in humans include coronal suture synostosis with rare pansynostosis, hypertelorism, severe ocular proptosis, strabismus, hypoplastic maxilla, and relative mandibular prognathism [11, 29]. Notably, high rates of stylohyoid ligament calcification and vertebral fusions, as well as the occasional fusion of limb joints, have also been reported [30–32]. Mice carrying the classic Crouzon syndrome associated Fgfr2C342Y mutation were initially reported to have characteristics similar to those of Crouzon syndrome patients including a dome-shaped skull, wide set and proptotic eyes, premature fusion of cranial sutures, and a shortened maxilla [32]. Notably, fusions were also evident between the femur and tibia and between cervical vertebral arches in homozygous mutant mice. The homozygous mutant mice also lacked vertebral body ossification. These findings indicate that the Crouzon syndrome phenotype involves both the axial and appendicular skeletons. The findings also suggest that the bony fusions of Crouzon syndrome occur in the context of diminished eutopic bone formation.
We recently reported that the frontal bones of Crouzon Fgfr2C342Y/+ mice are diminished in bone volume and density when compared to those of wild type mice, and that while frontal bone cells isolated from the mutant mice exhibit increased osteoblastic gene during early stages of osteoblast differentiation, the cells are also diminished in their ability to differentiate into mature osteoblasts in vitro [5]. To provide further support for the idea that the Crouzon syndrome associated Fgfr2C342Y mutation leads to intrinsic changes in osteoblast differentiation and bone formation, we isolated cells from Crouzon mice and assayed their ability to differentiate into osteoblasts and form mineralized tissue, as compared to cells isolated from wild type mice. Because cell culture in a three-dimensional matrix supports a more physiologically relevant environment than more traditional two-dimensional cell culture methods [33], we assayed cells in traditional in vitro monolayer culture and in a three-dimensional collagenous matrix in vivo. Additionally, to determine if the appendicular skeleton is altered in Fgfr2C342Y/+ mice, we used microcomputed tomography to quantify parameters of tibial bone quality and quantity. Here we report that the Fgfr2C342Y mutation enhances expression of osteoblastic genes during early stages of differentiation while inhibiting expression of alkaline phosphatase enzyme (AP/Tnap/Alpl/Akp2) and mineralization during later differentiation, in two-dimensional in vitro cell culture. Here we also show that the Fgfr2C342Y mutation inhibits the ability of these cells to express Tnap enzyme and form mineralized tissue when allowed to differentiate in a three-dimensional matrix in vivo. Additionally, here we report for the first time that the long bones of Fgfr2C342Y/+ mice have significantly diminished cortical bone quality and quantity, when compared to those of wild type mice. Together, these results demonstrate that the Crouzon syndrome associated Fgfr2C342Y mutation causes cell autonomous abnormalities in osteoblast differentiation that include enhanced early differentiation but inhibited later expression of Tnap enzyme and mineralization by bone marrow stromal cells. Results also show that Crouzon syndrome is associated with significantly diminished appendicular bone volume and density.
2. Experimental Procedures
2.1. Animals
Fgfr2C342Y/+ and wild type mice were genotyped as previously described [29]. Briefly, DNA from tail digests was amplified by polymerase chain reaction using 5′-gagtaccatgctgactgcatgc-3′ and 5′-ggagaggcatctctgtttcaagacc-3′ primers. The reaction product was resolved by gel electrophoresis, yielding a 200 base pair band for wild type Fgfr2 and a 300 base pair band for Fgfr2C342Y. Fgfr2C342Y/+ and wild type mice on the Balb/C genetic background were utilized for cell isolations and for micro-CT analyses. NIH III nude mice were obtained from Charles River Laboratories International, Inc. (Wilmington, MA) and utilized as donor mice for subcutaneous implant experiments. All animal procedures were performed according to University of Michigan's University Committee on Use and Care of Animals.
2.2. Microcomputed Tomography
Tibias of Fgfr2C342Y/+ (n = 14) and wild type (n = 14) four-week-old mice were embedded in 1% agarose and scanned at the proximal metaphysis and the mid-diaphysis using a microcomputed tomography imaging system (Scanco µCT100, Scanco Medical, Bassersdorf, Switzerland). Scan settings were voxel size 12 µm, 70 kVp, 114 µA, 0.5 mm AL filter, and integration time of 500 ms. Density measurements were calibrated to the manufacturer's hydroxyapatite phantom. Analysis was performed using the manufacturer's evaluation software (Scanco µCT100, Scanco Medical, Bassersdorf, Switzerland) using a fixed global threshold of 28% (280 on a grayscale of 0–1000) to segment bone from nonbone. Micro-CT bone data was analyzed and is reported in accordance with the recommendations of Bouxsein et al., 2010 [34]. Statistical significance between groups was established by use of the Student's t-test.
2.3. Primary Cell Isolation
Bone marrow stromal cells were isolated from the long bones of four-week-old Crouzon mice and wild type littermates, as previously described [35]. Briefly, marrow cells were aspirated using a 25-gauge needle and a 5 mL syringe containing media. Marrow was flushed and cells were then dispersed by aspirating several times through a 22-gauge needle. Cells were cultured in αMEM supplemented with 20% fetal bovine serum (FBS) and 10,000 μg/mL penicillin/streptomycin for several days. Media was changed every three days until all suspension cells were removed and adherent cells were confluent.
2.4. In Vitro Osteoblast Differentiation
Cells were induced to differentiate in vitro by culture in media containing 50 μg/mL ascorbate for the indicated number of days. RNA was isolated using Trizol reagent (Invitrogen) following manufacturer protocols. mRNA levels were assayed by reverse transcription and real-time PCR. Real-time PCR was performed utilizing the murine tissue nonspecific alkaline phosphatase (TNAP) primer/probe set Mm00475834_m1, the murine bone sialoprotein (BSP) primer/probe set Mm00492555_m1, the murine osteocalcin (OCN) primer/probe set Mm03413826_mH, the murine runt related transcription factor 2 (Runx2) primer/probe set Mm00501578_m1, the murine collagen type I, alpha 1 (col1a1) primer/probe set Mm00801666_g1, the murine hypoxanthine phosphoribosyltransferase-1 (Hprt1) primer/probe set Mm01545399_m1, and Taqman Universal PCR Master Mix (Applied Biosystems). Real-time PCR was performed on a GeneAmp 7700 thermocycler (Applied Biosystems) and quantified by comparison to a standard curve. mRNA levels are reported after normalization to Hprt1 mRNA levels. Cells were induced to form mineral by addition of 10 mM β-glycerophosphate. Mineralized nodules were stained by Von Kossa. Briefly, cells were rinsed with phosphate-buffered saline, fixed with 100% ethanol and rehydrated in a graded ethanol series. Cells were then incubated in 5% AgNO3, rinsed with dH2O and exposed to light for 1 hour. For quantification, wells were scanned and densitometry was measured using NIH Image software. Tissue non-specific alkaline phosphatase (AP) enzyme activity was assayed using the colorimetric substrate, NBT/BCIP (Sigma). Cells were fixed in 70% ethanol for 10 minutes at room temperature, air dried, and incubated with substrate for 1 hour at 37C. Cells were then rinsed with dH2O, air dried, and visualized macroscopically for evidence of staining. For quantification, wells were scanned and densitometry was measured using NIH Image software. Statistical significance between genotypes for mRNA levels, quantification of mineralization, and quantification of AP enzyme activity was established by use of the Student's t-test.
2.5. Subcutaneous Implant Preparation and Analysis of Ossicles
Subcutaneous implants were prepared as previously described [36]. 4 × 107 bone marrow stromal cells were mixed with 0.01% NaOH in phosphate buffered saline and 1ml of rat tail collagen solution (BD Biosciences, San Jose, CA) on ice. The solution was aliquoted into glass tissue culture wells (Lab-Tek 16 well chamber slide system; Nalge Nunc International, Rochester, NY) and then incubated at 37°C for 1 hour to allow for gel hardening. Midline longitudinal incisions were made along the dorsal surface of each six-week-old NIH III mouse, and subcutaneous pockets were formed laterally, by gentle blunt dissection. A single implant was placed into each subcutaneous pocket, for a total of six implants per animal (Figure 1). Implants were removed eight weeks after implantation and analyzed for mineralized tissue formation by radiography (Faxitron MX-20, Faxitron Bioptics LLC, Tucson, AZ). All implants were imaged on the same film. Mineralized tissue of twelve implants from each genotype was quantified by densitometry (ImageJ, NIH). Statistical significance between genotypes was established by use of the Student's t-test. Implants were then homogenized for alkaline phosphatase measurements or decalcified and embedded in paraffin for histologic analysis by trichrome or hematoxylin and eosin staining. Alkaline phosphatase enzyme activity of implants was measured by homogenizing the implants in a solution containing 1.6 M MgCl2, 0.2 M Tris-Cl pH 8.1, and 1% triton X-100 followed by incubation of lysate with 7.5 mM of 4-nitrophenyl-phosphate at room temperature for 1 hour. Absorbance at 405 nm was measured and results were normalized to DNA content. Statistical significance between genotypes was established by use of the Student's t-test.
Figure 1.

Subcutaneous implant placement. This schematic shows the position of six implants that were placed subcutaneously, on the dorsal surface of immunodeficient mice.
3. Results
3.1. Animals
The Crouzon Fgfr2C342Y/+ mutant mice show a phenotype similar to that which was previously reported [5, 32, 37]. The mutant mice are slightly smaller in body size than their wild type littermates and exhibit craniofacial abnormalities associated with craniosynostosis including a dome-shaped skull, wide set and proptotic eyes, and severe midface hypoplasia.
3.2. In Vitro Osteoblast Differentiation
Analysis of mRNA levels demonstrates that bone marrow stromal cells isolated from Crouzon mice express significantly higher levels of Runx2 and tissue non-specific alkaline phosphatase enzyme (AP) at days 1 and 3 of differentiation, significantly higher levels of bone sialoprotein at day 3 of differentiation, and significantly higher levels of osteocalcin at days 1, 3, and 6 of differentiation (Figures 2(a), 2(b), 2(c), and 2(d)). This data indicates that the Fgfr2C342Y mutation enhances the expression of osteoblastic genes in bone marrow stromal cells during early stages of osteoblast differentiation. In contrast, Runx2 and tissue non-specific alkaline phosphatase mRNA levels were significantly lower in cells isolated from Crouzon mice at days 12 and 18 of differentiation (Figures 2(a) and 2(b)). Cells isolated from Crouzon mice also exhibited less alkaline phosphatase enzyme activity at day 18 of differentiation and mineralized to a diminished extent than cells isolated from wild type littermates (Figures 2(f) and 2(g)). In combination, this data suggests that the Fgfr2C342Y mutation may inhibit later stages of osteoblast differentiation and inhibit mineralization. Notably, collagen type 1, alpha 1 mRNA expression levels were not different between Crouzon and wild type cells at any stages of differentiation. This data suggests that the diminished mineralization by Crouzon cells is not likely due to diminished collagen expression. Together, these results indicate that while the Fgfr2C342Y mutation may enhance early osteoblast differentiation, it also appears to inhibit expression of tissue non-specific alkaline phosphatase and mineralization by more differentiated cells. The findings also indicate that the Fgfr2C342Y mutation induces autonomous abnormalities in osteoblast differentiation when bone marrow stromal cells are cultured in a conventional two-dimensional monolayer system in vitro.
Figure 2.
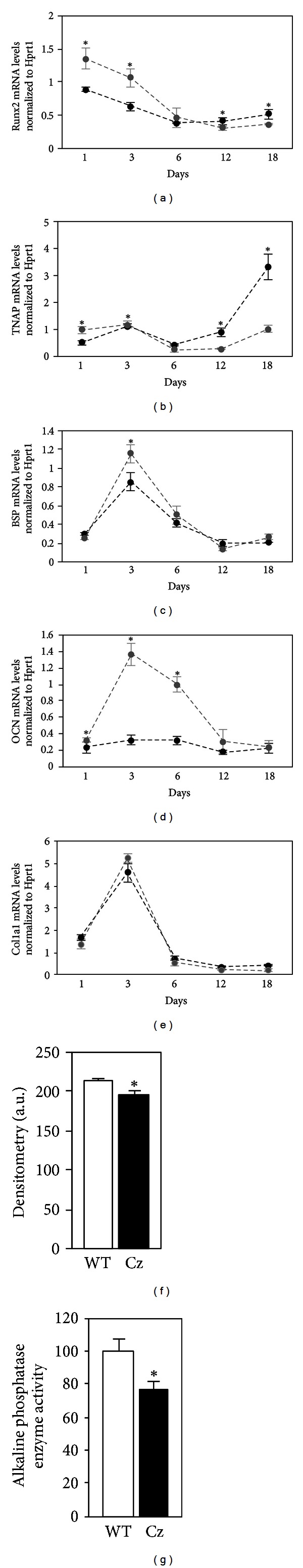
The Fgfr2C342Y mutant bone marrow stromal cells exhibit abnormal osteoblastic gene expression and diminished mineralization in vitro. Bone marrow stromal cells were isolated from Crouzon Fgfr2C342Y/+ (Cz) and wild type (WT) littermates and then cultured with ascorbate for the indicated number of days to induce osteoblast differentiation. Runx2, bone sialoprotein (BSP), osteocalcin (OCN), and tissue non-specific alkaline phosphatase (TNAP) and collagen type 1 alpha1 (col1a1) mRNA levels were measured by real-time PCR. Black lines represent wild type; grey lines represent Crouzon (a, b, c, d, and e). Results are presented as normalized to Hprt1. Cells were cultured with ascorbate and β-glycerophosphate to induce mineralization for 18 days (f). Mineralized nodules were stained by Von Kossa and quantified by densitometry. Cells were cultured with ascorbate for 18 days, and alkaline phosphatase (Tnap/Alpl/Akp2) enzyme activity was quantified by incubation of cells with a colorimetric substrate. Enzyme activity was quantified by densitometry (g). Results shown are means ± standard deviations from triplicate experiments for all data shown. *P < .05 between genotypes.
3.3. In Vivo Mineralized Tissue Formation
Because a three-dimensional matrix promotes a more physiologically relevant cellular state than conventional in vitro monolayer cell culture [33, 38–40], we next assayed the cells when allowed to differentiate in a collagenous matrix in vivo. In accordance with the in vitro data, bone marrow stromal cells isolated from Crouzon mice formed significantly less mineralized tissue than cells isolated from wild type mice, when allowed to differentiation in a three-dimensional collagenous matrix in vivo. Both trichrome (bone tissue stains deep blue and implanted collagen stains lighter blue) and H&E histologic stains (bone tissue stains pink and implanted collagen stains greyish-pink) indicate that the ossicles formed by Crouzon cells may contain less mineralized tissue than ossicles formed by wild type cells (Figure 3). Notably, the marrow of implants prepared using either Crouzon or wild type cells contains both hematopoietic and adipocytic cells (small empty ovals are adipocyte ghosts). This may indicate that the Fgfr2C342Y mutation does not influence hematopoietic or adipocytic cell differentiation, although more comprehensive analyses of these cells types in both heterozygous and homozygous mutant mice are required to definitely determine if this is the case. Radiographs of representative implants dissected eight weeks after implantation reveal apparently diminished radiodensity of ossicles prepared using cells isolated from the mutant, as compared to the wild type mice (Figure 4(a)). Quantification of mineralized tissue confirms that the mutant cell implants have significantly less mineralized tissue than the wild type cell implants (Figure 4(b)). Finally, alkaline phosphatase enzyme expression, which is essential for mineral deposition in collagenous tissues [41] was found to be significantly lower in implants prepared using mutant than wild type cells (Figure 4(c)). Together, these results demonstrate that the Fgfr2C342Y mutation inhibits alkaline phosphatase enzyme expression and the formation of mineralized tissue by osteoprogenitor cells when allowed to differentiate in a three-dimensional matrix in vivo.
Figure 3.
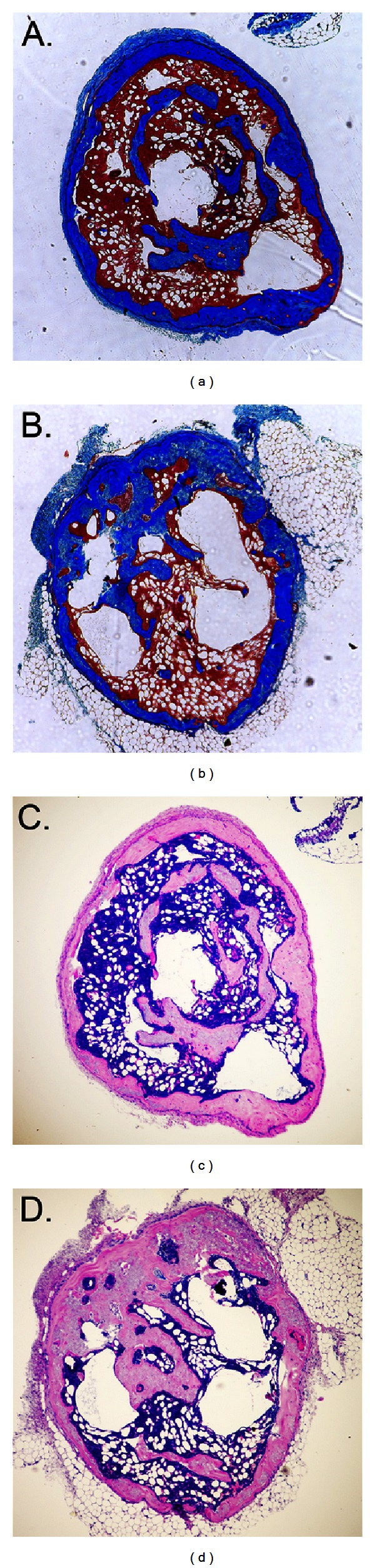
Histologic staining of implants. Ossicles formed eight weeks after subcutaneous implantation of cells mixed with a collagenous matrix were stained by trichrome (a and b) or by hematoxylin and eosin (c and d). Note the greater amount of deep blue (a) and light pink (c) staining in ossicles formed by wild type cells, indicative of greater bone formation by these cells. In comparison, note the greater amount of light blue (b) and greyish-pink (d) staining in ossicles formed by Crouzon cells, indicative of greater amounts of original implanted collagen. Also note that ossicles formed by either wild type or mutant cells contain bone marrow.
Figure 4.
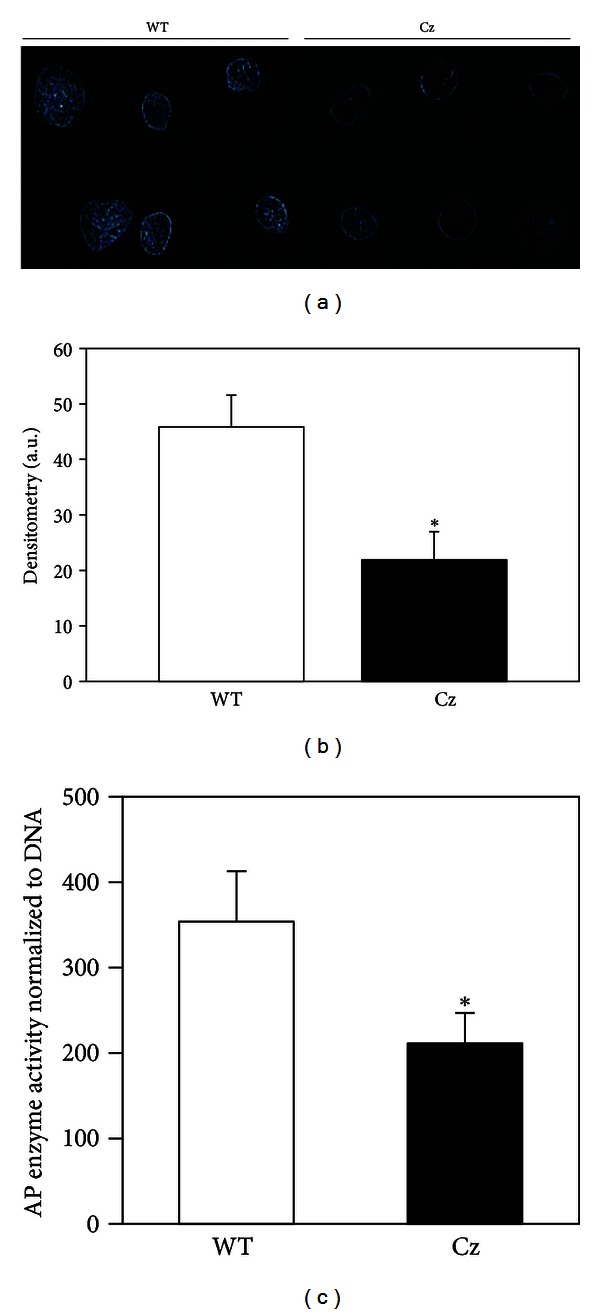
The Fgfr2C342Y mutation inhibits mineralized tissue formation in vivo. (a) Radiographic image of representative implants shows increased radiodensity of ossicles formed by wild type (WT), as compared to those formed by Crouzon (Cz) cells. (b) Quantification of radiodense tissue by densitometry confirms that implanted wild type cells formed significantly greater amounts of mineralized tissue compared to mutant cells (n = 12 implants per genotype). *P < .05 versus WT. (c) Homogenized implants formed by wild type cells also have significantly greater levels of alkaline phosphatase enzyme expression than homogenized implants formed by Crouzon cells (n = 3 implants per genotype). *P < .05 versus WT.
3.4. Microcomputed Tomographic Analysis of Long Bones
We recently showed that the frontal cranial bones of Fgfr2C342Y/+ mice are diminished in bone volume and density when compared to those of wild type mice [5]. To determine if bones of the appendicular skeleton are similarly affected, we utilized microcomputed tomography to analyze parameters of bone quality and quantity in tibias of four-week-old Fgfr2C342Y/+ and Fgfr2+/+ mice. Results demonstrate that Fgfr2C342Y/+ mice have significantly diminished tibial cortical bone volume/total volume, bone mineral density, and tissue mineral density (Figure 5), when compared to FGFR2+/+ mice. Trabecular measures of tibial bone volume/total volume, bone mineral density, and tissue mineral density were not different between Fgfr2C342Y/+ and Fgfr2+/+ mice (Figure 6). These results demonstrate that the Fgfr2C342Y/+ mutation is associated with decreased bone volume and density that is not limited to the craniofacial skeleton.
Figure 5.

Diminished cortical bone volume and density in the long bones of Fgfr2C342Y/+ mice. Micro-CT analyses demonstrate significantly diminished cortical bone volume/total volume, bone mineral density, and tissue mineral density in tibias of Fgfr2C342Y/+ (Cz) mice as compared to wild type (WT) mice. *P < .05 between genotypes.
Figure 6.
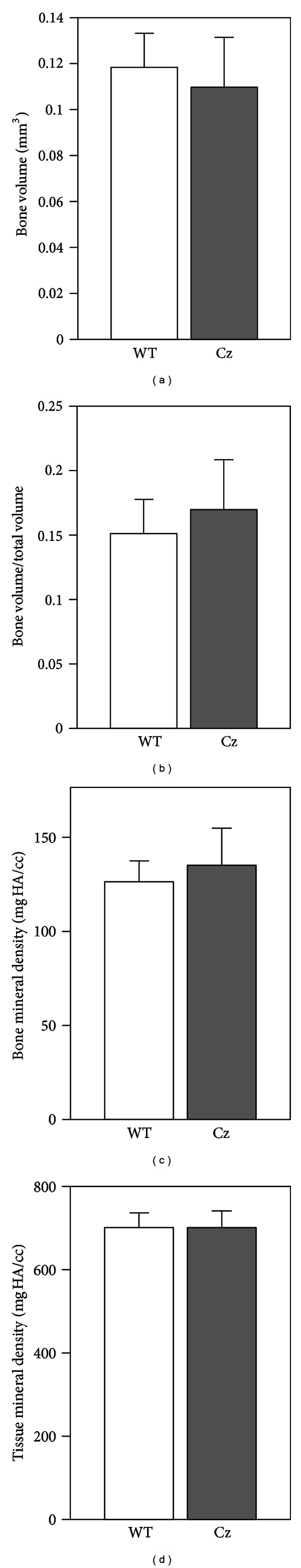
Similar trabecular bone volume and density in the long bones of Fgfr2C342Y/+ mice. Micro-CT analyses demonstrate no significant differences in bone volume, bone volume/total volume, bone mineral density, and tissue mineral density in tibias of Fgfr2C342Y/+ (Cz) mice as compared to wild type (WT) mice.
4. Discussion
The pathogenesis of craniosynostosis remains unknown and until this knowledge has been realized, the only treatment for craniosynostosis will remain that of surgical intervention. While it is not known if craniosynostosis results primarily from abnormalities in cranial bone cells, cranial suture cells and/or abnormalities in other cell types, mounting evidence does indicate that craniosynostosis occurs in the context of diminished cranial bone quantity and quality. Fgfr2S250W Apert mice and Fgfr3P244R Muenke mice were both previously shown to exhibit craniosynostosis in combination with lower levels of bone formation and/or mineralization when compared to wild type littermates [42, 43]. As noted previously, Fgfr2C342Y/C342Y Crouzon mice were originally characterized as having decreased ossification of vertebral bodies [32]. In addition, we recently reported that Fgfr2C342Y/+ mice on a BALB/c genetic background have diminished cranial bone volume and density when compared to wild type mice [5]. Abnormal BMP signaling in neural crest cells was also recently shown to cause diminished cranial bone volume and density in combination with craniosynostosis [44]. Taken together, these results appear to indicate that craniosynostosis is an abnormality involving excessive ectopic mineralization (bone formation at a temporally and spatially inappropriate location, such as the juvenile cranial suture) and not a disorder of excessive eutopic bone formation. In fact, craniosynostosis associated with several distinct genetic mutations (cited earlier) appears to occur in combination with diminished eutopic bone mass. This distinction is critical for the future development of biologically based therapeutics for the prevention and/or treatment of craniosynostosis.
Here we report that bone marrow stromal cells isolated from Crouzon Fgfr2C342Y/+ mice express significantly higher levels of some osteoblastic genes during early stages of differentiation but significantly lower levels of tissue non-specific alkaline phosphatase mRNA and enzyme activity, as well as diminished mineralization when cells are further differentiated in a 2D in vitro cell culture system. These results are in accordance with our previous report which showed that frontal bone cells isolated from Fgfr2C342Y/+ mice exhibited enhanced early osteoblastic differentiation but diminished later stage differentiation and a decreased tendency to form mineralized tissue, when compared to cells isolated from wild type mice in vitro [5]. That the Fgfr2C342Y mutation stimulates early osteoblast differentiation while inhibiting later maturation into fully functional osteoblasts could explain the apparently discrepant results found in the literature regarding effects of craniosynostosis-associated FGFR mutations on osteoblast differentiation. Our in vitro data, showing that Crouzon Fgfr2C342Y marrow stromal cells express higher levels of multiple osteoblastic genes than wild type cells during early stages of differentiation, is in accordance with previous reports which showed increased osteoblastic gene expression in cranial suture tissues. Previous in vivo analyses of the Fgfr2P253R/+ and Fgfr2S252W/+ mouse models of Apert syndrome revealed increased osteoblastic gene expression around the coronal suture [45, 46]. Mice carrying the P250R mutation in Fgfr1 associated with Pfeiffer syndrome were also previously shown to exhibit enhanced osteoblastic differentiation of cells within the sagittal suture [47]. Finally, newborn Fgfr2C342Y/+ mice were also previously shown to have increased Runx2 mRNA levels around the coronal suture, as compared to wild type littermates [32]. Increased expression of osteoblastic genes in suture tissues is well reconciled with our results showing increased osteoblastic gene expression in Fgfr2C342Y/+ bone marrow stromal cells during early stages of differentiation. In contrast, our data showing that Fgfr2C342Y marrow stromal cells express significantly lower levels of Runx2 and tissue non-specific alkaline phosphatase mRNA, as well as significantly diminished alkaline phosphatase enzyme activity and mineralization, is in accordance with previous studies showing that S252W, C342Y, and P253R craniosynostosis-associated mutations in FGFR2 inhibit osteoblast differentiation [48–50]. The data can also potentially explain the diminished eutopic bone mass that is seen in multiple mouse models of craniosynostosis, including the Fgfr2C342Y mouse model of Crouzon syndrome. Taken together, it appears that craniosynostosis syndrome-associated mutations in Fgfr's enhance or inhibit osteoblast differentiation in a cell type, environment, and differentiation stage dependent manner.
Importantly, two-dimensional cell culture on plastic does not well represent the environmental conditions that cells find in physiologic tissues, and it has been suggested that three-dimensional cell culture helps to “bridge the gap” between cultured cells and the in vivo environment [33]. Therefore, to increase confidence in our in vitro findings, we also mixed bone marrow stromal cells in a three dimensional collagenous matrix and allowed them to differentiate when implanted into donor mice. Results of these experiments showed that the Crouzon Fgfr2C342Y mutation inhibited bone formation and alkaline phosphatase enzyme expression, again supporting the idea that the Fgfr2C342Y mutation inhibits later stage osteoblast differentiation and bone formation. While our results also show significantly diminished bone volume and density in the long bones of Fgfr2C342Y mice, future studies are required to definitively establish that Crouzon bone marrow stromal cells are deficient in their ability to differentiate into fully functional osteoblasts and form bone in vivo. If correct, our results suggest that patients carrying craniosynostosis syndrome associated Fgfr mutations may be at higher risk for osteoporosis and/or slow repair of long bone fractures.
While the diminished cranial bone formation in these Fgfr-associated mouse models of craniosynostosis has not been previously considered as contributing to the development of craniosynostosis, it is interesting to note that craniosynostosis is also known to occur in other disorders of low bone mineralization. Mutations in the phosphate regulating protein Phex cause X-linked hypophosphatemic rickets involving low serum phosphate, defective bone mineralization and also craniosynostosis [51–53]. It is unknown how Phex mutations lead to craniosynostosis, but, similar to studies of human patients with Fgfr2-associated craniosynostosis [30], these patients also commonly have paradoxical heterotopic calcification of normally nonmineralizing tissues, such as tendons and ligaments [53]. Craniosynostosis is also seen in infantile hypophosphatasia due to inactivating mutations in the enzyme, tissue non-specific alkaline phosphatase (TNAP) [54–56]. These patients have severely deficient bone mineralization [57]. TNAP is an enzymatic generator of inorganic phosphate and an established essential promoter of tissue mineralization, but it is again unknown how diminished TNAP activity leads to craniosynostosis [41, 58]. Finally, craniosynostosis was also previously reported to occur secondary to antacid-induced infantile hypophosphatemia [59]. This data indicates that craniosynostosis occurs in multiple disorders involving dysregulated phosphate homeostasis and bone mineralization. Notably, signaling through Fgfr's was also previously shown to regulate expression of enzymes that control the local production of inorganic phosphate [35], and here we show that cells isolated from Crouzon mice express significantly lower levels of tissue non-specific alkaline phosphatase mRNA and significantly diminished alkaline phosphatase enzyme expression. Together, these results make it tempting to hypothesize that craniosynostosis may be promoted by abnormal tissue levels of phosphate leading to aberrant tissue mineralization.
In conclusion, this study demonstrates that the Crouzon syndrome associated C342Y mutation in Fgfr2 enhances early osteoblast differentiation but inhibits later differentiation of bone marrow stromal cells into fully functional osteoblasts when cultured in a conventional in vitro monolayer system and when allowed to differentiate in a three-dimensional matrix in vivo. This study also demonstrates that the long bones of Fgfr2C342Y mice have significantly diminished bone volume and density when compared to wild type littermates. Taken together, our results indicate that Crouzon cells have an intrinsic or cell autonomous defect in osteoblast differentiation and bone formation that includes cells of the appendicular skeleton. Future studies are required to determine if Crouzon syndrome patients are at increased risk for osteoporosis and/or poor repair of bony fractures due to this abnormal cell behavior.
Conflict of Interests
The authors have no relationship with the aforementioned companies and so have no conflict of interests to report.
Acknowledgment
This work was supported by NIDCR Grant R03DE021082.
References
- 1.Renier D, Lajeunie E, Arnaud E, Marchac D. Management of craniosynostoses. Child’s Nervous System. 2000;16(10-11):645–658. doi: 10.1007/s003810000320. [DOI] [PubMed] [Google Scholar]
- 2.Seruya M, Oh A, Boyajian MJ, Posnick JC, Keating RF. Treatment for delayed presentation of sagittal synostosis: challenges pertaining to occult intracranial hypertension—clinical article. Journal of Neurosurgery. 2011;8(1):40–48. doi: 10.3171/2011.4.PEDS1160. [DOI] [PubMed] [Google Scholar]
- 3.Morriss-Kay GM, Wilkie AOM. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. Journal of Anatomy. 2005;207(5):637–653. doi: 10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kreiborg S. Craniofacial growth in plagiocephaly and Crouzon syndrome. Scandinavian Journal of Plastic and Reconstructive Surgery. 1981;15(3):187–197. doi: 10.3109/02844318109103433. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, Na HK, Wang E, Hatch NE. Further analysis of the crouzon mouse: effects of the FGFR2C342Y mutation are cranial bone dependent. Calcified Tissue International. 2013;92(5):451–466. doi: 10.1007/s00223-013-9701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okajima K, Robinson LK, Hart MA, et al. Ocular anterior chamber dysgenesis in craniosynostosis syndromes with a fibroblast growth factor receptor 2 mutation. American Journal of Medical Genetics. 1999;85(2):160–170. doi: 10.1002/(sici)1096-8628(19990716)85:2<160::aid-ajmg11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 7.Stavrou P, Sgouros S, Willshaw HE, Goldin JH, Hockley AD, Wake MJC. Visual failure caused by raised intracranial pressure in craniosynostosis. Child’s Nervous System. 1997;13(2):64–67. doi: 10.1007/s003810050043. [DOI] [PubMed] [Google Scholar]
- 8.Abe H, Ikota T, Akino M, Kitami K, Tsuru M. Functional prognosis of surgical treatment of craniosynostosis. Child’s Nervous System. 1985;1(1):53–61. doi: 10.1007/BF00706732. [DOI] [PubMed] [Google Scholar]
- 9.Shah PS, Siriwardena K, Taylor G, et al. Sudden infant death in a patient with FGFR3 P250R mutation. American Journal of Medical Genetics A. 2006;140(24):2794–2796. doi: 10.1002/ajmg.a.31517. [DOI] [PubMed] [Google Scholar]
- 10.Cohen MM, Kreiborg S. Upper and lower airway compromise in the Apert syndrome. American Journal of Medical Genetics. 1992;44(1):90–93. doi: 10.1002/ajmg.1320440121. [DOI] [PubMed] [Google Scholar]
- 11.Rasmussen SA, Yazdy MM, Frías JL, Honein MA. Priorities for public health research on craniosynostosis: summary and recommendations from a Centers for Disease Control and Prevention-sponsored meeting. American Journal of Medical Genetics A. 2008;146(2):149–158. doi: 10.1002/ajmg.a.32106. [DOI] [PubMed] [Google Scholar]
- 12.Williams JK, Cohen SR, Burstein FD, Hudgins R, Boydston W, Simms C. A longitudinal, statistical study of reoperation rates in craniosynostosis. Plastic and Reconstructive Surgery. 1997;100(2):305–310. doi: 10.1097/00006534-199708000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nature Genetics. 1994;8(1):98–103. doi: 10.1038/ng0994-98. [DOI] [PubMed] [Google Scholar]
- 14.Schell U, Hehr A, Feldman GJ, et al. Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome. Human Molecular Genetics. 1995;4(3):323–328. doi: 10.1093/hmg/4.3.323. [DOI] [PubMed] [Google Scholar]
- 15.Wilkie AOM, Slaney SF, Oldridge M, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nature Genetics. 1995;9(2):165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- 16.Ibrahimi OA, Zhang F, Eliseenkova AV, Linhardt RJ, Mohammadi M. Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Human Molecular Genetics. 2004;13(1):69–78. doi: 10.1093/hmg/ddh011. [DOI] [PubMed] [Google Scholar]
- 17.Hatch NE. FGF signaling in craniofacial biological control and pathological craniofacial development. Critical Reviews in Eukaryotic Gene Expression. 2010;20(4):295–311. doi: 10.1615/critreveukargeneexpr.v20.i4.20. [DOI] [PubMed] [Google Scholar]
- 18.Andersen J, Burns HD, Enriquez-Harris P, Wilkie AOM, Heath JK. Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Human Molecular Genetics. 1998;7(9):1475–1483. doi: 10.1093/hmg/7.9.1475. [DOI] [PubMed] [Google Scholar]
- 19.Yu K, Herr AB, Waksman G, Ornitz DM. Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(26):14536–14541. doi: 10.1073/pnas.97.26.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neilson KM, Friesel RE. Constitutive activation of fibroblast growth factor receptor-2 by a point mutation associated with Crouzon syndrome. Journal of Biological Chemistry. 1995;270(44):26037–26040. doi: 10.1074/jbc.270.44.26037. [DOI] [PubMed] [Google Scholar]
- 21.Shukla V, Coumoul X, Wang RH, Kim HS, Deng CX. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nature Genetics. 2007;39(9):1145–1150. doi: 10.1038/ng2096. [DOI] [PubMed] [Google Scholar]
- 22.Eswarakumar VP, Özcan F, Lew ED, et al. Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(49):18603–18608. doi: 10.1073/pnas.0609157103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooper GM, Durham EL, Cray JJ, Siegel MI, Losee JE, Mooney MP. Tissue interactions between craniosynostotic dura mater and bone. Journal of Craniofacial Surgery. 2012;23(3):919–924. doi: 10.1097/SCS.0b013e31824e645f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ang BU, Spivak RM, Nah HD, Kirschner RE. Dura in the pathogenesis of syndromic craniosynostosis: fibroblast growth factor receptor 2 mutations in dural cells promote osteogenic proliferation and differentiation of osteoblasts. Journal of Craniofacial Surgery. 2010;21(2):462–467. doi: 10.1097/SCS.0b013e3181cfe9a0. [DOI] [PubMed] [Google Scholar]
- 25.Slater BJ, Kwan MD, Gupta DM, Amasha RR, Wan DC, Longaker MT. Dissecting the influence of regional dura mater on cranial suture biology. Plastic and Reconstructive Surgery. 2008;122(1):77–84. doi: 10.1097/PRS.0b013e318177478c. [DOI] [PubMed] [Google Scholar]
- 26.Mangasarian K, Li Y, Mansukhani A, Basilico C. Mutation associated with Crouzon syndrome causes ligand-independent dimerization and activation of FGF receptor-2. Journal of Cellular Physiology. 1997;172(1):117–125. doi: 10.1002/(SICI)1097-4652(199707)172:1<117::AID-JCP13>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 27.Lomri A, Lemonnier J, Hott M, et al. Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. Journal of Clinical Investigation. 1998;101(6):1310–1317. [PMC free article] [PubMed] [Google Scholar]
- 28.Holmes G, Rothschild G, Roy UB, Deng CX, Mansukhani A, Basilico C. Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Developmental Biology. 2009;328(2):273–284. doi: 10.1016/j.ydbio.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Online Mendelian Inheritance in Man. MIM Number. 123500. Baltimore, Md, USA: Johns Hopkins University; 2011, http://omim.org/entry/123500. [Google Scholar]
- 30.Kreiborg S. Crouzon Syndrome. A clinical and roentgencephalometric study. Scandinavian Journal of Plastic and Reconstructive Surgery and Hand Surgery. 1981;18:1–198. [PubMed] [Google Scholar]
- 31.Proudman TW, Moore MH, Abbott AH, David DJ. Noncraniofacial manifestations of Crouzon's disease. Journal of Craniofacial Surgery. 1994;5(4):218–222. doi: 10.1097/00001665-199409000-00004. [DOI] [PubMed] [Google Scholar]
- 32.Eswarakumar VP, Horowitz MC, Locklin R, Morriss-Kay GM, Lonai P. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(34):12555–12560. doi: 10.1073/pnas.0405031101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pampaloni F, Reynaud EG, Stelzer EHK. The third dimension bridges the gap between cell culture and live tissue. Nature Reviews Molecular Cell Biology. 2007;8(10):839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- 34.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. Journal of Bone and Mineral Research. 2010;25(7):1468–1486. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 35.Hatch NE, Li Y, Franceschi RT. FGF2 stimulation of the pyrophosphate-generating enzyme, PC-1, in pre-osteoblast cells is mediated by RUNX2. Journal of Bone and Mineral Research. 2009;24(4):652–662. doi: 10.1359/JBMR.081213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krebsbach PH, Gu K, Franceschi RT, Rutherford RB. Gene therapy-directed osteogenesis: BMP-7-transduced human fibroblasts form bone in vivo . Human Gene Therapy. 2000;11(8):1201–1210. doi: 10.1089/10430340050015248. [DOI] [PubMed] [Google Scholar]
- 37.Perlyn CA, DeLeon VB, Babbs C, et al. The craniofacial phenotype of the Crouzon mouse: analysis of a model for syndromic craniosynostosis using three-dimensional microCT. Cleft Palate-Craniofacial Journal. 2006;43(6):740–747. doi: 10.1597/05-212. [DOI] [PubMed] [Google Scholar]
- 38.Birgersdotter A, Sandberg R, Ernberg I. Gene expression perturbation in vitro—a growing case for three-dimensional (3D) culture systems. Seminars in Cancer Biology. 2005;15(5):405–412. doi: 10.1016/j.semcancer.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 39.Gurkan UA, Kishore V, Condon KW, Bellido TM, Akkus O. A scaffold-free multicellular three-dimensional in vitro model of osteogenesis. Calcified Tissue International. 2011;88(5):388–401. doi: 10.1007/s00223-011-9467-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baraniak PR, McDevitt TC. Scaffold-free culture of mesenchymal stem cell spheroids in suspension preserves multilineage potential. Cell and Tissue Research. 2012;347(3):701–711. doi: 10.1007/s00441-011-1215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murshed M, Harmey D, Millán JL, McKee MD, Karsenty G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes and Development. 2005;19(9):1093–1104. doi: 10.1101/gad.1276205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Li D, Li C, Engel A, Deng CX. A Ser250Trp substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone. 2003;33(2):169–178. doi: 10.1016/s8756-3282(03)00222-9. [DOI] [PubMed] [Google Scholar]
- 43.Twigg SRF, Healy C, Babbs C, et al. Skeletal analysis of the Fgfr3 P244R mouse, a genetic model for the muenke craniosynostosis syndrome. Developmental Dynamics. 2009;238(2):331–342. doi: 10.1002/dvdy.21790. [DOI] [PubMed] [Google Scholar]
- 44.Komatsu Y, Yu PB, Kamiya N, et al. Augmentation of Smad-dependent BMP signaling in neural crest cells causes craniosynostosis in mice. Journal of Bone and Mineral Research. 2012 doi: 10.1002/jbmr.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holmes G, Rothschild G, Roy UB, Deng CX, Mansukhani A, Basilico C. Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Developmental Biology. 2009;328(2):273–284. doi: 10.1016/j.ydbio.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin L, Du X, Li C, et al. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone. 2008;42(4):631–643. doi: 10.1016/j.bone.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 47.Zhou YX, Xu X, Chen L, Li C, Brodie SG, Deng CX. A Pro250Arg substitution in mouse Fgfr1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures. Human Molecular Genetics. 2000;9(13):2001–2008. doi: 10.1093/hmg/9.13.2001. [DOI] [PubMed] [Google Scholar]
- 48.Fragale A, Tartaglia M, Bernardini S, et al. Decreased proliferation and altered differentiation in osteoblasts from genetically and clinically distinct craniosynostotic disorders. American Journal of Pathology. 1999;154(5):1465–1477. doi: 10.1016/S0002-9440(10)65401-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mansukhani A, Bellosta P, Sahni M, Basilico C. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. Journal of Bone and Mineral Research. 2000;149:1297–1308. doi: 10.1083/jcb.149.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Online Mendelian Inheritance in Man. MIM Number. 307800. Baltimore, Md, USA: Johns Hopkins University; 2011, http://omim.org/entry/307800. [Google Scholar]
- 51.Roy WA, Iorio RJ, Meyer GA. Craniosynostosis in vitamin D-resistant rickets. A mouse model. Journal of Neurosurgery. 1981;55(2):265–271. doi: 10.3171/jns.1981.55.2.0265. [DOI] [PubMed] [Google Scholar]
- 52.Sabbagh Y, Jones AO, Tenenhouse HS. PHEXdb, a locus-specific database for mutations causing X-linked hypophosphatemia. Human Mutation. 2000;16:1–6. doi: 10.1002/1098-1004(200007)16:1<1::AID-HUMU1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 53.Liang G, Katz LD, Insogna KL, Carpenter TO, MacIca CM. Survey of the enthesopathy of X-linked hypophosphatemia and its characterization in Hyp mice. Calcified Tissue International. 2009;85(3):235–246. doi: 10.1007/s00223-009-9270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Online Mendelian Inheritance in Man. MIM Number. 171760. Baltimore, Md, USA: Johns Hopkins University; 2009, http://omim.org/entry/171760. [Google Scholar]
- 55.Wenkert D, Benigno M, Mack K, McAlister W, Mumm S, Whyte M. Hypophosphatasia: prevalence of clinical problems in 175 pediatric patients. Proceedings of the American Society for Bone and Mineral Research 31st Annual Meeting; 2009; Denver, Colo, USA. Abstract A09001674. [Google Scholar]
- 56.Whyte MP. Physiological role of alkaline phosphatase explored in hypophosphatasia. Annals of the New York Academy of Sciences. 2010;1192:190–200. doi: 10.1111/j.1749-6632.2010.05387.x. [DOI] [PubMed] [Google Scholar]
- 57.Mornet E. Hypophosphatasia. Orphanet Journal of Rare Diseases. 2007;2:p. 40. doi: 10.1186/1750-1172-2-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Narisawa S, Fröhlander N, Millán JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasi. Developmental Dynamics. 1997;208:432–446. doi: 10.1002/(SICI)1097-0177(199703)208:3<432::AID-AJA13>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 59.Shetty AK, Thomas T, Rao J, Vargas A. Rickets and secondary craniosynostosis associated with long-term antacid use in an infant. Archives of Pediatrics and Adolescent Medicine. 1998;152(12):1243–1245. doi: 10.1001/archpedi.152.12.1243. [DOI] [PubMed] [Google Scholar]