

Abstract

We describe and apply a scaffold-focused virtual screen based upon scaffold trees to the mitotic kinase TTK (MPS1). Using level 1 of the scaffold tree, we perform both 2D and 3D similarity searches between a query scaffold and a level 1 scaffold library derived from a 2 million compound library; 98 compounds from 27 unique top-ranked level 1 scaffolds are selected for biochemical screening. We show that this scaffold-focused virtual screen prospectively identifies eight confirmed active compounds that are structurally differentiated from the query compound. In comparison, 100 compounds were selected for biochemical screening using a virtual screen based upon whole molecule similarity resulting in 12 confirmed active compounds that are structurally similar to the query compound. We elucidated the binding mode for four of the eight confirmed scaffold hops to TTK by determining their protein–ligand crystal structures; each represents a ligand-efficient scaffold for inhibitor design.
Introduction
Scaffold hopping is a technique used to identify compounds with similar activity to known bioactive compounds that also contain a new core structure. Several excellent reviews summarize computational methods for the identification of novel scaffolds.1−3 Scaffold hopping may be employed to move into uncharted chemical space to avoid, for example, undesirable pharmacokinetic properties, toleration issues, or crowded IP space.1−3 When applied to virtual screening, scaffold hopping can be defined as either ligand- or structure-based. A recent survey of prospective virtual screening studies shows that although more structure-based methods have been published, ligand-based methods identify compounds that are, on average, more potent.4 Ligand-based methods utilize information from known bioactive ligands to identify compounds with similar biological activity; for example, similarity searches5 based on the principle that structurally similar compounds have similar activity6 have frequently yielded scaffold hops.7−9 Descriptors for ligand-based similarity searches such as chemically advanced template search (CATS)10 have been specifically designed to identify scaffold hops. A recent review summarizes descriptors suitable for scaffold hopping.3
In order to expand the hit matter identified in our medicinal chemistry programs and broaden the chemical space available in hit follow-up, we set out to develop a ligand-based virtual screening method in which the similarity search is focused on the core scaffold of the query compound rather than on the whole molecule as implemented in previously described similarity methods.7,8 In our method compounds with scaffolds similar to the query compound are identified from large compound libraries and diverse examples of each scaffold are selected. For the efficient identification of core scaffolds in large compound libraries, we required a high-throughput data set-independent objective method. The scaffold tree11 is an example of such a method that fragments molecules by iteratively removing rings until only one ring remains; the order in which the rings are removed is based upon a set of prioritization rules. A molecule represented by the scaffold tree will have n + 1 levels (Figure 1a), where level n is the original molecule, level n – 1 is the Murcko framework12 of the molecule, and level 0 is the single remaining ring after all other rings have been removed. We have previously shown that level 1 of the scaffold tree is a useful medicinal chemistry representation of a molecular scaffold across fragment-like, lead-like, and drug-like chemical space.13
Figure 1.

(a) Query compound 1 and its scaffold tree fragmentation including the level 1 scaffold (A). (b) Tautomers/rotamers (1a and 1b) of compound 1.
In this article, we describe the application of level 1 of the scaffold tree to a scaffold-focused virtual screen and, in a prospective validation of this methodology, identify novel TTK (monopolar spindle kinase 1, MPS1) inhibitor scaffolds from a library of over two million compounds. In addition, we compare this scaffold-focused methodology to a whole molecule virtual screening protocol widely used in the literature.7,8 A total of 198 compounds selected from the scaffold-focused and whole molecule-based virtual screens were purchased for biochemical testing against TTK, and the binding modes of a set of four confirmed hit compounds were determined using protein crystallography versus TTK.
TTK was chosen as a target protein due to its well-validated and critical role in the spindle assembly checkpoint signal, a biological function originally identified by a genetic screen in budding yeast.14 Subsequently, the TTK gene has been shown to encode an essential dual-specificity kinase15,16 conserved from yeast to humans.17 TTK activity peaks at the G2/M transition and is enhanced upon activation of the spindle checkpoint with nocodazole.18,19 The importance of TTK kinase activity in spindle checkpoint activation has inspired the search for small molecule TTK inhibitors as potential cancer therapeutics. First generation inhibitors of TTK have been extensively used to elucidate the function of TTK in mitosis,20−26 and subsequent publications have highlighted potent TTK inhibitors with potential for therapeutic use in cancer treatment.27−30 However, in common with many drug discovery campaigns targeting protein kinases, extensive exploration of chemical space is often required to discover chemical series with the potential to fulfill all the in vitro and in vivo requirements of therapeutic agents. Furthermore, it is essential to explore the novelty and diversity of hit matter to increase the chances of success in a drug discovery program.
In summary, the work presented here identifies fragment-like and lead-like TTK hit matter from scaffold-focused and whole molecule-based virtual screens, respectively, and demonstrates that the scaffold-focused method has the potential to identify active compounds that are more structurally differentiated from the query compound compared to those selected using a whole molecule similarity searching method.
Methods
Query Compound and Compound Library
As our query compound, we used compound 1 (Figure 1a), a potent TTK inhibitor from our in-house drug discovery program with an IC50 of 24.1 nM (±12.6 nM, n = 19). Figure 1a shows the scaffold tree fragmentation for query compound 1; we used level 1 of the scaffold tree (A, Figure 1a) as the query scaffold for our virtual screen. Compound 1 was used as a complete molecule in the query for the comparative whole molecule virtual screen using literature 2D and 3D similarity searches.7,8
The Institute of Cancer Research compound collection used for virtual screening consists of commercially available compounds from 11 vendors: Acros,31 Asinex,32 ChemBridge,33 ChemDiv,34 Enamine,35 InterBioScreen,36 Key Organics,37 Life Chemicals,38 Maybridge,39 Specs,40 and Tocris.41 Libraries were downloaded from each of these vendors in May 2010. This database was then filtered using in-house filters to remove compounds with AlogP42 greater than 6, more than 35 heavy atoms and compounds that contain toxicophores. This resulted in a collection of 2,431,176 unique compounds.
To prepare the compound library for the virtual screen, we applied the Lipinski rule of 5 filters43 as implemented in Pipeline Pilot 7.0:44 2,221,074 compounds remained. All compounds that have A as their level 1 scaffold (46 compounds) were removed from the compound collection. This left a library containing 2,221,028 compounds hereafter referred to as the Compound Library (Figure 2). The level 1 scaffold of each compound in the Compound Library was determined using the scaffold tree11,12 by applying the Linear Fragmentation function in MOE.45 Each unique level 1 scaffold was kept while also retaining the identity of all compounds represented by that scaffold. This gave 103,110 unique level 1 scaffolds hereafter referred to as the Scaffold Library (Figure 2). The Compound Library and Scaffold Library were used for the whole molecule and scaffold-focused virtual screens, respectively, with the aim of selecting a total of 200 molecules (see below).
Figure 2.

Scaffold-focused virtual screen (solid arrows) and whole molecule-based virtual screen (dashed arrows) as described in the Methods section.
Similarity Methods
Both the scaffold-focused and whole molecule-based virtual screens use a two-dimensional (2D) and three-dimensional (3D) similarity search. The 2D similarity was calculated in Pipeline Pilot 7.0 using the Fingerprint Similarity component44 with ECFP_4 fingerprints46 and the Tanimoto coefficient.47,48 ECFP_4 fingerprints are extended-connectivity fingerprints; this is a 2D method that describes the identity and connectivity of atoms in a molecule. For each atom in a molecule, a substructure of up to four bonds in diameter is described with the atom in question at the center. Scaffolds or whole molecules were then ranked by their Tanimoto coefficient. 3D Similarity was evaluated using the Rapid Overlay of Chemical Structures (ROCS) software.49 ROCS is a 3D method that matches the shape of a molecule to the shape of the query molecule. ROCS also incorporates pharmacophoric features in assessing overlays such that the TanimotoCombo score in ROCS measures the similarity of the matched shapes as well as the matched pharmacophoric features. ROCS requires a 3D compound input; therefore, OMEGA50 was used to generate a maximum of 50 conformers for each scaffold or molecule. During the ROCS similarity search, scaffolds or whole molecules were represented as a multiple conformer molecule to allow ROCS to compare all conformers of the query to all conformers of the library scaffolds or whole molecules and output the single best overlay. The TanimotoCombo parameter in ROCS was used to score and rank the overlays.
Mean Pairwise Similarity (MPS) was used to evaluate the similarity of compounds retrieved using the whole molecule and scaffold-focused virtual screens. MPS is a quantitative measure of chemical diversity across a set of compounds; here, we used MDL Public Keys51 and the Tanimoto coefficient to calculate the similarity between every pair of compounds in each set of active compounds and then calculated the mean of these similarities. MPS can range between 0 and 1 with a low value indicating a set of diverse compounds and a high value indicating a set of highly similar compounds.
Scaffold-Focused Virtual Screen
Both 2D and 3D scaffold similarity searches were performed between the query scaffold and the Scaffold Library. For each search, the Scaffold Library was then ranked from the most similar to the least similar scaffold. Up to five compounds represented by the top-ranked scaffolds were retained to adequately sample the chemical space representative of at least 20 scaffolds. This process was repeated down the ranked list of scaffolds until 100 compounds had been selected (Figure 2).
2D Scaffold Similarity Search
The list of scaffolds ranked by their Tanimoto coefficient to query A was interrogated from the most similar to the least similar. Pipeline Pilot 7.0 was used to retrieve up to five compounds representing each prioritized scaffold. These compounds were selected using the Diverse Molecules component in Pipeline Pilot 7.0 such that selected compounds have a diverse range of molecular weight to ensure that both fragment-like and substituted lead-like representations of the scaffold were included.
3D Scaffold Similarity Search
We used OMEGA50 to generate a maximum of 50 conformers for the query scaffold and each level 1 scaffold in the library; 838 library scaffolds failed in OMEGA. For scaffold A, only one conformer was generated in OMEGA. The TanimotoCombo parameter to query A in ROCS was used to rank the scaffolds, and up to five compounds were retrieved for each level 1 scaffold as described for the 2D similarity search.
We selected a total of 105 compounds from 28 unique level 1 scaffolds. Compounds represented by the top 16 scaffolds from both the 2D and 3D similarity searches were selected for purchase and experimental validation (four scaffolds were in the top 16 for both 2D and 3D searches). Ten compounds were no longer available from vendors; therefore, a similarity search using ECFP_4 fingerprints was used to select analogs of these 10 compounds that contained the same level 1 scaffold. Of these 10 compounds, two could not be replaced (no available compounds with the same scaffold); therefore, 103 compounds were ordered representative of 27 unique level 1 scaffolds. A further five compounds were unavailable after ordering from vendors. Therefore, 98 compounds from 27 unique level 1 scaffolds were received for testing; 48 compounds came from highly ranked level 1 scaffolds in the 2D search, 28 came from highly ranked level 1 scaffolds in the 3D search, and 22 came from six level 1 scaffolds that ranked highly in both 2D and 3D searches. Of these 98 compounds, 44 had a molecular weight less than 300 Da and were classed as fragment-like.
Whole Molecule Virtual Screen
The whole molecule virtual screen was based on previously published 2D and 3D similarity searching methods shown to have scaffold hopping ability in the literature.7,8 A similarity search was performed between the query compound 1 and the Compound Library. Hit compounds were ranked by their similarity to query 1, and a set number of compounds from the top-ranked list were selected for biological testing (Figure 2). Both 2D and 3D similarity to query molecule 1 was calculated, consistent with the scaffold-focused virtual screen protocol described above.
For the 3D similarity search, a maximum of 50 conformers were generated for each compound in the Compound Library using OMEGA; 1909 library compounds failed in OMEGA. The conformation of compound 1 was obtained from its co-crystal structure with the kinase domain of TTK. Two pyrazole tautomers/rotamers (1a and 1b, Figure 1b) could not be clearly distinguished in the co-crystal structure; therefore, we considered both forms for the whole molecule virtual screen while retaining the overall conformation of compound 1 from its co-crystal structure. While the two forms are identical when represented by a 2D fingerprint, this is not the case when compound 1 is represented in a specific 3D conformation. Therefore, we used both forms for the 3D similarity searches (ROCS searches were performed using 1a and 1b as independent queries). For both 2D and 3D searches, the Compound Library was ranked by TanimotoCombo.
The top 50 compounds from the 2D similarity searches and the top 54 compounds from the 3D similarity searches were selected for purchase, giving a total of 102 unique compounds (two compounds were present in both the 2D and 3D similarity searches). Of these compounds, 15 were no longer available from vendors and were replaced with the next most similar compound from the respective 2D or 3D similarity search. A further two compounds were unavailable after ordering from the vendors giving a total of 100 compounds for testing from the whole molecule-based virtual screen. Of the 100 compounds selected, 14 had a molecular weight less than 300 Da and were classed as fragment-like.
Prospective Validation
A total of 98 and 100 compounds selected using our scaffold-focused virtual screen and the comparator whole molecule virtual screen, respectively, were tested in a biochemical TTK assay at a single concentration. Active compounds were confirmed by IC50 determination and co-crystal structures were obtained for four active compounds from the scaffold-focused virtual screen (Experimental Section).
Results and Discussion
We present here a prospective study to compare a scaffold-focused virtual screen with a comparator whole molecule virtual screen. The comparator method we used has previously been shown to be useful for the identification of active compounds with structurally different scaffolds to a query compound using 2D and 3D similarity searches.7,8
A total of 98 compounds selected using our scaffold-focused virtual screen and 100 compounds selected using the comparator whole molecule virtual screen were tested in a biochemical TTK assay (Experimental Section). All compounds were tested at a concentration of 40 μM, and compounds defined as fragment-like (molecular weight <300 Da) were also tested at 400 μM in recognition of the higher concentration required to detect ligand-efficient fragments in a biochemical screen. Compounds with a percentage inhibition ≥50% at either concentration were confirmed by IC50 determination. IC50 values and ligand efficiencies52 for compounds displaying ≥50% inhibition at 40 or 400 μM are shown in Table 1 for the scaffold-focused virtual screen and Table 2 for the whole molecule virtual screen.
Table 1. Biochemical Assay Results, IC50, and Ligand Efficiencies (LE) for Compounds Derived from the Scaffold-Focused Virtual Screen, with ≥50% Inhibition at 40 or 400 μMa.
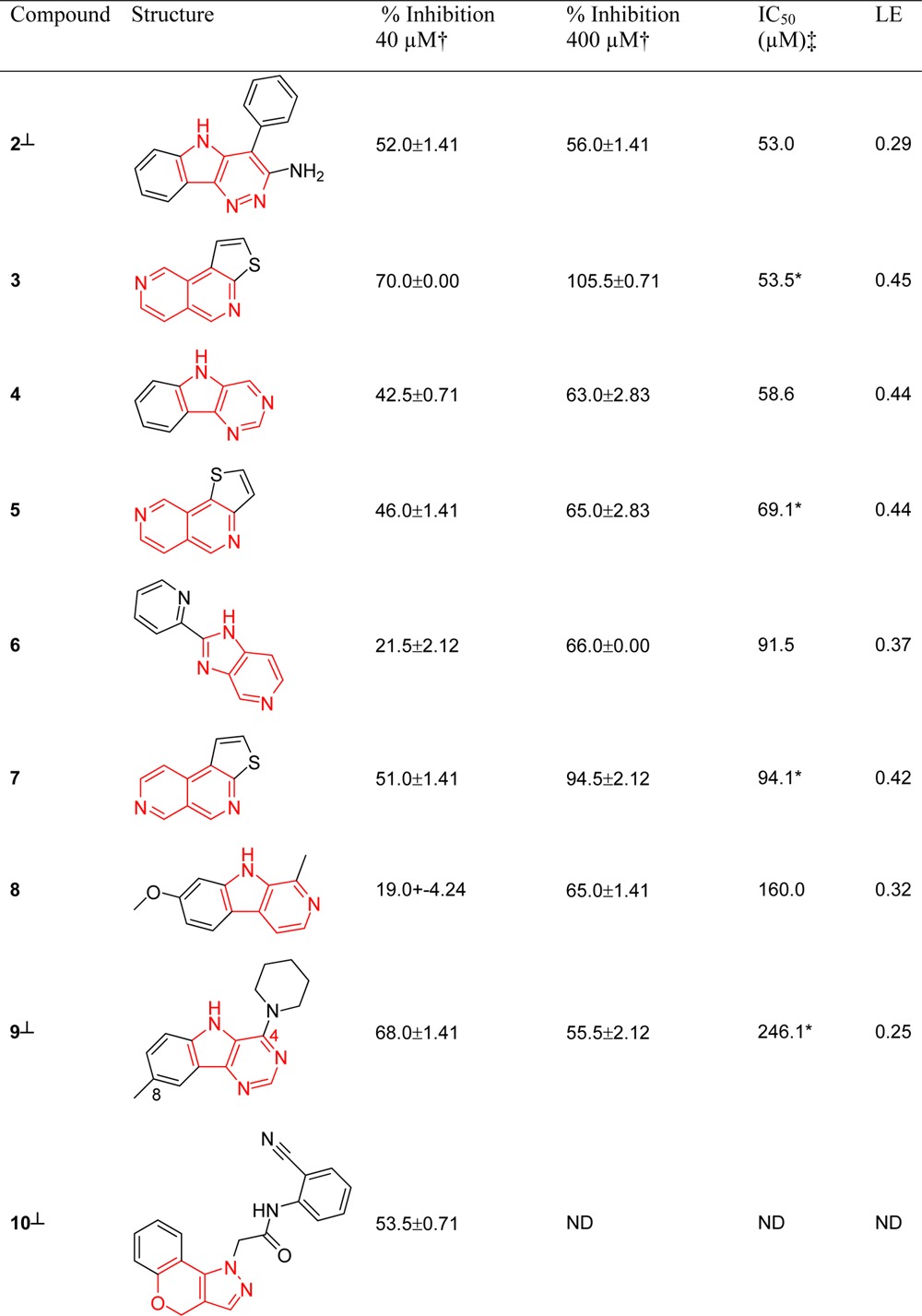
ND = not determined.
Single point assays were performed in duplicate; percent inhibition values shown are the mean of the two results reported with the standard deviation.
IC50 experiments were performed in duplicate on the same assay plate at 1% and 10% DMSO. An asterisk indicates compounds tested at 10% DMSO; all other compounds were tested at 1% DMSO.
Percent inhibition and IC50 values do not correlate for these compounds due to poor solubility at higher concentrations. The level 1 scaffold for each compound is highlighted in red.
Table 2. Biochemical Assay Results IC50 and Ligand Efficiencies (LE) for Compounds Derived from the Whole Molecule Virtual Screen, with ≥50% Inhibition at 40 or 400 μMa.
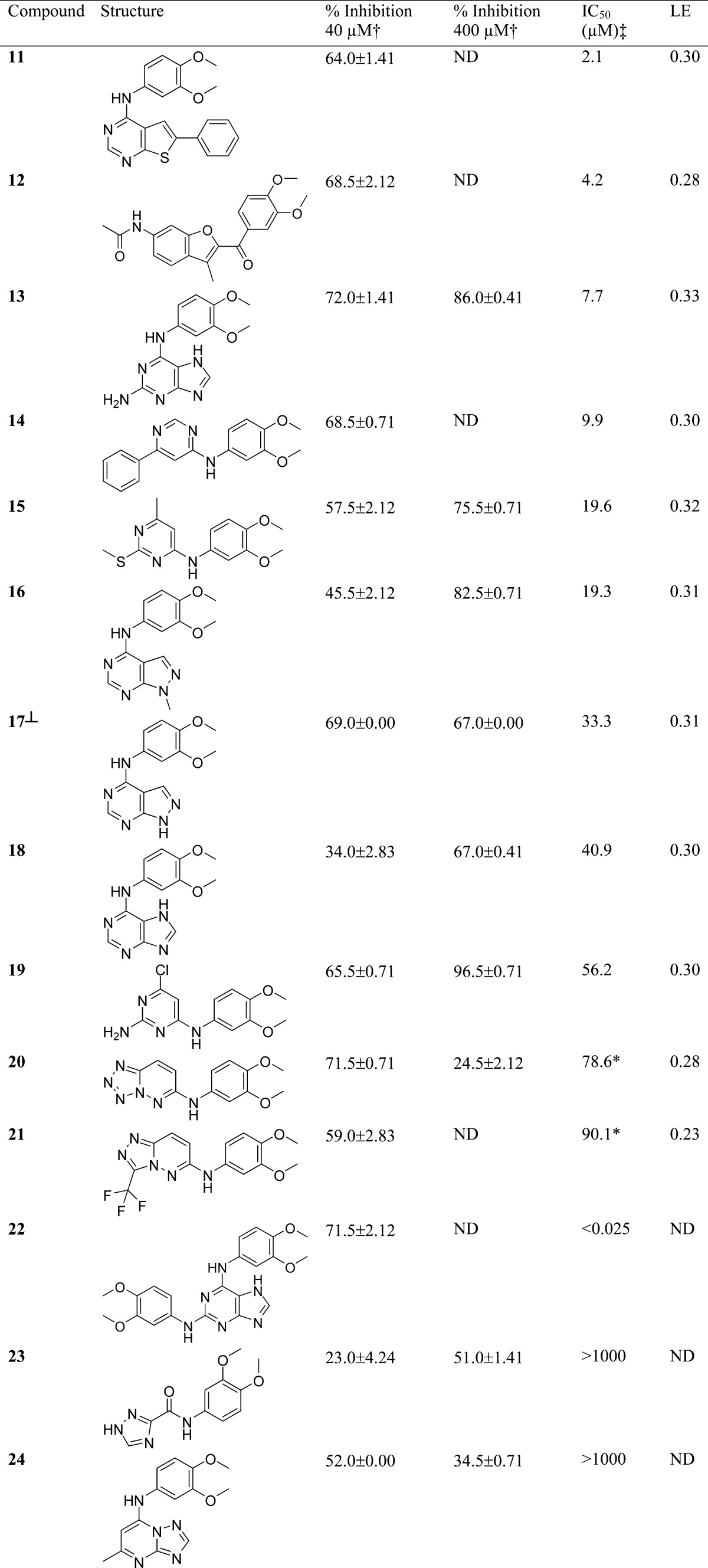
ND = not determined.
Single point assays were performed in duplicate; percent inhibition values shown are the mean of the two results reported with the standard deviation.
IC50 experiments were performed in duplicate on the same assay plate at 1% and 10% DMSO. An asterisk indicates compounds tested at 10% DMSO; all other compounds were tested at 1% DMSO.
Percent inhibition and IC50 values do not correlate for these compounds due to poor solubility at higher concentrations.
Of the nine compounds selected using the scaffold-focused virtual screen that exhibited ≥50% inhibition of TTK, eight were confirmed by subsequent IC50 determination (compounds 2–9), all of which are classed as fragment-like with IC50 values in the range 53.0–246.1 μM (Table 1). Of the 14 compounds selected using the whole molecule virtual screen that exhibited ≥50% inhibition of TTK, 12 were confirmed by subsequent IC50 determination with IC50 values ranging from <25 nM to 90.1 μM (Table 2); six of these compounds were classed as fragment-like. More fragment-like hits were discovered using the scaffold-focused virtual screen (eight from eight) than the whole molecule virtual screen (six from twelve). We attribute this finding to the selection of five diverse compounds from each top-scoring scaffold using molecular weight as a key parameter, which therefore ensures the selection of fragment-like exemplar molecules of the level 1 scaffold for screening, if they are present in the Compound Library. We propose that fragment-like molecules identified using the scaffold-focused virtual screening method contain a core scaffold required for TTK binding, whereas more substituted analogs of the core scaffold may lose activity as the appended substituents are not necessarily optimized for binding to TTK. To explore this hypothesis, we determined protein co-crystal structures of fragment-like hit compounds to further understand their binding modes (vide infra).
Five scaffolds discovered using the scaffold-focused method have ligand efficiencies >0.35 compatible with useful medicinal chemistry starting points (compounds 3–7, Table 1).52 Thienonapthyridines 3, 5, and 7 have no reported kinase activity nor biochemical activity against any other target to our knowledge; we also found no reported compounds with associated biochemical data containing 3, 5, and 7 as substructures. Compounds 2 and 6 also have no reported kinase activity; however, compounds containing the pyrimido-indole substructure 4 have been reported as potential inhibitors of LIMK.53 Compound 9 contains the same core ring system as 4 and has no literature-reported activity against kinases. Compound 8 is the β-carboline alkaloid harmine, which has previously been reported as an inhibitor of the dual specificity kinase Dyrk1A.54
Compounds 2–5 and 7–10 (Table 1) are tricyclic examples of their corresponding bicyclic level 1 scaffold. We observed that bicyclic level 1 scaffolds of compounds 2–5 and 7–10 (Table 1) are predominantly represented by tricyclic exemplars in the Compound Library; thus, it is not surprising that a preponderance of tricyclic compounds is selected. The prevalence of tricyclic exemplars for these level 1 scaffolds in the Compound Library is likely a result of their synthetic chemistry accessibility. The lipophilic gatekeeper residue of TTK (Met 602) may also favor an enrichment of lipophilic tricyclic hits. Thus, the propensity for tricyclic hit compounds is likely a function of the Compound Library composition (prioritized level 1 scaffolds are highly represented by tricyclic compounds in the Compound Library) and the binding site topology of TTK (lipophilic gatekeeper residue) rather than the virtual screen protocol itself.
The whole molecule virtual screen identified a higher proportion of high molecular weight compounds (six out of twelve) compared to the scaffold-focused virtual screen (zero from eight). We propose that, because the whole molecule similarity search was performed using compound 1, and similar compounds are more likely to be of similar molecular weight to the query molecule, then higher molecular weight hit compounds are more likely to be discovered. We also observed that active compounds identified using the whole molecule virtual screen frequently contain identical substructures to those present in query compound 1. For example, the 3,4-dimethoxyphenyl moiety is present in all 12 confirmed hits, and the 3,4-dimethoxyphenylamine moiety is present in 11 of the confirmed hits and appears twice in compound 22. Interestingly, compound 22 has an IC50 comparable to the query compound 1 (IC50 < 25 nM), and its 2,6-diaminopurine core is claimed within the scope of a published TTK patent from Myriad pharmaceuticals.55
To assess the scaffold-hopping ability of the two virtual screens described here, we examined the hit rate, ratio of unique scaffolds to compounds tested (N/M), ratio of unique active scaffolds to active compounds (NA/MA), and MPS across both sets of active compounds (Table 3). The hit rate for the whole molecule screen (12%) is higher than the scaffold-focused virtual screen (8.2%). We postulate that the whole molecule virtual screen has a higher observed hit rate because it identifies compounds that are highly similar to the query and are more likely to have similar biochemical activity. In addition, we have previously demonstrated that compound libraries comparable to the one used in this study have low scaffold diversity, especially when analyzed using level 1 scaffolds;13 we therefore propose that the lower hit rate of our level 1 scaffold-based similarity search may also reflect a lack of scaffold diversity in the Compound Library.
Table 3. Number of Compounds and Unique Scaffolds Discovered Using the Scaffold-Focused and Whole Molecule Virtual Screena.
| M | N | N/M | MA | NA | NA/MA | Hit Rate | MPS molecule | MPS scaffold | |
|---|---|---|---|---|---|---|---|---|---|
| scaffold-focused | 98 | 27 | 0.28 | 8 | 6 | 0.75 | 8.2% | 0.60 ± 0.19 | 0.76 ± 0.13 |
| 2D ECFP_4 | 70 | 17 | 0.24 | 8 | 6 | 0.75 | 11.4% | 0.60 ± 0.19 | 0.76 ± 0.13 |
| 3D ROCS | 50 | 16 | 0.32 | 1 | 1 | 1 | 2.0% | N/A | N/A |
| whole molecule | 100 | 44 | 0.44 | 12 | 8 | 0.67 | 12% | 0.74 ± 0.10 | 0.53 ± 0.18 |
| 2D ECFP_4 | 50 | 22 | 0.44 | 10 | 6 | 0.60 | 20% | 0.77 ± 0.08 | 0.61 ± 0.15 |
| 3D ROCS tautomer 1 | 37 | 22 | 0.59 | 2 | 2 | 1 | 5.4% | 0.61 ± 0.00 | 0.37 ± 0.00 |
| 3D ROCS tautomer 2 | 35 | 19 | 0.54 | 1 | 1 | 1 | 2.9% | N/A | N/A |
M = number of compounds tested, N = number of unique scaffolds present in the compounds tested, MA = number of active compounds, NA = number of unique active scaffolds. Actives include only compounds confirmed by IC50 determination. MPS molecule = mean pairwise similarity of active compounds with the standard deviation. MPS scaffold = mean pairwise similarity of active scaffolds with the standard deviation.
Comparison of the hit rate for the two similarity methods used for each virtual screen indicates that ECFP_4 2D fingerprints significantly outperform ROCS for both the scaffold-focused and whole molecule virtual screens (Table 3). A possible reason for this difference is that while a single conformer of the query compound (1) was used, multiple conformations were generated using OMEGA50 for each member of the interrogated Compound Library. Thus the well-documented “conformer problem” introduces additional conformations of each library compound, many of which may not be biologically relevant, and this may be responsible for the poor performance of the 3D-similarity method in our hands.56 Although level 1 scaffolds tend to be planar ring systems, we anticipated added value in performing the 3D ROCS method on these flat scaffolds because ROCS also incorporates a matching of pharmacophoric features; however, ECFP_4 2D fingerprints also outperformed ROCS for the scaffold-focused virtual screen described here (Table 3).
The whole molecule virtual screen selected compounds with more unique scaffolds (N/M = 0.44) compared to the scaffold-focused virtual screen (N/M = 0.28). However, in the scaffold-focused virtual screen, we limited our selection to a maximum of five compounds for each top-scoring scaffold thereby restricting the ratio of unique scaffolds to compounds (minimum N/M = 0.20 for 100 compounds selected); not every top scoring scaffold has five representatives, thus a higher ratio was observed (N/M = 0.28). The ratio of scaffolds to active compounds (NA/MA) for the scaffold-focused and whole molecule virtual screens is 0.75 and 0.67, respectively, indicating that active compounds identified by both methods contain a variety of different scaffolds. The higher ratio (NA/MA = 0.75) for the scaffold-focused virtual screen indicates that this method identifies more actives with unique scaffolds than the whole molecule virtual screen, consistent with enhanced scaffold hopping potential despite a lower overall hit rate.
We considered the pairwise similarity (Figure 3) and MPS (Table 3) of both the whole molecule and of the scaffold of the hit molecules. We observed that, relative to the whole molecule virtual screen, the scaffold-focused virtual screen gave hit matter with higher pairwise similarity of the scaffold but lower pairwise similarity when considering the whole molecule (Figure 3). The higher MPS scaffold score for the scaffold-focused virtual screen (0.76, Table 3) compared to the whole molecule virtual screen (0.53, Table 3) is expected from our use of level 1 scaffolds to define the search space in this scaffold-focused method. The lower MPS molecule score for the scaffold-focused virtual screen (0.60, Table 3) versus the whole molecule virtual screen (0.74, Table 3) is consistent with the discovery of a more diverse compound hit set from the scaffold-focused virtual screen compared to the whole molecule virtual screen as illustrated by the prevalence of 3,4-dimethoxyphenyl and 3,4-dimethoxyphenylamine moieties in the hit matter from the whole molecule screen as described above.
Figure 3.

Box and whisker plots depicting the pairwise similarity for confirmed active compounds. SF molecule = pairwise similarity of active molecules from the scaffold-focused virtual screen. WM molecule = pairwise similarity of active molecules from the whole molecule virtual screen. SF scaffold = pairwise similarity of the level 1 scaffolds of active molecules from the scaffold-focused virtual screen. WM scaffold = pairwise similarity of the level 1 scaffolds of active molecules from the whole molecule virtual screen. All similarities are calculated using the Tanimoto coefficient and MDL public keys. Box plots for the scaffold-focused virtual screen are shown in blue and box plots for the whole molecule virtual screen are shown in red.
Both NA/MA and MPS consider the intraset diversity of the active compounds retrieved. To assess the similarity of active compounds to the query, we assessed compound and scaffold similarity to the query compound 1 (Figure 4). We observed that relative to the whole molecule virtual screen the scaffold-focused virtual screen gave hits with lower whole molecule similarity to the query compound 1 and higher scaffold similarity to the level 1 scaffold of query compound 1 (Figure 4) as expected for a method which applies level 1 scaffolds to define the search space.
Figure 4.

Box and whisker plots depicting the similarity to the query compound 1 of compounds selected by virtual screening. SF molecule = similarity of compounds selected using the scaffold-focused virtual screen to the query molecule. WM molecule = similarity of compounds selected using the whole molecule virtual screen to the query molecule. SF scaffold = similarity of the level 1 scaffolds of compounds selected using the scaffold focused virtual screen to the query molecule. WM scaffold = similarity of the level 1 scaffolds of compounds selected using the whole molecule virtual screen to the query molecule. Box plots for the scaffold-focused virtual screen are shown in blue and box plots for the whole molecule virtual screen are shown in red.
To explore further these relationships, we plotted compound activity versus whole molecule similarity to query compound 1 as determined by the Tanimoto coefficient (Figures 5 and 6). Close analogs to the original query with high activity lie in the upper right quadrant. Dissimilar compounds from the query that retain activity lie in the upper left quadrant, desired scaffold hopping space.2 Similarity versus activity plots for compounds tested at 40 and 400 μM (Figures 5 and 6, respectively) show a clear separation between compounds selected using the scaffold-focused and whole molecule virtual screens. Compounds selected using the scaffold-focused virtual screen tend to lie on the left-hand side of the plot (i.e., they have a low similarity the query compound 1), whereas compounds selected using the whole molecule virtual screen tend to lie on the right-hand side of the plot (i.e., they are similar to the query compound 1). All compounds selected by the whole molecule virtual screen with a percent inhibition greater than 50% have a similarity greater than 0.6 to the query compound. Thus, the whole molecule virtual screen selects compounds similar to the original query compound 1 with a high hit rate (12%) as expected for this type of method and consistent with the hypothesis that compounds similar to the query compound are more likely to have similar activity.6 However, the majority of compounds selected by the scaffold-focused method with a percent inhibition greater than 50% have a similarity less than 0.5 to the query compound 1, demonstrating that the scaffold-focused virtual screen can identify active compounds that are more structurally differentiated from the original query compound (i.e., scaffold hops), albeit with a lower hit rate (8.2%).
Figure 5.

Activity of all compounds selected with scaffold-focused and whole molecule-based virtual screening against TTK at 40 μM versus their similarity to the query compound 1 calculated using the Tanimoto coefficient and MDL public keys. Numbers indicate which compound the respective point represents. Hit compounds located in the upper right quadrant have high structural similarity to query compound 1 and high activity. Hit compounds in the upper left quadrant have low structural similarity to query compound 1 and high activity.
Figure 6.

Activity of fragment-like compounds selected with scaffold-focused and whole molecule fragment virtual screening against TTK at 400 μM versus their similarity to the query compound 1 calculated using the Tanimoto coefficient and MDL public keys. Numbers indicate which compound the respective point represents. Hit compounds located in the upper right quadrant have high structural similarity to query compound 1 and high activity. Hit compounds in the upper left quadrant have low structural similarity to query compound 1 and high activity.
In summary, we have applied level 1 of the scaffold tree as a high-throughput data set-independent method to identify the core scaffolds from a large (2,221,028) Compound Library and conducted both a scaffold-focused and whole molecule-based prospective virtual screen. The scaffold-focused protocol identifies active hit compounds in a TTK biochemical screen that are more structurally differentiated from the query compound 1 in comparison to the literature-based whole molecule similarity searching method. We classed all eight confirmed hits (compounds 2–9) from the scaffold-focused protocol as fragment-like (molecular weight < 300 Da), whereas six of the twelve confirmed hits from the whole molecule-based virtual screen (compounds 13, 15–19) were classed as fragment-like. We propose that the scaffold-focused method described here, where up to five diverse exemplars of each hit scaffold are selected for biochemical screening, is capable of identifying structurally differentiated and ligand-efficient core scaffolds that serve as useful fragment-like medicinal chemistry starting points and which may not be represented in current fragment libraries. We observed that more substituted lead-like derivatives of the fragment-like hit compounds 2–9, although selected for screening, were not detected as confirmed hits, which we attribute to the incompatibility of the appended substituents with the biochemical target under investigation (vide infra). This scaffold-focused approach may be considered as an example of scaffold hopping, although we recognize that this method does not retain information about the substitution pattern of the original query molecule.
Binding Mode Determination of Hit Matter by Crystallography
In recognition of the desirability of protein–ligand crystal structures to educate the efficient follow-up of fragment-like hit matter and our wish to determine the mode of binding of hits identified with the scaffold-focused virtual screen, we sought co-crystal structures of confirmed hits with TTK. To this end, apo crystals of TTK kinase domain were soaked in buffer solution containing the respective inhibitor (2–9). X-ray data were collected for all crystals, and the structures were solved using molecular replacement. We successfully obtained co-crystal structures of TTK with fragment-like hit compounds 3, 4, 5, and 7. For crystals soaked with compounds 2, 6, 8, and 9, no electron density was observed in the ATP binding site, indicating that the compounds were not bound. A summary of crystallographic analysis is presented in Table 4, and the binding modes of compounds 3, 4, 5, and 7 are illustrated in Figure 7 with Fo–Fc electron density omit maps surrounding the ligand shown in green wire-mesh contoured at 3σ. In all four structures, the activation loop of TTK is disordered, possibly due to the presence of a PEG molecule bound to Lys553, a residue which would usually coordinate with Glu571 to help stabilize the activation loop (Lys553 is not shown in Figure 7 for clarity).
Table 4. Data Collection and Refinement Statisticsa.
| PDB | 4BHZ | 4BI0 | 4BI1 | 4BI2 |
| Compound | 3 | 4 | 5 | 7 |
| space group | I222 | I222 | I222 | I222 |
| Lattice Constants | ||||
| a (Å) | 71.16 | 71.23 | 70.91 | 70.69 |
| b (Å) | 111.68 | 111.97 | 111.82 | 111.68 |
| c (Å) | 113.28 | 113.78 | 113.57 | 112.16 |
| Data Collection | ||||
| resolution range (Å) | 56.64–2.85 | 56.89–2.84 | 56.78–2.70 | 56.08–3.11 |
| highest resolution shell | (3.00–2.85) | (2.99–2.84) | (2.85–2.70) | (3.28–3.11) |
| unique reflections | 10886 (1573) | 11081 (1582) | 12760 (1827) | 8291 (1191) |
| completeness (%) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) |
| multiplicity | 4.2 (4.3) | 4.1 (4.1) | 4.0 (4.1) | 4.3 (4.4) |
| Rmerge (%) | 6.7 (52.4) | 7.9 (48.8) | 7.2 (52.1) | 7.9 (51.5) |
| I/σ(I) | 9.3 (1.5) | 7.8 (1.5) | 7.2 (1.5) | 8.2 (1.5) |
| mean (I/ σ(I)) | 13.9 (2.8) | 12.2 (2.8) | 10.6 (2.4) | 11.5 (2.7) |
| mean mosaicity (deg) | 0.71 | 0.49 | 0.78 | 0.89 |
| Refinement | ||||
| R factor (%) | 18.7 | 18.4 | 18.8 | 19.4 |
| R free (%) | 23.0 | 22.36 | 22.8 | 22.6 |
| no. amino acids | 251 | 254 | 252 | 253 |
| no. waters | 7 | 15 | 21 | 3 |
| no. ligands | 1 | 1 | 1 | 1 |
| no. PEG | 2 | 2 | 4 | 1 |
| no. ethylene glycol | 1 | 1 | 4 | 2 |
| R.m.s. deviation | ||||
| bond length (Å) | 0.010 | 0.010 | 0.010 | 0.010 |
| bond angles (deg) | 1.15 | 1.18 | 1.20 | 1.17 |
| Ramachandran plot | ||||
| favored (%) | 96.7 | 96.4 | 96.7 | 95.6 |
| forbidden (%) | 0.0 | 0.4 | 0.0 | 0.0 |
Values in parentheses are highest shell values. The wavelength for data collection was 0.8726 Å. All data was collected on April 13, 2011, on the ID23eh2 beamline at ESRF.
Figure 7.

(a) X-ray structure of 3 bound to TTK at 2.85 Å. PDB: 4BHZ. (b) X-ray structure of 4 bound to TTK at 2.84 Å. PDB: 4BI0. (c) X-ray structure of 5 bound to TTK at 2.70 Å. PDB: 4BI1. (d) X-ray structure of 7 bound to TTK at 3.11 Å. PDB: 4BI2. The electron density shown in green wire mesh is the Fo–Fc electron density omit map contoured at 3σ. TTK is shown in purple and residues near to the ligand are shown in cylinder representation with carbon atoms in purple and are labeled. Hydrogen bonds are indicated by black dashed lines.
Thienonaphthyridine 3, 5, and 7 all bind to the hinge region of TTK via a hydrogen bond from the nitrogen of the central pyridine ring to the amine of Gly605 in the hinge, a key interaction also present for the pyridine nitrogen of the 1H-pyrrolo[3,2-c]pyridine scaffold present in query compound 1. We also observe evidence for a putative aromatic C–H hydrogen bond from the adjacent C5 position of the thienonaphthyridine template to the carbonyl group of Glu603. Additional hydrophobic packing observed with leucine and isoleucine residues at the base of the pocket (Leu654, Ile663, and Ile586) and the methionine gatekeeper (Met602) likely contribute to the binding affinity. Thieno[2,3-c][2,6]naphthyridine 3 and thieno[3,2-c][2,6]naphthyridine 5 are both exemplars of the 2,6-naphthyridine level 1 scaffold, while thieno[2,3-c][2,7]naphthyridine 7 is an exemplar of the 2,7-naphthyridine level 1 scaffold; no analogs of compounds 3, 5, or 7 bearing additional substitution were available for biochemical screening. In the case of the 5H-pyrimido[5,4-b]indole 4, a hydrogen bond is formed between the backbone amine of Gly605 and the N1-pyrimidine nitrogen of the tricyclic scaffold with a putative weak hydrogen bond observed from the C9–H to the backbone carbonyl of Glu603. Additional hydrophobic packing between the phenyl ring of the 5H-pyrimido[5,4-b]indole and the lipophilic methionine gatekeeper side chain (Met602) likely contributes to the binding affinity. While a small lipophilic methyl substituent in the C8 position and a piperidine ring at the C4 position of the core scaffold are tolerated in the weakly active hit 9, (IC50 = 246 μM, Table 1), the combination of a more polar halogen at C8 with larger substituents at the C4 position render compounds with the same 5H-pyrimido[5,4-b]indole scaffold inactive (see compounds 4a and 4b, Table S1, Supporting Information). These observations are consistent with the binding pose depicted in Figure 7b where a polar C8 halogen substituent would buttress against the lipophilic gatekeeper residue (Met602) and where bulky and extended substituents from the C4 position may be incompatible with binding site topology through steric or electronic clash with the β-sheet extending from Glu605. We also observed that more highly functionalized derivatives of the hit compounds 6 and 8 (namely, compounds 6a–6c and 8a–8d, Table S1, Supporting Information) were inactive. These observations are consistent with the argument that larger more highly substituted exemplars of a ligand-efficient core scaffold may mask its activity if the substituent groups are incompatible with binding pocket topology and alternative binding modes are not tolerated.57
In summary, the crystal structures described here (Figure 7) demonstrate that the binding modes of a selection of the fragment-like hits discovered by scaffold-focused virtual screening. In one case (compound 4), the observed binding mode is consistent with the lack of activity of 4,8-disubstituted exemplars of the core scaffold, which were also retrieved as diverse exemplars of the scaffold from the scaffold-focused virtual screening protocol. Similarly, substituted derivatives of hit compounds 6 and 8 also masked the activity inherent to the core scaffold.
Conclusions
We have experimentally validated a scaffold-focused virtual screen based upon level 1 of the scaffold tree and compared its performance to a whole molecule virtual screen that uses methods previously shown to identify scaffold hops. We applied both methods to identify novel inhibitors of the dual specificity kinase TTK from a vendor collection of 2,221,028 compounds. We purchased 98 and 100 compounds selected with the scaffold-focused and whole molecule virtual screen, respectively, and tested these compounds in a TTK biochemical assay. The scaffold-focused and whole molecule virtual screen gave hit rates of 8.2% and 12%, respectively. We postulate that the whole molecule virtual screen has an observed higher hit rate because it identifies compounds that are highly similar to the query compound which are, therefore, more likely to have similar biochemical activity. In addition, we have previously demonstrated low scaffold diversity across exemplified medicinal chemistry space including comparable compound libraries to the one used in this study13 and postulate that the lower hit rate of our level 1 scaffold-based similarity search reflects this lack of scaffold diversity in these chemical libraries. Despite a lower hit rate, the scaffold-focused virtual screen identifies hit compounds containing scaffolds that are significantly structurally different from the original query. These hits have been confirmed by IC50, and the binding mode of selected hits has been determined by protein–ligand crystallography. Confirmed scaffold hops include the thienonaphthyridine class that have no previously reported biological activities and the 5H-pyrimido[5,4-b]indole class where we demonstrate that C4 and C8 disubstituted exemplars also selected in the virtual screen are inactive, consistent with the argument that highly substituted exemplars of an interesting core scaffold may mask activity if the substituents are incompatible with binding pocket topology and alternative binding modes are not tolerated.
The scaffold-focused virtual screen presented here, which uses similarity of level 1 scaffolds derived from query and library compounds as the selection criteria, has the potential to identify ligand-efficient bioactive compounds as potential starting points for hit-to-lead evaluation and which are more structurally differentiated from the query compound compared to those selected using a whole molecule similarity search. This scaffold-focused approach may be considered as an example of scaffold hopping, although we recognize that this method does not retain information from the substitution pattern of the query molecule. We are currently exploring methods that incorporate increased scaffold information to improve the selection of scaffolds with appropriate substitution patterns from compound libraries.
Experimental Section
Purchased Compounds
All compounds were purchased as solids. All compounds were dissolved in 100% (v/v) DMSO in Thermo Scientific Abgene Storage Tubes (Thermo Fisher Scientific, Inc., Waltham, MA, U.S.A.). Compounds with a molecular weight less than 300 Da were stored at 100 mM, and all other compounds were stored at 10 mM. Compound solutions were directly transferred from Abgene tubes into 384 well plates. The Abgene tubes and compound plates were stored under nitrogen in a FluidX StoragePod (FluidX, Nether Alderley, U.K.) until needed. Compounds were transferred from the 384 well compound plates into assay plates and diluted to assay concentrations using the Labcyte Echo Liquid Handling System (Labcyte Inc., Sunnydale, CA, U.S.A.).
All active compounds were confirmed to have >95% purity by LC-MS. A list of compounds purchased and vendors can be found in Table S1 of the Supporting Information.
LC-MS
LC-MS CHROMASOLV solvents, formic acid, or alternative eluent modifiers were purchased from Sigma Aldrich (Poole, U.K.) unless otherwise stated. LC-MS measurements were conducted on the 384 well plates described above. The 0.1 μL standard injections (with needle wash) of the sample were made onto a Purospher STAR RP-18 end-capped column (3 μm, 30 mm × 4 mm, encased in LiChroCART assembly, Merck KGaA, Darmstadt, Germany).
Chromatographic separation at 30 °C was carried out using a 1200 Series HPLC (Agilent, Santa Clara, CA, U.S.A.) over a 4 min gradient elution (Fast4 min m) from 90:10 to 10:90 water:methanol (both modified with 0.1% formic acid) at a flow rate of 1.5 mL/min. UV–vis spectra were acquired at 254 nm on a 1200 Series diode array detector (Agilent, Santa Clara, CA, U.S.A.).
The post-column eluent flow from the diode array detector was split with 90% sent to waste. The remainder was infused into a 6520 Series Q-ToF mass spectrometer fitted with an ESI/APCI MultiMode ionization source (Agilent, Santa Clara, CA, U.S.A.). LC eluent and nebulizing gas was introduced into the grounded nebulizer with spray direction orthogonal to the capillary axis. A total of 2 kV was applied to the charging electrode to generate a charged aerosol. The aerosol was dried by infrared emitters (200 °C) and heated drying gas (8 L/min of nitrogen at 300 °C, 40 psi), producing ions by ESI. Aerosol and ions were transferred by nebulizing gas to the APCI zone where infrared emitters vaporized solvent and analyte. A corona discharge was produced between the corona needle and APCI counter electrode by applying a current of 4 μA, ionizing the solvent to transfer charge to analyte molecules, producing ions by APCI. ESI and APCI ions simultaneously entered the transfer capillary along which a potential difference of 4 kV was applied. The fragmentor voltage was set at 175 V and skimmer at 60 V. Signal was optimized by AutoTune.m. Profile mass spectrometry data was acquired in positive ionization mode over a scan range of m/z 130–950 (scan rate 1.0) with reference mass correction at m/z 322/048121 (hexamethoxyphosphazene), 622.02896 (hexakis (2,2-difluoroethoxy)phosphazene), and 922.0098 ((1H, 1H, 3H-tetrafluoropentoxy)phosphazene). Raw data was processed using Agilent MassHunter Qualitative Analysis B.03.01.
Biochemical Assay
The TTK kinase activity was measured in a mobility shift microfluidics assay with the Caliper LabChip EZReader II (Caliper Life Sciences, Hopkinton, MA, U.S.A.). For the initial percent inhibition compound screen, all compounds were tested at 40 μM, and fragment-like compounds (MW < 300 Da) were also tested at 400 μM. A 2.14 μM stock of GST-tagged recombinant human full-length TTK protein kinase from Invitrogen (Invitrogen, Life Technologies Ltd., Paisley, U.K.) and custom synthesized peptide from Pepceuticals (Pepceuticals Ltd., Enderby, U.K.) with sequence 5-FAM-DHTGFLTEYVATR-amide were used. The reaction mixture contained compound at the desired concentration, 5 μM peptide, 10 μM ATP (Km of ATP = 10 μM), assay buffer (final concentration: 10 mM HEPES pH 7.0, 0.004% (w/v) NaN3, 0.002% (w/v) BSA, and 20 μM orthovanadate), 10 mM MgCl2, 1 mM DTT, and 1% (v/v) DMSO. For every 10 mL of stock buffer, one Roche EDTA free complete mini-protease inhibitor cocktail tablet (Roche, Basel, Switzerland) was added. TTK was used at a final concentration of 12.5 nM. The assay plate was sealed and incubated at room temperature for 60 min. The assay was stopped by addition of stop buffer which contained 100 mM HEPES (pH 7.3), 20 mM EDTA, and 0.05% (v/v) Brij-35. The assay plate was then analyzed using the EZReader II. Z′ for compounds at 40 μM was 0.48, and Z′ for compounds at 400 μM was 0.61.
IC50 determination was carried out with the same protocol as above but with a 3 nM final concentration of TTK. Compounds were run in duplicate on the same assay plate. Compound 1 was used as a positive control for compounds with MW ≥ 300 Da, the positive control for compounds with MW < 300 Da was 4-(6-((3-acetamidophenyl) amino)-H-pyrrolo[3,2,-c]pyridine-2-yl)-N,N-dimethylbenzamide. An eight point dilution curve was used to determine IC50 by fitting percent inhibitions at each concentration to a four parameter logistic fit. Compounds insoluble in 1% (v/v) DMSO were testing in same protocol at 10% (v/v) DMSO. We determined that the substrate turnover of TTK was unaffected by buffer concentrations containing up to 10% (v/v) DMSO (data not shown).
Crystallization and Structure Determination
TTK was purified and crystallized as described previously.26 Plasmids for expression of TTK were received from Stefan Knapp, Structural Genomics Consortium, Oxford, U.K. TTK kinase domain (residues 519–808) was expressed in E. coli BL21 AI competent cells (Invitrogen, Life Technologies Ltd., Paisley, U.K.). Cells were cultured in LB media containing kanamycin selection antibiotic and induced with 0.5 mM IPTG and 1g/L l-arabinose at an OD280 of 0.7. Cells were then incubated overnight at 18 °C with shaking at 225 rpm. Cells were harvested and resuspended in lysis buffer containing 50 mM HEPES pH 7.5, 500 mM NaCl, 10 mM imidazole, 5% (v/v) glycerol, lysozyme, and 1 Roche EDTA free complete protease inhibitor cocktail tablet (Roche, Basel, Switzerland) per 3 L of buffer. TTK was purified using Ni–NTA nickel affinity resin and washed in buffer containing 30 mM imidazole. TTK was then eluted from the resin with 250 mM imidazole. Eluate was concentrated using a Vivaspin concentrator with a 30 kDa molecular weight cutoff (GE Healthcare, Chalfont St. Giles, U.K.) and further purified on gel filtration column Superdex 200 16/60 (GE Healthcare, Chalfont St. Giles, U.K.) equilibrated in buffer containing 10 mM HEPES pH 7.5, 150 mM NaCl, 5 mM DTT, and 5 mM EDTA. The sample was concentrated to a final concentration of 8.9 mg/mL, snap frozen on dry ice, and stored at −80 °C until required.
Purified TTK was crystallized at 18 °C using the sitting drop vapor diffusion method with drops composed of 2 μL protein (8.9 mg/mL) and 2 μL reservoir solution placed over 200 μL reservoir solution of H2O/PEG300 (30–45% (v/v)) in 48 well plates. Crystals typically grew in 72 h.
Protein crystals were soaked in hanging drop plates in 4 μL drops composed of 10 mM compound (compounds 2–4 and 6–8), 25 mM compound (compound 8), or 50 mM compound (compound 5) and reservoir solution placed over 400 μL reservoir solution of 35% (v/v) PEG300, 0.1 mM HEPES pH 7.5, and 10% (v/v) DMSO in 15 well plates. Plates were incubated for 24 h at 18 °C. Protein crystals were briefly transferred to cryoprotectant solution, containing 40% (v/v) PEG300, 0.1 M HEPES pH 7.5, and 20% (w/v) ethylene glycol, prior to flash freezing in liquid nitrogen.
X-ray data were collected at the ESRF synchrotron Grenoble, France, at beamline ID23eh2. Crystals belonged to the space group I222 and diffracted to a resolution between 2.7 and 3.11 Å. Data were integrated and merged using MOSFLM58,59 and SCALA.58 The structures were solved by molecular replacement using PHASER58,60 and a publicly available TTK structure (PDB: 2ZMC)61 with ligand and water molecules removed as the molecular replacement model. The protein–ligand structures were manually rebuilt in COOT62 and refined with BUSTER63 in iterative cycles. Ligand restraints were generated with grade64 and Mogul.65 The quality of the structures was assessed with MOLPROBITY.66 Co-crystal structures of TTK with compounds 3, 4, 5, and 7 were successfully obtained. For crystals soaked with compounds 2, 6, 8, and 9, no electron density was observed in the protein binding site indicating that the compounds were not bound. Table 4 contains the data collection and refinement statistics.
Acknowledgments
S.R.L. is supported by The Institute of Cancer Research. N.B. and J.B. are supported by Cancer Research U.K. Grant C309/A11566. We thank Meirion Richards for carrying out LCMS. We thank the Hit Discovery and Structure Design team at The Institute of Cancer Research for their assistance with TTK protein production and biological assays, in particular Rosemary Burke, Jessica Schmitt, Kathy Boxall, Yvette Newbatt, and Amy Simpson. We are grateful to Stefan Knapp, Structural Genomics Consortium, Oxford, U.K., for the generous gift of expression plasmids for TTK. We thank Nora Cronin and the staff of ESRF beam time ID23eh2 for their support during data collection.
Supporting Information Available
Table S1. Chemical structures, vendor, and vendor ID of the 198 compounds selected from the scaffold-focused and whole molecule virtual screens. All active compounds (those with a compound number from the main paper) were confirmed >95% pure by LCMS. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
S. R. Langdon, N. Brown, and J. Blagg devised the study and wrote the manuscript. S. R. Langdon conducted all the research. R. L. M. van Montfort and I. M. Westwood supervised X-ray crystallography.
The authors declare no competing financial interest.
Supplementary Material
References
- Böhm H.-J.; Flohr A.; Stahl M. Scaffold hopping. Drug Discovery Today: Technol. 2004, 1, 217–224. [DOI] [PubMed] [Google Scholar]
- Brown N.; Jacoby E. On scaffolds and hopping in medicinal chemistry. Mini-Rev. Med. Chem. 2006, 6, 1217–1229. [DOI] [PubMed] [Google Scholar]
- Langdon S. R.; Ertl P.; Brown N. Bioisosteric replacement and scaffold hopping in lead generation and optimization. Mol. Inf. 2012, 29, 366–385. [DOI] [PubMed] [Google Scholar]
- Ripphausen P.; Nisius B.; Peltason L.; Bajorath J. Quo vadis, virtual screening? A comprehensive survey of prospective applications. J. Med. Chem. 2010, 53, 8461–8467. [DOI] [PubMed] [Google Scholar]
- Willett P. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar]
- Johnson M. A.; Maggiora G. M.. Concepts and Applications of Molecular Similarity; Wiley Inter-Science, New York, 1990. [Google Scholar]
- Gardiner E. J.; Holliday J. D.; O’Dowd C.; Willett P. Effectiveness of 2D fingerprints for scaffold hopping. Future Med. Chem. 2011, 3, 405–415. [DOI] [PubMed] [Google Scholar]
- Rush T. S.; Mosyak L.; Grant A. J.; Nicholls A. A shape-based 3-D scaffold hopping method and its application to a bacterial protein–protein interaction. J. Med. Chem. 2005, 48, 1489–1495. [DOI] [PubMed] [Google Scholar]
- Schneider G.; Schneider P.; Renner S. Scaffold-hopping: How far can you jump?. QSAR Comb. Sci. 2006, 25, 1162–1171. [Google Scholar]
- Schneider G.; Neidhart W.; Giller T.; Schmidt G. Scaffold-hopping by topological pharmacophore search: A contribution to virtual screening. Angew. Chem., Int. Ed. 1999, 38, 2894–2896. [PubMed] [Google Scholar]
- Schuffenhauer A.; Ertl P.; Roggo S.; Wetzel S.; Koch M. A.; Waldmann H. The scaffold tree: Visualization of the scaffold universe by hierarchical scaffold classification. J. Chem. Inf. Model. 2007, 47, 47–58. [DOI] [PubMed] [Google Scholar]
- Bemis G. W.; Murcko M. A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–1893. [DOI] [PubMed] [Google Scholar]
- Langdon S. R.; Brown N.; Blagg J. Scaffold diversity of exemplified medicinal chemistry space. J. Chem. Inf. Model. 2011, 51, 2174–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss E.; Winey M. The Saccharomyces cerevisiae spindle pole body duplication gene MPS1 is part of a mitotic checkpoint. J. Cell Biol. 1996, 132, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauzé E.; Stoelcker B.; Luca F. C.; Weiss E.; Schutz A. R.; Winey M. Yeast spindle pole duplication gene MPS1 encodes an essential dual specificity protein kinase. EMBO J. 1995, 14, 1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poch O.; Schwob E.; de Fraipont F.; Camasses A.; Bordonné R.; Martin P. R. RPK1, an essential yeast protein kinase involved in the regulation of the onset of mitosis, shows homology to mammalian dual-specificity kinases. Mol. Gen. Genet. 1994, 243, 641–653. [DOI] [PubMed] [Google Scholar]
- Mills G. B.; Schmandt R.; McGill M.; Amendola A.; Hill M.; Jacobs K.; May C.; Rodericks A. M.; Campbell S.; Hogg D. Expression of TTK, a novel human protein kinase, is associated with cell proliferation. J. Biol. Chem. 1992, 267, 16000–16006. [PubMed] [Google Scholar]
- Stucke V. M.; Sillié H. H.; Arnaud L.; Nigg E. A. Human Mps1 kinase is required for the spindle assembly checkpoint but not for centrosome duplication. EMBO J. 2002, 21, 1723–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. T.; Chan G. K.; Hittle J. C.; Fujii G.; Lees E.; Yen T. J. Human MPS1 kinase is required for mitotic arrest induced by the loss of CENP-E from kinetochores. Mol. Biol. Cell 2003, 14, 1638–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorer R. K.; Tallarico J. A.; Wong W. H.; Mitchinson T. J.; Murray A. W. A small-molecule inhibitor of Mps1 blocks the spindle-checkpoint response to a lack of tension on mitotic chromosomes. Curr. Biol. 2005, 15, 1070–1078. [DOI] [PubMed] [Google Scholar]
- Schmidt M.; Budirahardia Y.; Klompmaker R.; Medema R. H. Ablation of the spindle assembly checkpoint by a compound targeting Mps1. EMBO Rep. 2005, 6, 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan W.; Cleveland D. W. A chemical tool box defines mitotic and interphase roles for Mps1 kinase. J. Cell Biol. 2010, 190, 21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt L.; Tighe A.; Santaguida S.; White A. M.; Jones C. D.; Musacchio A.; Green S.; Taylor S. S. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J. Cell Biol. 2010, 190, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S.; Tighe A.; D’Alise A. M.; Taylor S. S.; Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J. Cell Biol. 2010, 190, 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J.; George K. A; Terret M. E; Zhang C.; Shokat K. M; Jallepalli P. V. Mps1 directs the assembly of Cdc20 inhibitory complexes during interphase and mitosis to control M phase timing and spindle checkpoint signaling. J. Cell Biol. 2010, 190, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N.; Jelluma N.; Filippakopoulos P.; Soundararajan M.; Manak M. S.; Kwon M.; Choi H. G.; Sim T.; Deveraux Q. L.; Rottmann S.; Pellman D.; Shah J. V.; Kops G. J.; Knapp S.; Gray N. S. Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nat. Chem. Biol. 2010, 6, 259–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldarelli M.; Angiolini M.; Disingrini T.; Donati D.; Guanci M.; Nuvoloni S.; Posteri H.; Quartieri F.; Silvagni M.; Colombo R. Synthesis and SAR of new pyrazolo[4,3-h]quinazoline-3-carboxamide derivatives as potent and selective MPS1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 4507–4511. [DOI] [PubMed] [Google Scholar]
- Colombo R.; Caldarelli M.; Mennecozzi M.; Giorgini M. L.; Sola F.; Cappella P.; Perrera C.; Depaolini S. R.; Rusconi L.; Cucchi U.; Avanzi N.; Bertrand J. A.; Bossi R. T.; Pesenti E.; Galvani A.; Isacchi A.; Colotta F.; Donati D.; Moll J. Targeting the mitotic checkpoint for cancer therapy with NMS-P715, an inhibitor of MPS1 kinase. Cancer Res. 2010, 70, 10255–10264. [DOI] [PubMed] [Google Scholar]
- Kusakabe K.; Ide N.; Daigo Y.; Itoh T .; Higashino K.; Okano Y.; Tadano G.; Tachibana Y.; Sato Y.; Inoue M.; Wada T.; Iguchi M.; Kanazawa T.; Ishioka Y.; Dohi K.; Tagashira S.; Kido Y.; Sakamoto S.; Yasuo K.; Maeda M.; Yamamoto T.; Higaki M.; Endoh T.; Ueda K.; Shiota T.; Murai H.; Nakamura Y. Diaminopyridine-based potent and selective Mps1 kinase inhibitors binding to an unusual flipped-peptide conformation. ACS Med. Chem. Lett. 2012, 3, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardif K. D.; Rogers A.; Cassiano J.; Roth B. L.; Cimbora D. M.; McKinnon R.; Peterson A.; Douce T. B.; Robinson R.; Dorweiler I.; Davis T.; Hess M. A.; Ostanin K.; Papac D. I.; Baichwal V.; McAlexander I.; Willardsen J. A.; Saunders M.; Christophe H.; Kumar D. V.; Wettstein D. A.; Carlson R. O.; Williams B. L. Characterization of the cellular and antitumor effects of MPI-0479605, a small-molecule inhibitor of the mitotic kinase Mps1. Mol. Cancer Ther. 2011, 10, 2267–2275. [DOI] [PubMed] [Google Scholar]
- Acros Organics. http://www.acros.be/ (accessed December 14, 2010).
- Asinex. http://www.asinex.com/ (accessed December 14, 2010).
- ChemBridge. http://www.chembridge.com/index.php (accessed December 14, 2010).
- ChemDiv. http://eu.chemdiv.com/ (accessed December 14, 2010).
- Enamine. http://www.enamine.net/ (accessed December 14, 2010).
- InterBioScreen. http://www.ibscreen.com/ (accessed December 14, 2010).
- Key Organics. http://www.keyorganics.co.uk/ (accessed December 14, 2010).
- Life Chemicals. http://www.lifechemicals.com/ (accessed December 14, 2010).
- Maybridge. http://www.maybridge.com/ (accessed December 14, 2010).
- Specs. http://www.specs.net (accssed December 14, 2010).
- Tocris Bioscience. http://www.tocris.com/ (accessed December 14, 2010).
- Ghose A. K.; Crippen G. M. Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure-activity relationships I. Partition coefficients as a measure of hydrophobicity. J. Comput. Chem. 1986, 7, 565–577. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- Pipeline Pilot 7.0; Accelrys, Inc.: San Diego, CA. http://accelrys.com/ (accessed October 18, 2012). [Google Scholar]
- MOE 2009.10; Chemical Computing Group: Montreal, Quebec, Canada. http://www.chemcomp.com/ (accessed October 18, 2012). [Google Scholar]
- Rogers D.; Hahn M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 724–754. [DOI] [PubMed] [Google Scholar]
- Jaccard P. Distribution de la flore alpine dans le bassin des Dranses et dans quelques rėgions voisines. Bulletin del la Sociėtė Vaudoise des Sciences Naurelles 1901, 37, 241–272. [Google Scholar]
- Tanimoto T. T. IBM Internal Report; November 17, 1957.
- ROCS. OpenEye, Scientific Software. http://www.eyesopen.com/ (accessed October 18, 2012). [Google Scholar]
- OMEGA. OpenEye, Scientific Software. http://www.eyesopen.com/ (accessed October 18, 2012). [Google Scholar]
- Durant J. L.; Leland B. A.; Henry D. R.; Nourse J. G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Sleebs B. E.; Levit A.; Street I. P.; Falk H.; Hammonds T.; Wong A. C.; Charles M. D.; Olson M. F.; Baell J. B. Identification of 3-aminothieno[2,3-b]pyridine-2-carboxamides and 4-aminobenzothieno[3,2-d]pyrimidines as LIMK1 inhibitors. MedChemComm 2011, 2, 977–981. [Google Scholar]
- Adayev T.; Wegiel J.; Hwang Y. W. Harmine is an ATP-competitive inhibitor for dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A). Arch. Biochem. Biophys. 2011, 507, 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D. V.; McAlexander I. A.; Bursavich M. G.; Hoarau C.; Slattum P. M.; Gerrish D. A.; Lockman J. W.; Judd W. R.; Saunders M.; Parker D. P.; Zigar D. F.; Kim I. C.; Willardsen J. A.; Yager K. M.; Shenderovich M. D.; Williams B. L.; Tardif K. D.. Preparation of Purine Derivatives as Inhibitors of TTK Protein Kinase Production, Patent WO2010/111406 A2, 2010.
- Scior T.; Bender A.; Tresadern G.; Medina-Franco J. L.; Martínez-Mayorga K.; Langer T.; Cuanalo-Contreras K.; Agrafiotis D. K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881. [DOI] [PubMed] [Google Scholar]
- Hann M. M.; Leach A. R.; Harper G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A. G. W.; Powell H. R.. Processing Diffraction Data with Mosflm, in Evolving Methods for Macromolecular Crystallography; Read R. J., Sussman J. L., Eds.; Springer: New York, 2007; Vol. 245, pp 41–51. [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu M. L.; Chavas L. M.; Douglas K. T.; Eyers P. A; Tabernero L. Crystal structure of the catalytic domain of the mitotic checkpoint kinase Mps1 in complex with SP600125. J. Biol. Chem. 2008, 283, 21495–21500. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G.; Blanc E.; Brandl M.; Flensburg C.; Keller P.; Paciorek W.; Roversi P.; Sharff A.; Smart O. S.; Vonrhein C.; Womack T. O.. BUSTER, version 1.11.2; Global Phasing Ltd.: Cambridge, U.K, 2011. [Google Scholar]
- Smart O. S.; Womack T. O.; Sharff A.; Flensburg C.; Keller P.; Paciorek W.; Vonrhein C.; Bricogne G.. Grade, version 1.1.1; Global Phasing Ltd.: Cambridge, U.K.. http://www.globalphasing.com (accessed October 18, 2012). [Google Scholar]
- Bruno I. J.; Cole J. C.; Lommerse J. P.; Rowland R. S.; Taylor R.; Verdonk M. L. Isostar: A library of information about non-bonded interactions. J. Comput.-Aided Mol. Des 1997, 11, 525–537. [DOI] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B.; Snoeyink J.; Richardson J. S.; Richardson D. C. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.