

Abstract
Ring-fused 2-pyridones, termed pilicides, are small synthetic compounds that inhibit pilus assembly in uropathogenic E. coli. Their biological activity is clearly dependent upon a carboxylic acid functionality. Here we present the synthesis and biological evaluation of carboxylic acid isosteres, including e.g. tetrazoles, acyl sulfonamides and hydroxamic acids, of two lead 2-pyridones. Two independent biological evaluations show that acyl sulfonamides and tetrazoles significantly improve pilicide activity against uropathogenic E. coli.
Keywords: 2-pyridone, Bioisostere, Antimicrobial, Pili, Escherichia coli, Pilicide
A vast number of pathogenic microbes assemble virulence factors in the form of rod-like surface organelles via the chaperone-usher pathway.1, 2 Due to its common abundance and high conservation, the chaperone-usher pathway constitutes an attractive target in the development of new strategies directed against Gram-negative infectious diseases, which are reemerging as a global health problem.
The infectious ability of uropathogenic E. coli (UPEC) depends on adhesive surface organelles called pili/fimbriae. These virulence factors comprise both type 1 and P pili, which allow bacterial adherence and colonization of the bladder3, 4 and kidney5, 6, respectively. In addition to being important in the initial stages of urinary tract infections, type 1 pili are also involved in the formation of intracellular bacterial communities (IBCs), where bacteria enter a biofilm-like state and thus evade innate defenses and traditional antibiotic treatment.5, 7–9
We have used UPEC as a model pathogen to design10 small synthetic compounds, pilicides, which regulate pilus assembly.11, 12 These substituted 2-pyridones bind to periplasmic chaperones (PapD/FimC) responsible for the folding13 and transport of pilus subunits to the outer membrane, and thus interfere with the essential chaperone function in the assembly mechanism. Thereby, pilicides can either partially inhibit or completely block pili formation. Importantly, the targeted chaperones have a high level of structural preservation among pathogens utilizing this assembly/secretion machinery.2 Thus, pilicides have the potential of wide-range activity and, indeed, inhibit the formation of both types of UPEC pili i.e., type 1 and P pili encoded by the pap and the fim gene clusters, respectively.
Based on their mode of action, the applicability of pilicides is anticipated to be versatile. First, they can be directly applied to down-regulate pilus assembly and thus, potentially function as chemical tools to study the role of pili in important disease processes.14, 15 Secondly, the utility of these molecules to gain insight in molecular details of the chaperone-usher pathway has been recognized.11 Furthermore, considering the problems encountered with UPEC being persistent to antibiotics and innate defenses, an appealing therapeutic strategy would be a synergistic use of pilicides and traditional antibacterial agents. Altogether, the utility of pilicides ranges from gaining insight into the chaperone-usher pathway at a molecular level to potential future therapeutic applications as novel antibacterial agents that target bacterial virulence.16, 17 We have previously demonstrated that a carboxylic acid functionality on the 2-pyridone framework is important for chaperone affinity and essential for the ability to prevent pilus assembly in E. coli.11, 18, 19 In this paper we further investigate the role of the carboxylic acid functionality using functional group isosterism. This strategy is commonly applied in the development of lead compounds to address e.g. affinity, target specificity and pharmacokinetic properties.20, 21 Two active pilicides have been transformed into a total of twelve derivatives, including well-known carboxylic acid isosteres such as tetrazoles22, acyl sulfonamides23–25 and hydroxamic acids20. The generated set of potential pilicides has been evaluated for chaperone (PapD) affinity and the ability to disrupt P pilus biogenesis in E. coli.
Twelve derivatives of two active pilicides (1 and 2, Fig. 1A) have been synthesized and evaluated. Replacement of the carboxylic acid functionality has been performed, generating a set of eight isosteres comprising tetrazoles, acyl sulfonamides and hydroxamic acids (Fig. 1B). Since primary amides served as precursors in the synthetic route to the tetrazoles, these derivatives were also included in the biological evaluation.
Figure 1.
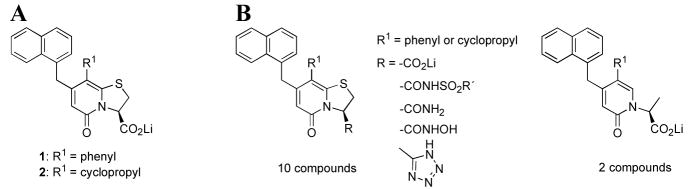
(A) Ring-fused 2-pyridones substituted with a carboxylic acid (1 and 2) inhibit pilus biogenesis in E. coli.11, 18, 19 (B) Acyl sulfonamides, hydroxamic acids, tetrazoles and amides have been synthesized and evaluated as isosteres of 1 and 2. Ring-opened analogues were also included to study the effects of increased flexibility of the carboxylate functionality.
In addition, ring-opened derivatives were synthesized to establish SARs regarding the rigidity of the scaffold and the flexibility of the carboxylic acid. Furthermore, a tolerance for the ring-opened analogues would encourage screening of pilicide activity among more simple generic structures. In addition, such 2-pyridone derivatives could be synthesized via complementary synthetic pathways.
The ring-fused 2-pyridone methyl esters 3 and 4 were first synthesized according to published procedures that had previously been developed in our laboratories.26, 27 From 3 and 4, the synthesis of the hydroxamic acids 5 and 6 was straightforward and proceeded in quantitative yields using hydroxylamine in H2O/MeOH (Scheme 1).
Scheme 1.

a) NH2OH (50 wt% aq.) in MeOH, rt (quant.). (b) First 0.1 M LiOH (aq.) in MeOH:THF (4:1), rt (quant.), then Amberlite® IR120+. (c) ) First N,N′-carbonyldiimidazole in CH2Cl2, preactivation for 1 hour and 45 minutes at rt, then PhSO2NH2 or MeSO2NH2, MW 80 °C, 8h (60–90%) or MW 150 °C, 55 min for 10 (95%).
The acyl sulfonamides 9–12 were obtained from the carboxylic acids 7 and 8, which were first synthesized from 3 and 4 using alkaline hydrolysis (Scheme 1). Activation of the carboxylic acids 7 and 8 was performed using N,N′-carbonyldiimidazole (CDI),24 which after addition of benzene- or methyl sulfonamide and microwave (MW) induced heating at 80 °C for 8 h, rendered the four acyl sulfonamides 9–12 in 60–90% yields (Scheme 1). However, the reaction time was long and the yield of compound 10 was modest (60%), therefore an attempt to increase the yield and shorten the reaction time was performed. Fortunately, increasing the temperature to 150 °C rendered compound 10 in excellent 95% yield after 55 min of microwave heating.
The synthesis of the tetrazoles was undertaken from methyl esters 3 and 4 via the amide intermediates 13 and 14 (Scheme 2). The amides were then transformed to tetrazoles 15 and 16 using SiCl4 and NaN3 in refluxing acetonitrile according to a published procedure.28 A number of unidentified byproducts were also formed under these reaction conditions, which resulted in poor yields of the tetrazoles. However, the obtained amounts of 15 and 16 were sufficient to allow screening and, thus, determination of their potential value as bioisosteres.
Scheme 2.
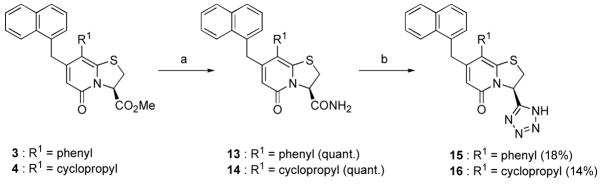
(a) NH3(g) in MeOH, 40 °C (quant.) (b) NaN3, SiCl4, MeCN, reflux (14–18%)
Desulfurization into the ring-opened derivatives 17 and 18 was accomplished using Raney-Nickel® in refluxing MeOH (Scheme 3). Portion-wise addition of Raney-Nickel® was needed for complete conversion of the starting material and the reaction was carefully monitored by LC-MS to avoid any undesired over-reduction of the 2-pyridone ring. The lithium carboxylates 19 and 20 were obtained after alkaline hydrolysis of 17 and 18 (Scheme 3).
Scheme 3.
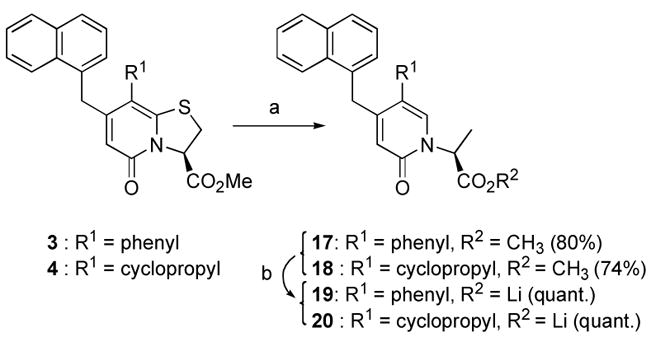
(a) Raney-Nickel® in MeOH, reflux (74–80%) (b) 0.1 M LiOH (aq.) in MeOH:THF (4:1), rt (quant.).
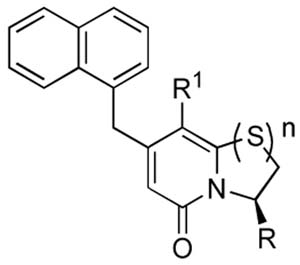
The synthesized set of isosteres (5, 6, 9–16, 19 and 20) and their parent lead compounds (1 and 2) were evaluated as pilicides. More specifically, both their ability to bind to the chaperone PapD in vitro and their ability to inhibit pilus biogenesis in E. coli were evaluated. The cyclopropyl series of derivatives was first ranked for chaperone (PapD) binding affinity using relaxation-edited 1H-NMR spectroscopy (Table 1, Relative PapD affinity). Ranking of binding affinity for small molecules that bind to a large protein can be performed using relaxation-edited 1H-NMR spectroscopy.29, 30 The methodology relies on the fact that a signal originating from a small molecule decreases in intensity when the molecule binds to the protein (given that an appropriate spin-lock filter is applied). The observed reduction in signal intensity (as compared to a non-binding reference) reflects the binding strength and thus allows ranking of affinities. However, the reduced intensity can vary between protons situated at different positions on a molecule (as a result of varying flexibility or differences in specific protein interactions). Thus, ranking is only valid when it is based on a signal that originates from a proton being represented in all compounds. As for most screening techniques, the assumption of a common binding site for the compounds is a prerequisite for affinity ranking. We have previously described and applied relaxation-edited 1H-NMR spectroscopy when ranking series of compounds for chaperone binding10, 18, 19 and this technique was now used also for the cyclopropyl-series of isosteres (Table 1).
Table 1.
Ranking of affinities for chaperone PapD using relaxation-edited 1H-NMR spectroscopy (Relative PapD affinity) and evaluation of inhibitory activity of P pilus biogenesis in HB101/pPAP5 at a concentration of 1.8 mM compound (HA-titer).
| ||||||
|---|---|---|---|---|---|---|
| Entry | Compound | R1 | R | n | Relative PapD affinity (%)a | HA-titerd |
| 1 | 1 | phenyl | -CO2Li | 1 | -b | -e |
| 2 | 2 | cyclopropyl | -CO2Li | 1 | 71 | 8 |
| 3 | 11 | phenyl | -CONHSO2Ph | 1 | -b | 4g |
| 4 | 12 | cyclopropyl | -CONHSO2Ph | 1 | 100 | 4 |
| 5 | 9 | phenyl | -CONHSO2Me | 1 | -b | 4 |
| 6 | 10 | cyclopropyl | -CONHSO2Me | 1 | 62c | 2 |
| 7 | 15 | phenyl | -tetrazole | 1 | -b | 2g |
| 8 | 16 | cyclopropyl | -tetrazole | 1 | 67 | 4 |
| 9 | 5 | phenyl | -CONHOH | 1 | -b | 128f |
| 10 | 6 | cyclopropyl | -CONHOH | 1 | 52 | 64f,g |
| 11 | 13 | phenyl | -CONH2 | 1 | -b | 64f |
| 12 | 14 | cyclopropyl | -CONH2 | 1 | 40 | 64 f |
| 13 | 19 | phenyl | -CO2Hh | 0 | -b | 32 |
| 14 | 20 | cyclopropyl | -CO2Li | 0 | 33c | 32 |
| 15 | None | 64 | ||||
Pilicide:PapD, 25:95 μM. 6% RSD as determined from triplicates with compound 2.
Only the R1-cyclopropyl derivatives were evaluated for PapD binding affinity.
The reduced intensity was dependent on observed proton; 62 or 78% for 10 and 33 or 52% for 20. The tabulated value originates from protons with identical positioning on the naphthyl unit.
Representative HA-titers (the highest dilutions that still provides hemagglutination) for duplicate runs.
Not evaluated.
Precipitated.
Possible growth defect.
Previous studies have shown that there are no significant difference in biological effect between the carboxylic acid and the corresponding lithium carboxylate.31
Binding data clearly showed that the affinity of the lead compound 2 for the chaperone PapD could be retained or even improved after isosteric replacement of the carboxylic acid. The benzene sulfonamide 12 was the best binder (entry 4, 100% relative PapD affinity) resulting in a completely abolished signal from the compound. The methyl sulfonamide 10 and the tetrazole 16 displayed comparable affinities to 2 (entries 6 and 8 vs entry 2). Decreased affinities were seen for the hydroxamic acid 6, amide 14 and also for the ring-opened analogue 20 (entries 10, 12 and 14). In addition, the reduced binding of 20 compared to the parent, bicyclic lead 2 was central since it demonstrated the importance of having the carboxylate positioned on the rigid scaffold. Interestingly, an increased line-width was observed for one of the naphthyl protons on compounds 10 and 20. This could indicate a different binding mode or site for these compounds. However, considering the potency of 10 (see below), a different binding site seems unlikely for this compound. Altogether, although a reduced binding affinity was observed for some isosteres, all compounds bound to PapD in a range that from previous studies was known to be adequate for inhibitory activity on pilus biogenesis.
Next, all compounds were evaluated in a hemagglutination (HA)-assay for their ability to inhibit P pilus biogenesis in E. coli (Table 1, HA-titer).11 P pili expressing E. coli (HB101/pPAP5) were cultured in the presence of 1.8 mM compound and the degree of piliation could subsequently be assessed by the ability of pilicide treated bacteria to agglutinate erythrocytes (Table 1, HA-titer). The reported HA-titer is the highest dilution of bacteria that still provides hemagglutination and, thus, a high HA-titer denotes a high abundance of pili on the bacterial surface. Rewardingly, the HA-data clearly demonstrated an improved ability to block P pili formation for the tetrazoles 15 and 16 and the acyl sulfonamides 9–12 (entries 3–8 vs entry 1 and 2, HA-titer). Neither the hydroxamic acids 5 and 6 nor the primary amides 13 or 14 were active (entries 9–12). A slight effect was seen for the ring-opened derivatives 19 and 20 (entries 13 and 14), but there was an evident loss in activity compared to the bicyclic lead 2 (entry 2).
Finally, the best compounds from the HA-titers and NMR-studies were further evaluated in a biofilm assay11 both to confirm their biological effect but also to be able to see if they behave in a dose dependent manner (Figure 2). The results show that the compounds could be suspended to a concentration as low as 22 μM, with a dose dependent behavior, and still displaying significant biofilm inhibition.
Figure 2.

Compounds 10 and 16 were evaluated in a biofilm assay where they showed a dose-dependent pattern with an estimated IC50 around 30–40 μM for compound 10 and 65–75 μM for 16. The corresponding carboxylic acid substituted pilicide 2 displayed a 45 % inhibition at 400 μM. The figure shows the average inhibition of duplicate runs and the standard deviations are shown in error bars.
Overall, the reduced activity seen for the ring opened compounds 19 and 20 shows that there is an apparent central role of the ring-fused 2-pyridone scaffold, which inspires to continued investigations of this entity. Furthermore, sterically demanding isosteres appear to be accepted, which encourage further fine-tuning of this position, for example with various, substituted acyl sulfonamides. Even though activity in whole cell-based assays is often difficult to relate to direct binding affinity, a correlation can be seen for the evaluated isosteres. The fact that the hydroxamic acids 5 and 6 and primary amides 13 and 14 were inactive may be attributed to their poor solubility in the HA-assay.
In conclusion, the importance of a rigid, ring-fused scaffold has been clearly demonstrated for carboxylic acid functionalized 2-pyridones that inhibit pilus biogenesis in E. coli. Furthermore, tetrazoles and acyl sulfonamides have been shown to be potent bioisosteres of carboxylic acids for 2-pyridone based pilicides. Compared to a parent carboxylic acid functionalized lead compound, the tetrazole and acyl sulfonamides displayed improved or retained binding affinity for the chaperone PapD. In addition, their ability to block P pilus assembly in E. coli was significantly enhanced as determined by hemagglutination and their ability to inhibit pili dependent biofilm formation. The increased potencies, particularly for the sterically more demanding isosteres, open up possibilities to further investigate this position from a medicinal chemistry perspective.
Supplementary Material
Acknowledgments
We thank the Swedish Natural Science Research Council and the Knut and Alice Wallenberg foundation for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sauer FG, Remaut H, Hultgren SJ, Waksman G. Biochim Biophys Acta. 2004;1694(1–3):259–267. doi: 10.1016/j.bbamcr.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Hung D, Hultgren SJ. J Struct Biol. 1998;124:201–220. doi: 10.1006/jsbi.1998.4049. [DOI] [PubMed] [Google Scholar]
- 3.Dodson KW, Pinkner JS, Rose T, Magnusson G, Hultgren SJ, Waksman G. Cell. 2001;105(6):733–743. doi: 10.1016/s0092-8674(01)00388-9. [DOI] [PubMed] [Google Scholar]
- 4.Roberts JA, Marklund BI, Ilver D, Haslam D, Kaack MB, Baskin G, Louis M, Mollby R, Winberg J, Normark S. Proc Natl Acad Sci USA. 1994;91(25):11889–11893. doi: 10.1073/pnas.91.25.11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mulvey MA, Lopez-Boado YS, Wilson CL, Roth R, Parks WC, Heuser J, Hultgren SJ. Science. 1998;282(5393):1494–1497. doi: 10.1126/science.282.5393.1494. [DOI] [PubMed] [Google Scholar]
- 6.Langermann S, Palaszynski S, Barnhart M, Auguste G, Pinkner JS, Burlein J, Barren P, Koenig S, Leath S, Jones CH, Hultgren SJ. Science. 1997;276(5312):607–611. doi: 10.1126/science.276.5312.607. [DOI] [PubMed] [Google Scholar]
- 7.Soto SM, Smithson A, Horcajada JP, Martinez JA, Mensa JP, Vila J. Clin Microbiol Infect. 2006;12(10):1034–1036. doi: 10.1111/j.1469-0691.2006.01543.x. [DOI] [PubMed] [Google Scholar]
- 8.Justice SS, Hung C, Theriot JA, Fletcher DA, Anderson GG, Footer MJ, Hultgren SJ. Proc Natl Acad Sci USA. 2004;101(5):1333–1338. doi: 10.1073/pnas.0308125100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Science. 2003;301(5629):105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 10.Svensson A, Larsson A, Emtenäs H, Hedenström M, Fex T, Hultgren SJ, Pinkner JS, Almqvist F, Kihlberg J. ChemBioChem. 2001;2(12):915–918. doi: 10.1002/1439-7633(20011203)2:12<915::AID-CBIC915>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 11.Pinkner JS, Remaut H, Buelens F, Miller E, Aberg V, Pemberton N, Hedenstrom M, Larsson A, Seed P, Waksman G, Hultgren SJ, Almqvist F. Proc Natl Acad Sci USA. 2006;103(47):17897. doi: 10.1073/pnas.0606795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aberg V, Almqvist F. Org Biomol Chem. 2007;5(12):1827. doi: 10.1039/b702397a. [DOI] [PubMed] [Google Scholar]
- 13.Bann JG, Pinkner JS, Frieden C, Hultgren SJ. Proc Natl Acad Sci USA. 2004;101(50):17389. doi: 10.1073/pnas.0408072101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aberg V, Fallman E, Axner O, Uhlin BE, Hultgren SJ, Almqvist F. Mol BioSyst. 2007;3(3):214. doi: 10.1039/b613441f. [DOI] [PubMed] [Google Scholar]
- 15.Andersson M, Axner O, Uhlin BE, Almqvist F, Fallman E. ChemPhysChem. 2008;9(2):221. doi: 10.1002/cphc.200700389. [DOI] [PubMed] [Google Scholar]
- 16.Clatworthy AE, Pierson E, Hung DT. Nat Chem Biol. 2007;3(9):541. doi: 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 17.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. Nat Rev Microbiol. 2008;6(1):17. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Åberg V, Sellstedt M, Hedenström M, Pinkner JS, Hultgren SJ, Almqvist F. Bioorg Med Chem. 2006;14(22):7563. doi: 10.1016/j.bmc.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 19.Åberg V, Hedenström M, Pinkner JS, Hultgren SJ, Almqvist F. Org Biomol Chem. 2005;3(21):3886. doi: 10.1039/b509376g. [DOI] [PubMed] [Google Scholar]
- 20.Lima ML, Eliezer JB. Curr Med Chem. 2005;12:23. [Google Scholar]
- 21.Patani GA, LaVoie EJ. Chem Rev. 1996;96(8):3147. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- 22.Herr RJ. Bioorg Med Chem. 2002;10(11):3379. doi: 10.1016/s0968-0896(02)00239-0. [DOI] [PubMed] [Google Scholar]
- 23.Rönn R, Sabnis YA, Gossas T, Åkerblom E, Danielson UH, Hallberg A, Johansson A. Bioorg Med Chem. 2006;14(2):544. doi: 10.1016/j.bmc.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 24.Johansson A, Poliakov A, Åkerblom E, Wiklund K, Lindeberg G, Winiwarter S, Danielson UH, Samuelsson B, Hallberg A. Bioorg Med Chem. 2003;11(12):2551. doi: 10.1016/s0968-0896(03)00179-2. [DOI] [PubMed] [Google Scholar]
- 25.Chakravarty PK, Naylor EM, Chen A, Chang RSL, Chen TB, Faust KA, Lotti VJ, Kivlighn SD, Gable RA, Zingaro GJ, Schorn TW, Schaffer LW, Broten TP, Siegl PKS, Patchett AA, Greenlee WJ. J Med Chem. 1994;37(24):4068. doi: 10.1021/jm00050a002. [DOI] [PubMed] [Google Scholar]
- 26.Emtenäs H, Taflin C, Almqvist F. Mol Diversity. 2003;7:165. doi: 10.1023/b:modi.0000006800.46154.99. [DOI] [PubMed] [Google Scholar]
- 27.Emtenäs H, Alderin L, Almqvist F. J Org Chem. 2001;66(20):6756. doi: 10.1021/jo015794u. [DOI] [PubMed] [Google Scholar]
- 28.Fang XQ, Bandarage UK, Wang T, Schroeder JD, Garvey DSi. Synlett. 2003;4:489. [Google Scholar]
- 29.Stebbins JL, Jung DW, Leone M, Zhang XK, Pellecchia M. J Biol Chem. 2006;281(24):16643. doi: 10.1074/jbc.M600318200. [DOI] [PubMed] [Google Scholar]
- 30.Hajduk PJ, Olejniczak ET, Fesik SW. J Am Chem Soc. 1997;119(50):12257. [Google Scholar]
- 31.Emtenäs H, Åhlin K, Pinkner JS, Hultgren SJ, Almqvist F. J Comb Chem. 2002;4(6):630. doi: 10.1021/cc020032d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.