

Abstract
Background
Homozygous null mutation of the fibroblast growth factor receptor 2IIIb (Fgfr2IIIb) gene in mice results in 42% of embryos developing duodenal atresias. Retinaldehyde dehydrogenase 2 (Raldh2, a gene critical for the generation of retinoic acid) is expressed in the mouse duodenum during the temporal window when duodenal atresias form. Raldh2 is critical for the normal development of the pancreatoduodenal region; therefore, we were interested in the effect of a Raldh2 mutation on duodenal atresia formation. To test this, we rendered Fgfr2IIIb−/− embryos haploinsufficient for the Raldh2 and examined these embryos for the incidence and severity of duodenal atresia.
Methods
Control embryos, Fgfr2IIIb−/− mutants, and Fgfr2IIIb−/−; Raldh2+/− mutants were harvested at embryonic day 18.5, genotyped, and fixed overnight. Intestinal tracts were isolated. The type and severity of duodenal atresia was documented.
Results
A total of 97 Fgfr2IIIb−/− embryos were studied; 44 had duodenal atresias, and 41 of these presented as type III. In the 70 Fgfr2IIIb−/−; Raldh2+/− embryos studied, a lesser incidence of duodenal atresia was seen (15 of 70; P = .0017; Fisher exact test). Atresia severity was also decreased; there were 12 embryos with type I atresias, 3 with type II atresias, and 0 with type III atresias (P < 2.81E–013; Fisher exact test).
Conclusion
Haploinsufficiency of Raldh2 decreases the incidence and severity of duodenal atresia in the Fgfr2IIIb−/− model. The ability to alter defect severity through manipulation of a single gene in a specific genetic background has potentially important implications for understanding the mechanisms by which intestinal atresias arise.
The etiology of intestinal atresia remains undetermined. Atresias have been ascribed to a number of events, including in utero vascular accidents,1–3 mechanical compression,4 and disruptions in notochord development.5,6 In the case of the duodenum, atresias have been hypothesized to result from a failure of the intestinal lumen to recanalize after a period of exuberant endodermal growth.7
Over the last decade, genetic animal models of intestinal atresia have been developed. The best characterized of these genetic models result from homozygous mutation of either fibroblast growth factor 10 (Fgf10) or its cognate receptor, fibroblast growth factor receptor 2IIIb (Fgfr2IIIb) in mice.8,9 The homozygous mutation of either of these genes results in nonviable progeny at embryonic day (E) 18.5, in which 100% of the embryos have colonic atresias9,10 and 38% to 42% have duodenal atresias.8,11 Approximately 92% of the duodenal atresias observed in the Fgf10 mouse model are type III (gap in the intestine and mesentery), and 8% are type I (intestinal tube is intact externally but has a blockage in the lumen).8 In the Fgfr2IIIb model, the formation of colonic and duodenal atresia is preceded by an increase level of apoptosis in the endoderm.12 Interestingly, unlike human duodenal development, normal development of the mouse duodenum proceeds without a solid core phase in the lumen.12 These results suggest that in the Fgfr2IIIb mouse model, the formation of duodenal and colonic atresias result from the loss of endoderm during a critical phase in development, as opposed to a failure in recanalization of the lumen as proposed originally.7
We observed previously that retinaldehyde dehydrogenase 2 (Raldh2) is expressed in the duodenum during the temporal window of atresia formation (E 11.0–E 13.5).13 This gene encodes for an enzyme that catalyzes the final step of vitamin A conversion to its active metabolite, which is retinoic acid. Recent work has shown that this gene is critical for normal development in the pancreatic–duodenal region because homozygous mutations in Raldh2 result in disruptions in pancreatic development. These embryos fail to progress beyond E 9.5 in development, suggesting that Raldh2 is a critical gene in the development of numerous systems during embryogenesis.14
We hypothesized that haploinsufficiency of Raldh2 would affect the severity of duodenal atresias in the Fgfr2IIIb−/− model. In this model, 93% of the atresias are type III defects; however, the penetrance ranges from 38% to 42%.8,12 Therefore, if haploinsufficiency of Raldh2 were to increase the severity of the defects, we would expect to see a significant increase in atresia incidence. If haploinsufficiency of Raldh2 were to have the opposite effect, then we would expect both the severity and the incidence of atresias to decrease. To test this, we generated compound mutants using the hypoxanthine-guanine phosphoribosyltransferase-Cre (Hprt-Cre) system with conditional alleles for both Fgfr2IIIb and Raldh2.11 We observed that haploinsufficiency of Raldh2 decreased both the severity and incidence of duodenal atresia in comparison to mutant controls.
METHODS
Animals
Institutional Animal Care and Use Committee approval for these studies was obtained from the University of Wisconsin School of Medicine and Public Health (P.F.N. protocol #M02258). All animals were maintained in a clean facility with unlimited access to fresh food and water under a 12-hour alternating light/dark cycle.
Generation of mutant fetuses
The use of Hprt-Cre technology15 has been described previously.11 Fgfr2IIIb−/− mutants were generated by mating Fgfr2IIIb c/c males to Fgfr2III−/+; Hprt-Cre m/+ females. Fgfr2IIIb−/−; Raldh2+/− embryos were generated by mating Fgfr2IIIb c/c; Raldh2 c/c males to Fgfr2III−/+; Raldh2+/+; Hprt-Cre m/+ females. Primer sets for genotyping have been described previously.16
Whole mount in situ hybridization
Wild-type embryos were harvested at E 12.5 into cold phosphate-buffered saline and fixed overnight in 4% paraformaldehyde at 4°C. Fixed samples were dissected and dehydrated to 100% methanol through a series of escalating methanol/phosphate-buffered saline–Tween steps and stored at −20°C. The hybridization process included incubation at 68°C with Raldh2 antisense probes.13,17 Photographs were taken under a stereoscopic dissecting microscope.
Morphologic studies
Embryos were harvested at E 18.5, and whole mount photographs were taken under a stereoscopic dissecting microscope. The thorax was opened and the embryos were fixed overnight in 4% paraformaldehyde at 4°C. On day 2, the gut was dissected out and photographs of the stomach, duodenum, cecum, and colon were obtained. The incidence and severity of colonic, duodenal atresia was determined.
Three-dimensional reconstruction
An Fgfr2III−/−; Raldh2+/− mutant with a type I atresia was fixed overnight in Bouin’s fixative at 4°C, dehydrated through a series of escalating ethanol/phosphate-buffered saline steps, embedded in paraffin, and cut into 7-μm thick slices. Sections were floated onto slides in a 50°C water bath, dewaxed, and stained with hematoxylin–eosin. Photomicrographs were taken at 40× using a standard light microscope (Olympus BX-41; Olympus, Tokyo, Japan). Photomicrographs of sequential hematoxylin–eosin-stained sections were uploaded into the Amira Program (Visage Imaging, San Diego, CA). Uploaded images were aligned with the “auto align” tool and labeled using the “segmentation editor.” After this procedure, a 3-dimensional structure was configured using the “surface generator” tool to show the external contour of the embryo, the extent of the notochord, and the external surfaces of the intestines. A final rendering was generated as a JPEG file at 600 pixels per square inch and imported into Adobe Photoshop (Adobe, San Jose, CA) for labeling.11
RESULTS
Expression of Raldh2 during mouse development
We examined the expression of Raldh2 in the developing mouse using in situ hybridization. At E 11.5, Raldh2 was expressed robustly in the neural tube, limb buds, genital tubercle, duodenum (Fig 1, A; orange arrow), and intestine distal to the duodenum (Fig 1, A; green arrow), but was excluded from the stomach (Fig 1, A; red arrow) and the segment of the gut distal to the bend in the intestine—a region that includes the cecum and colon (area above the yellow arrow and below the stained intestine proximal to the bend; Fig 1, A). As development progressed through E 13.5, this expression pattern persisted. There was little to no expression in the stomach (Fig 1, B) or colon (Fig 1, C; yellow arrow); expression in the duodenum (Fig 1, B and C; orange arrow) and small intestine (Fig 1, C; green arrow) remained robust.
Fig 1.
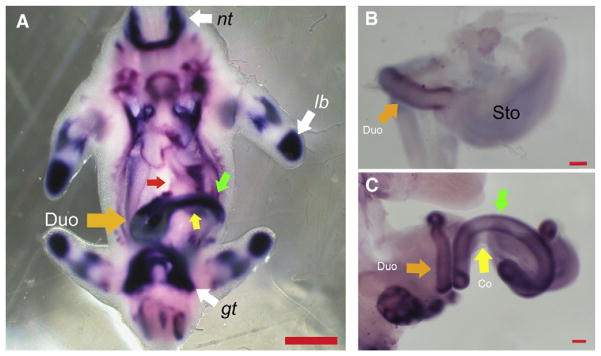
Whole mount in situ for retinaldehyde dehydrogenase 2 (Raldh2) in wild-type mouse embryos. (A) Mouse embryo at embryonic day (E) 11.5. Raldh2 is expressed in the neural tube (nt), limb buds (lb), and genital tubercle (gt); expression is robust in the duodenal region (orange arrow) and the intestine immediately distal to this (green arrow), but is excluded from the stomach (red arrow) and colon (region above the yellow arrow and below the stained intestine). Mouse stomach (B) and intestine (C) at E 13.5. Expression of Raldh2 persists in the duodenum (B and C; orange arrow) and small intestine (C; green arrow); Raldh2 continues to be excluded from the stomach (B) and colon (C; yellow arrow). Measure bar in A indicates 1 mm. Measure bars in B and C indicate 100 μm.
The effect of haploinsufficiency of Raldh2 in Fgfr2IIIb−/− embryos
Fgfr2IIIb−/− embryos present with a number of defects at birth, which include the near absence of all 4 limbs (100% penetrance), microgastria (100% penetrance), duodenal atresia (42% penetrance), and cecal and colonic atresia (both 100% penetrance). These defects form during the time window (E9.5–13.5) when Raldh2 is expressed in development. Based on the observed pattern of Raldh2 expression in wild-type embryos, we predicted that haploinsufficiency of Raldh2 in Fgfr2IIIb−/− embryos would affect the severity and possibly the incidence of limb defects and duodenal atresia. We predicted that there would be no effect on the microgastria or cecal or colonic phenotypes. To examine this, we generated Fgfr2IIIb−/−; Raldh2+/+ and Fgfr2IIIb−/−; Raldh2+/− embryos and conducted morphologic studies.
Limb development
Externally Fgfr2IIIb−/−; Raldh2+/− embryos appeared similar to Fgfr2IIIb−/−; Raldh2+/+ embryos (Fig 2; compare D–E to G–I). They had defects of both the limbs and eyelids and were smaller than their normal littermates (Fig 2, A–C). The severity of the hind limb defects, however, was decreased compared to Fgfr2IIIb−/−; Raldh2+/+ embryos (Fig 2; compare F to I). Whereas the majority of Fgfr2III−/−; Raldh2+/+ embryos had no hind limbs, haploinsufficiency of Raldh2 in Fgfr2IIIb−/−; Raldh2+/− embryos yielded one-third without limbs, one-third with hind limb development that proceeded to a point beyond 1 joint, and one-third developed 2 joints (Table I). This shift toward a less severe hind limb phenotype was statistically significant (P < .02; Fisher exact test).
Fig 2.

Morphology of fibroblast growth factor receptor 2IIIb/retinaldehyde dehydrogenase 2 (Fgfr2IIIb−/−; Raldh2+/−) mutants at embryonic day 18.5. (A) Fgfr2IIIb+/+; Raldh2+/− littermate controls have normal external morphology, including the eyelids (B) and limbs (C). (D) Fgfr2IIIb−/−; Raldh2+/+ mutants have eyelid (E) and severe limb defects (F). (G) Fgfr2IIIb−/−; Raldh2+/− mutants closely resemble Fgfr2IIIb−/−; Raldh2+/+ mutants with eyelid defects (H). Hind limb defects (I), although still present, are less severe (compare I to F). Measure bars indicate 1 cm.
Table I.
Severity of hind limb defect*
| Genotype | No limbs | 1 joint | ≥2 joints |
|---|---|---|---|
| Fgfr2IIIb−/−; Raldh2+/+ | 14 | 2 | 1 |
| Fgfr2IIIb−/−; Raldh2+/− | 5 | 5 | 5 |
P < .02.
Fgfr2IIIb, Fibroblast growth factor receptor 2IIIb; Raldh2, retinaldehyde dehydrogenase 2.
Stomach and colon development
Fgfr2III−/−; Raldh2+/− embryos exhibited a microgastria phenotype, similar to Fgfr2IIIb−/−; Raldh2+/+ mutants (Fig 3; compare B and C to littermate control in A). Likewise, there was no effect on the incidence of the colonic atresia phenotype (Table II). The severity of colonic atresia was unaltered because all Fgfr2IIIb−/−; Raldh2+/− mutants manifested a type III colonic atresia (Fig 4; compare B and C to littermate control in A). Fgfr2IIIb−/−; Raldh2+/− mutants also had an atretic cecal phenotype (Fig 4, C; green arrow), and severity and penetrance of this defect was unaltered compared to that of Fgfr2III−/−; Raldh2+/+ mutants (Table II). The lack of any change in the severity of these defects was consistent with the exclusion of Raldh2 expression from these regions during gut development and in accordance with our predictions.
Fig 3.

Morphology of the stomach in fibroblast growth factor receptor 2IIIb/retinaldehyde dehydrogenase 2 (Fgfr2IIIb−/−; Raldh2+/−) mutants at embryonic day 18.5. (A) Fgfr2IIIb+/+; Raldh2+/− control littermates have normal external morphology of the stomach. Both Fgfr2IIIb+/+; Raldh2+/+ (B) and Fgfr2IIIb−/−; Raldh2+/− mutants (C) and have microgastria, indicating that haploinsufficiency of Raldh2 has no effect on this defect.
Table II.
Incidence of cecal and colonic atresia
| Genotype | n | Atresia type
|
% | |
|---|---|---|---|---|
| Cecal | Colonic | |||
| Fgfr2IIIb−/−; Raldh2+/+ | 20 | 20 | 20 | 100 |
| Fgfr2IIIb−/−; Raldh2+/− | 20 | 20 | 20 | 100 |
Fgfr2IIIb, Fibroblast growth factor receptor 2IIIb; Raldh2, retinaldehyde dehydrogenase 2.
Fig 4.

Morphology of the colon in fibroblast growth factor receptor 2IIIb/retinaldehyde dehydrogenase 2 (Fgfr2IIIb−/−; Raldh2+/−) mutants at embryonic day 18.5. (A) Fgfr2IIIb+/−; Raldh2+/− control littermates have normal morphology of the colon. Both Fgfr2IIIb−/−; Raldh2+/+ (B) and Fgfr2IIIb−/−; Raldh2+/− mutants (C) develop a colonic atresia. Yellow arrow in (A) indicates colon and the atretic end of the proximal colon in (B) and (C). Green arrow in (A) indicates the cecum and the atretic cecum in (C).
Duodenum
In contrast to the colon, we observed a decrease in the incidence of duodenal atresia in Fgfr2IIIb−/−; Raldh2+/− embryos from 45% to 21% as compared to Fgfr2IIIb−/−; Raldh2+/+ embryos (P < .002; Table III). Fgfr2IIIb−/−; Raldh2+/− embryos also had a dramatic shift toward a less severe atresia phenotype (P < .001; Fig 5, A–D). No type III atresias were observed in Fgfr2IIIb−/−; Raldh2+/− embryos (Table IV), 80% of the atretic defects were type I and 20% were type II.
Table III.
Incidence of duodenal atresia*
| Genotype | Normal | Atresia | % |
|---|---|---|---|
| Fgfr2IIIb−/−; Raldh2+/+ | 53 | 44 | 45.36 |
| Fgfr2IIIb−/−; Raldh2+/− | 55 | 15 | 21.43 |
P < .0017.
Fgfr2IIIb, Fibroblast growth factor receptor 2IIIb; Raldh2, retinaldehyde dehydrogenase 2.
Fig 5.
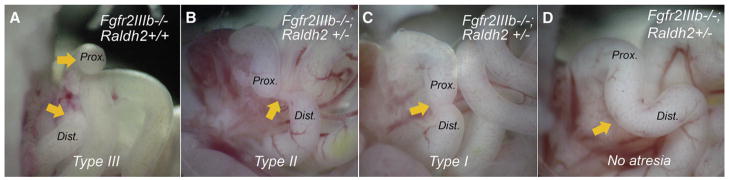
Haploinsufficiency of retinaldehyde dehydrogenase 2 (Raldh2) reduces the severity of duodenal atresia in fibro-blast growth factor receptor 2IIIb (Fgfr2IIIb−/−) mutants from (A) type III atresias in an Fgfr2IIIb−/−; Raldh2+/+ mutant and (B) type II atresia, (C) type I atresia, and (D) no atresia in Fgfr2IIIb−/−; Raldh2+/− mutants. Orange arrows in (A) indicate proximal and distal ends of atresia of duodenum. In (B), the arrow indicates the solid cord between the proximal and distal ends. In (C), the arrow indicates stenosis between ends and in (D) indicates the region where the atresia would normally form.
Table IV.
Severity of duodenal atresia*
| Genotype | No atresia | Type I | Type II | Type III |
|---|---|---|---|---|
| Fgfr2IIIb−/−; Raldh2+/+ | 53 | 3 | 0 | 41 |
| Fgfr2IIIb−/−; Raldh2+/− | 55 | 12 | 3 | 0 |
P < 2.81E–13.
Fgfr2IIIb, Fibroblast growth factor receptor 2IIIb; Raldh2, retinaldehyde dehydrogenase 2.
We were unable to distinguish between a true type I atresia (external narrowing of the intestinal tube with luminal occlusion) from stenosis (external narrowing of the intestinal tube without occlusion) based on morphologic appearance. We therefore generated a 3-dimensional reconstruction of a type I defect from hematoxylin–eosin-stained sections using Amira Imaging technologies (Visage Imaging; Fig 6, A). We then subtracted the mesoderm (Fig 6, B) and the endoderm (Fig 6, C), leaving only a cast of the lumen. This approach revealed that in some cases, these type I defects are stenoses in which luminal continuity is maintained.
Fig 6.
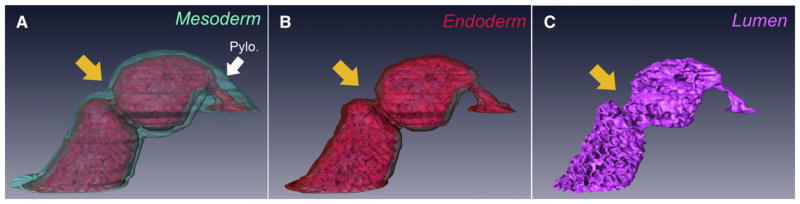
A 3-dimensional reconstruction of a type I atresia (orange arrow) from a fibroblast growth factor receptor 2IIIb/ retinaldehyde dehydrogenase 2 (Fgfr2IIIb−/−; Raldh2+/−) embryo at embryonic day 18.5. In (A), the mesoderm (aquamarine) and endoderm (red) are present. In (B), the mesoderm has been digitally subtracted from the reconstruction, leaving the endoderm. In (C), the endoderm has been digitally subtracted from the reconstruction, leaving a cast of the duodenal lumen (pink) which shows luminal continuity. The pylorus is indicated by the white arrowhead.
DISCUSSION
We report for the first time that the severity and incidence of intestinal atresia can be manipulated by the mutation of a specific gene. Haploinsufficiency of Raldh2 in Fgfr2IIIb−/− mutants converts the duodenal atresias in these mutants from type III defects to type II and type I defects. In addition, the incidence of atresia was decreased by >50% with this approach. We also observed a similar shift toward a less severe phenotype in hind limb defects. By contrast, in anatomic regions where Raldh2 was not expressed (the cecum, colon, and stomach), defect severity and penetrance remained unchanged. Our findings suggest that the decreased penetrance and severity of duodenal atresia in the models is a direct result of a decrease in Raldh2 expression in the duodenal region.
Currently, the mechanism of duodenal atresia formation is unknown. In mice, formation of this defect is preceded by apoptosis within the endoderm at E 10.5,12 followed by the disappearance of the endoderm in the atretic region (also termed the atretic precursor) by E 11.5 and involution of the mesoderm in the absence of further apoptosis. Formation of this defect goes to completion by E 13.5.12,13 It is unclear where in this sequence of events alterations in retinoic acid signaling intervene to limit the severity of atresia defects. Based on our 3-dimensional reconstruction of a type I atresia, the lumen of the duodenum can be preserved. This observation suggests that haploinsufficiency of Raldh2 decreases the loss of endoderm in the duodenum, possibly by limiting the regional extent and/or intensity of apoptosis. We are currently conducting studies to map the timing and extent of apoptosis in the duodenum of both Fgfr2IIIb−/− and Fgfr2IIIb−/−; Raldh2+/− embryos at E 10.5 to determine if this is the case.
Our results have important implications in terms of understanding the etiology of atresia formation. In the past, it has been thought that intestinal atresias of varying degrees of severity arose from distinctly separate mechanisms. Therefore, the mechanism that gave rise to type III defects was considered different from that leading to type I defects or stenosis. Our ability to alter the severity of these atresias in the Fgfr2IIIb−/− model from type III defects to an intestinal stenosis through the manipulation of a single gene argues against this theory. In fact, our data suggest that there are multiple cellular and molecular events involved in the formation of these defects that determine their incidence and spectrum of severity.
Acknowledgments
The authors would like to thank Dr Herbert Chen for sponsoring this work at the 2012 Central Surgical Association meeting.
Supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (1K08DK087854-03), the Society for Surgery of the Alimentary Tract, the American Pediatric Surgical Association, and the Department of Surgery at the University of Wisconsin.
Footnotes
Presented at the 69th Annual Meeting of the Central Surgical Association, March 1–3, 2012, Madison, Wisconsin.
References
- 1.Barnard CN. The genesis of intestinal atresia. Surg Forum. 1957;7:393–6. [PubMed] [Google Scholar]
- 2.Barnard CN, Louw JH. The genesis of intestinal atresia. Minn Med. 1956;39:745. [PubMed] [Google Scholar]
- 3.Louw JH, Barnard CN. Congenital intestinal atresia; observations on its origin. Lancet. 1955;269:1065–7. doi: 10.1016/s0140-6736(55)92852-x. [DOI] [PubMed] [Google Scholar]
- 4.Dalla Vecchia LK, Grosfeld JL, West KW, Rescorla FJ, Scherer LR, Engum SA. Intestinal atresia and stenosis: a 25-year experience with 277 cases. Arch Surg. 1998;133:490–6. doi: 10.1001/archsurg.133.5.490. [DOI] [PubMed] [Google Scholar]
- 5.Dawrant MJ, Giles S, Bannigan J, Puri P. Adriamycin produces a reproducible teratogenic model of gastrointestinal atresia in the mouse. Pediatr Surg Int. 2008;24:731–5. doi: 10.1007/s00383-008-2138-4. [DOI] [PubMed] [Google Scholar]
- 6.Gillick J, Mortell A, Dawrant M, Giles S, Bannigan J, Puri P. The Adriamycin rat/mouse model and its importance to the paediatric surgeon. Pediatr Surg Int. 2008;24:113–8. doi: 10.1007/s00383-007-2035-2. [DOI] [PubMed] [Google Scholar]
- 7.Tandler J. Zur Entwicklungsgeschichte des menschlichen Duodenum in fruhen Embryonalstadien [in German] Morphol Jahrb. 1900;29:187–216. [Google Scholar]
- 8.Fairbanks TJ, Kanard R, Del Moral PM, Sala FG, De Langhe S, Warburton D, et al. Fibroblast growth factor receptor 2 IIIb invalidation—a potential cause of familial duodenal atresia. J Pediatr Surg. 2004;39:872–4. doi: 10.1016/j.jpedsurg.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 9.Fairbanks TJ, Kanard RC, Del Moral PM, Sala FG, De Langhe SP, Lopez CA, et al. Colonic atresia without mesenteric vascular occlusion. The role of the fibroblast growth factor 10 signaling pathway. J Pediatr Surg. 2005;40:390–6. doi: 10.1016/j.jpedsurg.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 10.Fairbanks TJ, Sala FG, Kanard R, Curtis JL, Del Moral PM, De Langhe S, et al. The fibroblast growth factor pathway serves a regulatory role in proliferation and apoptosis in the pathogenesis of intestinal atresia. J Pediatr Surg. 2006;41:132–6. doi: 10.1016/j.jpedsurg.2005.10.054. [DOI] [PubMed] [Google Scholar]
- 11.Nichol PF, Botham R, Saijoh Y, Reeder AL, Zaremba KM. A more efficient method to generate null mutants using Hprt-Cre with floxed alleles. J Pediatr Surg. 2011;46:1711–9. doi: 10.1016/j.jpedsurg.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Botham RA, Franco M, Reeder AL, Lopukhin A, Shiota K, Yamada S, et al. Formation of duodenal atresias in fibroblast growth factor receptor 2IIIb−/− mouse embryos occurs in the absence of an endodermal plug. J Pediatr Surg. 2012;47:1369–79. doi: 10.1016/j.jpedsurg.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nichol PF, Tyrrell JD, Saijoh Y. Retinaldehyde dehydrogenase 2 is down-regulated during duodenal atresia formation in Fgfr2IIIb−/− mice. J Surg Res. 2012;175:82–7. doi: 10.1016/j.jss.2011.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z, Dollé P, Cardoso WV, Niederreither K. Retinoic acid regulates morphogenesis and patterning of posterior foregut derivatives. Dev Biol. 2006;297:433–45. doi: 10.1016/j.ydbio.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 15.Tang SH, Silva FJ, Tsark WM, Mann JR. A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis. 2002;32:199–202. doi: 10.1002/gene.10030. [DOI] [PubMed] [Google Scholar]
- 16.De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127:483–92. doi: 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- 17.Rossant J, Zirngibl R, Cado D, Shago M, Giguére V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991;5:1333–44. doi: 10.1101/gad.5.8.1333. [DOI] [PubMed] [Google Scholar]