

Abstract
Runt-related transcription factor 3 (RUNX3) is a tumor suppressor gene, its inactivation due to hypermethylation related to carcinogenesis. The aim of this study was to investigate the effects of 5-aza-2′-deoxycytidine (5-Aza-CdR) on cell proliferation and apoptosis by demethylation of the promoter region and restoring the expression of RUNX3 in the breast cancer MCF-7 cell line. MCF-7 cells were cultured with different concentrations (0.4–102.4 μmol/L) of 5-Aza-CdR in vitro. MTT assay was used to determine the proliferation of MCF-7 cells. Flow cytometry and Hoechst staining were used for analyzing cell apoptosis. The methylation status and expression of RUNX3 in mRNA and protein levels were measured by methylation-specific polymerase chain reaction (PCR [MSP]), reverse transcription (RT)-PCR, and Western blot. It was shown that the RUNX3 gene downregulated and hypermethylated in MCF-7 cells. 5-Aza-CdR induced demethylation, upregulated the expression of RUNX3 on both mRNA and protein levels in cancer cells, and induced growth suppression and apoptosis in vitro in a dose- and time-dependent manner. The results demonstrate that RUNX3 downregulation in breast cancer is frequently due to hypermethylation, and that 5-Aza-CdR can inhibit cell proliferation and induce apoptosis by eliminating the methylation status of RUNX3 promoter and restoring its expression.
Keywords: breast cancer, RUNX3, methylation, apoptosis
Introduction
Breast cancer is one of the most frequently diagnosed cancers and also the leading cause of cancer death in women. Breast cancer accounted for 23% of the total new cases of cancer and nearly 14% of the total cancer deaths in 2008.1 In the People’s Republic of China, female breast cancer ranks as the sixth most common cause of cancer mortality and accounts for 5.90% of all female deaths from cancer.2 The mortality rate increased by 99.99% in the past 30 years.2 Various factors have been found to relate to the development and progression of breast cancer, including activation of oncogenes, inactivation of tumor suppressor genes, exposure to hormones, and others. Runt-related transcription factor 3 (RUNX3) is located on 1p36. It is 30.31 kb in size, composed of five exons, and a known regulator of major developmental pathways, and has recently been reported as a candidate tumor suppressor related to apoptosis.3 Its inactivation due to DNA hypermethylation has been found in various types of solid tumor, including pancreatic,4 bladder,5 gastric,6 colorectal,7 lung,8 and others. Kim et al9 and Suzuki et al10 first proved that RUNX3 hypermethylation was a characteristic of breast cancers. Its hypermethylation has been seen in both primary tumors and in the adjacent normal breast tissue, indicating that hypermethylation of RUNX3 is an early event in breast carcinogenesis.11
As an epigenetic mechanism of gene silencing of tumor suppressors, DNA hypermethylation can be reversed by DNA methylation inhibitors.12 5-aza-2′-deoxycytidine (5-Aza-CdR), a nucleoside analog, and specific DNA methyltransferase inhibitors have shown antineoplastic activity in patients with leukemia and myelodysplastic syndrome.13,14 Furthermore, its potential to increase chemotherapy sensitivity in cancer cells in vitro has also been proved.15,16 Though much progress has been achieved in methylation-related research, few reports are available related to RUNX3 inactivation due to in breast cancer and how to reverse RUNX3 hypermethylation to restore its biological effect as a tumor suppressor. In the present study, the demethylative effect of 5-Aza-CdR and biological effect of RUNX3 gene in the breast cancer MCF-7 cell line were investigated.
Materials and methods
Reagents
The following reagents were used: fetal bovine serum ([FBS] Gibco; Life Technologies, Carlsbad CA, USA); Roswell Park Memorial Institute (RPMI) medium (Gibco RPMI 1640; Life Technologies); 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide ([MTT] Sigma-Aldrich, St Louis, MO, USA); propidium iodide ([PI] Sigma-Aldrich); dimethyl sufloxide ([DMSO] Sigma-Aldrich); annexin V-fluorescein isothiocyanate (FITC)/PI apoptosis detection kit (BD Biosciences, San Jose, CA, USA); RUNX3 monoclonal antibody (Santa Cruz Biotechnology Inc, Dallas, TX, USA); TRIzol Reagent (Life Technologies); reverse transcription (RT)-polymerase chain reaction (PCR) kit (Ampliqon, Odense, Denmark); 5-Aza-CdR (Sigma-Aldrich); and Hoechst 33258 (Sigma-Aldrich).
Cell line and cell culture
Breast cancer MCF-7 cell line was purchased from Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, People’s Republic of China) and cultured in RPMI 1640 enriched with 1% penicillin/streptomycin and 10% FBS. Cell culture plates were maintained in humidified incubators at 37°C in a 5% CO2 atmosphere. The cells were harvested for analysis by trypsinization.
MTT assay
The viability of the MCF-7 cells was assessed using MTT assay as described previously17 After treatment, cells were seeded in a 96-well plate at a density of 1 × 104 cells/well, cultured for 12 hours, then treated with different concentrations (0.4, 1.6, 6.4, 25.6, 102.4 μmol/L) 5-Aza-CdR for 0–72 hours. At the end of the treatment, MTT 50 μg/10μL was added and the cells were incubated for another 4 hours. After removal of the supernatant, 200 μL DMSO was added to each well. After shaking the plate for 10 minutes, cell viability was assessed by measuring the absorbance at 490 nm using an enzyme-labeling instrument (ELX800, Bio-Tek, Winooski, VT, USA); all measurements were performed three times.
Flow cytometry (FCM) analysis for apoptosis
The apoptotic cells were determined using FCM analysis by annexin V-FITC and PI combined staining as previously described.18 The samples were washed with PBS two times and adjusted to 1 × 106 cells /mL. The suspensions (100 μL) were added to each Falcon tube (12 mm × 75 mm, polystyrene round-bottom), and then the cells were stained with 40 μg/mL annexin V-FITC (10 μL) and 20 μg/mL PI (10 μL). After incubation for 20 minutes in the dark at room temperature, 4°C PBS (400 μL) was added to each tube and analyzed under FCM (BD Biosciences) within 30 minutes. These assays were done three times.
Detection of apoptosis with Hoechst staining
MCF-7 cells were harvested by centrifugation at 800 rpm for 5 minutes, washed with PBS, and fixed with 1% glutaraldehyde for 1 hour at room temperature. Fixed cells were washed with PBS and stained with 200 μM Hoechst 33258 for 10 minutes, and changes in the nuclei of cells, after staining with Hoechst 33258, were observed using a fluorescence microscope (Olympus BX60; Olympus Corp, Tokyo, Japan).
Cell cycle analysis by FCM
FCM analysis was used to evaluate the cell-cycle phase distribution of MCF-7 cells. After the cells became adherent, 5-Aza-CdR at concentrations of 0.4, 1.6, 6.4, 25.6, and 102.4 μmol/L, was added with incubation for 48 hours. Cells were harvested and fixed in 70% ice-cold ethanol and incubated overnight at 4°C. The fixed cells were washed with PBS, then resuspended in 1 mg/mL ribonuclease ([RNase] Sigma-Aldrich), stained with 50 μg/mL PI and incubated at 37°C for 30 minutes in the dark. The stained cells were analyzed for DNA content by a FACScan FCM (BD, Franklin Lakes, NJ, USA) and cell-cycle phase distributions were analyzed with Cell Quest™ Pro software (BD Biosciences).
Methylation-specific PCR (MSP) and RT-PCR
Methylation of RUNX3 was determined by MSP, and non-methylation of RUNX3 by RT-PCR.19 A 24-hour MSP kit (QIA-GEN, Hilden, Germany) was used to determine the methylation status of the RUNX3 gene promoter in genomic DNA extracted from MCF-7 cells. Pre-denatured for 15 minutes at 95°C, denatured for 20 seconds at 95°C, renaturated for 40 seconds at 60°C, and extended 60 seconds at 72°C. Final elongation was performed for 3 minutes at 72°C. The primer sequence for methylation was 5'-TTACGAGGGGCGGTCGTACGC GGG-3′ and antisense for methylation was 5′-AAAACGAC CGACGCGAACGCCTCC-3′ (220 bp). The primer sequence for non-methylation was 5′-TTA TGAGGGGTGGTTG-TATGTGGG-3′ and antisense for non-methylation was 5′-AAAACAACC AACACAAACACCTCC-3′ (212 bp). The amplified PCR products were visualized under 2% agarose gel electrophoresis and an ethidium bromide imaging system. Water was substituted for DNA as a blank control.
Western blot analysis
As previously described20 after treatment with different concentrations of 5-Aza-CdR for 48 hours, cells were harvested and prepared by suspending 1 × 106 cells in 100 mL whole-cell lysis buffer, then disrupted by sonication and extracted by radioimmunoprecipitation assay (RIPA) lysis buffer 72 hours after transfection. Forty micrograms of total protein was added to each well and loaded onto 12% sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) then electrotransferred to Amershand Hybond ECL nitrocellulose membrane (GE Healthcare, Little Chalfont, UK ) in Tris-glycine buffer at 100 Volts for 1.5 hours.
Membranes were incubated overnight at 4°C with anti-RUNX3 monoclonal antibody (dilution: 1/400) and anti-β-actin monoclonal antibody (dilution: 1/200), respectively. After being washed with PBS at room temperature, it was incubated with secondary antibodies (dilution: 1/1000) for 2 hours, then washed with PBS twice and colored by ECL. It was visualized for the relative expression of protein in gray scale by Image-Pro Plus software (v 6.0; Media Cybernetics, Rockville, MD, USA) according to the manufacturer’s instructions. The protein expression intensity of RUNX3 was calculated by the ratio of the photodensity of RUNX3 and β-actin.
Statistical analysis
All data were expressed as mean ± standard error of the mean (SEM). Statistical analyses were performed with one-way analysis of variance (ANOVA) using the statistical software SPSS (v 13.0; IBM Corporation, Armonk, NY, USA). P-values less than 0.05 were considered statistically significant.
Results
5-Aza-CdR inhibits proliferation in breast cancer MCF-7 cells
In the present study, MCF-7 cells were treated with different concentrations of 5-Aza-CdR. MTT assay was used to examine the inhibiting effect of 5-Aza-CdR on MCF-7 cell growth. The effects of 5-Aza-CdR (0.4, 1.6, 6.4, 25.6, and 102.4 μmol/L) on cell growth after 72 hours are shown in Figure 1. The inhibitory rate of 5-Aza-CdR was as high as 58.63% ± 4.82% when the cells were treated with 102.4 μmol/L 5-Aza-CdR for 72 hours. MTT assay showed that 5-Aza-CdR significantly suppressed the proliferation of MCF-7 cells in a dose- and time-dependent manner, with cell numbers markedly reduced compared to control.
Figure 1.
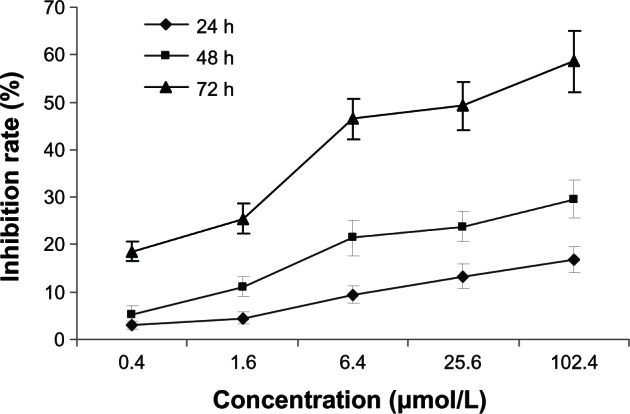
Growth-inhibiting effects of 5-Aza-CdR on MCF-7 cells.
Notes: MCF-7 cells were treated with different concentrations of 5-Aza-CdR for 0–72 hours. Cell viability was determined by MTT assay, performed in triplicate. Dose- and time-dependent inhibition of cell growth could be observed after 72 hours (P < 0.05, one-way ANOVA).
Abbreviations: 5-Aza-CdR, 5-aza-2′-deoxycytidine; ANOVA, analysis of variance; MTT, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; h, hours.
FCM analysis for cell apoptosis
To demonstrate the apoptosis-inducing effect of 5-Aza-CdR, FCM analysis was used with annexin V-FITC and PI combined staining. Apoptosis was observed after treatment with different concentrations of 5-Aza-CdR for 48 hours.
Viable apoptotic cells were rarely seen in the normal control group, while in the 5-Aza-CdR groups, apoptotic cell numbers gradually increased in a time-dependent manner (Figure 2). The apoptotic rates in the blank control group and 0.4, 1.6, 6.4, 25.6, and 102.4 μmol/L 5-Aza-CdR groups were 4.32% ± 1.15%, 9.26% ± 2.24%, 16.53% ± 4.12%, 25.35% ± 4.98%, 37.64% ± 6.32%, and 53.56% ± 7.83%, respectively.
Figure 2.
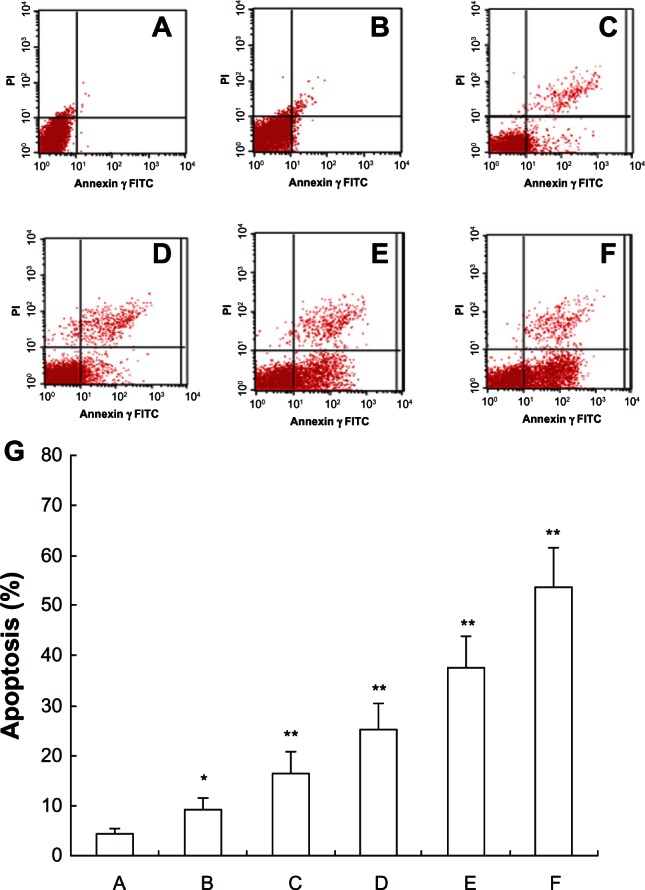
FCM analysis for apoptosis after treatment by annexin V-FITC and PI staining on MCF-7 cells. After treatment with different doses of 5-Aza-CdR for 48 hours, apoptosis induction was observed. Apoptotic cells were differentiated from viable or necrotic ones by combined application of annexin V-FITC and PI. This assay was performed in triplicate. (A) Blank control group; (B) 0.4 μmol/L 5-Aza-CdR group; (C) 1.6 μmol/L 5-Aza-CdR group; (D) 6.4 μmol/L 5-Aza-CdR group; (E) 25.6 μmol/L 5-Aza-CdR group; (F) 102.4 μmol/L 5-Aza-CdR group. (G) comparison of all groups.
Notes: *P < 0.05; **P < 0.01 versus control group.
Abbreviations: 5-Aza-CdR, 5-aza-2′-deoxycytidine; FCM, flow cytometry; FITC, fluorescein isothiocyanate; PI, propidium iodide.
Measurement of apoptosis of MCF-7 cells by Hoechst 33258 staining
To confirm the apoptosis-inducing effect of 5-Aza-CdR, we observed the cells with Hoechst 33258 staining using a fluorescence microscope. The dye stains condense chromatin of apoptotic cells more brightly than chromatin of normal cells. As shown in Figure 3, few apoptotic cells were observed in the control group (Figure 3A), while morphological changes of cell apoptosis, including condensation of chromatin and nuclear fragmentation, were observed in the 5-Aza-CdR-treated groups (Figure 3B–D). The percentages of apoptotic cells in the control group and 0.4, 6.4, and 102.4 μmol/L 5-Aza-CdR-treated groups were 2.52% ± 1.37%, 6.65% ± 2.52%, 18.75% ± 4.68%, and 49.32% ± 6.08%, respectively. Furthermore, apoptotic cell numbers gradually increased in a dose-dependent manner.
Figure 3.
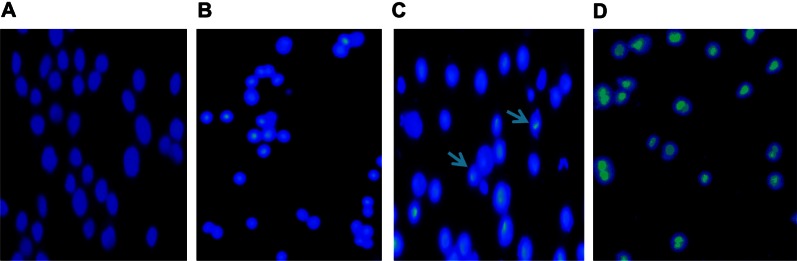
Cell apoptosis observed by Hoechst 33258 staining using a fluorescence microscope (200×). Values represent mean ± SD from three independent experiments. (A) Blank control group; (B) 0.4 μmol/L 5-Aza-CdR group; (C) 6.4 μmol/L 5-Aza-CdR group; (D) 102.4 μmol/L 5-Aza-CdR group.
Notes: The arrows represent the apoptotic cells.
Abbreviations: 5-Aza-CdR 5-aza-2′-deoxycytidine; SD, standard deviation.
5-Aza-CdR arrests the cell cycle
We analyzed the effects of 5-Aza-CdR on the cell-cycle distributions using FCM. As shown in Figure 4, in the 5-Aza-CdR-treated groups, the percentage of cells at the S phase significantly decreased, while increasing at the G2/M phase. The results suggest that 5-Aza-CdR can induce cell-cycle arrest at the G2/M phase in MCF-7 cells.
Figure 4.

Effects of 5-Aza-CdR on the cell cycle of MCF-7 cells by FCM.
Notes: The cell cycle distributions in MCF-7 cells were determined by PI staining and FCM analysis after treatment with 0.4–102.4 μmol/L 5-Aza-CdR for 48 hours. A = Blank control group; B = 0.4 μmol/L 5-Aza-CdR group; C = 1.6 μmol/L 5-Aza-CdR group; D = 6.4 μmol/L 5-Aza-CdR group; E = 25.6 μmol/L 5-Aza-CdR group; F = 102.4 μmol/L 5-Aza-CdR group. Results presented were representative of three independent experiments. *P < 0.05; **P < 0.01 versus control group.
Abbreviations: 5-Aza-CdR 5-aza-2′-deoxycytidine; FCM, flow cytometry; G0/G1, gap0/gap1 phase. G2/M, gap2/mitosis phase; PI, propidium iodide; S, synthesis phase.
5-Aza-CdR inhibits RUNX3 methylation in MCF-7 cells with MSP assay
To confirm whether 5-Aza-CdR inhibits RUNX3 methylation in MCF-7 cells, the methylation of RUNX3 was determined using MSP assay. MSP results show that the promoter regions of RUNX3 were heavily methylated in MCF-7 cells. However, after 5-Aza-CdR treatment, the levels of unmethylated RUNX3 increased gradually along with increasing doses of 5-Aza-CdR in MCF-7 cells (Figure 5) (P < 0.05).
Figure 5.
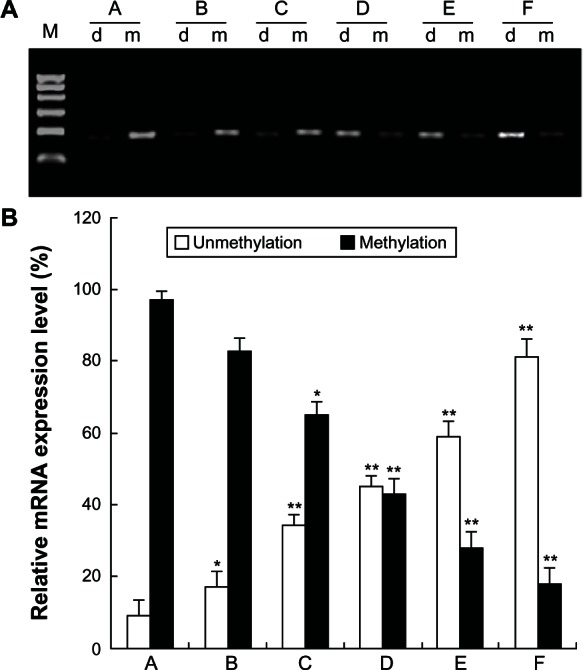
Effect of 5-Aza-CdR on RUNX3 demethylation in MCF-7 cells. MCF-7 cells were treated with various concentrations of 5-Aza-CdR (0.4, 1.6, 6.4, 25.6, and 102.4 μmol/L) for 48 hours, and then RUNX3 methylation was identified by MSP assay. This assay was done in triplicate. (A) RUNX3 methylation in each group. (B) Relative expression level of RUNX3 methylation. A = Control group; B = 0.4 μmol/L 5-Aza-CdR group; C = 1.6 μmol/L 5-Aza-CdR group; D = 6.4 μmol/L 5-Aza-CdR group; E = 25.6 μmol/L 5-Aza-CdR group; F = 102.4 μmol/L 5-Aza-CdR group.
Notes: Values represent means ± SEM. *P < 0.05; **P < 0.01 versus blank control group.
Abbreviations: 5-Aza-CdR, 5-aza-2′-deoxycytidine; MSP, methylation-specific polymerase chain reaction; RUNX3, runt-related transcription factor 3; SEM, standard error of the mean.
5-Aza-CdR increases the expression of RUNX3 in both mRNA and protein levels
To examine whether 5-Aza-CdR increases the expression of RUNX3, we tested the mRNA and protein expression with RT-PCR and Western blot analysis. Compared with the control group, RUNX3 mRNA expression significantly increased in the 5-Aza-CdR-treated group (Figure 6A) (P < 0.05). At the same time, Western blot results showed that RUNX3 protein expression also significantly increased in the 5-Aza-CdR-treated group (Figure 6B) (P < 0.05). Furthermore, the enhancement effects were in a dose-dependent manner (Figure 6C) (P < 0.05).
Figure 6.

Effects of 5-Aza-CdR on RUNX3 mRNA and protein expression in MCF-7 cells. MCF-7 cells were treated with various concentrations of 5-Aza-CdR (0.4, 1.6, 6.4, 25.6, and 102.4 μmol/L) for 48 hours. The mRNA and protein expression of RUNX3 were determined by RT-PCR (A) and Western blot analysis (B) respectively. These assays were done in triplicate. 1 = Control group; 2 = 0.4 μmol/L 5-Aza-CdR group; 3 = 1.6 μmol/L 5-Aza-CdR group; 4 = 6.4 μmol/L 5-Aza-CdR group; 5 = 25.6 μmol/L 5-Aza-CdR group; 6 = 102.4 μmol/L 5-Aza-CdR group. (C). Relative expression level of RUNX3. Values represent means ± SEM.
Notes: *P < 0.05; **P < 0.01 versus blank control group.
Abbreviations: 5-Aza-CdR, 5-aza-2′-deoxycytidine; bp, base pair; GAPDH, phosphoglyceraldehyde dehydrogenase; RT-PCR, reverse transcription polymerase chain reaction; RUNX3, runt-related transcription factor 3; SEM, standard error of the mean.
Discussion
Downregulation of the tumor suppressor RUNX3 is related to a variety of solid cancers. It was first reported by Li et al that the expression of RUNX3 was downregulated in both gastric carcinoma cell lines and gastric carcinoma tissues.19 Up to now, it was reported that the RUNX3 gene was downregulated in a variety of malignancies, including breast cancer.11,21 It has been reported that RUNX3 was downexpressed in 50% of breast cancer cells (n = 19), and hypermethylation of RUNX3 was observed in 52% of primary breast cancers (n = 44) and negatively expressed in the matched adjacent breast epithelium.22 Jiang et al reported that the expression of RUNX3 in breast cancer was much lower than that in normal breast tissues and breast fibroadenoma, and associated with a more favorable prognosis with reduced recurrence and better survival rates in breast cancer patients.23 Chen et al demonstrated that the protein level of RUNX3 was overexpressed in normal breast tissues and widely downregulated in more than 85% of breast cancers.24 In vitro, RUNX3 stable overexpression significantly suppressed the invasive capability of MDA-MB-231 breast cancer cells.24 RUNX3 inactivation is considered to be an early event in breast cancer progression, and the decrease of expression starts to appear in ductal carcinoma in situ, and remains at a similar frequency in invasive ductal carcinoma.11,21 These studies provide evidence of RUNX3 as a tumor suppressor gene in breast cancer, and silencing of tumor suppressor genes confers a selective proliferative advantage to corresponding cells, mediates invasiveness, and facilitates metastasis.
RUNX3 is inactivated in breast cancer by multiple mechanisms, including reduced copy number, promoter hypermethylation, and protein mislocalization.21–24 Epigenetic investigations have reported that DNA hypermethylation plays an important role in the inactivation of RUNX3.25,26 In this study, MSP showed that the promoter region of RUNX3 was heavily methylated in MCF-7 breast cancer cells, while unmethylated RUNX3 was scarcely expressed, indicating that hypermethylation may contribute to the downregulation of RUNX3, in accordance with prior results reported by other authors.11,21–24
With the understanding of hypermethylation in cancer, many methods have been tested to reverse the DNA methylative status so as to improve the treatment efficiency for cancer.27 5-Aza-CdR, a specific DNA methyltransferase inhibitor, has been approved for the treatment of cancer,28 and has demonstrated excellent results.14 In myelodysplastic syndrome treatment, 5-Aza-CdR induced a high response rate at optimal doses and has been shown to prolong survival in clinical research.14,29 In breast cancer, 5-Aza-CdR has shown antineoplastic characteristics and the ability to induce apoptosis, increasing the chemosensitivity and radiosensitivity in cancer cell lines.15,16,30 In this study, RT-PCR and Western blot results show that 5-Aza-CdR treatment can significantly reverse the hypermethylation status of the Runx3 promoter region and promote expression of the RUNX3 gene in both mRNA and protein levels in MCF-7 cancer cell lines in a dose-dependent manner, indicating that demethylation by 5-Aza-CdR contributes to the reactivation of RUNX3, thus resulting in an increase of apoptosis and G2/M cell arrest and a decrease of proliferation of MCF-7 cells in a dose- and time-dependent manner. These findings suggest that demethylation may restore the tumor suppressor activity of RUNX3.
Conclusion
The RUNX3 gene was found to be hypermethylated in MCF-7 cells. 5-Aza-CdR induces demethylation and reactivates the RUNX3 gene to restore its anticancer ability in the breast cancer MCF-7 cell line..
Acknowledgments
This study was supported by the Fundamental Research Funds for the Central Universities, People’s Republic of China; 2013 International Cooperative Project of Shaanxi province, People’s Republic of China; and The Science and Technology Foundation of Shanxi Province, People’s Republic of China (No 2011 K03-[08]).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Zheng Y, Wu CX, Wu F. [Status and trends of breast cancer mortality in Chinese females.] Zhonghua Yu Fang Yi Xue Za Zhi. 2011;45:150–154. Chinese. [PubMed] [Google Scholar]
- 3.Blyth K, Cameron ER, Neil JC. The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer. 2005;5:376–387. doi: 10.1038/nrc1607. [DOI] [PubMed] [Google Scholar]
- 4.Nomoto S, Kinoshita T, Mori T, et al. Adverse prognosis of epigenetic inactivation in RUNX3 gene at 1p36 in human pancreatic cancer. Br J Cancer. 2008;98:1690–1695. doi: 10.1038/sj.bjc.6604333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim EJ, Kim YJ, Jeong P, Ha YS, Bae SC, Kim WJ. Methylation of the RUNX3 promoter as a potential prognostic marker for bladder tumor. J Urol. 2008;180:1141–1145. doi: 10.1016/j.juro.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Ito K, Chuang LS, Ito T, et al. Loss of Runx3 is a key event in inducing precancerous state of the stomach. Gastroenterology. 2011;140:1536–1546. doi: 10.1053/j.gastro.2011.01.043. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe T, Kobunai T, Yamamoto Y, et al. Predicting ulcerative colitis-associated colorectal cancer using reverse-transcription polymerase chain reaction analysis. Clin Colorectal Cancer. 2011;10:134–141. doi: 10.1016/j.clcc.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 8.Yanagawa N, Tamura G, Oizumi H, et al. Promoter hypermethylation of RASSF1A and RUNX3 genes as an independent prognostic prediction marker in surgically resected non-small cell lung cancers. Lung Cancer. 2007;58:131–138. doi: 10.1016/j.lungcan.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Kim TY, Lee HJ, Hwang KS, et al. Methylation of RUNX3 in various types of human cancers and premalignant stages of gastric carcinoma. Lab Invest. 2004;84:479–484. doi: 10.1038/labinvest.3700060. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki M, Shigematsu H, Shames DS, et al. DNA methylation-associated inactivation of TGFbeta-related genes DRM/Gremlin, RUNX3, and HPP1 in human cancers. Br J Cancer. 2005;93:1029–1037. doi: 10.1038/sj.bjc.6602837. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Subramaniam MM, Chan JY, Soong R, et al. RUNX3 inactivation by frequent promoter hypermethylation and protein mislocalization constitute an early event in breast cancer progression. Breast Cancer Res Treat. 2009;113:113–121. doi: 10.1007/s10549-008-9917-4. [DOI] [PubMed] [Google Scholar]
- 12.Tycko B. Cancer epigenetics and targeted therapies. Oncology (Williston Park) 2011;25:228–231. [PubMed] [Google Scholar]
- 13.Yang AS, Doshi KD, Choi SW, et al. DNA methylation changes after 5-aza-2′-deoxycytidine therapy in patients with leukemia. Cancer Res. 2006;66:5495–5503. doi: 10.1158/0008-5472.CAN-05-2385. [DOI] [PubMed] [Google Scholar]
- 14.Kantarjian H, Oki Y, Garcia-Manero G, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109:52–57. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 15.Mirza S, Sharma G, Pandya P, Ralhan R. Demethylating agent 5-aza-2-deoxycytidine enhances susceptibility of breast cancer cells to anticancer agents. Mol Cell Biochem. 2010;342:101–109. doi: 10.1007/s11010-010-0473-y. [DOI] [PubMed] [Google Scholar]
- 16.Sandhu R, Rivenbark AG, Coleman WB. Enhancement of chemotherapeutic efficacy in hypermethylator breast cancer cells through targeted and pharmacologic inhibition of DNMT3b. Breast Cancer Res Treat. 2012;131:385–399. doi: 10.1007/s10549-011-1409-2. [DOI] [PubMed] [Google Scholar]
- 17.Dai ZJ, Gao J, Ma XB, et al. Up-regulation of hypoxia inducible factor-1α by cobalt chloride correlates with proliferation and apoptosis in PC-2 cells. J Exp Clin Cancer Res. 2012;31:28. doi: 10.1186/1756-9966-31-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai ZJ, Gao J, Ma XB, et al. Antitumor effects of rapamycin in pancreatic cancer cells by inducing apoptosis and autophagy. Int J Mol Sci. 2013;14:273–285. doi: 10.3390/ijms14010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li QL, Ito K, Sakakura C, et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109:113–124. doi: 10.1016/s0092-8674(02)00690-6. [DOI] [PubMed] [Google Scholar]
- 20.Zhong ZQ, Song MM, He Y, Cheng S, Yuan HS. Knockdown of Ezrin by RNA interference reverses malignant behavior of human pancreatic cancer cells in vitro. Asian Pac J Cancer Prev. 2012;13:3781–3789. doi: 10.7314/apjcp.2012.13.8.3781. [DOI] [PubMed] [Google Scholar]
- 21.Park SY, Kwon HJ, Lee HE, et al. Promoter CpG island hypermethylation during breast cancer progression. Virchows Arch. 2011;458:73–84. doi: 10.1007/s00428-010-1013-6. [DOI] [PubMed] [Google Scholar]
- 22.Lau QC, Raja E, Salto-Tellez M, et al. RUNX3 is frequently inactivated by dual mechanisms of protein mislocalization and promoter hypermethylation in breast cancer. Cancer Res. 2006;66:6512–6520. doi: 10.1158/0008-5472.CAN-06-0369. [DOI] [PubMed] [Google Scholar]
- 23.Jiang Y, Tong D, Lou G, Zhang Y, Geng J. Expression of RUNX3 gene, methylation status and clinicopathological significance in breast cancer and breast cancer cell lines. Pathobiology. 2008;75:244–251. doi: 10.1159/000132385. [DOI] [PubMed] [Google Scholar]
- 24.Chen W, Salto-Tellez M, Palanisamy N, et al. Targets of genome copy number reduction in primary breast cancers identified by integrative genomics. Genes Chromosomes Cancer. 2007;46:288–301. doi: 10.1002/gcc.20411. [DOI] [PubMed] [Google Scholar]
- 25.Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 26.Schübeler D. Epigenomics: methylation matters. Nature. 2009;462:296–297. doi: 10.1038/462296a. [DOI] [PubMed] [Google Scholar]
- 27.Issa JP. Cancer prevention: epigenetics steps up to the plate. Cancer Prev Res (Phila) 2008;1:219–222. doi: 10.1158/1940-6207.CAPR-08-0029. [DOI] [PubMed] [Google Scholar]
- 28.Gore SD, Jones C, Kirkpatrick P. Decitabine. Nat Rev Drug Discov. 2006;5:891–892. doi: 10.1038/nrd2180. [DOI] [PubMed] [Google Scholar]
- 29.Kantarjian HM, O’Brien S, Huang X, et al. Survival advantage with decitabine versus intensive chemotherapy in patients with higher risk myelodysplastic syndrome: comparison with historical experience. Cancer. 2007;109:1133–1137. doi: 10.1002/cncr.22508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J, Zhou JY, Tainsky MA, Wu GS. Evidence that tumor necrosis factor-related apoptosis-inducing ligand induction by 5-Aza-2′- deoxycytidine sensitizes human breast cancer cells to adriamycin. Cancer Res. 2007;67:1203–1211. doi: 10.1158/0008-5472.CAN-06-2310. [DOI] [PubMed] [Google Scholar]