

Abstract
The epidermal growth-factor receptor (EGFR) is overexpressed in the majority of epithelial ovarian cancers and promotes cell proliferation, migration and invasion, and angiogenesis, as well as resistance to apoptosis. This makes EGFR an attractive therapeutic target in this disease. A number of strategies to block EGFR activity have been developed, including small-molecular-weight tyrosine kinase inhibitors such as erlotinib. Erlotinib has been evaluated as a single agent in recurrent ovarian cancer, as well as in combination with chemotherapeutic agents in the first-line and recurrent settings, and in combination with the antiangiogenic agent bevacizumab in the recurrent setting, as well as in the maintenance setting after completion of first-line chemotherapy. Unfortunately, erlotinib has shown only minimal efficacy as a single agent, and it has not enhanced the effects of chemotherapy or bevacizumab when combined with these agents. Ongoing and future studies of erlotinib and other agents blocking EGFR will need to define mechanisms resulting in resistance to such interventions, and to validate biomarkers of response to identify patients most likely to benefit from such approaches.
Keywords: ovarian cancer, epidermal growth factor, epidermal growth-factor receptor, erlotinib, tyrosine kinase inhibitor
Introduction
Ovarian cancer is the fifth-leading cause of death in women in Canada, the US, and Europe. The majority of patients, except those with surgically resected disease of low stage and grade, have a need for effective postoperative systemic treatment.1,2 Platinum containing combination chemotherapy has been standard for nearly two decades, and paclitaxel plus carboplatin has become the most widely accepted first-line regimen on the basis of several randomized trials.3–6 Despite the efficacy of the combination of platinum/paclitaxel chemotherapy in advanced ovarian carcinoma, over 75% of patients with stage III/IV disease ultimately relapse and die from their disease. Treatment after relapse is dependent upon initial response to therapy and the interval between initial therapy and relapse for platinum-sensitive patients. This usually involves either carboplatin as a single agent, or in combination with paclitaxel, gemcitabine, or liposomal doxorubicin for platinum-sensitive disease. For platinum-resistant patients, this usually involves treatment with single-agent topotecan, doxorubicin (free or liposome-encapsulated), etoposide, gemcitabine, melphalan, or consideration of investigational agents.7
EGF and EGFR biology and role in ovarian cancer
New therapies with a novel mechanism of action with activity in this disease setting are clearly needed. Recently, attention has turned from classical cytotoxic agents to those new drugs that target molecular pathways of relevance in malignancy. One such molecular target is the epidermal growth-factor receptor (EGFR). EGFR is one of four known related members of a family of growth-factor receptors that are important mediators of cell growth, differentiation, and survival: human epidermal growth factor receptor type 1 (HER1; EGFR or ErbB1), HER2 (neu or ErbB2), HER3 (ErbB3), and HER4 (ErbB4). EGFR and its ligands, epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-α) are important in cell proliferation, as well as motility, adhesion, invasion, survival, and angiogenesis.8 Structurally, the EGFR family consists of an extracellular ligand-binding domain, a single transmembrane-spanning region, and an intracellular region containing the kinase domain (Figure 1).
Figure 1.

Epidermal growth factor (EGF) receptor structure.
Notes: Ligand binding of EGF leads to receptor dimerization, resulting in receptor autophosphorylation. This results in activation of a number of downstream signaling pathways. Autophosphorylation of the receptor is blocked by erlotinib. A, extracellular ligand-binding domain; B, transmembrane-spanning domain; C, intracellular domain containing the kinase domain; P, phosphorylation group.
Abbreviations: MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositide 3-kinase.
More than 30 ligands that bind to the EGFR family in humans have been identified, including EGF and EGF-like ligands, TGF-α, and heregulins (also known as neuregulins).9 The EGFR binding partner appears to depend on several properties. These include the proportion of EGFR family members in the membrane, as well as the type and proportion of ligand,10,11 and cell lineage. EGF and TGF-α are the main endogenous ligands for EGFR. EGFR is activated upon ligand binding, which results in a conformational change in the extracellular domain, leading to homo- or heterodimerization with another EGFR family member, activation of tyrosine kinases, followed by receptor autophosphorylation and activation and propagation of signaling cascade, promoting growth. The major signaling pathways activated by EGFR dimerization intracellularly are the Ras/Raf/mitogen-activated protein kinase pathway, which regulates specific intranuclear transcription factors, thus inducing cell migration and proliferation, the signal transducer and activator of transcription (STAT) proteins pathway, which induces oncogenesis and tumor progression mainly through constitutive activation of STAT-3 and STAT-5, the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, which regulates cell growth, apoptosis resistance to chemotherapy, as well as tumor invasion and migration, and the Src kinase pathway, which plays a fundamental role in the regulation of cell proliferation, migration, adhesion, and tumor angiogenesis (Figure 1).12–14
EGFR family members can also be activated by other signaling proteins independently of addition of exogenous EGFR ligands. These include other receptor tyrosine kinases (RTKs), such as the insulin-like growth factor-1 receptor (IGF-1R)15,16 and tyrosine kinase receptor B,17 as well as other types of receptors, such as G protein-coupled receptors (GPCRs),18 the leptin receptor,19 and adhesion proteins, such as E-cadherin20 and integrins.21 While the details of EGFR transactivation upon cross talk are not yet fully elucidated, transactivation has been shown to occur by a variety of mechanisms. There is evidence that EGFR can be transactivated by IGF-1R by direct binding.22 Additionally, EGFR transactivation by GPCR has been shown to occur intracellularly, such as by activation of Src upon GPCR stimulation,23 as well as extracellularly, such as by GPCR activation by gastrin-releasing peptide.24 Lysophosphatidic acid (LPA) GPCR-induced ectodomain shedding of proheparin-binding EGF also activates EGFR.25 LPA-mediated signaling is of particular importance in ovarian cancer, as abnormalities in LPA metabolism and function likely contribute to initiation and progression of ovarian cancer.26–28 Additionally, tyrosine kinase receptor B may also play a role in ovarian cancer, as its activation has been shown to enhance migration and proliferation and to suppress anoikis in human ovarian cancer cells.17,29
EGFR is widely expressed in the surface of mammalian epithelial cells, fibroblasts, gliocytes, keratinocytes, and other cell types. Using an EGFR gene-knockout mouse model, it has been demonstrated that EGFR plays a physiologically favorable role during embryonic and postnatal development.30–32 The EGF pathway is also critical in the control of ovulation. Luteinizing hormone induction of EGF-like growth factors and activation of EGFR signaling is essential for ovulation of mature oocytes.33,34
EGFR expression, mutation, and dysregulation
EGFR plays a pivotal role in tumorigenesis, and its expression strongly affects the outcomes of cancer patients in the clinic.9 Overexpression of EGFR and its ligands leads to malignant transformation.35 EGFR expression reported by various groups in malignant ovarian tumors appears to be highly variable, with expression detected by immunohistochemistry (IHC) ranging from 4% to 100%.36–44 These differences are likely secondary to differences in reagents, experimental procedures, and study design.36 In relation to the development of ovarian cancer, EGFR and its ligands are important in regulating the growth of the ovarian surface epithelium. Alterations of the receptor and its ligands result in a disruption in normal growth-regulatory pathways.36 The presence of both TGF-α and EGFR in ovarian cancer cells suggests that an autocrine growth pathway may be implicated.45 Accumulating evidence suggests dysregulation of EGFR may contribute to the malignancy of ovarian and other tumors through promotion of cell proliferation, migration and invasion, and angiogenesis, as well as resistance to apoptosis.46–49 EGFR has also been found to act as a strong prognostic indicator in ovarian cancers, with increased expression being associated with reduced recurrence-free or overall survival (OS) rates.50–52 Berchuck et al demonstrated that in ovarian cancer specimens with EGFR expression, survival was significantly reduced compared to EGFR-negative specimens, and patients without EGFR expression had a median survival of 40 months compared to 26 months in patients with EGFR-expressing tumors.53
The EGFR gene is mapped to chromosome 7 (7p12.3–p12.1). It consists of 28 exons and spans over 190 kb. EGFR gene amplification or protein overexpression occurs across all epithelial ovarian cancer histotypes.54,55 Increased EGFR expression has been associated with high tumor grade,41,54,56 high cell-proliferation index,41 aberrant p53 expression,41 and poor patient outcome.41,56 The expression of EGFR, phosphorylated AKT, or phosphorylated ERK does not show any significant association with histological subtypes. However, overexpression of pAKT is correlated with progression-free survival (PFS) in ovarian cancer patients, based on their stage of disease and the degree of tumor differentiation.57
Polymorphisms of EGFR may affect the biology of ovarian cancer. Araújo et al58 examined the effect of the A61G polymorphism (substitution of G for A at position 61). They found a decreased risk for developing ovarian cancer in the GG carriers compared to the AA carriers (odds ratio 0.46, confidence interval 0.25–0.81; P = 0.010). Garcia et al reported an association with response to lapatinib and a polymorphism in EGFR exon 20 (2361 G > A, Q787Q).59
The most common EGFR mutation is the type III deletion mutation (EGFRvIII) characterized by elimination of exons 2–7, causing an in-frame deletion of 801 base pairs in the extracellular domain coding sequence, which frequently occurs in malignant gliomas, breast cancer, non-small-cell lung cancer and other types of cancer.60,61 The truncation of extracellular domain leads to constitutive activation of the receptor.62 These activating mutations of EGFR are found exclusively in tumor cells. This type of alteration thus constitutes an optimal target for cancer therapy, and various medical agents have been developed and undergone clinical trials. With respect to ovarian cancer, studies assessing EGFRvIII expression show conflicting results. Moscatello et al demonstrated that EGFRvIII alteration was present in 73% (24/32) of ovarian carcinomas.63 Lassus et al were unable to detect such mutations in EGFR in serous ovarian carcinoma.41 Steffensen et al were also unable to detect such mutations. None of the tissues from 225 patients with normal, benign, borderline, or malignant ovarian cancers were positive for the EGFRvIII mutation, either at the mRNA level or at the protein level.64 The consensus at this time is that EGFRvIII mutations are rare in ovarian cancer and do not contribute significantly to the malignant phenotype in this disease.
Targeting EGFR in ovarian cancer
While several strategies have been developed to block EGFR activity, two types of inhibitors are currently used in the clinic: (1) monoclonal antibodies, and (2) small-molecule tyrosine kinase inhibitors (TKIs).48,65 The focus of this paper will be on the small-molecular-weight TKI erlotinib (OSI-774, R 1415, CP 358774, NSC 718781; Tarceva®).
Erlotinib is an HER1/EGFR TKI. It is a quinazolinamine with the chemical name N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine. Erlotinib hydrochloride has the molecular formula C22H23N3O4 ⋅ HCl and a molecular weight of 429.9066 (Figure 2).
Figure 2.
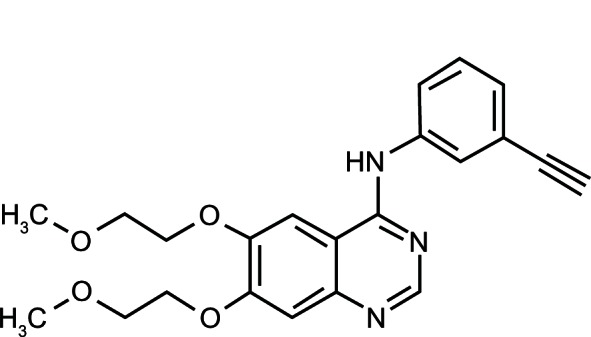
Erlotinib – chemical structure.
Notes: Chemical name, N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4-quinazolinamine, monohydrochloride; United States Adopted Name, erlotinib hydrochloride; other names, NSC 718781, CP-358, OSI-774; molecular formula, C22H23N3O4; molecular weight, 393.4 (free base).
Mechanism of action
Erlotinib is an orally active, potent, selective inhibitor of the EGFR tyrosine kinase.66 Erlotinib inhibits the human EGFR tyrosine kinase with an IC50 of 2 nM (0.786 ng/mL) in an in vitro enzyme assay and reduces EGFR autophosphorylation in intact tumor cells, with an IC50 of 20 nM (7.86 ng/mL).66 This inhibition is selective for EGFR tyrosine kinase in assays of isolated tyrosine kinases, and cellular assays. Erlotinib inhibits EGF-dependent proliferation of cells at submicromolar concentrations and blocks cell-cycle progression in the G1 phase.
Erlotinib reversibly binds to the adenosine triphosphate-binding site of EGFR and completely inhibits autophosphorylation by EGFR tyrosine kinase. This results in blockage of downstream EGFR signal-transduction pathways, cell-cycle arrest, and inhibition of angiogenesis. However, the mechanism of clinical antitumor action of erlotinib is not fully characterized.66
Erlotinib is administered orally. It is absorbed slowly, with peak plasma concentrations occurring 3–4 hours after dosing, with a mean bioavailability of 60%. Bioavailability is significantly improved by administration with food (to approximately 100%) with the mean area under the curve (AUC) increasing by approximately 33% when given with food.66 The increase in bioavailability from administration with food is substantial and increases the risk of drug-related side effects; therefore, erlotinib should be given on an empty stomach. Time to reach steady-state plasma concentration is 7–8 days, and its half-life is about 36 hours. Erlotinib is metabolized in the human liver primarily by cytochrome P450 (CYP)3A4 but also by CYP1A2, and to a minor extent by CYP2C8 (66). Extrahepatic metabolism by CYP3A4 in the intestine, CYP1A1 in the lung, and CYP1B1 in tumor tissue is thought to contribute to the metabolic clearance of erlotinib. Excretion is predominantly via the feces (83%), with renal elimination of the drug and metabolites accounting for 8% of the administered dose. Less than 2% of a dose is eliminated as unchanged drug.66
Clinical trials of EGFR inhibition in ovarian carcinoma
Erlotinib single-agent trials
A single-arm phase II study was conducted by Gordon et al to evaluate erlotinib (150 mg/day) as a treatment option in 34 patients with platinum-resistant or -refractory ovarian cancer.67 All tumors were confirmed to be EGFR protein expression-positive by IHC. Erlotinib demonstrated limited activity for ovarian cancer patients, with an objective response rate of 6% (all partial responses [PRs]), and an additional 14 patients had stable disease that lasted longer than 2 months. Median time to disease progression was 62 days, and the median survival was 8 months. Survival was significantly longer in women who developed a rash.67
Erlotinib in combination with chemotherapy
There is preclinical evidence suggesting that EGFR TKIs potentiate the antitumor effects of cytotoxic agents, including carboplatin.68 Preliminary evidence in an in vitro fibroblast model indicates that EGFR TKIs may beneficially modulate drug resistance, and that EGFR may be causal in the development of resistance to platinum.69
There have been several reports of studies in which standard chemotherapeutic agents were combined with erlotinib in the treatment of ovarian carcinoma. A phase Ib study of erlotinib in combination with carboplatin (AUC 5) and docetaxel (75 mg/m2), followed by erlotinib (75 to 100 mg/day orally) every 21 days in women with chemotherapy-naive ovarian cancer demonstrated an objective response rate of 52% (12/23 patients).70 EGFR aberration or positivity was not an inclusion criterion. The response rate of the erlotinib + docetaxel + carboplatin combination therapy was slightly lower than that of the docetaxel + carboplatin therapy previously conducted by the same group (52% versus 59%).71
In a phase II study of newly diagnosed patients with advanced ovarian cancer, 56 patients were treated with paclitaxel (175 mg/m2) and carboplatin (AUC 6) every three cycles, plus erlotinib (150 mg daily). The objective was to increase the pathologic complete response rate (pCR); however, this was achieved in only eight of 28 patients (29%) after optimal cytoreduction (<1 cm residual disease) and three of 23 patients (13%) who were suboptimally debulked. Tumor specimens were analyzed for EGFR amplification in 20 patients, but no statistically significant correlation was observed between amplification status and response. The addition of erlotinib to carboplatin–paclitaxel did not improve the likelihood of achieving a pCR compared to historical controls.72
Hirte et al investigated the effect of adding erlotinib (150 mg/day) to carboplatin chemotherapy (AUC 5 every 21 days) in 50 ovarian cancer patients who previously had received platinum-based drugs, with 33 in the platinum-sensitive arm and 17 in the platinum-resistant arm.73 In the platinum-sensitive arm, there were three (10%) complete responses (CRs) and 14 (47%) PRs, for an overall response rate (ORR) of 57%. In the platinum-resistant arm, there were no CRs and one PR, for an ORR of 7%. For platinum-sensitive patients with EGFR-positive tumors, there were twelve responses (60% ORR), and in the platinum-resistant arm, the only responding patient was EGFR-positive.73 The combination could be safely administered, and the toxicities were those expected from this combination. However, there was no evidence that erlotinib enhanced the response rate in the platinum-sensitive patients, nor was erlotinib able to reverse resistance to platinum in the platinum-resistant arm.73
Erlotinib in combination with targeted agents
Erlotinib has also been tested in combination with the vascular endothelial growth factor-neutralizing antibody bevacizumab (Avastin) in a phase II trial of patients with recurrent or refractory ovarian cancer.74 Patients were treated with bevacizumab (15 mg/kg intravenously) every 21 days, and with erlotinib (150 mg/day) orally continuously. EGFR aberration or positivity was not required for inclusion in the study. EGFR status was examined by EGFR positivity via IHC and activating mutations in exons 19 and 21 via polymerase chain reaction amplification and sequencing. The ORR was 15% (2/13 patients, one CR and one PR).74 No EGFR mutations were detected, and one patient demonstrated EGFR positivity, but this patient was unresponsive to erlotinib and bevacizumab therapy. The addition of erlotinib did not appear to be associated with an improvement over bevacizumab therapy alone, and there were two incidents of fatal gastric perforations with the combination.74
Chambers et al treated 40 patients with platinum-refractory or -resistant recurrent ovarian cancer with erlotinib (150 mg/day orally) and bevacizumab (10 mg/kg intravenously) every 2 weeks until disease progression.75 Nine (23.1%) of 39 evaluable patients had a response (median duration of 36 weeks, one CR and eight PRs), and ten (25.6%) patients had stable disease, for a disease-control rate of 49%. The authors concluded that bevacizumab plus erlotinib in this patient population was clinically active and well tolerated, but that erlotinib did not appear to contribute to efficacy.75
Erlotinib as maintenance therapy
Recently, a phase III clinical trial randomizing patients to erlotinib versus observation following first-line therapy with no evidence of disease progression was completed.76 A total of 835 patients received six to nine cycles of platinum-based chemotherapy every 3 weeks, and were eligible if they showed no signs of disease progression at the end of chemotherapy. They were then randomly assigned into two arms: one group received 150 mg of maintenance erlotinib daily for 2 years, and the other group was observed. As a secondary analysis, IHC and fluorescence in situ hybridization (FISH) analyses were conducted in 330 patients to determine the predictive value of IHC and FISH for EGFR and EGFR mutations. The primary end point was PFS, with secondary end points of OS, quality of life, and complications. After 24 months of accrual, there were not enough events to reach the study’s end point, and patient accrual was stopped. PFS was 12.7 months for patients treated with erlotinib and 12.4 months for observed patients (P = 0.916). OS for the two groups was 51 months for patients treated with erlotinib and 59 months for observed patients (P = 0.603). Subsequent analyses of the data looked at the relationship between EGFR-mutation status and PFS. Among patients treated with erlotinib, 318 had mutations in EGFR, KRAS, NRAS, BRAF, or PI3KCA; however, there was no significant relationship between PFS and the development of rash during erlotinib treatment, and no differences based on International Federation of Gynecology and Obstetrics stage, age, or response at the end of first-line chemotherapy. There were no subgroups identified that might benefit from erlotinib maintenance therapy after first-line chemotherapy for ovarian cancer.76
Resistance mechanisms
An understanding of the mechanisms leading to resistance of EGFR inhibitors could help patients likely to respond to therapy and could help identify other agents that could be combined with such inhibitors. A number of mechanisms may allow cancer cells to become resistant to EGFR inhibitors. Resistance may be present at the onset of treatment (intrinsic) or may develop over time (acquired).77 At a molecular level, mechanisms of resistance to EGFR therapy include production of EGFR-activating ligands, receptor mutations, constitutive activation of downstream pathways, and activation of alternative signaling pathways.77,78 The downstream cellular signals transduced by EGFR are mediated by several other kinases whose activity is usually dependent on activation by EGFR (Figure 1). If any of these enzymes become mutated, this can lead to a constitutively active pathway. Regardless of EGFR blockade, this constitutively active pathway will remain active, which can result in an EGFR inhibitor-resistant phenotype.
Other mechanisms proposed include resistance to autophagic cell death upon increased EGFR expression via stabilization of the facilitated glucose transporter sodium/glucose cotransporter 1 (SGLT1)79 and inflammation.80 SGLT1 can transport glucose upstream of a glucose gradient, enabling cells to accumulate higher glucose concentrations than their environment.79 Since the increased SGLT1 stability is dependent on EGFR expression and not its activity,79 agents that target EGFR activity but not its expression are likely to be ineffective. Another potential mechanism of EGFR-inhibitor resistance is inflammation, which allows cancer cells to induce phosphorylation of mitogen-activated protein kinase, allowing a bypass of EGFR activation.80 The sequence or timing of multidrug administration may be important in how efficacious drug combinations are. Proliferation of an esophageal squamous epithelial cancer cell line possessing autocrine EGFR activity was either inhibited or enhanced depending on whether a cytotoxic drug (platinum derivative or taxane) was administered before or after an EGFR inhibitor.81 Although many of these mechanisms have been described in other cancer types, the relevance for these mechanisms in ovarian cancer is currently not clear.
Discussion
There are a number of factors that may explain the relative lack of activity of erlotinib in ovarian cancer. There is a lack of validated biomarkers for response to such TKIs, and to date the only known predictors of response are the activating mutations in the EGFR kinase domain, and these do not appear to play a significant role in the biology of ovarian cancer. Although the EGFR pathway appears to play an important role in the biology of ovarian cancer, in particular driving cellular processes linked to ovarian tumor development, tumor-cell survival, and metastasis, it is not clear how this can best be taken advantage of for therapeutic benefit. As a single agent, erlotinib has demonstrated minimal therapeutic activity in the first-line setting. Even though EGFR is overexpressed in most ovarian cancers, it does not appear that blocking signaling of the receptor alters sensitivity to platinum-based chemotherapy. In the first-line setting, combining erlotinib with platinum-based chemotherapy resulted in high response rates, although it is not clear in these nonrandomized studies whether these agents enhanced the activity of the chemotherapy or not. This also reflects the experience in the recurrent-disease setting. Nor does erlotinib appear to have efficacy in maintenance of response postchemotherapy in the first-line setting. And lastly, combining erlotinib with the angiogenesis inhibitor bevacizumab does not appear to have enhanced efficacy compared to bevacizumab alone.
One key goal in applying erlotinib to ovarian and other cancers will be to identify patients most likely to benefit from such a targeted therapy and to validate biomarkers of response.82,83 Clearly, a better understanding of in vivo efficacy, improved predictive biomarkers of response, and an understanding of the molecular resistance pathways for EGFR antagonists is needed in ovarian cancer. Given that concurrent activation of multiple signaling pathways and pathway cross talk occurs in tumor cells, inhibition of multiple pathways has been proposed as a strategy to improve the impact of targeted therapeutics.82 As such, the latest approaches in clinical trials, in a variety of tumors, are to combine the EGFR antagonists with inhibitors of other related or downstream signaling pathways. The impact on biologic endpoints in vivo will be critical to assess the mechanisms of action of these combined therapies. Ongoing research continues to identify new and more effective inhibitors of EGFR activity and novel approaches to target antitumor therapies via EGFR. Exploiting EGFR to target and deliver drugs or imaging agents to tumor cells shows promise in preclinical models.84 Although the clinical application of EGFR antagonists and EGFR-targeted therapies to ovarian cancer treatment has not kept pace with their application in other tumors, such as lung and colorectal cancers, what is learned from using these agents in other diseases could well be applied to the benefit of ovarian cancer patients.
Table 1.
Clinical trials of erlotinib in ovarian cancer
| Study reference | Phase | Number of patients | Therapy | Patient population | Response |
|---|---|---|---|---|---|
| Gordon et al67 | II | 34 | Erlotinib 150 mg/day | Platinum-refractory HER1/EGFR-positive | RR – 6% (2/34) PR – 6% (2/34) |
| Vasey et al70 | Ib | 23 | Carboplatin AUC 5 + docetaxel 75 mg/m2 q3 weekly Erlotinib 50–100 mg/day | First-line therapy | RR – 52% (12/23) CR – 22% (5/23) PR – 30% (7/23) |
| Blank et al72 | II | 56 | Carboplatin AUC 6 + paclitaxel 175 mg/m2 q3 weekly Erlotinib 150 mg/day | First-line therapy |
Pathological CR Optimal cytoreduction – 29% (8/28) Suboptimal cytoreduction – 13% (3/23) |
| Hirte et al73 | II | 50 | Carboplatin AUC 6 + paclitaxel 175 mg/m2 q3 weekly Erlotinib 150 mg/day | Recurrent platinum-sensitive or -resistant disease Up to 2 prior therapies |
Platinum-sensitive RR – 57% CR – 10% (3/33) PR – 47% (14/33) Platinum-resistant RR – 7% (1/17) CR – 0% PR – 7% (1/17) |
| Nimeiri et al74 | II | 13 | Bevacizumab 15 mg/kg q3 weeks Erlotinib 150 mg/day | Platinum-resistant or platinum-refractory | RR – 15% (2/13) CR – 7.5% (1/13) PR – 7.5% (1/13) |
| Chambers et al75 | II | 40 | Bevacizumab 10 mg/kg q2 weeks Erlotinib 150 mg/day | Platinum-resistant or platinum-refractory | RR – 23% (9/39) CR – 3% (1/39) PR – 20% (8/39) |
| Vergote et al76 | III | 835 | Maintenance postchemotherapy Erlotininb 150 mg/day versus placebo | Post-first-line chemotherapy |
Overall survival Erlotinib – 51 months Placebo – 59 months (P = 0.60) Progression-free survival Erlotinib – 12.7 months Placebo – 12.4 months (P = 0.90) |
Abbreviations: RR, response rate; CR, complete response; PR, partial response; AUC, area under the curve; HER1, human epidermal growth factor receptor type 1; EGFR, epidermal growth-factor receptor; q, every.
Footnotes
Disclosure
The author reports no conflicts of interest in this work.
References
- 1.Monga M, Carmichael JA, Shelley WE, et al. Surgery without adjuvant chemotherapy for early epithelial ovarian carcinoma after comprehensive surgical staging. Gynecol Oncol. 1991;43(3):195–197. doi: 10.1016/0090-8258(91)90018-z. [DOI] [PubMed] [Google Scholar]
- 2.Vermorken JB, Pecorelli S. Clinical trials in patients with epithelial ovarian cancer: past, present and future. Eur J Surg Oncol. 1996;22(5):455–466. doi: 10.1016/s0748-7983(96)92737-7. [DOI] [PubMed] [Google Scholar]
- 3.McGuire WP, Hoskins WJ, Brady MF, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334(1):1–6. doi: 10.1056/NEJM199601043340101. [DOI] [PubMed] [Google Scholar]
- 4.Piccart MJ, Bertelsen K, James K, et al. Randomized Intergroup trial of cisplatin–paclitaxel versus cisplatin–cyclophosphamide in women with advanced epithelial ovarian cancer: three year results. J Natl Cancer Inst. 2000;92(9):699–708. doi: 10.1093/jnci/92.9.699. [DOI] [PubMed] [Google Scholar]
- 5.du Bois A, Luck HJ, Bauknecht T. First-line chemotherapy with epirubicin, paclitaxel, and carboplatin for advanced ovarian cancer: a phase I/II study of the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group. J Clin Oncol. 1999;17(1):46–51. doi: 10.1200/JCO.1999.17.1.46. [DOI] [PubMed] [Google Scholar]
- 6.Ozols RF, Bundy BN, Greer BE, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21(17):3194–3200. doi: 10.1200/JCO.2003.02.153. [DOI] [PubMed] [Google Scholar]
- 7.Fung-Kee-Fung M, Oliver T, Elit L, Oza A, Hirte HW, Bryson P. Optimal chemotherapy treatment for women with recurrent ovarian cancer. Curr Oncol. 2007;14(5):195–208. doi: 10.3747/co.2007.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther. 1999;82(2–3):241–250. doi: 10.1016/s0163-7258(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 9.Yarden Y. The EGFR family and its ligands in human cancer: signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37( Suppl 4):S3–S8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 10.Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer. 2001;8(1):11–31. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- 11.Zaczek A, Brandt B, Bielawski KP. The diverse signaling network of EGFR, HER2, HER3 and HER4 tyrosine kinase receptors and the consequences for therapeutic approaches. Histol Histopathol. 2005;20(3):1005–1015. doi: 10.14670/HH-20.1005. [DOI] [PubMed] [Google Scholar]
- 12.Arteaga C. Targeting HER1/EGFR: a molecular approach to cancer therapy. Semin Oncol. 2003;30(3 Suppl 7):3–14. [PubMed] [Google Scholar]
- 13.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8(4):945–954. [PubMed] [Google Scholar]
- 14.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7(7):505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 15.Adams TE, McKern NM, Ward CW. Signalling by the type 1 insulin-like growth factor receptor: interplay with the epidermal growth factor receptor. Growth Factors. 2004;22(2):89–95. doi: 10.1080/08977190410001700998. [DOI] [PubMed] [Google Scholar]
- 16.Jones HE, Gee JMW, Hutcheson IR, Knowlden JM, Barrow D, Nicholson RI. Growth factor receptor interplay and resistance in cancer. Endocr Rel Cancer. 2006;13( Suppl 1):S45–S51. doi: 10.1677/erc.1.01275. [DOI] [PubMed] [Google Scholar]
- 17.Qiu L, Zhou C, Sun Y, et al. Crosstalk between EGFR and TrkB enhances ovarian cancer cell migration and proliferation. Int J Oncol. 2006;29(4):1003–1011. [PubMed] [Google Scholar]
- 18.Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 2001;20(13):1594–1600. doi: 10.1038/sj.onc.1204192. [DOI] [PubMed] [Google Scholar]
- 19.Shida D, Kitayama J, Mori K, Watanabe T, Nagawa H. Transactivation of epidermal growth factor receptor is involved in leptin-induced activation of Janus-activated kinase 2 and extracellular signal-regulated kinase 1/2 in human gastric cancer cells. Cancer Res. 2005;65(20):9159–9163. doi: 10.1158/0008-5472.CAN-05-0598. [DOI] [PubMed] [Google Scholar]
- 20.Andl CD, Rustgi AK. No one-way street: crosstalk between E-cadherin and receptor tyrosine kinase (RTK) signaling: a mechanism to regulate RTK activity. Cancer Biol Ther. 2005;4(1):28–31. doi: 10.4161/cbt.4.1.1431. [DOI] [PubMed] [Google Scholar]
- 21.Cabodi S, Moro L, Bergatto E, et al. Integrin regulation of epidermal growth factor (EGF) receptor and of EGF dependent responses. Biochem Soc Trans. 2004;32(3):438–442. doi: 10.1042/BST0320438. [DOI] [PubMed] [Google Scholar]
- 22.Riedemann J, Takiguchi M, Sohail M, Macaulay VM. The EGF receptor interacts with the type 1 IGF receptor and regulates its stability. Biochem Biophys Res Commun. 2007;355(3):707–714. doi: 10.1016/j.bbrc.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 23.Hur EM, Park YS, Lee BD, et al. Sensitization of epidermal growth factor-induced signaling by bradykinin is mediated by c-Src: implications for a role of lipid microdomains. J Biol Chem. 2004;279(7):5852–5860. doi: 10.1074/jbc.M311687200. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q, Thomas SM, Lui VW, et al. Phosphorylation of TNF-α converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc Natl Acad Sci USA. 2006;103(18):6901–6906. doi: 10.1073/pnas.0509719103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyamoto S, Yagi H, Yotsumoto F, Kawarabayashi T, Mekada E. Heparin-binding epidermal growth factor-like growth factor as a novel targeting molecule for cancer therapy. Cancer Sci. 2006;97(5):341–347. doi: 10.1111/j.1349-7006.2006.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu S, Murph MM, Lu Y, et al. Lysophosphatidic acid receptors determine tumorigenicity and aggressiveness of ovarian cancer cells. J Natl Cancer Inst. 2008;100(22):1630–1642. doi: 10.1093/jnci/djn378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3(8):582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 28.Fang X, Schummer M, Mao M, et al. Lysophosphatidic acid is a bioactive mediator in ovarian cancer. Biochim Biophys Acta. 2002;1582(1–3):257–264. doi: 10.1016/s1388-1981(02)00179-8. [DOI] [PubMed] [Google Scholar]
- 29.Yu X, Liu L, Cai B, He Y, Wan X. Suppression of anoikis by the neurotrophic receptor TrkB in human ovarian cancer. Cancer Sci. 2008;29(3):543–552. doi: 10.1111/j.1349-7006.2007.00722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miettinen PJ, Berger JE, Meneses J, et al. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376(6538):337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- 31.Sibilia M, Wagner EF. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science. 1995;269(5221):234–238. doi: 10.1126/science.7618085. [DOI] [PubMed] [Google Scholar]
- 32.Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation. 2007;75(9):770–787. doi: 10.1111/j.1432-0436.2007.00238.x. [DOI] [PubMed] [Google Scholar]
- 33.Hsieh M, Thao K, Conti M. Genetic dissection of epidermal growth factor receptor signaling during luteinizing hormone-induced oocyte maturation. PLoS One. 2011;6(6):e21574. doi: 10.1371/journal.pone.0021574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jamnongjit M, Gill A, Hammes SR. Epidermal growth factor receptor signaling is required for normal ovarian steroidogenesis and oocyte maturation. Proc Natl Acad Sci USA. 2005;102( 45):16257–16262. doi: 10.1073/pnas.0508521102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19(3):183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 36.Lafky JM, Wilken JA, Baron AT, Maihle NJ. Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim Biophys Acta. 2008;1785(2):232–265. doi: 10.1016/j.bbcan.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Bartlett JM, Langdon SP, Simpson BJ, et al. The prognostic value of epidermal growth factor receptor mRNA expression in primary ovarian cancer. Br J Cancer. 1996;73(3):301–306. doi: 10.1038/bjc.1996.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischer-Colbrie J, Witt A, Heinzl H, et al. EGFR and steroid receptors in ovarian carcinoma: comparison with prognostic parameters and outcome of patients. Anticancer Res. 1997;17(1B):613–619. [PubMed] [Google Scholar]
- 39.Skirnisdottir I, Sorbe B, Seidal T. The growth factors HER-2/neu and EGFR, their relationship, and their effects on the prognosis in early stage (FIGO I–II) epithelial ovarian carcinoma. Int J Gynecol Cancer. 2001;11(2):119–129. doi: 10.1046/j.1525-1438.2001.011002119.x. [DOI] [PubMed] [Google Scholar]
- 40.Alper O, Bergmann-Leitner ES, Bennett TA, Hacker NF, Stromberg K, Stetler-Stevenson WG. Epidermal growth factor receptor signaling and the invasive phenotype of ovarian carcinoma cells. J Natl Cancer Inst. 2001;93(18):1375–1384. doi: 10.1093/jnci/93.18.1375. [DOI] [PubMed] [Google Scholar]
- 41.Lassus H, Sihto H, Leminen A, et al. Gene amplification, mutation, and protein expression of EGFR and mutations of ERBB2 in serous ovarian carcinoma. J Mol Med. 2006;84(8):671–681. doi: 10.1007/s00109-006-0054-4. [DOI] [PubMed] [Google Scholar]
- 42.Stadlmann S, Gueth U, Reiser U, et al. Epithelial growth factor receptor status in primary and recurrent ovarian cancer. Mod Pathol. 2006;19(4):607–610. doi: 10.1038/modpathol.3800575. [DOI] [PubMed] [Google Scholar]
- 43.Lee CH, Huntsman DG, Cheang MCU, et al. Assessment of Her-1, Her-2, and Her-3 expression and Her-2 amplification in advanced stage ovarian carcinoma. Int J Gynecol Pathol. 2005;24(2):147–152. doi: 10.1097/01.pgp.0000152026.39268.57. [DOI] [PubMed] [Google Scholar]
- 44.Nielsen JS, Jakobsen E, Hølund B, Bertelsen K, Jakobsen A. Prognostic significance of p53, Her-2, and EGFR overexpression in borderline and epithelial ovarian cancer. Int J Gynecol Cancer. 2004;14(6):1086–1096. doi: 10.1111/j.1048-891X.2004.14606.x. [DOI] [PubMed] [Google Scholar]
- 45.Morishige KI, Kurachi H, Amemiya K, et al. Evidence for the involvement of transforming growth factor α and epidermal growth factor receptor autocrine growth mechanism in primary human ovarian cancers in vitro. Cancer Res. 1991;51(19):5322–5328. [PubMed] [Google Scholar]
- 46.Tsujioka H, Yotsumoto F, Shirota K, et al. Emerging strategies for ErbB ligand-based targeted therapy for cancer. Anticancer Res. 2010;30(8):3107–3112. [PubMed] [Google Scholar]
- 47.Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res. 2001;7(10):2958–2970. [PubMed] [Google Scholar]
- 48.Arteaga CL. Overview of epidermal growth factor receptor biology and its role as a therapeutic target in human neoplasia. Semin Oncol. 2002;29(5 Suppl 14):3–9. doi: 10.1053/sonc.2002.35642. [DOI] [PubMed] [Google Scholar]
- 49.Arteaga CL. The epidermal growth factor receptor: from mutant oncogene in nonhuman cancers to therapeutic target in human neoplasia. J Clin Oncol. 2001;19(Suppl 18):32s–40s. [PubMed] [Google Scholar]
- 50.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37( Suppl 4):S9–S15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 51.Ilekis JV, Connor JP, Prins GS, Ferrer K, Niederberger C, Scoccia B. Expression of epidermal growth factor and androgen receptors in ovarian cancer. Gynecol Oncol. 1997;66(2):250–254. doi: 10.1006/gyno.1997.4764. [DOI] [PubMed] [Google Scholar]
- 52.Owens OJ, Stewart C, Brown I, Leake RE. Epidermal growth factor receptors (EGFR) in human ovarian cancer. Br J Cancer. 1991;64(5):907–910. doi: 10.1038/bjc.1991.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berchuck A, Rodriguez GC, Kamel A, et al. Epidermal growth factor receptor expression in normal ovarian epithelium and ovarian cancer. I. Correlation of receptor expression with prognostic factors in patients with ovarian cancer. Am J Obstet Gynecol. 1991;164(2):669–674. doi: 10.1016/s0002-9378(11)80044-x. [DOI] [PubMed] [Google Scholar]
- 54.Dimova I, Zaharieva B, Raitcheva S, Dimitrov R, Doganov N, Toncheva D. Tissue microarray analysis of EGFR and erbB2 copy number changes in ovarian tumors. Int J Gynecol Cancer. 2006;16(1):145–151. doi: 10.1111/j.1525-1438.2006.00286.x. [DOI] [PubMed] [Google Scholar]
- 55.Lin CK, Chao TK, Yu CP, Yu MH, Jin JS. The expression of six biomarkers in the four most common ovarian cancers: correlation with clinicopathological parameters. APMIS. 2009;117(3):162–175. doi: 10.1111/j.1600-0463.2008.00003.x. [DOI] [PubMed] [Google Scholar]
- 56.Brustmann H. Epidermal growth factor receptor expression in serous ovarian carcinoma: an immunohistochemical study with galectin-3 and cyclin D1 and outcome. Int J Gynecol Pathol. 2008;27(3):380–389. doi: 10.1097/PGP.0b013e31815d060d. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka Y, Terai Y, Tanabe A, et al. Prognostic effect of epidermal growth factor receptor gene mutations and the aberrant phosphorylation of Akt and ERK in ovarian cancer. Cancer Biol Ther. 2011;11(1):50–57. doi: 10.4161/cbt.11.1.13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Araújo AP, Ribeiro R, Pereira D, et al. Ovarian cancer and genetic susceptibility: association of A61G polymorphism in the EGF gene. Exp Biol Med. 2009;234(3):241–245. doi: 10.3181/0805-RM-146. [DOI] [PubMed] [Google Scholar]
- 59.Garcia AA, Sill MW, Lankes HA, et al. A phase II evaluation of lapatinib in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a gynecologic oncology group study. Gynecol Oncol. 2012;124(3):569–574. doi: 10.1016/j.ygyno.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ullrich A, Coussens L, Hayflick JS, et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermal carcinoma cells. Nature. 1984;309(5968):418–425. doi: 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- 61.Pedersen MW, Meltorn M, Damstrup L, Poulsen HS. The type III epidermal growth factor receptor mutation, biological significance and potential target for anti-cancer therapy. Ann Oncol. 2001;12(6):745–760. doi: 10.1023/a:1011177318162. [DOI] [PubMed] [Google Scholar]
- 62.Chu CT, Everiss KD, Wikstrand CJ, Batra SK, Kung HJ, Bigner DD. Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII) Biochem J. 1997;324(Pt 3):855–861. doi: 10.1042/bj3240855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moscatello DK, Holgado-Madruga M, Godwin AK, et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995;55(23):5536–5539. [PubMed] [Google Scholar]
- 64.Steffensen KD, Waldstrom M, Olsen DA, et al. Mutant epidermal growth factor receptor in benign, borderline, and malignant ovarian tumors. Clin Cancer Res. 2008;14(11):3278–3282. doi: 10.1158/1078-0432.CCR-07-4171. [DOI] [PubMed] [Google Scholar]
- 65.Palayekar MJ, Herzog TJ. The emerging role of epidermal growth factor receptor inhibitors in ovarian cancer. Int J Gynecol Cancer. 2008;18(5):879–890. doi: 10.1111/j.1525-1438.2007.01144.x. [DOI] [PubMed] [Google Scholar]
- 66.Iyer R, Bharhuar A. A review of erlotinib – an oral selective epidermal growth factor receptor tyrosine kinase inhibitor. Expert Opin Pharmacother. 2010;11(2):311–320. doi: 10.1517/14656560903551283. [DOI] [PubMed] [Google Scholar]
- 67.Gordon AN, Finkler N, Edwards RP, et al. Efficacy and safety of erlotinib HCl, an epidermal growth factor receptor (HER1/EGFR) tyrosine kinase inhibitor, in patients with advanced ovarian carcinoma: results from a phase II multicenter study. Int J Gynecol Cancer. 2005;15(5):785–792. doi: 10.1111/j.1525-1438.2005.00137.x. [DOI] [PubMed] [Google Scholar]
- 68.Sirotnak FM, Zakowski MF, Miller VA, Scher HI, Kris MG. Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin Cancer Res. 2000;6(12):4885–4892. [PubMed] [Google Scholar]
- 69.Cullinane C, Kleinschmidt M, Webster LK. Antitumour activity of ZD1839 in combination with cisplatin in NIH 3T3 cells expressing human epidermal growth factor receptor. Proc Am Assoc Cancer Res. 2000;41:482. [Google Scholar]
- 70.Vasey PA, Gore M, Wilson R, et al. A phase Ib trial of docetaxel, carboplatin and erlotinib in ovarian, fallopian tube and primary peritoneal cancers. Br J Cancer. 2008;98(11):1774–1780. doi: 10.1038/sj.bjc.6604371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vasey PA, Jayson GC, Gordon A, et al. Phase III randomized trial of docetaxel-carboplatin versus paclitaxel carboplatin as first-line chemotherapy for ovarian carcinoma. J Natl Cancer Inst. 2004;96(22):1682–1691. doi: 10.1093/jnci/djh323. [DOI] [PubMed] [Google Scholar]
- 72.Blank SV, Christos P, Curtin JP, et al. Erlotinib added to carboplatin and paclitaxel as first-line treatment of ovarian cancer: a phase II study based on surgical reassessment. Gynecol Oncol. 2010;119(3):451–456. doi: 10.1016/j.ygyno.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hirte H, Oza A, Swenerton K, et al. A phase II study of erlotinib (OSI-774) given in combination with carboplatin in patients with recurrent epithelial ovarian cancer. Gynecol Oncol. 2010;118(3):308–312. doi: 10.1016/j.ygyno.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 74.Nimeiri HS, Oza AM, Morgan RJ, et al. Efficacy and safety of bevacizumab plus erlotinib for patients with recurrent ovarian, primary peritoneal, and fallopian tube cancer: a trial of the Chicago, PMH, and California Phase II Consortia. Gynecol Oncol. 2008;110(1):49–55. doi: 10.1016/j.ygyno.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chambers SK, Clouser MC, Baker AF, et al. Overexpression of tumor vascular endothelial growth factor A may portend an increased likelihood of progression in a phase II trial of bevacizumab and erlotinib in resistant ovarian cancer. Clin Cancer Res. 2010;16(21):5320–5328. doi: 10.1158/1078-0432.CCR-10-0974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vergote IB, Joly F, Katsaros D, et al. Randomized phase III study of erlotinib versus observation in patients with no evidence of disease progression after first-line platin-based chemotherapy for ovarian carcinoma: A GCIG and EORTC-GCG study. J Clin Oncol. 2012;30(Suppl):LBA5000. doi: 10.1200/JCO.2013.50.5669. [DOI] [PubMed] [Google Scholar]
- 77.Morgillo F, Bareschino MA, Bianco R, Tortora G, Ciardiello F. Primary and acquired resistance to anti-EGFR targeted drugs in cancer therapy. Differentiation. 2007;75(9):788–799. doi: 10.1111/j.1432-0436.2007.00200.x. [DOI] [PubMed] [Google Scholar]
- 78.Bianco R, Damiano V, Gelardi T, Daniele G, Ciardiello F, Tortora G. Rational combination of targeted therapies as a strategy to overcome the mechanisms of resistance to inhibitors of EGFR signaling. Curr Pharm Des. 2007;13(33):3358–3367. doi: 10.2174/138161207782360564. [DOI] [PubMed] [Google Scholar]
- 79.Weihua Z, Tsan R, Huang WC, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13(5):385–393. doi: 10.1016/j.ccr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krysan K, Lee JM, Dohadwala M, et al. Inflammation, epithelial to mesenchymal transition, and epidermal growth factor receptor tyrosine kinase inhibitor resistance. J Thorac Oncol. 2008;3(2):107–110. doi: 10.1097/JTO.0b013e3181630ece. [DOI] [PubMed] [Google Scholar]
- 81.Morelli MP, Cascone T, Troiani T, et al. Sequence dependent antiproliferative effects of cytotoxic drugs and epidermal growth factor receptor inhibitors. Ann Oncol. 2005;16(Suppl 4):iv61–iv68. doi: 10.1093/annonc/mdi910. [DOI] [PubMed] [Google Scholar]
- 82.Bast RC, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9(6):415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer. 2009;9(3):167–181. doi: 10.1038/nrc2583. [DOI] [PubMed] [Google Scholar]
- 84.Sampson JH, Akabani G, Archer GE, et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro Oncol. 2008;10(3):320–329. doi: 10.1215/15228517-2008-012. [DOI] [PMC free article] [PubMed] [Google Scholar]