

Abstract
Introduction
A major barrier to the advancement of therapeutic nanomedicines has been the non-target toxicity caused by the accumulation of the drug delivery systems in organs associated with the reticuloendothelial system, particularly the liver and spleen. Herein, we report the development of peptide based metabolically active linkers (MALs) that are enzymatically cleaved by cysteine cathepsin B and S, two proteases highly expressed in the liver and spleen. The overall goal of this approach is to utilize the MALs to lower the non-target retention and toxicity of radiolabeled drug delivery systems, thus resulting in higher diagnostic and radiotherapeutic efficacy.
Methods
In this study three MALs (MAL0, MAL1 and MAL2) were investigated. MAL1 and MAL2 are composed of known substrates of cathepsin B and S, respectively, while MAL0 is a non-cleavable control. Both MAL1 and MAL2 were shown to undergo enzymatic cleavage with the appropriate cathepsin protease. Subsequent to conjugation to the HPMA copolymer and radiolabeling with 177Lu, the peptide-polymer conjugates were renamed 177Lu- metabolically active copolymers (177Lu-MACs) with the corresponding designation 177Lu-MAC0, 177Lu-MAC1 and 177Lu-MAC2.
Results
In vivo evaluation of the 177Lu-MACs was performed in a HPAC human pancreatic cancer xenograft mouse model. 177Lu-MAC1 and 177Lu-MAC2 demonstrated 3.1 and 2.1 fold lower liver retention, respectively, compared to control (177Lu-MAC0) at 72 h post-injection. With regard to spleen retention, 177Lu-MAC1 and 177Lu-MAC2 each exhibited a nearly fourfold lower retention, relative to control, at the 72 h time point. However, the tumor accumulation of the 177Lu-MAC0 was two to three times greater than 177Lu-MAC1 and 177Lu-MAC2 at the same time point. The MAL approach demonstrated the capability of substantially reducing the non-target retention of the 177Lu-labeled HPMA copolymers.
Conclusions
While further studies are needed to optimize the pharmacokinetics of the 177Lu-MACs design, the ability of the MAL to significantly decrease non-target retention establishes the potential this avenue of research may have for the improvement of diagnostic and radiotherapeutic drug delivery systems.
Keywords: Cathepsins, Lutetium-177, HPMA Copolymers, Cleavable Linkers and Pancreatic Cancer
INTRODUCTION
Over the last three decades, biomedical researchers have seen the dramatic emergence of polymeric micro- and nanoparticle drug delivery technologies for the treatment of human disease.[1–3] These drug delivery systems include an array of paradigms such as: liposomes, polymer micelles, dendrimers and polymer conjugates. The development of polymeric diagnostic and therapeutic drug delivery systems offer several advantages over traditional drug delivery including the ability to control both the rate and location of drug release. However, one of the main disadvantages of these micro- and nanoparticle based systems has been opsonization and uptake by macrophages (e.g. Kupffer cells) of the mononuclear phagocyte system (MPS).[3, 4] The hallmark of this MPS uptake is the significant accumulation of the drug delivery system in organs containing MPS cells, specifically the liver and spleen, which can lead to significant toxicity. The development of PEGylation strategies to create “stealth” micro- and nanoparticle systems has contributed to a decrease in MPS accumulation thereby increasing the longevity of the polymeric drug delivery system in the bloodstream.[3] Unfortunately, even with the development of PEGylated techniques, a substantial portion of these polymeric drug delivery systems are still being sequestered by MPS tissues.
N-(2-hydroxypropyl) methacrylamide (HPMA) copolymers have been extensively investigated as a drug delivery vehicle for a variety of chemotherapeutics.[2, 5] While most of the focus on HPMA polymer conjugates has centered on chemotherapeutics, there have been published reports by a variety of investigators regarding the development of diagnostic nuclear imaging and radiotherapeutic agents.[6–14] To date, the majority of these radiolabeled HPMA copolymer conjugates have centered on radioisotopes of iodine (e.g. 131I and 123I). However, recent work by Line, Ghandehari and co-workers, has employed various radiometals (e.g. 99mTc, 111In and 90Y) common to clinical nuclear medicine.[9, 10, 12, 13, 15] The bulk of the HPMA copolymer conjugates investigated has focused on copolymers that were below the renal excretion threshold (i.e. ~45 kDa) in order to reduce MPS accumulation. While HPMA copolymer conjugates with molecular weights higher than the renal excretion thresholds have demonstrated significantly enhanced tumor delivery, the corresponding substantial increase in MPS tissue retention has largely made this approach undesirable for diagnostics and therapeutic agent development.[5]
Cysteine cathepsins are predominantly lysosomal proteases that have been found to have diverse functions, but are primarily attributed to protein catabolism.[16] The function of cathepsin B has mainly been associated with the turnover of both intracellular and extracellular (through endocytosis) proteins in the lysosomal compartment. Tissue expression of cysteine cathepsins has been shown to be variable largely depending on the function of the protease. In some cases such as cathepsin B, the protease is expressed ubiquitously in almost every tissue.[2, 17] However, the highest levels of cathepsin B activity have been found in the liver, spleen, thyroid and kidneys for both murine and humans.[18, 19] For humans the activity of these tissues are 3 –10 fold higher than other non-target tissues. Conversely, the expression of some cysteine cathepsins, for instance cathepsin S, is selectively expressed in only a small group of cells. Cathepsin S is thought to be an integral protease in the degradation of antigenic proteins as part of the Major Histocompatibility Complex Class II pathway in antigen presenting cells.[17, 20] As a consequence of the critical role of cathepsin S in human immune response, the expression of cathepsin S has been shown to reside primarily in immune cells.[21, 22] Cathepsin B and S are both highly expressed in tissues that are associated with MPS uptake and retention of drug delivery systems.
Protease cleavable linkers are attractive for use in drug delivery due to the typically high serum stability and specificity of the substrate-linkers.[23] In polymer based drug delivery, Kopeèek and others have demonstrated the utility of protease cleavable linkers, specifically cathepsin B cleavable linkers, to increase in vivo drug delivery to tumor tissues.[2, 5] However, this technique has also been exploited to decrease retention of radioimmunoconjugates in non-target tissues (e.g. liver) without significantly affecting tumor retention thereby increasing the therapeutic index of the agent.[24] One of the research focuses of our laboratory has been the design of protease cleavable linkers for the development of diagnostic and radiotherapeutic drug delivery systems. The impetus for the development of linkers that are substrates for cathepsin B and S is the high level of expression of these proteases in MPS tissues. The goal of this research is to create metabolically active linkers (MALs) that will increase the efficacy of the diagnostic or radiotherapeutic drug delivery systems by reducing the MPS accumulation. Herein, we describe our initial efforts in the development of MALs using 177Lu-radiolabeled HPMA copolymers as our model system.
MATERIALS AND METHODS
Chemicals and Supplies
All chemicals were used without further purification unless otherwise noted. Water was deionized and purified (18.2 MΩ-cm) by means of a Milli-Q water filtration system (Millipore Corp., U.S.). Acetonitrile (HPLC Grade), formic acid (HPLC Grade), NN, -dimethylformamide (DMF, Peptide Synthesis Grade), dichloromethane (DCM, Peptide Synthesis Grade), N-methylpyrrolidone (NMP, Peptide Synthesis Grade), trifluoroacetic acid (TFA, Peptide Synthesis Grade), L-ascorbic acid, ethylene glycol, tin(II) chloride, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), bovine serum albumin (BSA), sodium dodecyl sulfate (SDS), sodium bicarbonate, glutathione, epidermal growth factor (EGF), 2-mercaptoethanol, 0.9% sodium chloride (NaCl) solution (irrigation USP), Dulbecco’s Modified Eagle Medium (DMEM), and phosphate buffered saline (PBS) were obtained from Fisher Scientific (U.S.). HPMA and N-(3-aminopropyl) methacrylamide (APMA) hydrochloride were purchased from Polysciences (U.S.). Fluorescein isothiocyanate (FITC), N-hydroxysuccinimide (NHS), N, N′-dicyclohexylcarbodiimide (DCC), 4,4′-Azobis(4-cyanovaleric acid) (V-501), 4-cyano-4-(phenylcarbono-thioylthio)pentanoic acid (CTP), thioanisole, 3,6-dioxa-1,8-octanedithiol (DODT), triisopropylsilane (TIS), ethylenediaminetetraacetic acid (EDTA), ninhydrin, glycine, sodium acetate, insulin, transferrin and hydrocortisone were obtained from Sigma-Aldrich (U.S.). 5-(3-(methacryloylaminopropyl) thioureidyl) fluorescein (APMA-FITC) was synthesized in our laboratory.[25] 1-[(1-(Cyano-2-ethoxy-2-oxoethylideneaminooxy) dimethylaminomorpholino)] uronium hexafluorophosphate (COMU), fluorenylmethyloxycarbonyl (Fmoc)-protected natural amino acids, H-Pro-2-Trt resin and H-Phe-2-ClTrt resin were purchased from Nova Biochem (U.S.). 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) was purchased from Macrocyclics (U.S.). Lutetium-177 trichloride was obtained from PerkinElmer (U.S.). Cysteine cathepsin B and S as well as the corresponding activity assays kits were obtained from BioVision (U.S.). The human pancreatic adenocarcinoma (HPAC) CRL-2119 cell line was purchased from American Type Culture Collection (ATCC) (U.S.). Matrigel™ was obtained from BD Biosciences (U.S.). Ham’s F12 medium, l-glutamine, Human AB serum and sodium pyruvate were obtained from Mediatech (U.S.). Fetal bovine serum (FBS) and Roswell Park Memorial Institute (RPMI) 1640 media was purchased from Invitrogen/GIBCO (U.S.). Non-essential amino acids (NEAA) were purchased from Hyclone Laboratories, Inc. (U.S.). Recombinant Human Macrophage-Colony Stimulating Factor (rhM-CSF) was obtained from R&D Systems (U.S.). Female Institute of Cancer Research severe combined immunodeficient (ICR SCID) mice, aged 5–6 weeks at time of arrival, were purchased from Taconic Farms (U.S.).
Equipment
The peptides were synthesized by solid phase peptide synthesis (SPPS) on a Liberty microwave peptide synthesizer from CEM (U.S.). Reversed phase-high performance liquid chromatography/mass spectrometry (RP-HPLC/MS) analyses were performed on a Waters (U.S.) e2695 system equipped with a Waters 2489 absorption detector and a Waters Qtof Micro electrospray ionization mass spectrometer. Evaluation and purification of radiolabeled conjugates were performed on a Waters 1515 binary pump equipped with a Waters 2489 absorption detector and a Bioscan (U.S.) Flow Count radiometric detector system. A Phenomenex (U.S.) Jupiter 10μ Proteo 250 × 10 mm semiprep column was used for the purification of bulk amounts of peptides. A GE HealthCare (U.S.) ÄKTA Fast Protein Liquid Chromatography (FPLC) system equipped with a Superose 6 column (HR 10/30) was used for HPMA copolymer molecular weight estimates. A Sephadex™ LH-20 size exclusion resin obtained from GE HealthCare was used for chromatographic separations. For the purification and metabolism studies of the 177Lu-radioconjugates, an Agilent (U.S.) PL aquagel-OH 7.5 × 300 mm, 8 μm column was employed. Purification and concentration of radioconjugates was performed using Amicon Ultra-4 Centrifugal Filter (10 kDa & 30 kDa) Devices from Millipore (U.S.). Gamma decay detection of 177Lu-radioconjugates for biodistribution studies were accomplished using a PerkinElmer 1480 WIZARD® 3″ Automatic Gamma Counter. UV spectroscopy was performed on a Biochrom WPA Biowave II+ (U.K.). Fluorometer measurements were made on a Molecular Devices (U.S.) SpectraMax M3. Flow cytometry analysis was performed by the UNMC Cell Analysis Facility using a Becton, Dickinson and Company FACSCalibur flow cytometer. All animal protocols conformed to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, and were approved by the Institutional Animal Care and Use Committee at the University of Nebraska Medical Center.
Solid-Phase Peptide Synthesis
The MAL1 and MAL2 peptides were synthesized on a H-Phe-2-ClTrt and H-Pro-2-ClTrt SPPS resin, respectively, using a microwave peptide synthesizer. The resin (containing 0.25 mmole of peptide anchors) was deprotected using piperidine resulting in the formation of the primary amine. The carboxylic acids of the Fmoc protected amino acids (1 mmol) were activated using COMU and conjugated to the primary amines of the growing peptide on the resin. The process of deprotection, activation and conjugation was repeated until the desired peptide was synthesized. The selective cleavage of the peptide from the 2-chlorotrityl resin anchor was achieved by reaction with 1% TFA in dry DCM and subsequent filtration. The combined crude filtrates were dried by a rotary concentrator. Purification of the peptides was performed using a semi-preparative Proteo C12 column with a flow rate of 5.0 mL/min. HPLC solvents consisted of H2O containing 0.1% formic acid (solvent A) and acetonitrile containing 0.1% formic acid (solvent B). For both peptides, the initial gradient of 70 % A: 30 % B linearly decreased to 60 % A: 40 % B over a 15 minute time period. At the end of the run time for all HPLC experiments, the column was flushed with the gradient 5 % A: 95 % B and re-equilibrated to the starting gradient. The yields after purification were 0.042 mmole, 16.9% (n = 1) for MAL1 and 0.090 mmole, 37.5% (n = 1) for MAL2. Both peptides were determined to be ≥ 95% purity as characterized by LC/MS before in vitro or in vivo use.
Cathepsin Cleavage Assay of MALs
For MAL1 and MAL2, 1.51 and 1.22 μmole, respectively, were each weighed and placed into a 1 mL centrifuge tube. Each peptide was dissolved in 100 μl of buffer solution containing 100 mM sodium acetate, 1 mM EDTA and 5 mM DTT. Solutions of cathepsin B and S were prepared at a concentration of 25 ng/μL using the buffer solution. The pH of the solution for dissolution of both the peptide and protease was adjusted to 5.0 and 6.0 for cathepsin B and S assays, correspondingly. In the appropriate tube, 2 and 50 μl of the cathepsin B and S, respectively, were added. The cleavage assay was allowed to proceed for 96 hours at room temperature and was evaluated every 24 hours by LC/MS. Evaluation of the peptides was performed using an analytical Proteo C12 column with a flow rate of 1.5 mL/min. HPLC solvents consisted of H2O containing 0.1% formic acid (solvent A) and acetonitrile containing 0.1% formic acid (solvent B). For both peptides, the initial gradient of 70 % A: 30 % B linearly decreased to 60 % A: 40 % B over a 15 minute time period. At the end of the run time for all HPLC experiments, the column was flushed with the gradient 5 % A: 95 % B and re-equilibrated to the starting gradient. Cleavage estimates were determined by integration of the peptides and metabolite peaks from the total ion chromatogram generated by the LC/MS Waters MassLynx software.
Synthesis and Characterization of the HPMA Copolymer
The HPMA copolymerization was accomplished using the reversible addition-fragmentation chain transfer polymerization reaction with V-501 as the initiator and CTP as the chain transfer agent. The feed ratio of the polymerization consisted of 98.75:0.85:0.40 mole% HPMA, APMA and APMA-FITC, respectively. The polymerization was run at a 1 M total monomer concentration in aqueous acetic buffer (pH = 5.2, 0.27 M acetic acid and 0.73 M sodium acetate) with a molar ratio of total monomer: chain transfer agent: initiator of 2340:3:1. Prior to proceeding with the polymerization reaction, the glass ampule and contents were purged with argon for 20 min. Subsequently, the ampule was flame sealed and allowed to heat at 70 °C in an oil bath for 14 hours. After cooling the HPMA copolymer was evaporated to dryness, dissolved in methanol and purified by size exclusion chromatography using an LH-20 packing material with methanol as the eluent. The conversion rate was 71% as determined by weight after purification. The weight average molecular weight and polymer dispersity of copolymers were determined by FPLC equipped with UV and RI detectors. The FPLC measurements were carried out on Superose 6 column (HR 10/30) with PBS (pH 7.3) as the eluent. The average molecular weights of the copolymers were estimated using HPMA copolymer standards with narrow polydispersity indices.
Quantification of the Amine Content in the HPMA Copolymers
Quantitation of the primary amine content of the HPMA copolymer was assessed using a slightly modified ninhydrin assay reported by York and co-workers.[26] A ninhydrin solution was prepared by dissolving 5.61 mmole of ninhydrin in 25 mL ethylene glycol. A SnCl2 solution was made by dissolving 0.177 mmole of SnCl2.2H2O in 25 mL of 0.2 M citrate buffer. A ninhydrin-SnCl2 solution, of the desired quantity, was prepared by mixing the two solutions together in a 1:1 volumetric ratio. For the purpose of the creation of a calibration curve, a solution of 3 mM glycine was dissolved in 10 mL DI H2O and seven standard solutions were prepared by aliquoting amounts from 7.5 – 45 μL of this glycine solution into separate tubes. A 3 mM HPMA copolymer solution was prepared in DI H2O. For the unmodified HPMA copolymer, MAC0, MAC1 and MAC2 the sample size used were 0.0171, 0.0176, 0.0173 and 0.010 μmole, respectively. For each HPMA copolymer solution, an aliquot of 7.5 μL was added to a sample tube. To both the standard and sample tubes, 200 μL of the ninhydrin-SnCl2 solution was added. The sample and standard solutions were heated to 100 °C for 2 h and then allowed to cool to room temperature. The volumes of all solutions were adjusted to 3 mL using DI H2O and analyzed by UV spectroscopy at 570 nm. The amine content of the HPMA copolymers was determined based upon the calibration curve generated by the glycine standards. Amine content in the unmodified HPMA copolymer, MAC0, MAC1 and MAC2 was found to be 70.4, 11.5, 19.1 and 11.7 μmol/g of polymer (n = 1 for each copolymer), correspondingly. Conjugation efficiencies were calculated by determination of the difference in the amine content between the MAC and the unmodified HPMA copolymer.
Synthesis of the MACs
The HPMA copolymer (0.292 μmole) was dissolved in 500 μl of DMF. The amine content in 0.292 μmole of HPMA copolymer was 7.04 μmol (1 equiv). In a 10 mL round bottom flask, 20.3 (35.4 μmol, 5.0 equiv), 20.0 (16.2 μmol, 2.3 equiv) or 22.8 mg (16.2 μmol, 2.3 equiv) of MAL0, MAL1 or MAL2 was dissolved in 500 μl of DMF. The solution was cooled to 0 °C and NHS and DCC were added to the flask in the same equivalent amount as the MAL. The solution was stirred at 0 °C for 3 hours. At the end of that time, the HPMA copolymer was added and stirred for another 2 hours at 0 °C. Subsequently, the reaction was allowed to warm to room temperature and continued for an additional 18 hours. The precipitate generated from the reaction was filtered off and the filtrate was evaporated to dryness. The residue was dissolved in methanol followed by size exclusion chromatography using an LH-20 packing material. Evaporation of the high molecular weight fraction afforded light yellow HPMA copolymer-peptide conjugates. Based on the determined conjugation efficiencies from the ninhydrin assays, the percent recovery (n =1) of MAC0 was 77.6 (0.233 μmole), MAC1: 45.3 (0.140 μmole) and MAC2: 50.0% (0.157 μmole).
Deprotection of the orthogonal protecting groups was accomplished using standard peptide cleavage conditions. Briefly, to a 0.0875 μmole sample of the MAC0, MAC1 and MAC2 was added 4 mL of a cleavage cocktail at 0 °C consisting of a 1:1:1:37 volumetric ratio of DODT, water, TIS and TFA. For the MAC2, the cleavage cocktail used was a 1:1:1:0.75:46.25 volumetric ratios of DODT, water, TIS, thioanisole, and TFA. Initially, the solution was stirred for 15 min at 0 °C. Subsequently, the cleavage reaction was allowed to warm up to room temperature and was continued for an additional 3 hours. At the end of that time, the solution was evaporated and the residue dissolved in methanol followed by size exclusion chromatography using LH-20 packing material with methanol as the eluent. The collected high molecular weight fractions were evaporated to dryness to afford the deprotected copolymer-peptide conjugates. Based on the determined conjugation efficiencies, the percent recoveries (n = 1) of MAC0, MAC1 and MAC2 were 80.2 (0.0697 μmole), 100 (0.0863 μmole) and 93.4 % (0.0808 μmole), respectively.
Radiolabeling of the HPMA Copolymers
The radiolabeling of the MACs was accomplished by heating the copolymer at 90 °C for 1 h in the presence of 74 MBq (2mCi) of 177LuCl3. After cooling the resulting 177Lu-radiolabeled MACs were purified by radio-SEC-HPLC. Isolation of the 177Lu-MACs was accomplished using an isocratic mobile phase consisting of PBS with 0.02 mM EDTA at pH 7.4. The linear flow rate was 0.80 mL/min. UV analysis was performed at a wavelength of 494 nm. After collection of the peak-purified 177Lu-radiolabeled MACs, 0.113 mmole of L-ascorbic acid was added as a radiolysis inhibitor. The purified 177Lu-radioconjugate was concentrated using an Amicon Ultra-4 Centrifugal Filter and washed with 5 mL of deionized water. The purified 177Lu-radiconjugates were diluted with sterile saline to 0.37 MBq (10 μCi) per 100 μL. The radiochemical purity of all radioconjugates was re-evaluated before administration and found to be ≥ 95 % pure as determined by the radio-HPLC.
Cathepsin Cleavage Assay of Radiolabeled Copolymers
Briefly, 0.291 nmole of each purified 177Lu-radiolabeled MACs (90~120 μCi) was incubated in a buffer with the indicated cathepsin in Eppendorf tubes at 25 ºC. The buffer solution consisted of 50 mM sodium acetate, 1 mM EDTA and 5 mM DTT, and pH was adjusted 5.0 for cathepsin B and 6.0 for cathepsin S to maximize the enzyme activity. For the MAC0, the copolymer was incubated in the presence of either cathepsin B (5 ng/μl) or cathepsin S (10 ng/μl) in 400 μl of buffer solution. MAC1 and MAC2 were incubated with cathepsin B (5 ng/μl) and cathepsin S (10 ng/μl), respectively, in 400 μl buffer. After every 24 hours, a 100 μl aliquot of the copolymer solution was analyzed by radio-SEC-HPLC to determine the relative amount of cathepsin cleavage product. The analysis utilized an isocratic gradient with a mobile phase consisting of PBS with 0.02 mM EDTA at pH 7.4. The radio-SEC-HPLC analysis was carried out utilizing a linear flow rate of 0.80 ml/min and a UV wavelength of 494 nm.
Human Serum Stability of Radiolabeled Copolymers
Briefly, 0.291 nmole of purified 177Lu-MAC0, 177Lu- MAC1 and 177Lu-MAC2 was incubated with 1 mL of human AB serum at 37 °C. After 72 hours, the stability was determined by centrifuge filtration using an Amicon Ultra Ultracel 10 kDa filter. A 100μl aliquot was loaded in the filter and diluted with 4 mL DI water, centrifuged and washed with an additional 4 mL DI water. The percent stability of 177Lu-radiolabeled copolymers was calculated based on the radioactivity of the copolymers remaining in the 10 kDa filter and the total radioactivity added.
Macrophage Differentiation and Cell Culture
Human monocytes were harvested from normal donors in the UNMC Elutriation Core Facility. Upon receipt, monocytes were immediately plated into T75 tissue culture flasks at a concentration of 20 x106 cells in 20 mL of macrophage differentiation medium DMEM containing 10% FBS, 1% Penicillin/Streptomycin, 2 mM l-glutamine, 1% sodium-pyruvate, 1% NEAA and 500 U rhM-CSF. Media was half-exchanged every 2 to 3 days for 7–10 days, at which time, visual confirmation of differentiation was made and media was changed to macrophage maintenance medium (differentiation medium without the rhM-CSF). Cells were maintained at 37 °C at 5% CO2 in air.
The HPAC cells were cultured in our laboratory, as per ATCC protocols, in a 1:1 mixture of DMEM and Ham’s F12 medium containing 14.3 mM sodium bicarbonate, 2.5 mM L-glutamine, 15 mM HEPES and 0.5 mM sodium pyruvate supplemented with 0.350 μM insulin, 0.0625 μM transferrin, 0.110 μM hydrocortisone, 1.562 nM epidermal growth factor and 5% FBS. Cells were incubated at 37 °C at 5% CO2 in air.
Cathepsin B and S Activity Assays
The cathepsin B and S activity in macrophage and HPAC cell lines was determined using a commercial cathepsin B and S activity assay kit. The method is simplified as follows: 2 million macrophage and HPAC cells each were subjected to 500 μl cell lysis buffer. After 10-minute incubation on ice, the cell lysate was centrifuged at 3000 rpm for 5 min. For each individual assay, 50 μl of the supernatant was transferred to a 96-well plate to which 50 μl of reaction buffer and 2 μl of either cathepsin B or S substrate was added. For the control group, another 2 μl of cathepsin B or S inhibitor was added. The reaction was allowed to proceed at 37 °C for one hour. Samples were read using a fluorometer with a 400-nm excitation filter and a 505-nm emission filter. Activities correlating with the specified cathepsin protease were determined by subtraction of the RFU measurements of the samples with and without inhibitor. Each individual assay was replicated 10 times.
Internalization Study of MACs by Flow Cytometry
Macrophage and HPAC cells were seeded in 6-well plates at a density of 5 × 105 cells per well and incubated in media overnight (37 °C, 5% CO2). Cells were then incubated with three MACs (0.1 mg/mL) at 37 °C for the indicated time periods. After incubation, the media was removed. Cells were harvested and washed with PBS three times followed immediately by flow cytometry analysis. Each sample analysis involved the evaluation of 1.0 × 104 cells. The collected data was averaged and reported as the mean fluorescent intensity for each sample.
Biodistribution Studies
Biodistribution studies were carried out using SCID mice with HPAC xenografts. The inoculation media was prepared using a 1:1 volumetric ratio of suspended HPAC cells to Matrigel™ to obtain a final concentration of 2 × 106 HPAC cells per 100 μl. The mice received a 100 μl subcutaneous inoculation into each flank. Upon achieving sufficient tumor size, (10–20 mm in diameter) the mice were considered viable for biodistribution studies. The mice (average weight, 25g) were injected with a 0.37 MBq (10 μCi) intravenous bolus of the purified 177Lu-MAC. The mice were sacrificed and their tissues excised at 24, 48 and 72 h time points post-injection. The excised tissues were weighed and the 177Lu activity measured in each tissue using a gamma counter. Aliquots of the injected dose were counted as a reference standard for the calculation of % ID/g values.
Statistical Analysis
Data is presented as mean ± SEM. Evaluation of significance between the three groups of data was accomplished using a non-parametric Kruskal Wallis test. If the overall p-value from the Kruskal Wallis test was < 0.05, pairwise comparisons were made between the three groups using a non-parametric Mann Whitney test. In all cases, the reported p-values are adjusted and an adjusted p-value ≤ 0.10 was considered statistically significant.
RESULTS
Design and Synthesis of the Metabolically Active Linkers (MALs)
The MALs used in synthesis of the metabolically active copolymers (MACs) are depicted in Figure 1. For MAL1 and MAL2, the peptide sequences are composed of three parts: the radiometal chelation system, the clearance sequence, and the metabolic sequence. Both MAL1 and MAL2 contain the same chelation system (DO3A) and clearance sequence (G-(D)S(tBu)-(D)S(tBu)), but differ in the metabolic sequence. For MAL1 the metabolic sequence (G-G-G-F) is a known substrate for the cathepsin B protease whereas the metabolic sequence for MAL2 (P-M-G-L-P) is a known substrate for cathepsin S.[27, 28] In the studies described herein, MAL0 is simply the DO3A chelation system and is the control for our experiments. The MAL0 contains no amino acids and would not be expected to undergo any proteolytic degradation. All of the MALs were prepared with intact orthogonal protecting groups in order to carry out subsequent conjugation reactions with the HPMA copolymer. The synthesis of MAL1 and MAL2 was accomplished using SPPS. Detachment of the protected MALs from the resin support was accomplished with dilute acid (1% TFA) using 2-chlorotrityl anchor chemistry. The crude MALs were isolated and peak purified (n = 1) yielding 16.9% for MAL1 and 37.5 % for MAL2 of isolated, purified material (based on a 0.25 mmol scale synthesis). The RP-HPLC retention times and the mass spectrometric characterization of the MALs are depicted in Table 1.
Figure 1.

Schematic of the design of MALs.
Table 1.
MALs characterization.
| MAL Peptide | Sequence | Formula | Rt (min) RP-HPLC | Mass Expected [M+H]+ | Mass Observed [M+H]+ |
|---|---|---|---|---|---|
| MAL0 | DOTA(tBu)3 | C28H52N4O8 | -- | 573.4 | 573.2 |
| MAL1 | DO3A(tBu)3-G- (D)S(tBu)-(D)S(tBu)-G- G-G-F | C59H99N11O17 | 6.5 | 1234.7 | 1234.5 |
| MAL2 | DO3A(tBu)3-G- (D)S(tBu)-(D)S(tBu)-P- M-G-L-P | C67H118N12O18S | 9.8 | 1411.8 | 1411.5 |
In Vitro Metabolism of MALs
In order to confirm that the MAL1 and MAL2 will undergo proteolytic cleavage, the un-metalated, protected MALs were incubated in the presence of the appropriate cathepsin protease. LC/MS analyses (n = 1) of the metabolism of MAL1 and MAL2 by cathepsin B and S, correspondingly, are displayed in Figure 2. Under the gradient conditions employed, the MAL1 and MAL2 peptides exhibited retention times of 5.7 and 8.9 min, respectively. For MAL1 a significant metabolite peak was observed at a retention time of 4.3 min. At 24 and 48 h post incubation, LC/MS analyses revealed that 56 and 78 %, respectively, of the original MAL1 peptide had been metabolized. Evaluation of the MAL1 metabolite mass (1030.5 Da) corresponds to the site of proteolytic cleavage being between the first and second glycine residues from the C-terminus (GG┼GF). With respect to the MAL2 peptide, a metabolite was observed with a retention time of 7.0 min. Over the course of the study, the amount of MAL2 peptide metabolized increased from 11 to 80 %. The mass of the metabolite corresponded with the cleavage site being located between the glycine and leucine residues (DO3A-G-(D)S-(D)S-P-M-G┼L-P). Overall, these studies demonstrate that both MAL1 and MAL2 are proteolytically active and that the rate of metabolism of MAL2 by cathepsin S was slower relative to the cathepsin B cleavage of MAL1.
Figure 2.

Evaluation of cathepsin B and S cleavage of MAL1 and MAL2, correspondingly. (A) Total ion chromatograms of the cleavage of MAL1 by cathepsin B at 24 and 48 h post-incubation. (B) Total ion chromatograms of the cleavage of MAL2 by cathepsin S at 24, 48 and 72 h post-incubation. (C) Depicts the enzymatic cleavage sites of MAL1 and MAL2.
Design and Synthesis of the Metabolically Active Copolymers
The scheme for the synthesis of the MACs is depicted in Figure 3. The HPMA copolymer consisted of HPMA, APMA and APMA-FITC monomers which were polymerized, using reversible addition-fragmentation chain transfer (RAFT) polymerization, with a feed composition of 98.75:0.85:0.40, respectively. Assessment of the synthesized HPMA copolymer by FPLC revealed a molecular weight of 343 kDa with a polydispersity of 1.6. Evaluation by quantitative ninhydrin analysis revealed that 121% of the APMA monomer was incorporated into the HPMA copolymer which yielded a total amine content of 70.4 μmol/g (n = 1) of polymer.
Figure 3.

Synthetic scheme for the synthesis of the 177Lu-MAC conjugates. For MAC0, MAC1 and MAC2, X = Y = null, X = GSS, Y = GGGF and X = GSS, Y = PMGLP, respectively.
Conjugation of the MALs to the HPMA copolymer was accomplished using standard coupling chemistry. The preparation of protected peptides allowed for chemo-selective coupling of the C-terminal carboxylic acid with the aliphatic amine of the HPMA copolymer. Once the MALs were conjugated to the HPMA copolymer, the HPMA copolymers were termed MAC0, MAC1 and MAC2 corresponding to the conjugated MAL. Deprotection of the peptide components of the MACs were carried out using a standard cleavage cocktail containing predominantly trifluoroacetic acid. The deprotection of the MACs was verified by evaluation of MAC0 before and after deprotection using NMR (supporting information). The signal for the t-butyl groups of the DO3A was eliminated after the deprotection reaction corresponding to the loss of the t-butyl groups. With this information in hand, the cleavage conditions used for MAC0 were determined to be sufficient for the deprotection of MAC1 and MAC2. After deprotection the coupling yield of the MACs were analyzed by quantitative ninhydrin analyses. Ninhydrin analysis gave an 83.6, 72.9 and 83.4 % coupling efficiency (n = 1) for MAC0, MAC1 and MAC2, respectively.
Synthesis, Cathepsin Cleavage and Serum Stability of 177Lu-labeled-MACs
The 177Lu radiolabeled MACs were prepared using relatively mild conditions. Incubation of the MACs with 177LuCl3 for 1 hour at 90 °C gave relatively moderate to good radiochemical yields of 60 – 85 % (n = 3). Radiolabeled yields were found to be dependent, to some extent, on the HPMA copolymer concentration. A more thorough investigation of the parameters that influence the radiolabeling efficiency of MACs is on-going. The ability of the synthesized 177Lu-MACs to undergo Cathepsin B and S cleavage was investigated over a 72 h period, Figure 4. During the course of the investigation, the non-cleavable control (177Lu-MAC0) was found to be stable in the presence of Cathepsin B and S with negligible cleavage as determined by radio-SEC-HPLC. However, 177Lu-MAC1 and 177Lu-MAC2 demonstrated significant cleavage as shown by the generation of the low-molecular weight radiometabolite after 72 h of incubation. The percentage of the generated radiometabolite was 8 and 26 % (n = 1) for 177Lu-MAC1 and 177Lu-MAC2, respectively. The stability of the 177Lu-labeled-MACs in human serum was investigated by incubation at 37 °C for a 72 h period. Over the course of the study, the percentage of intact 177Lu-labeled-MACs remained greater than 98%.
Figure 4.

Cathepsin B and S cleavage of 177Lu-MAC0, 177Lu-MAC1 and 177Lu-MAC2 as determined by radio-SEC-HPLC at 72 h post-incubation.
Cathepsin Activity and MAC Uptake in Macrophage and HPAC Cell Lines
In the investigation of the in vitro properties of the MACs, differentiated human macrophage and HPAC cancer cell lines were used as in vitro models of the MPS and pancreatic cancer. The cathepsin B and S activity in both of these cell lines were investigated by fluorometric analysis, depicted in Figure 5. In a cell to cell comparison, the differentiated macrophage demonstrated substantially higher activities of both cathepsin proteases relative to the HPAC cell line. The protease activity in the macrophage was 70 and 10 fold higher for cathepsin B and S, respectively, relative to the HPAC cell line.
Figure 5.

Evaluation of cathepsin B and S activity between macrophage and HPAC cell lines. Data is presented as mean ± S.D. from n=10 experiments.
The cellular uptake of the MACs in differentiated macrophage and the HPAC cell line was investigated. Utilizing flow cytometry analysis, the relative uptake of the MACs in terms of mean fluorescence units is depicted in Figure 6. Over the 48 hour incubation time, all of the MACs investigated demonstrated steady increase in macrophage uptake. In comparison, the HPAC cell line revealed little uptake of the MACs over the time period studied.
Figure 6.
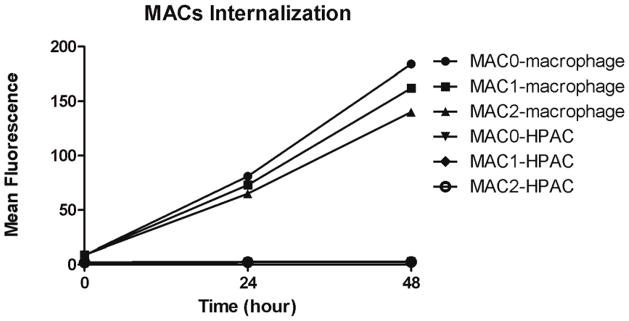
Mean fluorescence of MAC0, MAC1 and MAC2 after 0 hour, 24 hours and 48 hours incubation in macrophages and HPAC cells measured by flow cytometry.
Biodistribution Studies of 177Lu labeled MACs in HPAC Xenograft SCID Mice
A preliminary investigation of the in vivo tumor targeting and clearance properties of the MACs was performed in a SCID mouse model bearing HPAC tumor xenografts. The results of the biodistribution studies of the three 177Lu-MACs at 24 and 72 h post-injection are given in Table 2. At 24 h post-injection, the blood retention of the 177Lu-MAC1 and 177Lu-MAC2 was, on average, lower than the control (177Lu-MAC0). All of 177Lu-MACs demonstrated a 2 fold decrease in blood retention by the 72 h p.i. time point. At 24 h post-injection, the liver accumulation of the 177Lu-MAC1 and 177Lu-MAC2 was 3.44 ± 0.82 and 5.29 ± 0.98 %ID/g, correspondingly, which was lower than the 7.27 ± 2.64 %ID/g of 177Lu-MAC0 (p = 0.34). The liver retention of the 177Lu-MAC0 increased to a final accumulation of 11.36 ± 1.65 %ID/g at 72 h post-injection. Conversely, the liver retention of the MACs that utilize cleavable peptides, 177Lu-MAC1 and 177Lu-MAC2, remain statistically the same with 3.61 ± 0.63 and 5.46 ± 0.44 %ID/g, respectively, at the 72 h time point. At the 24 h timepoint, the spleen retention of 177Lu-MAC1 and 177Lu-MAC2 were lower on average than the 177Lu-MAC0. However, by the 72 h timepoint, the 177Lu-MAC0 demonstrated a substantial increase in spleen retention of 21.46 ± 4.41 %ID/g. In comparison, 177Lu-MAC1 and 177Lu-MAC2 exhibited a significantly lower retention of 5.63 ± 1.22 (p = 0.06) and 5.67 ± 0.75 %ID/g (p = 0.03), respectively, which corresponds to a substantial 4 fold reduction in spleen accumulation at the analogous time point. With regards to tumor retention, the control exhibited higher accumulation at all timepoints investigated. At the 24h time point, the tumor uptake of 1.18 ± 0.25 and 1.64 ± 0.18 %ID/g 177Lu-MAC1 and 177Lu-MAC2 was substantially lower than the 4.09 ± 1.41 %ID/g of the 177Lu-MAC0 (p = 0.16). By 72 h post-injection, the 177Lu-MAC0 tumor accumulation increased by 61% to 6.58 ± 0.93. For 177Lu-MAC1 and 177Lu-MAC2, the tumor retention increased to 2.33 ± 0.30 and 3.49 ± 0.28 %ID/g, representing an increase of 125 and 113%, correspondingly, at the 72 h time point.
Table 2.
Biodistribution studies of 177Lu-MAC0, 177Lu-MAC1 and 177Lu-MAC2 at 24 and 72 h post-injection in HPAC tumor-bearing SCID mice. Data is represented as %ID/g.
| 24 hours | 72 hours | |||||
|---|---|---|---|---|---|---|
| Tissues | Lu-MAC0b | Lu-MAC1a | Lu-MAC2c | Lu-MAC0 b | Lu-MAC1b | Lu-MAC2c |
| Blood | 28.50 ± 6.59 | 14.85 ± 1.23 | 17.76 ± 1.57 | 13.40 ± 1.71 | 8.54 ± 1.76 | 8.89 ± 0.65 |
| Bladder | 9.09 ± 3.49 | 1.63 ± 0.94 | 3.11 ± 0.82 | 7.15 ± 2.61 | 2.18 ± 0.25 | 3.61 ± 0.34 |
| Small Int. | 1.36 ± 0.42 | 0.85 ± 0.15 | 0.97 ± 0.12 | 1.25 ± 0.21 | 0.51 ± 0.09 | 0.67 ± 0.06 |
| Large Int. | 1.48 ± 0.38 | 0.84 ± 0.09 | 1.31 ± 0.17 | 1.25 ± 0.21 | 0.54 ± 0.09 | 1.06 ± 0.15 |
| Stomach | 1.02 ± 0.21 | 0.63 ± 0.07 | 0.80 ± 0.10 | 0.91 ± 0.14 | 0.37 ± 0.07 | 0.52 ± 0.04 |
| Pancreas | 2.16 ± 0.66 | 0.93 ± 0.22 | 1.35 ± 0.25 | 2.95 ± 0.55 | 1.10 ± 0.20 | 1.10 ± 0.13 |
| Spleen | 7.83 ± 3.32 | 5.09 ± 1.24 | 4.95 ± 0.49 | 21.46 ± 4.41 | 5.63 ± 1.22 | 5.67 ± 0.75 |
| Kidney | 5.22 ± 1.57 | 2.50 ± 0.59 | 3.34 ± 0.44 | 4.20 ± 0.58 | 1.93 ± 0.38 | 2.51 ± 0.17 |
| Tumor | 4.09 ± 1.41 | 1.18 ± 0.25 | 1.64 ± 0.18 | 6.58 ± 0.93 | 2.33 ± 0.30 | 3.49 ± 0.28 |
| Liver | 7.27 ± 2.64 | 3.44 ± 0.82 | 5.29 ± 0.98 | 11.36 ± 1.65 | 3.61 ± 0.63 | 5.46 ± 0.44 |
| Lung | 8.18 ± 2.60 | 3.81 ± 0.76 | 4.16 ± 0.37 | 5.56 ± 1.01 | 2.66 ± 0.46 | 3.56 ± 0.16 |
| Heart | 6.13 ± 1.75 | 2.37 ± 0.50 | 2.72 ± 0.30 | 5.00 ± 0.75 | 1.85 ± 0.38 | 2.26 ± 0.22 |
| Brain | 0.91 ± 0.23 | 0.30 ± 0.10 | 0.36 ± 0.04 | 0.57 ± 0.00 | 0.24 ± 0.05 | 0.27 ± 0.02 |
| Bone | 1.71 ± 0.51 | 0.85 ± 0.09 | 1.20 ± 0.16 | 3.06 ± 0.46 | 0.68 ± 0.12 | 1.02 ± 0.11 |
| Muscle | 1.36 ± 0.42 | 0.71 ± 0.13 | 0.75 ± 0.08 | 2.38 ± 0.55 | 1.37 ± 0.39 | 1.46 ± 0.15 |
The data is given in mean ± SEM.
N = 4,
N = 5,
N = 6.
DISCUSSION
For cancer diagnostics and therapeutics based on biopolymers such as HPMA copolymers, it is well known that increases in the molecular weight of the carrier typically results in an improvement in the enhanced permeability and retention (EPR) effect.[5, 29, 30] As a result of the increase in the EPR effect of the carrier, the drug delivery system will typically demonstrate an increased localization and retention in cancerous tissues. Conversely, an increase in the molecular weight of a biopolymer by and large causes a substantial increase in the MPS accumulation, particularly if the molecular weight of the carrier is above the renal excretion threshold. The MPS accumulation of HPMA copolymers, and other nanomedicine platforms, is a major hurdle in the development of more efficacious biopolymer systems and the translation of these systems to the clinic.[31, 32]
Our approach to this challenge is to develop MALs that preferentially hydrolyze in MPS cells in order to reduce the retention, and thus radiotoxicity, in non-target tissues. In our design peptide substrates of cathepsin B and S were chosen due to the high level of expression of these proteases in MPS tissues. As depicted in Figure 3, the MAL design is composed of three components: radiometal chelation system, metabolic sequence and clearance sequence. The DO3A chelation system was chosen because it has been shown to stabilize a variety of radiometals, including 177Lu, in preclinical and clinical investigations.[33, 34] The metabolic sequences are composed of known peptide substrates for the cathepsin B and S enzymes.[27, 28] It is expected that alterations of the metabolic sequence can be used to influence the rate of enzymatic cleavage and thus affect clearance of the radiometabolites from the tissues. While the chosen amino acid sequences have demonstrated selectivity for the corresponding protease relative to other lysosomal proteases, it is important to keep in mind that other proteases may contribute to the metabolism of these sequences in vivo. In order to better control the pharmacokinetics of the radiometabolite after cleavage, D-amino acids were employed in the design of the clearance sequence, G-D(S)-D(S), to prevent, or at least significantly reduce, metabolism. By preventing metabolism the pharmacokinetic properties of the generated radiometabolite can be better controlled to influence diffusion and thus biological clearance.[35]
The MALs were prepared with intact orthogonal protecting groups using standard SPPS techniques. Synthesis of the protected MALs was necessary for later chemoselective conjugation to the HPMA copolymers. Both MAL1 and MAL2, protecting groups intact, underwent incubation in the presence of the appropriate cathepsin protease. Over the course of the metabolism study, both of the MALs demonstrated conversion into a single metabolite. Both of these cleavage sites are in accordance with previous literature reports.[27, 36] From these initial investigations, the enzyme kinetics for cathepsin B for MAL1 appears to be significantly faster as compared to cathepsin S for MAL2.
Synthesis of the HPMA copolymer backbone was accomplished using the RAFT polymerization technique. The synthesized HPMA copolymer (Mw = 343 kDa, PDI = 1.6) is well above the renal threshold of 45 kDa. Conjugation of the MALs to the terminal amine of the APMA copolymer constituent was accomplished in fairly high yield as determine by ninhydrin assay. In order to ensure chemo-selectivity, MALs were prepared with intact orthogonal protection groups. The protecting groups prevented functional groups other than the desired C-terminal carboxylic acid from conjugating to the primary amine on the HPMA copolymer. Deprotection of the MACs was accomplished, as confirmed by NMR, in 3 hours using a standard SPPS cleavage cocktail. Radiolabeling of the MACs with 177Lu was achieved in good yield using standard radiolabeling conditions. Incubation of 177Lu-MAC1 and 177Lu-MAC2 in the presence of Cathepsin B and S, respectively, demonstrated, as expected, that a low-molecular weight radiometabolite is generated. Exposure of 177Lu-MAC0 (non-cleavable control) to either of the investigated cathepsin did not show any signs of radiometabolite generation. This data shows that the generation of the radiometabolite is a proteolytic process. In additional, this data confirms that the MAL can still be cleaved despite being attached to the HPMA copolymer support. Serum stability studies of the 177Lu-MACs revealed essentially no degradation (> 98% intact) suggesting that the 177Lu-MACs would not susceptible to extracellular blood proteases in vivo.
In our investigations of the in vitro properties of the MACs, we modeled the phagocytic cells of the MPS and pancreatic cancer cells utilizing differentiated human macrophage and the HPAC pancreatic cancer cell line, correspondingly. Evaluation of the cathepsin B and S activity of these cell lines revealed that both cathepsin B and S activity is substantially higher, 70 and 10 fold respectively, in the macrophage relative to the HPAC cell line. Additionally, analysis of the internalization rate of the MACs in these two cell lines demonstrated that macrophage undergo steady uptake of the MACs over the course of the study, while relatively little uptake was exhibited by the HPAC cancer cell line. These data imply that the in vivo uptake and metabolism of the MACs should be strongly favored in MPS cells relative to the pancreatic cancer cells. Given this, we would expect enhanced clearance from tissues containing MPS components (i.e., liver and spleen) in vivo.
Our initial investigation into the in vivo characteristics of the 177Lu-MACs was undertaken in a HPAC xenograft mouse model. All of the 177Lu-MACs investigated demonstrated extended blood circulation due to the molecular weight of the HPMA copolymer being substantially higher than the renal excretion threshold.[37] Interestingly, the blood retention of 177Lu-MAC1 and 177Lu-MAC2 demonstrate similar longevity in the blood pool and is approximately half of the value of the 177Lu-MAC0 at all reported time points. The reduced blood retention of the 177Lu-MAC1 and 177Lu-MAC2 relative to the control (177Lu-MAC0) strongly suggest that the HPMA copolymers with MALs underwent proteolytic cleavage in the blood. However, the in vitro serum stability studies of the 177Lu-MAC1 and 177Lu-MAC2 revealed no significant proteolysis. These data imply that the cleavage of the 177Lu-MAC1 and 177Lu-MAC2 is likely due to the phagocytic uptake and metabolism by cellular blood components. While long-term blood circulation of the 177Lu-MACs leads to better tumor targeting, the substantial blood retention of the 177Lu-MACs is detrimental to practical application of these agents for diagnostic and radiotherapeutic approaches. Utilizing these initial results, our future designs of the 177Lu-MACs will seek to provide faster blood clearance to better balance the circulation/tumor targeting time and the clearance rates needed for clinical applications.
The significant blood retention of the 177Lu-MACs overall led to overall high baseline radioactivity values observed in all analyzed tissues. When evaluating the biodistribution data, it is important to keep in mind the contribution to the tissue radioactivity given by blood residing in the tissue. It is undoubtedly true that the high blood activity of the 177Lu-MACs is contributing, to some extent, to the various tissues values, particularly those tissues that normally contain large blood volumes (e.g., liver and kidney). However, as we move from the 24 to the 72 h time point we observe substantial decreases (~2 fold) in blood retention and, at the same time, a significant increase in both liver, spleen and tumor retention. The increase in the liver, spleen and tumor retention, while during the same period blood activity reduces, leads us to the conclusion that increases observed in the biodistribution time points is mostly attributable to the tissue uptake and not background blood activities.
The MPS uptake of nanomedicine platforms is typified by significant accumulation of the delivery system in the liver and spleen. The liver accumulation of the 177Lu-MAC0 demonstrated an increase from 7.27 ± 2.64 to 11.36 ± 1.65 %ID/g over the time course of the experiment. In comparison 177Lu-MAC1 and 177Lu-MAC2 demonstrated lower average accumulation in the liver at every time point investigated. 177Lu-MAC1 and 177Lu-MAC1 demonstrated a 24 h post-injection liver retention of 3.44 ± 0.82 and 5.29 ± 0.98 %ID/g, but demonstrated nearly identical values at 72 h post-injection. At 72 h post-injection, 177Lu-MAC1 and 177Lu-MAC1 gave liver retention values that were 3.1 and 2.1 times lower than the control (p=0.024, both). With regard to spleen retention, the average spleen retention of 177Lu-MAC0 increased from 7.61 ± 1.37 to 21.46 ± 4.41 %ID/g over the time points investigated. For the 177Lu-MAC1 and 177Lu-MAC2, the HPMA copolymers demonstrated substantially lower spleen accumulation compared to control. This was particularly evident at the 72 h time point with spleen values 3.8 and 3.7 times lower (5.63 ± 1.22 and 5.67 ± 0.75 %ID/g) for 177Lu-MAC1 (p = 0.03) and 177Lu-MAC2 (p = 0.06), respectively. The substantial reduction in liver and spleen retention observed in these studies show that the MAL design is capable of enhancing clearance from the MPS tissues.
HPAC tumor accumulation of the 177Lu-MACs was achieved through passive targeting due to the EPR effect. Relative to 177Lu-MAC1 and 177Lu-MAC2, 177Lu-MAC0 demonstrated superior tumor retention at all of the time points studied, with the highest value being 6.58 ± 0.93 %ID/g at 72 h post-injection. At 72 h post-injection, 177Lu-MAC1 and 177Lu-MAC2 gave tumor activities of 2.33 ± 0.30%ID/g (p =0.024) and 3.49 ± 0.28 %ID/g (p =0.06), correspondingly, which were significantly lower than control. It is not necessarily clear why the tumor retention of the values of 177Lu-MAC1 and 177Lu-MAC2 are substantially lower than those of 177Lu-MAC0. Possibly, the reduced blood retention of the 177Lu-MAC1 and 177Lu-MAC2 or potential metabolism and clearance in the HPAC tumor could contribute to lower overall tumor retention. Unfortunately, the current biodistribution study does not unambiguously demonstrate the in vivo specificity and mechanism of the MACs. Future studies looking into the in vitro and in vivo metabolism of the 177Lu-MACs are planned and will hopefully shed more light with regard to the in vivo tumor targeting and retention properties of these agents.
CONCLUSIONS
In utilizing the MAL design, we seek to take advantage of the relative differences in cathepsin B and S expression as well as the lymphatic and vasculature structure of pancreatic cancer and non-target tissues leading to a decrease in non-target retention of various nanomedicine systems. Herein, we have reported the synthesis and purification of two MALs that are known substrates of cathepsin B and S. These MALs were shown to be metabolized by cathepsin B and S into single metabolites. The MALs were conjugated to the HPMA copolymer and radiolabeled with 177Lu. The synthesized 177Lu-MAC1 and 177Lu-MAC2 demonstrated a substantial decrease in long-term liver and spleen retention relative to the control (177Lu-MAC0). However, compared to the control, the MALs in 177Lu-MAC1 and 177Lu-MAC2 exhibited higher clearance from HPAC tumors. Our future work will focus on the optimization of the circulation time to achieve faster blood clearance of the MAC design to better suit clinical application. Additionally, further studies into the in vitro and in vivo behavior of the 177Lu-MACs are planned to elucidate the rates of uptake and metabolism in both MPS tissues and pancreatic tumors.
Acknowledgments
The authors gratefully acknowledge the National Cancer Institute (4 R00 CA137147), the National Center for Research Resources (5 P20 RR021937) and the National Institute of General Medical Sciences (8 P20 GM103480) for the funding and support of this research. The authors would also like to acknowledge Mr. Erik Moore and Dr. Surinder Batra for the use of the gamma counter and computer programs used to determine the biodistribution of the agents described in this paper. Lastly, we gratefully acknowledge the UNMC Elutriation Core for providing the human monocytes used in this manuscript.
ABBREVIATIONS
- MALs
metabolically active linkers
- MACs
metabolically active copolymers
- HPMA
N-(2-hydroxypropyl) methacrylamide
- MPS
mononuclear phagocyte system
- DOTA
1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid
- DO3A
1,4,7,10-tetraazacyclododecane-1,4,7-triacetic acid
- 177Lu
lutetium-177
- %ID
percent injected dose
- DCM
dichloromethane
- NMP
N-methylpyrrolidone
- DMF
NN, -dimethylformamide
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- BSA
bovine serum albumin
- SDS
sodium dodecyl sulfate
- EGF
epidermal growth factor
- DMEM
Dulbecco’s Modified Eagle Medium
- PBS
phosphate buffered saline
- APMA
N-(3-aminopropyl) methacrylamide
- FITC
fluorescein isothiocyanate
- NHS
N-hydroxysuccinimide
- DCC
N, N′-dicyclohexylcarbodiimide
- V-501
4,4′-Azobis(4-cyanovaleric acid)
- CTP
4-cyano-4-(phenylcarbono-thioylthio)pentanoic acid
- DODT
3,6-dioxa-1,8-octanedithiol
- TIS
triisopropylsilane
- EDTA
ethylenediaminetetraacetic acid
- APMA-FITC
5-(3-(methacryloylaminopropyl) thioureidyl) fluorescein
- COMU
1-[(1-(cyano-2-ethoxy-2-oxoethylideneaminooxy) dimethylaminomorpholino)] uronium hexafluorophosphate
- Fmoc
fluorenylmethyloxycarbonyl
- FBS
fetal bovine serum
- RPMI
roswell park memorial institute
- NEAA
non-essential amino acids
- rhM-CSF
recombinant human macrophage-colony stimulating factor
- ICR SCID
institute of cancer research severe combined immunodeficient
- SPPS
solid phase peptide synthesis
- FPLC
fast protein liquid chromatography
Footnotes
SUPPORTING INFORMATION
Information regarding NMR spectra of protected and deprotected MACO (Figure S1 and S2) and the flow cytometry data (Figures S3 and S4) is available free of charge via the internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blanco E, Kessinger CW, Sumer BD, Gao J. Multifunctional micellar nanomedicine for cancer therapy. Exp Biol Med (Maywood) 2009;234:123–31. doi: 10.3181/0808-MR-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2:347–60. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 3.Hamoudeh M, Kamleh MA, Diab R, Fessi H. Radionuclides delivery systems for nuclear imaging and radiotherapy of cancer. Adv Drug Deliv Rev. 2008;60:1329–46. doi: 10.1016/j.addr.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Kawaguchi T, Honda T, Nishihara M, Yamamoto T, Yokoyama M. Histological study on side effects and tumor targeting of a block copolymer micelle on rats. J Control Release. 2009;136:240–6. doi: 10.1016/j.jconrel.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Kopeèek J, Kopeèková P, Minko T, Lu Z. HPMA copolymer-anticancer drug conjugates: design, activity, and mechanism of action. Eur J Pharm Biopharm. 2000;50:61–81. doi: 10.1016/s0939-6411(00)00075-8. [DOI] [PubMed] [Google Scholar]
- 6.Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R, et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clin Cancer Res. 1999;5:83–94. [PubMed] [Google Scholar]
- 7.Pimm MV, Perkins AC, Strohalm J, Ulbrich K, Duncan R. Gamma scintigraphy of a 123I-labelled N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugate containing galactosamine following intravenous administration to nude mice bearing hepatic human colon carcinoma. Journal of drug targeting. 1996;3:385–90. doi: 10.3109/10611869608996829. [DOI] [PubMed] [Google Scholar]
- 8.Pimm MV, Perkins AC, Strohalm J, Ulbrich K, Duncan R. Gamma scintigraphy of the biodistribution of 123I-labelled N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugates in mice with transplanted melanoma and mammary carcinoma. Journal of drug targeting. 1996;3:375–83. doi: 10.3109/10611869608996828. [DOI] [PubMed] [Google Scholar]
- 9.Mitra A, Coleman T, Borgman M, Nan A, Ghandehari H, Line BR. Polymeric conjugates of mono- and bi-cyclic alpha(V)beta(3) binding peptides for tumor targeting. J Control Release. 2006;114:175–83. doi: 10.1016/j.jconrel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 10.Mitra A, Nan A, Papadimitriou JC, Ghandehari H, Line BR. Polymer-peptide conjugates for angiogenesis targeted tumor radiotherapy. Nucl Med Biol. 2006;33:43–52. doi: 10.1016/j.nucmedbio.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Lammers T, Kühnlein R, Kissel M, Subr V, Etrych T, Pola R, et al. Effect of physicochemical modification on the biodistribution and tumor accumulation of HPMA copolymers. J Control Release. 2005;110:103–18. doi: 10.1016/j.jconrel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 12.Mitra A, Mulholland J, Nan A, McNeill E, Ghandehari H, Line BR. Targeting tumor angiogenic vasculature using polymer-RGD conjugates. J Control Release. 2005;102:191–201. doi: 10.1016/j.jconrel.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 13.Line BR, Mitra A, Nan A, Ghandehari H. Targeting tumor angiogenesis: Comparison of peptide and polymer-peptide conjugates. J Nucl Med. 2005;46:1552–60. [PubMed] [Google Scholar]
- 14.Julyan PJ, Seymour LW, Ferry DR, Daryani S, Boivin CM, Doran J, et al. Preliminary clinical study of the distribution of HPMA copolymers bearing doxorubicin and galactosamine. J Control Release. 1999;57:281–90. doi: 10.1016/s0168-3659(98)00124-2. [DOI] [PubMed] [Google Scholar]
- 15.Mitra A, Nan A, Ghandehari H, McNeill E, Mulholland J, Line BR. Technetium-99m-labeled N-(2-hydroxypropyl) methacrylamide copolymers: Synthesis, characterization, and in vivo biodistribution. Pharm Res. 2004;21:1153–9. doi: 10.1023/b:pham.0000033001.49737.b7. [DOI] [PubMed] [Google Scholar]
- 16.Berdowska I. Cysteine proteases as disease markers. Clin Chim Acta. 2004;342:41–69. doi: 10.1016/j.cccn.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 17.Yasuda Y, Kaleta J, Brömme D. The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev. 2005;57:973–93. doi: 10.1016/j.addr.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 18.Shuja S, Sheahan K, Murnane MJ. Cysteine endopeptidase activity levels in normal human tissues, colorectal adenomas and carcinomas. Int J Cancer. 1991;49:341–6. doi: 10.1002/ijc.2910490305. [DOI] [PubMed] [Google Scholar]
- 19.Qian F, Bajkowski AS, Steiner DF, Chan SJ, Frankfater A. Expression of five cathepsins in murine melanomas of varying metastatic potential and normal tissues. Cancer Res. 1989;49:4870–5. [PubMed] [Google Scholar]
- 20.Petanceska S, Canoll P, Devi LA. Expression of rat cathepsin S in phagocytic cells. J Biol Chem. 1996;271:4403–9. doi: 10.1074/jbc.271.8.4403. [DOI] [PubMed] [Google Scholar]
- 21.Shi GP, Webb AC, Foster KE, Knoll JH, Lemere CA, Munger JS, et al. Human cathepsin S: chromosomal localization, gene structure, and tissue distribution. J Biol Chem. 1994;269:11530–6. [PubMed] [Google Scholar]
- 22.Villadangos JA, Ploegh HL. Proteolysis in MHC class II antigen presentation: who’s in charge? Immunity. 2000;12:233–9. doi: 10.1016/s1074-7613(00)80176-4. [DOI] [PubMed] [Google Scholar]
- 23.Christie RJ, Grainger DW. Design strategies to improve soluble macromolecular delivery constructs. Adv Drug Deliv Rev. 2003;55:421–37. doi: 10.1016/s0169-409x(02)00229-6. [DOI] [PubMed] [Google Scholar]
- 24.Kukis DL, Novak-Hofer I, DeNardo SJ. Cleavable linkers to enhance selectivity of antibody-targeted therapy of cancer. Cancer Biother Ra diopharm. 2001;16:457–67. doi: 10.1089/10849780152752065. [DOI] [PubMed] [Google Scholar]
- 25.Segal E, Pan H, Ofek P, Udagawa T, Kopeèková P, Kopeèek J, et al. Targeting Angiogenesis-Dependent Calcified Neoplasms Using Combined Polymer Therapeutics. PLoS One. 2009;4:e5233. doi: 10.1371/journal.pone.0005233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.York AW, Scales CW, Huang F, McCormick CL. Facile Synthetic Procedure for ω, Primary Amine Functionalization Directly in Water for Subsequent Fluorescent Labeling and Potential Bioconjugation of RAFT-Synthesized (Co)Polymers†. Biomacromolecules. 2007;8:2337–41. doi: 10.1021/bm700514q. [DOI] [PubMed] [Google Scholar]
- 27.Li M, Meares CF. Synthesis, metal chelate stability studies, and enzyme digestion of a peptide-linked DOTA derivative and its corresponding radiolabeled immunoconjugates. Bioconjug Chem. 1993;4:275–83. doi: 10.1021/bc00022a005. [DOI] [PubMed] [Google Scholar]
- 28.Lützner N, Kalbacher H. Quantifying cathepsin S activity in antigen presenting cells using a novel specific substrate. J Biol Chem. 2008;283:36185–94. doi: 10.1074/jbc.M806500200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: A royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15:457–64. doi: 10.1080/10611860701539584. [DOI] [PubMed] [Google Scholar]
- 30.Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol Biol. 2010;624:25–37. doi: 10.1007/978-1-60761-609-2_3. [DOI] [PubMed] [Google Scholar]
- 31.Owens DE, 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Nie S. Understanding and overcoming major barriers in cancer nanomedicine. Nanomedicine (Lond) 2010;5:523–8. doi: 10.2217/nnm.10.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Leon-Rodriguez LM, Kovacs Z. The synthesis and chelation chemistry of DOTA-peptide conjugates. Bioconjug Chem. 2008;19:391–402. doi: 10.1021/bc700328s. [DOI] [PubMed] [Google Scholar]
- 34.Kwekkeboom DJ, Teunissen JJ, Bakker WH, Kooij PP, de Herder WW, Feelders RA, et al. Radiolabeled somatostatin analog [Lu-177-DOTA(0), Tyr(3)] octreotate in patients with endocrine gastroenteropancreatic tumors. J Clin Oncol. 2005;23:2754–62. doi: 10.1200/JCO.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 35.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–92. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 36.Lutzner N, Kalbacher H. Quantifying cathepsin S activity in antigen presenting cells using a novel specific substrate. J Biol Chem. 2008;283:36185–94. doi: 10.1074/jbc.M806500200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seymour LW, Duncan R, Strohalm J, Kopeèek J. Effect of molecular weight (Mw) of N-(2-hydroxypropyl)methacrylamide copolymers on body distribution and rate of excretion after subcutaneous, intraperitoneal, and intravenous administration to rats. J Biomed Mater Res. 1987;21:1341–58. doi: 10.1002/jbm.820211106. [DOI] [PubMed] [Google Scholar]