

Abstract
microRNAs (miRNAs) are a class of small RNAs (sRNAs) of ~21 nucleotides (nt) in length processed from foldback hairpins by DICER-LIKE1 (DCL1) or DCL4. They regulate the expression of target mRNAs by base pairing through RNA-Induced Silencing Complex (RISC). In the RISC, ARGONAUTE1 (AGO1) is the key protein that cleaves miRNA targets at position ten of a miRNA:target duplex. The authenticity of many annotated rice miRNA hairpins is under debate because of their homology to repeat sequences. Some of them, like miR1884b, have been removed from the current release of miRBase based on incomplete information. In this study, we investigated the association of transposable element (TE)-derived miRNAs with typical miRNA pathways (DCL1/4- and AGO1-dependent) using publicly available deep sequencing datasets. Seven miRNA hairpins with 13 unique sRNAs were specifically enriched in AGO1 immunoprecipitation samples and relatively reduced in DCL1/4 knockdown genotypes. Interestingly, these species are ~21-nt long, instead of 24-nt as annotated in miRBase and the literature. Their expression profiles meet current criteria for functional annotation of miRNAs. In addition, diagnostic cleavage tags were found in degradome datasets for predicted target mRNAs. Most of these miRNA hairpins share significant homology with miniature inverted-repeat transposable elements (MITEs), one type of abundant DNA transposons in rice. Finally, the root-specific production of a 24 nt miRNA-like sRNA was confirmed by RNA blot for a novel EST that maps to the 3'-UTR of a candidate pseudogene showing extensive sequence homology to miR1884b hairpin. Our data are consistent with the hypothesis that TEs can serve as a driving force for the evolution of some MIRNAs, where co-opting of DICER-LIKE1/4 processing and integration into AGO1 could exapt transcribed TE-associated hairpins into typical miRNA pathways.
Keywords: MicroRNA, Transposable element, Repeat-associated small RNA, ARGONAUTE, DICER-LIKE
Introduction
Plant microRNAs (miRNAs) are 20- to 24-nucleotide (nt) small RNAs (sRNAs) that regulate gene expression epigenetically (Bartel 2009). They are processed from miRNA precursors with hairpin-like structures mainly by DICER-LIKE1 (DCL1). miRNAs guide RNA-Induced Silencing Complex (RISC)-directed cleavage or translational repression of target mRNAs and transcriptional silencing of target loci through base pairing (Voinnet 2009). ARGONAUTE1 (AGO1) is the catalytic engine of miRNA-programmed RISC, which loads functional sRNAs and “slices” cognate targets (Mallory and Vaucheret 2010). miRNAs have been proposed to evolve from inverted duplication of their founder (target) genes with protein-coding capacity (Allen et al. 2004), from random sequences (Felippes et al. 2008), or from repeats (Piriyapongsa and Jordan 2008). Another class of sRNAs, repeat-associated small interfering RNAs (rasiRNAs), originates from heterochromatic regions and interspersed repeats. In the Solanaceae, their biogenesis is dependent upon two RNase III family members, DCL3 and DCL4 (Kuang et al. 2009). RasiRNAs may constrain the proliferation of transposable elements (TEs) and other repeat sequences by limiting transcription (Lisch 2012; McCue and Slotkin, 2012). Reports have shown that rasiRNAs can act non-cell autonomously, interacting with AGO9 to specify cell fate in the germ line and developing gametophytes (Le Trionnaire et al. 2011, Olmedo-Monfil et al. 2010; Peng et al. 2012), and are associated with the regulation of gene expression and plant stress response (McCue et al. 2012; Nosaka et al. 2012). Less is known about the function of repeat-associated sRNAs in post-transcriptional gene silencing (PTGS) guided by AGO1. A recent report indicates that siRNAs are loaded into a cellular pool of AGO1 that is distinct from a miRNA-programmed AGO1 pool in planta (Schott et al., 2012).
Many miRNAs in rice have significant homology with transposable elements (TEs) however, their actual function(s) remain largely unknown. Weak expression, coupled with variable processing, may partly explain the relative lack of experimental support for TE-like miRNAs and other newly evolved miRNAs (Cuperus et al. 2011). Here we present a meta-analysis of deep sequencing datasets for repeat-associated sRNAs in Oryza sativa. We set up a series of custom filters aimed at looking for miRNAs that originate from repeats. Repeat-associated sRNAs were found to be largely dependent upon OsDCL1 and/or OsDCL4 for biogenesis. A substantial proportion of them were found to be enriched in OsAGO1 Immunoprecipitation (IP) samples, suggesting functional significance. Several typical miRNAs with homology to siRNA-like miRNAs and DNA transposons were identified, most of which are classified as miniature inverted-repeat transposable elements (MITEs). Target mRNAs bearing cleavage tags were found within degradome data sets, implicating miRNA-guided cleavage events. Our data provide evidence supporting the hypothesis that miRNAs can evolve from TEs, at least within the rice genome which contains an abundance of repeat elements. Two main determinants may exapt TE-associated sequences into miRNA pathways- one is the co-opting of OsDCL1 for miRNA biogenesis, while the other is functional association with OsAGO1.
Material and Methods
The repeat-masked rice genome and cDNA sequences were from the Rice Genome Annotation Project (RGAP, release 7, http://rice.plantbiology.msu.edu). miRNA sequences of Oryza sativa were downloaded from the miRBase database (release 18, http://microrna.sanger.ac.uk). Prediction of miRNA targets was conducted using the psRNATarget web interface with default parameters (Dai and Zhao 2011). Rice sRNA data sets used in this study included GSE18251 (for OsAGO1a, OsAGO1b, and OsAGO1c IPs) (Wu et al. 2009), GSE20748 (for OsDCL1IR-2, Osdcl3a-17 mutants) (Wu et al. 2010), and GSE22763 (for the Osdcl4-1 mutant) (Song et al. 2012). The rice degradome data sets analyzed were GSE18248 (Wu et al. 2009), GSE19050 (Zhou et al. 2010), and GSE17398 (Li et al. 2010).
Raw reads of sRNAs and degradome data were downloaded from the Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra). Adaptor sequences were removed by in-house Perl scripts. Sequence alignment to rice genome/transcriptome was performed with the Bowtie program (parameters: -a -v 0 --best --strata)(Langmead et al. 2009). The abundance of sRNAs or degradome sequence tags which matched more than one locus in the genome was repeat-normalized by dividing the number of reads with copy numbers. The normalized values are in the units of transcripts per ten million (TP10M) for sRNAs, or reads per ten million (RP10M) for degradome tags. By this method, we minimized the over-estimation for abundance of rasiRNAs. Degradome data sets analysis was performed as previously described (Addo-Quaye et al. 2008, German et al. 2008).
To look for novel miRNA-like hairpins, sRNA signatures of 17 nt from Massively Parallel Signature Sequencing database (MPSS) (Nobuta et al. 2007) were mapped to rice ESTs (www.ncbi.nlm.nih.gov) using the Bowtie program, demanding 100% sequence identity between them. The EST sequences (e.g. AU055776 from 260 to 508 nt) that contained predominant sRNA signatures were chosen for further analysis. Secondary structures were folded by the RNAfold program (Mathews et al. 1999) (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). Sequence alignment was done by the Clustal W program (Larkin et al. 2007).
For sRNA blots, total RNAs were extracted from 7-day-old rice seedlings (Oryza sativa spp. japonica cv. Nipponbare; available from the USDA-ARS Dale Bumpers National Rice Research Center, Almyra, AR), 3 month-old roots, leaves, stems and mature inflorescence (Stage “Inf 9,” 14–17 cm in length)(Barrera-Figueroa et al. 2012) by single-step method of Guanidinium isothiocyanate (Chomczynski and Sacchi 1987). There were no seeds in the inflorescence samples. The sRNA fraction was isolated from total RNA by precipitation with 1/5 volume of 8M Lithium Chloride. RNA blot analysis was performed as described (Williams et al. 2005, Xie et al. 2004), with 40 μg sRNA for each sample separated on a 17% PAGE-urea gel and a 21 nt DNA marker loaded to estimate sRNA sizes. DNA oligonucleotides were end-labeled with γ-32 P-ATP using T4 polynucleotide kinase and hybridized to the blots. The probe sequences were as follows: 5S rRNA, AGGACTTCCCAGGAGGTCACCC (Szymanski et al. 2002); complement to the most abundant AU055775-originated sRNA sequence, ACGGACGGTCAAACGTTAGAC.
Results
Some rasiRNAs are associated with OsDCL1 and OsAGO1s
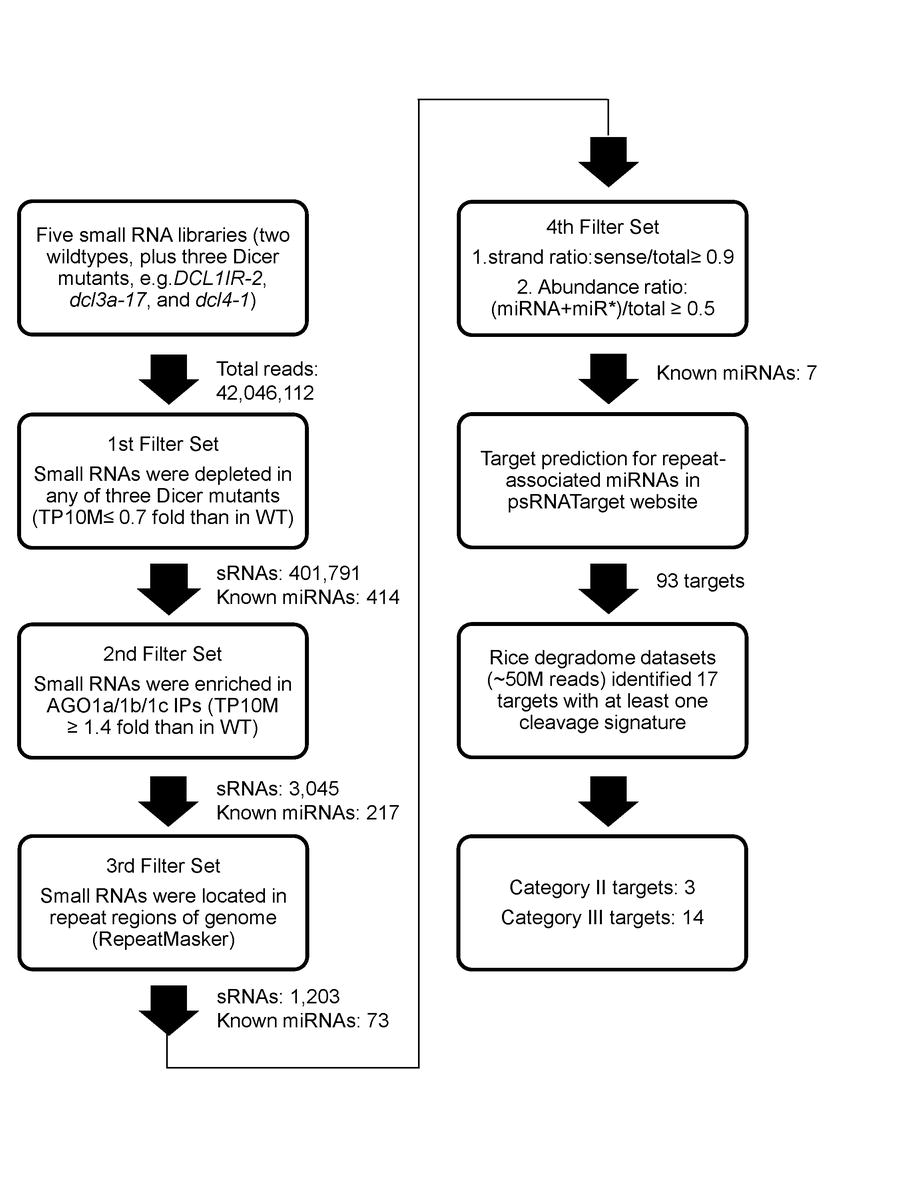
We took a systematic and unbiased approach for representing sRNA abundances from deep sequencing datasets (see “Methods” for details). Each unique sRNA was individually analyzed in different deep sequencing samples for relative enrichment or depletion. They were filtered for reduced relative abundance (≤ 0.75-fold) in sRNA biogenesis mutants OsDCL1IR-2, Osdcl3a-17, or Osdcl4-1 compared to corresponding wild-type controls. The abundance of 401,791 unique sRNAs appeared dependent upon altered function of at least one of the tested OsDCLs (Electronic Supplemental Material Figure 1, ESM Fig.1). At least one sequence read for sRNAs associated with precursor hairpins from 414 OsMIRNA genes, covering ~71% of the rice miRNAs listed in miRBase (release 18, data not shown).
Figure 1.

Target plots of predicted targets for repeat-associated miRNAs. Publically available rice degradome data were pooled together. The abundance of each sequencing “tag” was plotted as a function of its position on miRNA target transcript. Normalized abundance was presented (reads per ten million, RP10M). The “tags” matching ±1 positions of the expected miRNA cleavage site were combined and are shown as red vertical bars.
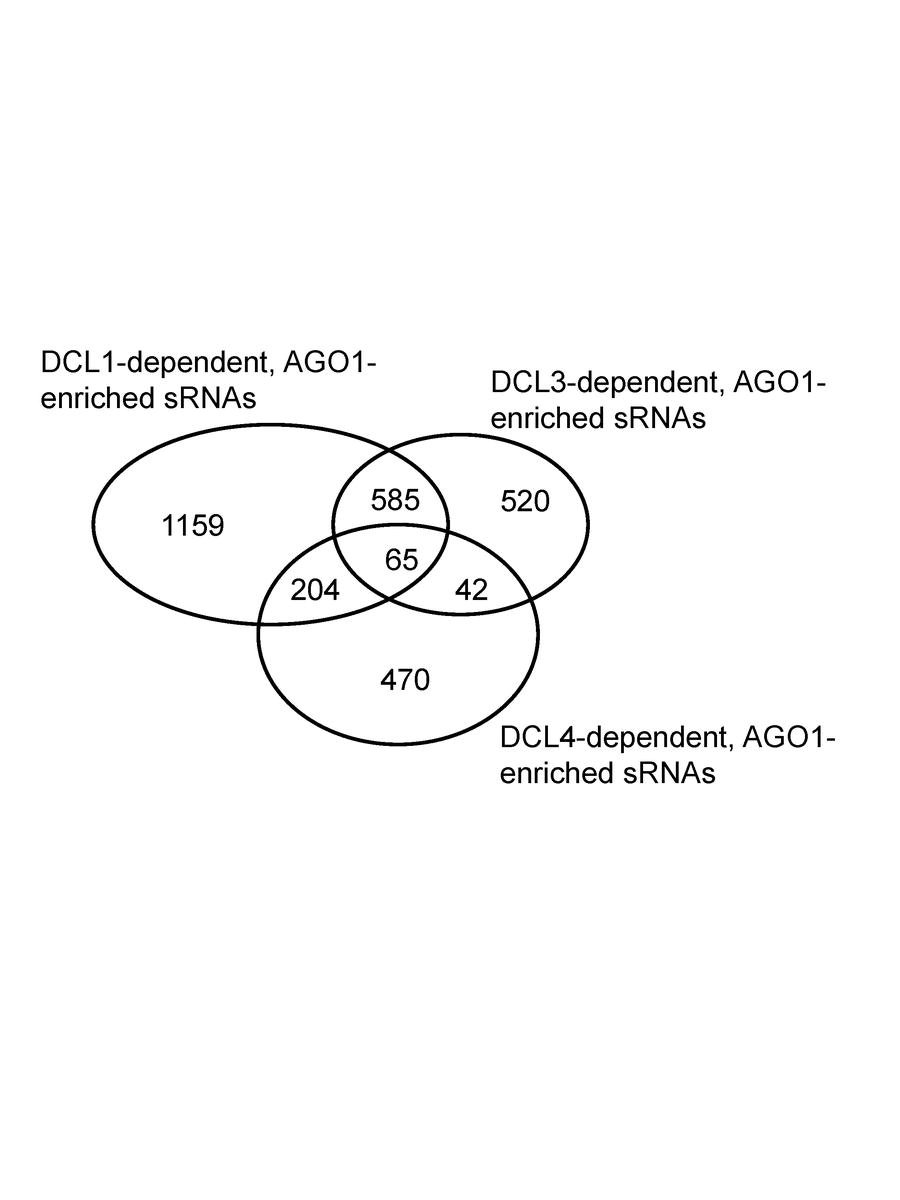
For the second filter, we required the preferential association of sRNAs with OsAGO1a–c (ESM Fig.1). 3,045 unique sRNAs were found to display an increased abundance in OsAGO1 IPs (≥1.4-fold) relative to total cell extract. 217 miRNA hairpins (representing 37% of all 591 OsMIRNAs) had mapped reads, a stringent criterion, with only ~1/3 of known miRNAs passing this filter. Among OsAGO1-enriched sRNAs, 1,159 were exclusively dependent upon normal OsDCL1 function, 520 sRNAs uniquely dependent upon wildtype OsDCL3 and 470 sRNAs associated only with normal OsDCL4 activity (ESM Fig.2). This suggests that OsDCL1 is the predominant DCLfor the production of functional sRNAs that are loaded into OsAGO1 complexes.
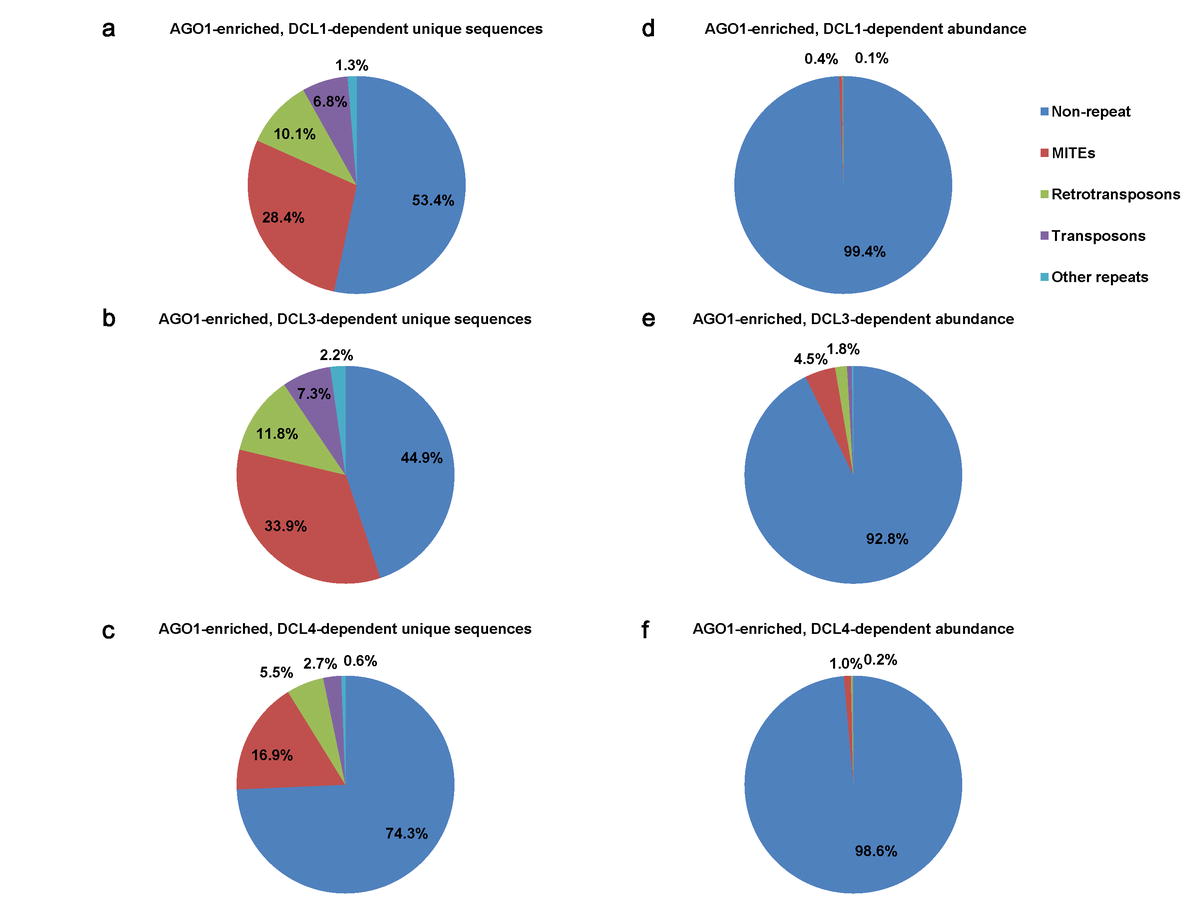
To check if OsDCL-dependent, OsAGO1-enriched sRNAs have origins in repeat sequences, we searched for associations with the rice genome repeat annotations (ESM Fig.1, the 3rd filter). The majority of DCL/AGO1- associated sRNAs were located in intergenic regions without apparent homology to repeats (namely “non-repeat”, ESM Fig.3). However, a large percentage (1,203/3,045 = 39.5% on average) of the unique sRNAs were found to be associated with repeats, among which 73 miRNA hairpins were represented (ESM Fig.1). There was a similar pattern for rasiRNA populations and abundance in each of the OsDCL datasets. MITEs were a major source for rasiRNAs, accounting for 17%–34% of unique sequences and 0.4%–4.5% of total abundance (ESM Fig.3). The particular enrichment of MITE-derived siRNAs is consistent with the genomic abundance of MITEs in rice (Jiang et al. 2004).
A handful of typical miRNAs are related to TEs
The 73 miRNA hairpins with repeat reads were examined for strand and abundance ratios to distinguish between siRNA-like miRNAs and typical miRNAs showing expression evidence that meets community standards for annotation (Jeong et al. 2011)(ESM Fig.1, the 4th filter). For typical miRNAs, the sRNAs derived from the miRNA encoding strand of the stem-loop should make up 90% or more of the total reads associated with the hairpin structure (strand ratio ≥ 0.9). In addition, the ratio of abundance between annotated miRNA:miRNA* duplex and all sRNAs matching the corresponding MIRNA locus (abundance ratio) should be ≥ 0.5. Seven miRNA hairpins with 13 unique sRNAs passed these filters (Table 1). From wild-type and OsAGO1 IP datasets, most of these hairpins had miRNA:miRNA* duplex abundance and strand ratios approaching unity and thus appeared as typical miRNAs, consistent with a recent analysis of rice sRNAs (Jeong et al. 2011).
Table 1.
Repeat-associated miRNAs from OsAGO1s-enriched, OsDCLs-dependent datasets.
| MIRNA | Enriched sequencea | Abundance (TP10M)b | Positionc | Size (nt) | Repeat class/familyi | Matching repeat | DCL dependency |
|---|---|---|---|---|---|---|---|
| MIR435 | TATCCGGTATTGGAGTTGAGG | 1268.1 | Mature | 21 | DNA/MULE-MuDR | Mermiteh | DCL1 |
| TTATCCGGTATTGGAGTTGA | 323.4 | Mature | 20 | DCL1/4 | |||
| MIR1850 | CCAAATTCCCAACTTTTCATC | 87.1 | Star | 21 | DNA | Unique | DCL3 |
| TTAGTTCACATCAATCTTCCT | 107.8 | Otherd | 21 | DCL4 | |||
| MIR1862d,e | TTTGTTTATTTTGGGACGGAG | 27.7 | Othere | 21h | DNA/TcMar-Stowaway | Stowaway47 | DCL4 |
| MIR1867 | TAGGACAGAGGGAGTAGATGT | 25.4 | Otherf | 21h | DNA/CMC-EnSpm | EnSpm-11 | DCL1 |
| TTTTTCTAGGACAGAGGGAGT | 125.1 | Otherg | 21h | DCL1/4 | |||
| MIR1868 | TACTTCCTCGTTTTCCGTAAA | 48.6 | Star | 21h | DNA/PIF-Harbinger | Wanderer | DCL3 |
| MIR1884b | TATGACGCTGTTGACTTTT | 7.7 | Otherd | 19h | DNA/TcMar-Stowaway | Stowaway2 | DCL1/3 |
| TATGACGCTGTTGACTTTTAGA | 62.3 | Otherd | 22h | DCL1 | |||
| TATGACGCTGTTGACTTTTA | 160.7 | Otherd | 20h | DCL3 | |||
| TATGACGCTGTTGACTTTTAG | 1515.7 | Otherd | 21h | DCL3/4 | |||
| MIR2872 | TTCGGTTTGTAGAATACCATC | 23.1 | Star | 21 | LTR/Gypsy | SZ-42_LTR | DCL1 |
Enriched sequences for each miRNA precursor in the pooled AGO1 datasets. Datasets include GSE18251 (Wu et al. 2009), GSE20748 (Wu et al. 2010), and GSE22763 (Song et al. 2012);
The normalized abundance (transcript per ten million, TP10M) in the pooled AGO1 datasets;
Mapping positions of each sequence on miRNA hairpin (mature: ±2nt of mature miRNA; star: ±2nt of miRNA*; other: any position other than mature and star);
5' end of these species were +4 nt off comparing to annotated mature miRNAs in miRBase;
5' end of these species were +6 nt off comparing to annotated mature miRNAs in miRBase;
5' end of these species were +9 nt off comparing to annotated mature miRNAs in miRBase;
5' end of these species were +3 nt off comparing to annotated mature miRNAs in miRBase;
Annotated mature miRNAs in miRBase are 24-nt long;
DNA, DNA transposons; TcM-Stowaway: parallel homologous groups Tourist class Mariner and Stowaway MITES; LTR, long terminal repeat retrotransposons.
The repeat-associated miRNAs found in our analysis are mostly ~21-nt long and start with a uridine at their 5'ends (Table 1), characteristics of typical miRNAs (Wu et al. 2009). This indicates the specificity of our custom filters to look for miRNAs originated from repeats. However, Wu and colleagues showed that miR435 and miR1884 were enriched by OsAGO1s IP, while miR1850, miR1862, miR1867, miR1868, and miR2872 were depleted from AGO1 IP samples using the annotated sequences for these 24-nt long miRNAs in miRBase (Wu et al. 2009). Since our filters were independent of current miRNA annotation, we were able to find that ~21-nt variants for miR1867 and miR1884b were actually enriched in OsAGO1 IP (Table 1). Their 5' ends were 3–4 nt offset downstream compared to the annotated cognate miRNAs. 5' end heterogeneity was also observed for other repeat-associated 21-nt miRNAs (4–9 nt offset). In line with this, probes against miR1862, miR1867 and miR1884b detected signals for 21-nt sRNA species on Northern blots, which were enriched in OsAGO1a/b IP samples (Wu et al. 2010). We also found that miR1850*, miR1868*, and miR2872* were specifically associated with OsAGO1a/b/c, instead of their corresponding mature miRNAs. Together, our data shows that repeat-associated miRNAs can produce 21-nt species with 5'-uridine and associate with OsAGO1s. It strongly suggests that they may function as typical miRNAs.
The repeat homologies for miRNAs found in this study are listed in Table 1. Of particular interest is the finding that six of the seven miRNAs possess significant homology with DNA transposons predicted by RepeatMasker. Among them, three have similarities with the short nonautonomous DNA-type TEs known as MITEs, specifically, miR1862d/e, miR1868, and miR1884b hairpins. This observation is generally in agreement with a recent report showing MITEs may be rich sources for miRNA origins (Barrera-Figueroa et al. 2012). These TE-derived miRNAs are mainly dependent on DCL1 for biogenesis (Table 1), however, OsDCL4 may also contribute to their production (Table I). The co-opting of DCL4 for miRNA processing, with an eventual shift to the use of DCL1 may facilitate the divergence of these hairpins from siRNA processing to a more typical miRNA pathway (Rajagopalan et al. 2006). Interestingly, some TE-derived miRNA sequences appear to depend on DCL3 (Table 1). The biological significance of this is unclear, but it has been reported that generation of MITE-derived siRNAs in the Solanaceae depend on DCL3 and DCL4 (Kuang et al. 2009).
Degradome datasets reveal targets for some TE-associated miRNAs
We searched for predicted targets of TE-associated miRNAs in the psRNATarget web interface (Dai and Zhao 2011) and found 93 targets (ESM Table 1). We then obtained ~50 million reads of all available rice degradome datasets from NCBI GEO databases (see “Methods”) to look for cleavage signatures. Of the 93 predicted targets, 17 had at least one sequencing tag carrying a 5' end that was precisely opposite the 10th nucleotide of the cognate miRNA, a characteristic of miRNA-mediated cleavage (ESM Table 2). As a control, diagnostic cleavage signatures were found for most validated miRNA targets (data not shown). According to previous reports (Addo-Quaye et al. 2008), miRNA targets can be grouped into three categories based on degradome datasets. For Category I targets, the sum of their degradome tag abundances from positions nine to eleven is higher than any other cleavage tag mapping to the transcript. Category II targets have degradome tag abundances on positions nine to eleven in the top 1/3 rank compared to any other tags on the target mRNAs. Category III targets are those which do not fall into the above two categories. Among the 17 candidate targets with cleavage tags for TE-associated miRNAs, three were Category II targets (Fig. 1a–c, see below), while the rest were Category III targets (Fig. 1, data not shown).
Examples of target degradome plots for TE-like miRNAs listed in Table 1 are presented in Figure 1. miR1850.1* has extensive complementarity with mRNA from Os09g26530.1, a gene of unknown function (ESM Table 1, Fig.1a). One of the predicted targets for 21-nt miR1867 variants is Os01g67370.2 encoding a pentatricopeptide repeat-containing protein (Fig. 1b). There is a G:A mismatch on position nine of this target:miRNA duplex, which suggests translational repression as a possible mode of action for this miRNA (Brodersen et al. 2008, Lanet et al. 2009). Nevertheless, significant degradome tags were found on positions nine to eleven of the Os01g67370.2 mRNA:miR1867 duplex. The Os01g67370.2 degradome tag abundances were in the top 1/3 rank compared to other tag abundances within the transcript, suggesting dominant cleavage events (Addo-Quaye et al. 2008). Os12g16350.13 transcripts (encoding an enoyl-CoA hydratase/isomerase family member) are predicted as a target for 21-nt miR1884b variants (Fig. 1c). We also found Os06g48240.1 as a Category III target for this miRNA, consistent with a recent report (Barrera-Figueroa et al. 2012). miR1867-other (a sRNA with +9 nt offset to mature miR1867, see Table 1) shares long complementary sequences with Os02g31100.1 transcripts encoding a lipopolysaccharide-induced tumor necrosis alpha factor (LITAF) domain-containing protein (Fig. 1d).
Interestingly, the complementary sites of the 21-nt miR1884b variants appear to be located within un-translated regions (UTRs) (ESM Table 2, data not shown). Predicted target sites within Os09g30454.1 and Os11g34460.2 mRNAs are in their 5' UTRs, while six other candidate targets have putative miRNA binding sites within their 3' UTRs. These miRNA complementary sites show extensive sequence homology with the reverse complement strands of both the miR1884b precursor and one of its closely related MITEs, ORSgTEMT01600413 (Fig. 2). This observation suggests that MIR1884b is recently evolved and its cognate UTR targets are likely derived from the same MITEs that have transposed to the UTRs. Consistent with this notion, miR1884b has only been described in rice (Zhu et al. 2008) and it also shows significant homology to Sorghum bicolor TE candystripe1 (p = 5×10−9; Plant Repeat Database, http://rice.plantbiology.msu.edu). Some siRNA-like miRNA precursors with homology to TEs also have long complementarities within 3' UTRs of their predicted targets (data not shown). Thus, we propose that transposition of TEs into UTR regions could subject their host genes to PTGS regulated by miRNAs and/or rasiRNAs generated from the same TE ancestors.
Figure 2.

Alignment of predicted targets in UTRs for 21-nt miR1884b variants and putative MITE sequence ORSgTEMT01600413. 100 nt upstream and downstream sequences flanking miRNA complementary sites in targets' 5' or 3' UTRs were extracted from rice reference genome and aligned with ClustalW (Larkin et al. 2007). The consensus nucleotides (> 90% identity) are indicated by boxes. miRNA complementary target sites are underlined. The reverse complement sequences of 5' half for the MITE ORSgTEMT01600413 is shown. 50 nt upstream genomic sequences are added in order to have equal length with miR1884b targets.
To test our hypothesis of TE-driven miRNA/siRNA evolution, we looked for novel hairpin-like sequences with extended homology to miR1884b by a de novo expression-based search. 17-nt Massively Parallel Signature Sequencing (MPSS) sRNAs from rice (Nobuta et al. 2007) were mapped to rice ESTs in GenBank that could fold into predicted hairpin structures. One EST (AU055776) mapped to the 3'-UTR of Os03g0227400 encoding a hypothetical glycoside hydrolase 17-like protein missing 152 amino acids from the N-terminus. The gene has a weak Kozak consensus at the next downstream AUG and a higher Ka/Ks (non-synonymous/synonymous substitutions) ratio (=0.53)(http://services.cbu.uib.no/tools/kaks) than that of the conserved domain of nearest homologues Os03g0722500 (=0.48) and At5g42720 (=0.36), suggesting it may be a recent pseudogene (Tanaka et al. 2008; Naito et al. 2009). The EST had abundant sense sRNAs mapping to the 5' arm of the predicted hairpin structure. Some other sRNAs with fewer reads were at the complementary position on the 3' arm, suggestive of miRNA:miRNA*-like duplexes (Fig. 3a). This differs from TE-derived sRNAs that are typically tiled along the full length of the source RNA. We performed an RNA blot analysis for the most abundant sRNA (from MPSS data) that matches AU055776. A strong 24-nt signal was found in roots, with moderate levels seen in leaves and seedlings, and weak or barely detectable levels found in stems and inflorescences, respectively (Fig. 3b). We were unable to detect a signal for 21-nt species, probably due to a low 21 nt to 24 nt ratios as reported for RNA blots of miR1862, miR1867, and miR1884b (Wu et al. 2010) (see Table I). Since we can only infer sRNA sequence from 17-nt long MPSS tags, the exact sRNA lengths generated from AU055776 and their expression levels is not clear. The reverse complement of EST AU055776 has strong homology with the TE-like miRNA hairpins miR806a, miR812a, miR818e and the reverse complement of miR1884 (Fig. 3c). Note that miR806a, miR812a and miR818e may represent siRNA-like miRNAs because they did not pass the filters for bona fide miRNAs (our data here and (Jeong et al. 2011)). Taken together, the finding of root-specific siRNAs for an EST with extended homology to miR1884 and numerous other TE-like miRNAs supports the hypothesis that TE may provide raw material for miRNA evolution.
Figure 3.

Analysis of TE-derived miRNA hairpin-like EST, AU055776. (a) hairpin structure for AU055776 predicted by the MFOLD program. Color-coded horizontal lines show the positions and abundances of MPSS sRNA signatures. TPQ, transcript per quarter million. (b) RNA blot for the most abundant sRNA from AU055776 inferred from MPSS data. 5S rRNA was used as loading control. A 21 nt DNA oligonucleotide marker (left lane) was run to estimate sRNA size. (c) Sequence alignment of AU055776 to cognate miRNA hairpins from miRBase. The ClustalW program was used for alignment. The reverse complement strands of AU055776 and miR1884 precursors are shown. The consensus nucleotides (> 90% identity) are indicated by boxes. Locations of annotated miRNAs are underlined for miR806a, miR812a, miR818e and miR1884a. For miR1884b, 21-nt variant found in this study is underlined. For AU055776, the probe sequence is underlined.
Discussion
The authenticity of some plant miRNAs has been questioned on the ground that they are repeat-related (Lu et al. 2008, Sunkar et al. 2008, Xue et al. 2009). However, these conclusions are based solely upon sRNA from total cell extracts (Li et al. 2011). Barrera-Figueroa et al. reported that 80 candidate miRNAs originated from repeats or TEs, including 24-nt miR1867 and miR1884 species (Barrera-Figueroa et al. 2012). Nevertheless, their analysis also relied on current annotation of mature miRNA and miRNA* position on hairpins. Our approach compared individual sRNA sequences across samples, independent of miRNA annotation and thus gives a more comprehensive and unbiased view of TE-associated miRNA origins. We focused our analysis on the relationships between TE-associated miRNA production and DCL gene function and on miRNAs that are associated with AGO1, both major specificity determinants for miRNA function. Our analysis identified a handful of repeat-related sequences that satisfy all functional criteria for typical miRNAs as described recently (Jeong et al. 2011). Intriguingly, we found that in most cases OsAGO1 IPs were enriched in miRNA variants that are offset from the corresponding annotated miRNA species by a few nucleotide at their 5' ends (Table 1). These offsets generated ~21-nt variants with a 5'-uridine (e.g. miR1867 and miR1884b), the sRNA species invariantly loaded into functional OsAGO1 (Wu et al. 2009). Consistent with a functional role for the miR1850* sRNAs that were found to be enriched in OsAGO1 IPs (Table 1), miR1850*-targeted cleavage products were identified within degradome datasets (Fig. 1). Altogether, our data support the hypothesis that functional miRNAs produced and processed similarly to typical miRNAs can originate from TEs. It is noteworthy that miR1884b has been removed from the current release of miRBase (release 19) because its pattern of reads was apparently more consistent with rasiRNAs than miRNAs, based on results from two deep sequencing experiments in rice embryogenic calli (Chen et al. 2011). Nevertheless, both our analysis and another group's data (Jeong et al. 2011) support miR1884b-3p and the 21 nt variant species (Table 1) as bona fide miRNAs. More importantly, we found functional evidence from degradome data for miR1884b activity. Thus, we argue that miR1884b should receive re-annotation in future releases of miRBase.
TEs may be one of the sources of repeats that drive the evolution of miRNAs. Several reports have provided evidence for this hypothesis in both plants and animals (Cai et al. 2012, Piriyapongsa and Jordan 2007, Piriyapongsa and Jordan 2008, Smalheiser and Torvik 2005). We report that several typical miRNA hairpins like miR1884b share significant similarities with TEs (Table 1). Interestingly, MITEs appears to have transposed into several UTRs, producing potential miR1884b target sites (Fig. 2). Extended sequence homology was found between the reverse complement strands of miR1884, miR806a/812a/818e precursors and their closely related MITEs, and miRNA target sites (Figs. 2 & 3c). Our data supports a relationship of convergent evolution for TE-like miRNAs and their target sites. Our findings are also consistent with the hypothesis that expressed TEs provide raw materials for RNA-hairpin formation, and for a subsequent siRNA-miRNA transition through sequence divergence. A model of dual-coding TEs has been proposed in which some TEs may encode both siRNAs and miRNAs as intermediates in the evolution of MIRNA genes (Piriyapongsa and Jordan 2008). However, many of the proposed dual-coding MIRNA genes produce mature miRNAs at a very low level, and they do not pass the thresholds of strand and abundance ratios associated with typical miRNAs (Jeong et al. 2011). This is consistent with a model for miRNA evolution that many newly evolved miRNAs undergo frequent birth and death, and such species may have no targets (Fahlgren et al. 2007, Rajagopalan et al. 2006). In other cases, sequence divergence may introduce bulges and/or gaps into hairpin structures. Such conformational changes may facilitate the processing of precursor RNAs by DCL1 in competition with DCL3 and/or DCL4. Subsequent linkage to target mRNAs by incorporation of these TE-associated miRNAs into AGO1 could finally provide the positive selection for their establishment into typical miRNA pathways.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Electronic Supplemental Material Figure 1. Pipeline for the identification of repeatassociated miRNAs from sRNA libraries and their targets from degradome data sets. Repeatassociated miRNAs were identified by a set of filters presented in the diagram and described in the text. Numbers of sRNAs and known miRNAs after each filter are shown.
Electronic Supplemental Material Figure 2. Venn diagram of OsAGO1-enriched sRNAs from different OsDCLs data sets. Numbers represent unique sRNA sequences from OsDCL1, OsDCL3, and OsDCL4, respectively, as well as shared sRNAs between sets.
Electronic Supplemental Material Figure 3. Comparison of repeat origins for sRNAs enriched in OsAGO1 IPs, while depleted in different OsDCLs.
Acknowledgements
The authors thank the High Performance Computing Center at Texas Tech University for use of the supercomputer cluster Hrothgar, Dr. Zhixin Xie for providing rice seeds, and Dr. Jeff Velten for critical reading of the manuscript. This work was supported by the National Institutes of Health grant GM077245 to C.D.R, and a Texas Tech University AT&T Chancellor's Fellowship to F.O-Y. The funders had no role in the study design, in the collection, analysis, or interpretation of data, or in the writing of the manuscript or decision to submit the manuscript for publication.
Reference
- Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol. 2008;18:758–762. doi: 10.1016/j.cub.2008.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen E, Xie Z, Gustafson AM, Sung GH, Spatafora JW, Carrington JC. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet. 2004;36:1282–1290. doi: 10.1038/ng1478. [DOI] [PubMed] [Google Scholar]
- Barrera-Figueroa BE, Gao L, Wu Z, Zhou X, Zhu J, Jin H, Liu R, Zhu JK. High throughput sequencing reveals novel and abiotic stress-regulated microRNAs in the inflorescences of rice. BMC Plant Biol. 2012;12:132. doi: 10.1186/1471-2229-12-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O. Widespread translational inhibition by plant miRNAs and siRNAs. Science. 2008;320:1185–1190. doi: 10.1126/science.1159151. [DOI] [PubMed] [Google Scholar]
- Cai Y, Zhou Q, Yu C, Wang X, Hu S, Yu J, Yu X. Transposable-element associated small RNAs in Bombyx mori genome. PLoS ONE. 2012;7:e36599. doi: 10.1371/journal.pone.0036599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CJ, Liu Q, Zhang YC, Qu LH, Chen YQ, Gautheret D. Genome-wide discovery and analysis of microRNAs and other small RNAs from rice embryogenic callus. RNA Biol. 2011;8:538–547. doi: 10.4161/rna.8.3.15199. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanatephenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Cuperus JT, Fahlgren N, Carrington JC. Evolution and functional diversification of MIRNA genes. Plant Cell. 2011;23:431–442. doi: 10.1105/tpc.110.082784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39:W155–159. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, Carrington JC. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS ONE. 2007;2:e219. doi: 10.1371/journal.pone.0000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felippes FF, Schneeberger K, Dezulian T, Huson DH, Weigel D. Evolution of Arabidopsis thaliana microRNAs from random sequences. RNA. 2008;14:2455–2459. doi: 10.1261/rna.1149408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, De Paoli E, Lu C, Schroth G, Meyers BC, Green PJ. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol. 2008;26:941–946. doi: 10.1038/nbt1417. [DOI] [PubMed] [Google Scholar]
- Jeong DH, Park S, Zhai J, Gurazada SG, De Paoli E, Meyers BC, Green PJ. Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell. 2011;23:4185–4207. doi: 10.1105/tpc.111.089045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N, Feschotte C, Zhang X, Wessler SR. Using rice to understand the origin and amplification of miniature inverted repeat transposable elements (MITEs) Curr Opin Plant Biol. 2004;7:115–119. doi: 10.1016/j.pbi.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Kuang H, Padmanabhan C, Li F, Kamei A, Bhaskar PB, Ouyang S, Jiang J, Buell CR, Baker B. Identification of miniature inverted-repeat transposable elements (MITEs) and biogenesis of their siRNAs in the Solanaceae: new functional implications for MITEs. Genome Res. 2009;19:42–56. doi: 10.1101/gr.078196.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanet E, Delannoy E, Sormani R, Floris M, Brodersen P, Crete P, Voinnet O, Robaglia C. Biochemical evidence for translational repression by Arabidopsis microRNAs. Plant Cell. 2009;21:1762–1768. doi: 10.1105/tpc.108.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Le Trionnaire G, Grant-Downton RT, Kourmpetli S, Dickinson HG, Twell D. Small RNA activity and function in angiosperm gametophytes. J Exp Bot. 2011;62:1601–1610. doi: 10.1093/jxb/erq399. [DOI] [PubMed] [Google Scholar]
- Li Y, Li C, Xia J, Jin Y. Domestication of transposable elements into microRNA genes in plants. PLoS ONE. 2011;6:e19212. doi: 10.1371/journal.pone.0019212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YF, Zheng Y, Addo-Quaye C, Zhang L, Saini A, Jagadeeswaran G, Axtell MJ, Zhang W, Sunkar R. Transcriptome-wide identification of microRNA targets in rice. Plant J. 2010;62:742–759. doi: 10.1111/j.1365-313X.2010.04187.x. [DOI] [PubMed] [Google Scholar]
- Lisch D. How important are transposons for plant evolution? Nat Rev Genet. 2012;14:49–61. doi: 10.1038/nrg3374. [DOI] [PubMed] [Google Scholar]
- Lu C, Jeong DH, Kulkarni K, Pillay M, Nobuta K, German R, Thatcher SR, Maher C, Zhang L, Ware D, Liu B, Cao X, Meyers BC, Green PJ. Genome-wide analysis for discovery of rice microRNAs reveals natural antisense microRNAs (nat-miRNAs) Proc Natl Acad Sci U S A. 2008;105:4951–4956. doi: 10.1073/pnas.0708743105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory A, Vaucheret H. Form, function, and regulation of ARGONAUTE proteins. Plant Cell. 2010;22:3879–3889. doi: 10.1105/tpc.110.080671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- McCue AD, Slotkin RK. Transposable element small RNAs as regulators of gene expression. Trends Genet. 2012;28:616–623. doi: 10.1016/j.tig.2012.09.001. [DOI] [PubMed] [Google Scholar]
- McCue AD, Nuthikattu S, Reeder SH, Slotkin RK. Gene expression and stress response mediated by the epigenetic regulation of a transposable element small RNA. PLoS Genet. 2012;8:e1002474. doi: 10.1371/journal.pgen.1002474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito K, Zhang F, Tsukiyama T, Saito H, Hancock CN, Richardson AO, Okumoto Y, Tanisaka T, Wessler SR. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature. 2009;461:1130–1134. doi: 10.1038/nature08479. [DOI] [PubMed] [Google Scholar]
- Nobuta K, Venu RC, Lu C, Belo A, Vemaraju K, Kulkarni K, Wang W, Pillay M, Green PJ, Wang GL, Meyers BC. An expression atlas of rice mRNAs and small RNAs. Nat Biotech. 2007;25:473–477. doi: 10.1038/nbt1291. [DOI] [PubMed] [Google Scholar]
- Nosaka M, Itoh JI, Nagato Y, Ono A, Ishiwata A, Sato Y. Role of transposon-derived small RNAs in the interplay between genomes and parasitic DNA in rice. PLoS Genet. 2012;8:e1002953. doi: 10.1371/journal.pgen.1002953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmedo-Monfil V, Duran-Figueroa N, Arteaga-Vazquez M, Demesa-Arevalo E, Autran D, Grimanelli D, Slotkin RK, Martienssen RA, Vielle-Calzada JP. Control of female gamete formation by a small RNA pathway in Arabidopsis. Nature. 2010;464:628–632. doi: 10.1038/nature08828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Chun J, Ai T-b, Tong Y-a, Zhang R, Zhao M-m, Chen F, Wang S-h. MicroRNA profiles and their control of male gametophyte development in rice. Plant Mol Biol. 2012;80:85–102. doi: 10.1007/s11103-012-9898-x. [DOI] [PubMed] [Google Scholar]
- Piriyapongsa J, Jordan IK. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS ONE. 2007;2:e203. doi: 10.1371/journal.pone.0000203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piriyapongsa J, Jordan IK. Dual coding of siRNAs and miRNAs by plant transposable elements. RNA. 2008;14:814–821. doi: 10.1261/rna.916708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006;20:3407–3425. doi: 10.1101/gad.1476406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott G, Mari-Ordonez A, Himber C, Alioua A, Voinnet O, Dunoyer P. Differential effects of viral silencing suppressors on siRNA and miRNA loading support the existence of two distinct cellular pools of ARGONAUTE1. EMBO J. 2012;31:2553–2565. doi: 10.1038/emboj.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalheiser NR, Torvik VI. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005;21:322–326. doi: 10.1016/j.tig.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Song X, Li P, Zhai J, Zhou M, Ma L, Liu B, Jeong DH, Nakano M, Cao S, Liu C, Chu C, Wang XJ, Green PJ, Meyers BC, Cao X. Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J. 2012;69:462–474. doi: 10.1111/j.1365-313X.2011.04805.x. [DOI] [PubMed] [Google Scholar]
- Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu JK. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008;8:25. doi: 10.1186/1471-2229-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski M, Barciszewska MZ, Erdmann VA, Barciszewski J. 5S Ribosomal RNA Database. Nucleic Acids Res. 2002;30:176–178. doi: 10.1093/nar/30.1.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Antonio BA, Kikuchi S, Matsumoto T, Nagamura Y, Numa H, Sakai H, Wu J, Itoh T, Sasaki T, Aono R, Fujii Y, Habara T, Harada E, Kanno M, Kawahara Y, Kawashima H, Kubooka H, Matsuya A, Nakaoka H, Saichi N, Sanbonmatsu R, Sato Y, Shinso Y, Suzuki M, Takeda J, Tanino M, Todokoro F, Yamaguchi K, Yamamoto N, Yamasaki C, Imanishi T, Okido T, Tada M, Ikeo K, Tateno Y, Gojobori T, Lin YC, Wei FJ, Hsing YI, Zhao Q, Han B, Kramer MR, McCombie RW, Lonsdale D, O'Donovan CC, Whitfield EJ, Apweiler R, Koyanagi KO, Khurana JP, Raghuvanshi S, Singh NK, Tyagi AK, Haberer G, Fujisawa M, Hosokawa S, Ito Y, Ikawa H, Shibata M, Yamamoto M, Bruskiewich RM, Hoen DR, Bureau TE, Namiki N, Ohyanagi H, Sakai Y, Nobushima S, Sakata K, Barrero RA, Sato Y, Souvorov A, Smith-White B, Tatusova T, An S, An G, OOta S, Fuks G, Fuks G, Messing J, Christie KR, Lieberherr D, Kim H, Zuccolo A, Wing RA, Nobuta K, Green PJ, Lu C, Meyers BC, Chaparro C, Piegu B, Panaud O, Echeverria M. The Rice Annotation Project Database (RAP-DB): 2008 update. Nucleic Acids Res. 2008;36:D1028–D1033. doi: 10.1093/nar/gkm978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136:669–687. doi: 10.1016/j.cell.2009.01.046. [DOI] [PubMed] [Google Scholar]
- Williams L, Carles CC, Osmont KS, Fletcher JC. A database analysis method identifies an endogenous trans-acting short-interfering RNA that targets the Arabidopsis ARF2, ARF3, and ARF4 genes. Proc Natl Acad Sci U S A. 2005;102:9703–9708. doi: 10.1073/pnas.0504029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Zhang Q, Zhou H, Ni F, Wu X, Qi Y. Rice MicroRNA effector complexes and targets. Plant Cell. 2009;21:3421–3435. doi: 10.1105/tpc.109.070938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y. DNA methylation mediated by a microRNA pathway. Mol Cell. 2010;38:465–475. doi: 10.1016/j.molcel.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, Jacobsen SE, Carrington JC. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004;2:E104. doi: 10.1371/journal.pbio.0020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue LJ, Zhang JJ, Xue HW. Characterization and expression profiles of miRNAs in rice seeds. Nucleic Acids Res. 2009;37:916–930. doi: 10.1093/nar/gkn998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Gu L, Li P, Song X, Wei L, Chen Z, Cao X. Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica) Front Biol. 2010;5:67–90. [Google Scholar]
- Zhu QH, Spriggs A, Matthew L, Fan LJ, Kennedy G, Gubler F, Helliwell C. A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res. 2008;18:1456–1465. doi: 10.1101/gr.075572.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Electronic Supplemental Material Figure 1. Pipeline for the identification of repeatassociated miRNAs from sRNA libraries and their targets from degradome data sets. Repeatassociated miRNAs were identified by a set of filters presented in the diagram and described in the text. Numbers of sRNAs and known miRNAs after each filter are shown.
Electronic Supplemental Material Figure 2. Venn diagram of OsAGO1-enriched sRNAs from different OsDCLs data sets. Numbers represent unique sRNA sequences from OsDCL1, OsDCL3, and OsDCL4, respectively, as well as shared sRNAs between sets.
Electronic Supplemental Material Figure 3. Comparison of repeat origins for sRNAs enriched in OsAGO1 IPs, while depleted in different OsDCLs.