

Abstract
Activation of hepatic stellate cells (HSCs) is crucial to the development of fibrosis in nonalcoholic fatty liver disease. Quiescent HSCs contain lipid droplets (LDs), whose depletion upon activation induces a fibrogenic gene program. Here we show that liver fatty acid-binding protein (L-Fabp), an abundant cytosolic protein that modulates fatty acid (FA) metabolism in enterocytes and hepatocytes also modulates HSC FA utilization and in turn regulates the fibrogenic program. L-Fabp expression decreased 10-fold following HSC activation, concomitant with depletion of LDs. Primary HSCs isolated from L-FABP−/− mice contain fewer LDs than wild type (WT) HSCs, and exhibit upregulated expression of genes involved in HSC activation. Adenoviral L-Fabp transduction inhibited activation of passaged WT HSCs and increased both the expression of prolipogenic genes and also augmented intracellular lipid accumulation, including triglyceride and FA, predominantly palmitate. Freshly isolated HSCs from L-FABP−/− mice correspondingly exhibited decreased palmitate in the free FA pool. To investigate whether L-FABP deletion promotes HSC activation in vivo, we fed L-FABP−/− and WT mice a high fat diet supplemented with trans-fatty acids and fructose (TFF). TFF-fed L-FABP−/− mice exhibited reduced hepatic steatosis along with decreased LD abundance and size compared to WT mice. In addition, TFF-fed L-FABP−/− mice exhibited decreased hepatic fibrosis, with reduced expression of fibrogenic genes, compared to WT mice.
Conclusion
L-FABP deletion attenuates both diet-induced hepatic steatosis and fibrogenesis, despite the observation that L-Fabp paradoxically promotes FA and LD accumulation and inhibits HSC activation in vitro. These findings highlight the importance of cell-specific modulation of hepatic lipid metabolism in promoting fibrogenesis in nonalcoholic fatty liver disease.
Keywords: Hepatic fibrosis, lipid droplets, lipogenesis, fructose, trans-fat
Nonalcoholic fatty liver disease (NAFLD) encompasses a spectrum of pathology ranging from simple steatosis to nonalcoholic steatohepatitis (NASH) and cirrhosis (1). Neutral lipid storage in hepatocytes, principally in the form of triglyceride, predisposes individuals to the subsequent development and progression of NASH (2) although much is still poorly understood regarding the metabolic regulation and clinical significance of distinctive storage pools of intrahepatic lipid. Among these intracellular storage compartments, lipid droplets (LDs) have emerged as a focal point of interest (3). LDs are specialized spherical organelles composed of a core of neutral lipids surrounded by proteins known as perilipins (Plins), which play key roles in regulating aspects of intracellular trafficking, signaling and cytoskeletal organization (4). Understanding the pathways that regulate metabolic flux in LDs is likely to provide insight into the mechanisms of lipid-mediated liver injury (5).
Hepatic stellate cells (HSCs) are the major effectors of hepatic fibrogenesis, characterized in their quiescent state by abundant LDs containing predominantly retinyl esters, triglyceride and cholesterol ester along with cholesterol, phospholipids, and fatty acids (FA) (6, 7). In the course of hepatic injury, quiescent HSCs undergo phenotypic changes including enhanced cell proliferation, loss of LDs, expression of α-smooth muscle actin (α-SMA), and excessive production of extracellular matrix (ECM). HSC activation is coupled with a reduction in lipogenesis and diminished expression of prolipogenic transcription factors, (8, 9), which together reflect an ‘anti-adipogenic’ program (10). However, the dynamic relationship between lipid metabolic pathways in hepatocytes and stellate cells is incompletely understood and the elements that regulate LD accumulation upon HSC activation remain obscure (11).
Mammalian intracellular fatty acid-binding proteins (FABPs) comprise a superfamily of lipid-binding proteins (12) involved in the uptake, transport and metabolism of FA and other lipid ligands. Liver Fabp (L-Fabp or Fabp1) is abundantly expressed in both hepatocytes and enterocytes and binds multiple ligands, including saturated FA and cholesterol (12). Germline L-Fabp−/− mice exhibit decreased hepatic triglyceride content (13) with altered FA uptake kinetics. In addition, L-Fabp−/− mice fed a high saturated fat, high cholesterol “Western” diet were protected against diet-induced obesity and hepatic steatosis, likely reflecting altered kinetics of saturated FA utilization (14, 15). Proteomic screens revealed L-Fabp to be overexpressed in obese subjects with simple steatosis, along with paradoxically decreased expression in the progressive versus mild forms of NASH (16).
Recent studies have validated new models of diet-induced NAFLD with fibrosis in murine models (17, 18), setting the stage for formal exploration of the role of candidate genes in the progressive forms of murine NAFLD. Here we explore a role for L-Fabp in lipid metabolism in both hepatocytes and stellate cells and report the impact of L-Fabp deletion in diet-induced HSC activation and hepatic fibrosis in vivo.
EXPERIMENTAL PROCEDURES
Animal Studies
C57BL/6J mice (Jackson Laboratory, Bar Harbor, MA) and congenic L-Fabp−/− mice (19) were used in all studies (see also Supplemental Methods). Hepatic steatosis with fibrosis was induced by feeding female mice a high trans-fat diet supplemented with high fructose corn syrup, modified from (18).
Isolation and culture of HSCs
HSCs were isolated by pronase-collagenase perfusion and density gradient centrifugation, with >90% purity (20). For lipidomics analysis, isolated HSC were subjected to a second gradient purification and frozen immediately at −80°C. Details of protein, immunohistochemical and lipidomics analyses are provided in Supplemental Methods. Conditions for culture and adenoviral transduction of passaged HSCs are detailed in Supplemental Methods.
Gene Expression Analysis
Total RNA was prepared and analyzed by Real Time PCR as previously described using primer pairs detailed in Supplemental Table 1
Statistical Analysis
Data are presented as mean ± standard error (SE) unless otherwise noted. Differences between means were evaluated using an unpaired two-sided Student’s t test (p<0.05 considered as significant, Microsoft Excel). Where appropriate, comparisons of multiple treatment conditions with controls were analyzed by ANOVA with the Dunnett’s test for post hoc analysis.
RESULTS
L-Fabp is expressed in quiescent HSCs and decreases upon activation
Surveying mRNA expression of Fabp family members in quiescent (day 1) and activated (day 7) primary mouse HSCs revealed L-Fabp to be the most abundantly expressed member and, unlike other Fabp family members (Figure 1A), decreased by >90% upon HSC activation, with a gradual decline in L-Fabp mRNA (Figure 1B) and protein (Figure 1C) abundance from 3 to 7 days of culture.. Freshly isolated HSCs from wild-type mice manifest abundant (oil-red-O staining) LDs, as expected (Figure 1D). By contrast, intracellular LDs were less abundant in freshly isolated HSCs from L-Fabp−/− mice (day 1) and almost undetectable by day 3. We also examined mRNA abundance of α-SMA and α(I)I collagen (αI(I)Col), as representative markers of HSC activation (8, 21). These mRNAs were barely detectable in freshly isolated HSCs from wild type mice, increasing ~5 fold after culture (Figure 1E, left panel), as expected (21). By contrast, expression of α-SMA and αI(I)Col mRNAs were readily detected in HSCs from L-Fabp−/− mice after one day of culture (Figure 1E, right panel), with continued upregulation after 7 days. Taken together, these findings suggest that L-Fabp may play a role in LD accumulation and activation of HSCs in vitro.
Figure 1. L-Fabp is expressed in quiescent HSCs and decreases upon activation.

(A) Relative expression of Fabp family member mRNAs in freshly isolated (Day 1) and activated (Day 7) C57BL/6J HSCs. Data are generated from pooled RNAs and are expressed relative to Fabp3 expression in Day 1 HSCs. Fabp3 was chosen because its expression does not change upon HSC activation. (B) Relative expression of L-Fabp mRNA in C57BL/6J HSCs after 1, 3 or 7 days of culture. Values are presented as mRNA fold change vs expression in Day 1 cells (mean ± standard deviation (SD), n=3). Asterisk indicates p<0.05 vs. Day 1. (C) Isolated C57BL/6J HSCs were cultured for 1–7 days and subjected to immunohistochemistry with antibodies to α-smooth muscle actin (α-SMA) and L-Fabp to monitor protein expression. Nuclei were visualized with DAPI staining. (D) HSCs were isolated from wild-type C57BL/6J and L-Fabp−/− mice and stained with Oil Red O after 1–3 days of culture to visualize neutral lipid droplets. Representative images from 10 views in three independent experiments are presented. (E) Real-time PCR quantitation of α-SMA and α(I)ICol mRNA in C57BL/6J and L-Fabp−/− HSCs after 1 or 7 days of culture. Values are presented as mRNA fold changes versus the level of expression of α-SMA or α(I)ICol in L-Fabp−/− HSC after one day of culture (mean ± SD, n=3), as levels of these mRNAs in were barely detectable Day 1 HSC from C57BL/6J mice. *p<0.05 vs. Day 1 L-Fabp−/− HSCs.
Ad-L-Fabp transduction augments TG and FA content in HSCs in vitro
Based on the coupled observations of a decline in L-Fabp expression with decreased lipid accumulation and increased activation of HSCs, we asked whether forced expression of L-Fabp would modulate lipid content and the patterns of FA utilization. Ad-L-Fabp transduction of cultured HSCs (Figure 2A) increased both cellular FA and TG content (Figure 2B–C) and revealed enrichment with palmitic acid (C16:0) as the major FA species (Figure 2D). These findings suggest that rescuing L-Fabp expression in cultured HSCs reverses lipid depletion and leads to enrichment in 16:0 FA. In line with these findings, the FA profile in freshly isolated HSCs from L-Fabp−/− mice revealed depletion of 16:0 with a shift to 18:0, 18:1 and 18:2 species in the free FA pool (Figure 2E). HSC triglyceride species, however, were comparable between the genotypes (Figure 2F). These findings demonstrate corresponding gain- and loss-of-function effects of L-Fabp on the FA profile in passaged HSCs transduced with Ad-L-Fabp and in freshly isolated HSCs from L-Fabp−/− mice, respectively, each approach revealing a role for L-Fabp in modulating palmitate abundance.
Figure 2. Ad-L-Fabp transduction augments TG and FA content in HSCs in vitro.


(Panel A–D) Passaged HSCs were transduced with no adenovirus (HSC ctr), adenovirus containing L-Fabp cDNA tagged with a FLAG peptide (Ad-L-Fabp), or adenovirus with LacZ (Ad-LacZ) as a mock control. After two days of culture, total RNA and whole cell extracts were prepared. For panels B–D, asterisks indicate p<0.05 vs. control HSC. (A). Western blotting analyses demonstrate expression of FLAG epitope-tagged L-Fabp and Lac Z in Ad-L-Fabp and Ad-Lac Z transduced cells, respectively. β-actin was used as an internal control for equal loading. (B). Biochemical measurement of cellular free fatty acid (FFA) content. Values are expressed as pmol/μg protein and presented as mean ± SD (n=3). (C). Biochemical quantitation of cellular triglyceride (TG) content. Values are expressed as pmol/μg protein (mean ± SD, n=3). (D). Identification of FFA by electrospray ionization mass spectrometry (ESI-MS). Only the major classes of FA are shown and values are expressed as μg/mg protein (means ± SD.) (n=3). (Panel E–F) Lipidomics analysis of FFA and TG species present in freshly isolated (Day 0) HSCs from C57BL/6J and L-Fabp−/− mice. Data represents average abundance of lipids in 2 pools of HSC per genotype, with each pool containing HSCs isolated from 5–8 mice. (E). Major FFA species in C57BL/6J and L-Fabp−/− HSCs, presented either as peak intensity normalized to total cellular protein (left panel) or relative abundance of each FFA species (right panel). (F). Average abundance of individual TG species presented as peak intensity normalized to cellular protein. TG species are identified as total number of carbons: total number of double bonds in the fatty acid chains.
Ad-L-Fabp transduction augments expression of prolipogenic genes and Plin5 in passaged HSCs
We next asked whether the changes in lipid content observed following Ad-L-Fabp transduction of cultured HSCs reflected changes in known master regulators of lipogenesis. Ad-LFabp transduction increased the expression of SREBP-1c, PPARγ and C/EBPα mRNA (Figure 3A) and protein (Figure 3B). The augmented lipid content observed following Ad-L-FABP transduction was associated with increased mRNA expression of the LD protein Plin5 (Figure 3C). Taken together, these results suggest that forced expression of L-Fabp upregulates expression of prolipogenic genes, which in turn increases lipid content in HSCs in vitro.
Figure 3. Ad-L-Fabp transduction augments expression of pro-lipogenic genes and Plin5 in passaged HSCs.

Passaged HSCs were transduced with Ad-L-Fabp, Ad-LacZ or mock transduced (HSC ctr). After two days of culture, total RNA and whole cell extracts were prepared. (A). Real-time PCR assays of mRNA of pro-lipogenic genes. Values are presented as mRNA fold changes vs control HSC (mean ± SD, n=3). *p<0.05 vs. untransduced HSC. (B). Western blot analyses of pro-lipogenic proteins. Blots shown are representative of three independent experiments. β-actin was used as an internal control for equal loading. (C). Real-time PCR assays of lipid droplet protein genes. Values are presented as mRNA fold changes (mean ± SD, n=3). *p<0.05 vs. untransduced HSC.
Ad-L-Fabp transduction reduces HSC activation in vitro
We next examined cellular proliferation and activation markers in HSCs cells following Ad-L-Fabp transduction. Ad-L-Fabp transduction reduced HSC proliferation compared to control (HSC ctr), or Ad-LacZ transduced HSCs (Figure 4A) and attenuated mRNA expression of genes related to HSC activation, including pro-fibrogenic type I and II transforming growth factor-beta receptors (TGF-βRI/II), CTGF, pro-mitogenic platelet-derived growth factor-beta receptor (PDGF-βR), as well as αI(I) collagen and α-SMA (Figure 4B). There was correspondingly decreased expression of cyclin D1 and anti-apoptotic Bcl-2, and increased expression of pro-apoptotic protein Bax in Ad-L-Fabp-transduced HSCs (Figure 4C), consistent with the observed decrease in cell proliferation. These findings collectively suggest that forced expression of L-Fabp in passaged HSCs reduces cell proliferation and decreases expression of genes related to stellate cell activation, implying that L-Fabp may play a role in regulating HSC activation in vivo. Taken together with the observation that Ad-L-Fabp rescue also augments HSC lipid content and LD formation, these observations imply a mechanistic link between cellular lipid storage and the maintenance of HSC quiescence, mediated at least in part through L-Fabp.
Figure 4. Ad-L-Fabp transduction reduces HSC activation in vitro.

Passaged HSCs were transduced with Ad-L-Fabp, Ad-LacZ or no adenovirus (HSC ctr), and harvested after two days of culture. (A). Cell growth measured by colorimetric MTS assays. Results are expressed as relative cell density compared to HSC ctr (mean ± SD, n=3). *p<0.05 vs. control HSC. (B). Real-time PCR assays of genes associated with HSC activation, showing reduced expression in cells transduced with Ad-L-Fabp. Values are presented as mRNA fold changes relative to control HSC (mean ± SD, n=3). *p<0.05 vs. control HSC. (C). Western blot analyses of proteins related to HSC activation. Blots shown are representative of three independent experiments. β-actin was used as a control for equal loading.
L-Fabp deletion attenuates hepatic steatosis in TFF-fed mice
Our earlier studies demonstrated that L-Fabp−/− mice are protected against diet induced hepatic steatosis when fed “Western” or high saturated fat diets (14, 15, 22). Because these diets do not produce fibrosis or inflammation in mice, we turned to a diet model in which hydrogenated fat, combined with fructose supplementation for 16 weeks induces hepatic steatosis with hepatocyte ballooning and fibrogenesis and is more representative of NAFLD (18). There was no significant difference in overall weight gain between the genotypes despite a subtle reduction in body weight in L-Fabp−/− mice (Table 1), but liver weight and liver/body weight ratio was significantly reduced in TFF-fed L-Fabp−/− mice compared to controls. Serum lipid levels were not significantly different, though serum cholesterol was slightly increased in TFF-fed L-Fabp−/− mice (Table 1).
Table 1.
Characterization of Mice Fed TFF Diet
| C57BL/6J (n=8) | L-Fabp−/− (n=11) | p value | |
|---|---|---|---|
| Body Weight (g) | 27.6 ± 0.5 | 26.0 ± 0.7 | ns |
| Weight Gain (g) | 9.6 ± 0.4 | 9.6 ± 0.5 | ns |
| Liver (g) | 1.84 ± 0.09 | 1.45 ± 0.06 | 0.002 |
| Liver/Body (%) | 6.85 ± 0.26 | 5.64 ± 0.12 | <0.001 |
| Fat (g) | 0.64 ± 0.07 | 0.55 ± 0.06 | ns |
| Fat/Body (%) | 2.35 ± 0.33 | 2.13 ± 0.16 | ns |
| Serum TG (mg/dL) | 14.1 ± 3.7 | 13.9 ± 1.9 | ns |
| Serum Chol (mg/dL) | 64.3 ± 3.9 | 75.5 ± 2.9 | 0.031 |
| Serum FFA (mmol/L) | 0.44 ± 0.07 | 0.51 ± 0.05 | ns |
| Serum Glucose (mg/dL) | 385 ± 17 | 365 ± 13 | ns |
| Serum ALT (IU/L) | 15.6 ± 4.1 | 9.2 ± 3.4 | ns |
| Serum β-HBA (μmol/L) | 246 ± 39 | 221± 26 | ns |
Histological evaluation revealed both macro- and micro-vesicular LDs in TFF-fed WT hepatocytes (Figure 5A, B). L-Fabp−/− mice by contrast contained significantly fewer LDs (Figure 5C), which were smaller (Figure 5D) and localized primarily in periportal regions. Biochemical analysis confirmed that hepatic TG content was reduced in L-Fabp−/− mice, with no difference in hepatic cholesterol, free cholesterol, phospholipid, or FA (Figure 5E). The decreased abundance of LDs in TFF fed L-Fabp−/− mice was accompanied by decreased expression of perilipin 4 (Plin4), perilipin 5 (Plin 5) and Cidec (Fsp27), (Figure 5F). These findings suggest that TFF-fed L-Fabp−/− mice exhibit reduced hepatic steatosis with attenuated LD formation compared to C57BL/6J control mice. There was no consistent change in the expression of genes mediating hepatic FA oxidation either by diet or genotype (Figure 5G) and both genotypes exhibited comparable upregulation of lipogenic genes in response to TFF feeding. We also examined the possibility that the shift in LD accumulation with TFF feeding reflected alterations in autophagy in L-Fabp−/− mice. We found that TFF feeding induced a significant change in the ratio of LC3II/LC3-I, implying increased autophagy (Figure 5H) but these changes were comparable in both genotypes (Figure 5I). Accordingly, the mechanisms underlying the attenuated accumulation of hepatic triglyceride likely reflect a combination of subtle shifts in FA utilization rather than changes in a single pathway.
Figure 5. L-Fabp deletion attenuates hepatic steatosis in TFF-fed mice.
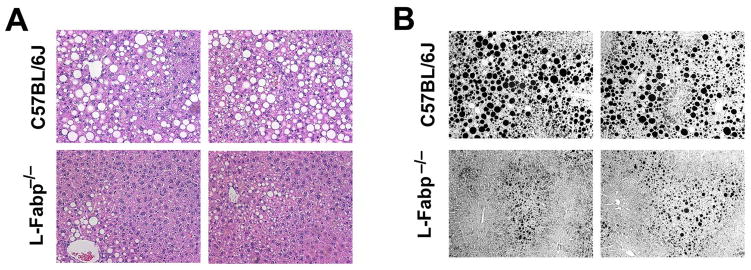


Livers were excised from wild-type C57BL/6J or L-Fabp−/− mice fed TFF diet for 16 weeks. (A, B). Hematoxylin and eosin (A) and Osmium tetroxide staining (B) of lipid droplets in liver tissues. Two representative images from each genotype are presented. (C–D). Calculation of the average number of lipid droplets (C) and average droplet size (D) in C57BL/6J and L-Fabp−/− mice fed TFF diet. Data were generated from 3 random fields per slide, 5–6 mice/genotype, and are shown as mean ± SE. Asterisks indicate p<0.05 vs C57BL/6J mice. (E). Biochemical quantitation of hepatic triglyceride (TG), cholesterol (Chol), free cholesterol (FC), phospholipid (PL) and free fatty acid (FFA) levels in livers of TFF-fed mice (n=8 C57BL/6; 11 L-Fabp−/− mice; * indicates p<0.05.) (F). Real-time PCR quantitation of perilipin (Plin)2, Plin3, Plin4, Plin5, Cidec and hypoxia-inducible protein-2 (HIG2) gene expression in livers of each genotype. Data are normalized to expression in C57BL/6J animals and are shown as mean ± SE. Asterisks indicate p<0.05 vs C57BL/6J controls; n=5 samples/genotype. (G). Real-time PCR assays of fatty acid oxidation (FAO, left panel) and lipogenic genes (right panel) in livers of C57BL/6J and L-Fabp−/− mice fed chow or TFF diet. Data are expressed relative to mRNA levels in chow-fed C57BL/6J controls (mean ± SE. n=5 mice/group). Asterisks indicate p<0.05 vs control. (H–I) Western blot analyses of LC3 expression in chow and TFF fed animals, showing both intact (LC3-I) and cleaved (LC3-II) isoforms (left panels). Relative abundance of each isform was quantitated and expressed as a ratio of cleaved (LC3-II) to uncleaved (LC3-I) protein (right panel). Expression of albumin is shown as a loading control (Figure H).
L-Fabp deletion attenuates hepatic fibrosis in TFF-fed mice
Since Ad-L-Fabp transduction attenuated the activation of HSCs in vitro, we reasoned that the development of hepatic fibrosis might be augmented in TFF-fed L-Fabp−/− mice, despite the reduction in hepatic triglyceride content. However, this was not the case. L-Fabp−/− mice exhibited reduced mRNA abundance of pro-fibrogenic genes, including tissue inhibitor of metalloproteinase 1 (TIMP1), connective tissue growth factor (CTGF), (αI(I)Col and α4(I)Col), with a trend towards decreased expression of α-SMA (Figure 6A). These findings were confirmed histologically with fewer collagen fibrils in TFF-fed L-Fabp−/− mice compared to controls (Figure 6B) and blinded evaluation revealed reduced fibrotic foci (Figure 6C). These results collectively demonstrate both attenuated steatosis and reduced fibrogenesis in TFF-fed L-Fabp−/− mice.
Figure 6. L-Fabp deletion attenuates hepatic fibrosis in TFF-fed mice.
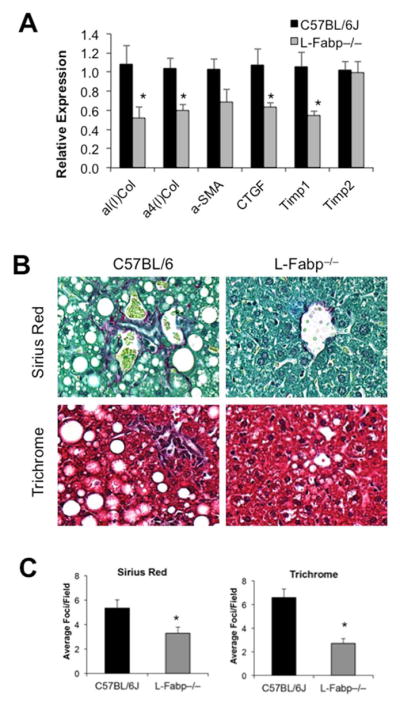
Liver tissue from C57BL/6J and L-Fabp−/− mice fed a TFF diet for 16 weeks was processed for histology or RNA isolation. (A). Real-time PCR analysis of pro-fibrogenic genes, including Type 1 and Type IV alpha collagens (α1(I) Col and α4(I) Col), alpha-smooth muscle actin (α-SMA), connective tissue growth factor (CTGF), and tissue inhibitor of metalloproteinase 1 and 2 (TIMP1 and TIMP2) in liver tissue from each genotype; n=7–8 per group. Asterisks indicate p<0.05 vs C57BL/6J controls. (B). Picro-Sirius Red and Trichrome staining of collagens in liver tissue from each group of mice. A representative image from each genotype is presented. (C). Blind calculation of fibrotic foci per field in Picro-sirius red (left panel) and Trichrome (right panel) stained slides. Data represent average number of fibrotic regions per field, with 10 fields counted per slide; mean ± SE. n=7 C57BL/6J, 8 L-Fabp−/− mice. Asterisks indicate p<0.05 vs C57BL/6J controls.
DISCUSSION
The central observations of this report demonstrate that L-Fabp plays a cell-specific role in regulating elements of lipid metabolism in murine hepatocytes and stellate cells, with implications for HSC activation in vitro and for the development and progression of diet induced NAFLD. The finding that L-Fabp mRNA is abundantly expressed in freshly isolated HSCs, with a coordinated decrease in mRNA expression after 3 days in culture, and that these changes are temporally related to LD depletion and HSC activation, along with reversal of these phenotypes upon Ad-L-Fabp transduction, collectively demonstrate a functional role for L-Fabp in both HSC lipid metabolism and HSC activation. The TFF feeding experiments extend earlier studies which demonstrated that L-Fabp−/− mice are protected against diet-induced obesity and hepatic steatosis (14). Those studies, however, did not address the critical issue of whether L-Fabp−/− mice exhibit alterations in hepatic fibrosis or other progressive forms of NAFLD. The current studies used a high trans-fat, high fructose diet to promote murine NAFLD with fibrosis (18), revealing that L-Fabp−/− mice not only exhibit attenuated steatosis with decreased LD accumulation but are also protected against steatosis-associated fibrogenesis. Several elements of these findings merit additional discussion.
Little is known about the expression of genes related to FA uptake and metabolic channeling during HSC activation. Earlier studies in freshly isolated rat HSCs revealed expression of mRNAs encoding Brain-Fabp (B-Fabp, Fabp7), L-Fabp, as well as retinol binding protein (Rbp), with decreased expression upon culture in vitro (23). The current findings in murine HSCs confirm some but not all of those findings (specifically, B-Fabp was undetectable in our hands) but also demonstrate that L-Fabp depletion temporally accompanies LD depletion from cultured wild-type murine HSCs and that HSCs isolated from L-Fabp−/− mice contain fewer LDs. Importantly, Ad-L-Fabp expression both increased the accumulation of FA and neutral lipid and also suppressed the expression of pro-fibrogenic genes in passaged HSCs. These findings imply that the expression of L-Fabp both promotes LD accumulation and also inhibits HSC activation in vitro. The underlying mechanisms and pathways remain to be defined, but we speculate that L-Fabp regulates the uptake and retention of lipid mediators and signaling molecules in HSCs, analogous to functions described for L-Fabp in liganding PPARα in isolated hepatocytes (24). Other studies have established a role for L-Fabp in the metabolic channeling of FA in enterocytes for complex lipid assembly (25). The findings in TFF-fed mice revealed a striking shift in LD accumulation in L-Fabp−/− mice with decreased expression of several LD-associated genes including Plin4, Plin5 and Cidec, each of which has been shown to be modulated as downstream targets of either Pparα (26) or Pparγ (27, 28) in murine liver.
Our a priori hypothesis, based on the role of L-Fabp in HSC activation in vitro, was that L-Fabp−/− mice would display enhanced susceptibility to high fat diet-induced liver injury and fibrosis. Instead we found that L-Fabp−/− mice exhibited reduced fibrogenesis which correlated with decreased hepatic steatosis. This discrepancy may reflect the complex intracellular crosstalk and lipid signaling that occurs in vivo between hepatocytes and HSCs and highlights the importance of in vivo models in understanding complex systems. Moreover, since germline deletion of L-Fabp has been shown to alter intestinal FA trafficking (12, 14, 15) it is unclear whether the absence of L-Fabp in the intestinal mucosa may also alter the progression of experimental NAFLD. Future studies using mice with tissue specific deletion of L-Fabp, particularly in hepatocytes or HSCs, will begin to address these issues.
The current findings also reflect elements of the apparent paradox previously noted in the lipid profiles of hepatocytes and HSCs and the pathogenesis and progression of steatohepatitis (11). On the one hand, NAFLD is characterized by the accumulation of lipid droplets in hepatocytes, an observation that drives the rationale for reversing hepatic steatosis as a therapeutic goal (1). On the other hand, the activation of HSCs is coupled to lipid droplet depletion (8), with reduced expression of pro-lipogenic genes (8, 10). This process of HSC activation has been referred to as an ‘anti-adipogenic’ phenomenon (9), similar to that described during adipocyte de-differentiation. Based upon these findings, potential strategies to attenuate HSC activation and decrease fibrogenesis include augmenting HSC lipid content with restoration of lipogenesis (10). Stated differently, the regulated accumulation of LDs within HSCs appears beneficial compared to LD accumulation in hepatocytes, specifically in terms of HSC activation and the development and progression of hepatic fibrosis (29).
Other examples exist for the apparently paradoxical cell-specific regulation of LDs and HSC activation. Specifically, adipose differentiation-related protein (Adrp/Plin2) is up-regulated in association with drug- and diet-induced hepatic steatosis (30);(31). Adrp−/− mice and mice treated with an antisense oligonucleotide (ASO) against Adrp both exhibit decreased hepatic steatosis when fed a high fat diet (32, 33) and Adrp−/− mice demonstrate improved insulin resistance and decreased hepatic steatosis when crossed into the Lepob/ob background (34). These observations together imply that hepatocyte Adrp/Plin2 might augment hepatic steatosis and potentially promote liver injury. Conversely, up-regulation of Adrp was demonstrated in HSCs upon retinol and palmitate supplementation, which in turn inhibited HSC activation with down-regulation of fibrogenic genes (35). Those findings are of particular interest in view of the current demonstration that palmitate abundance was attenuated in freshly isolated HSCs from L-Fabp−/− mice. While the source of free palmitate in HSCs is yet to be completely understood, our findings raise the possibility that the attenuated LD abundance in HSCs from L-Fabp−/− mice may reflect a corresponding decrease in retinyl palmitate. We were unable to detect HSC retinyl esters directly using our lipidomic assays, likely reflecting the detection limit with the available material, although other investigators have successfully quantitated retinyl esters in murine HSCs (36).
Another example of the divergence in cell-specific modulation of lipid metabolism and HSC activation is found in Pparγ. Basal expression of PPARγ in the liver is relatively low (37), yet PPARγ is highly expressed in steatotic liver in obese mice (38) and in human subjects (39). Although some studies suggest an anti-steatotic role for Pparγ (40, 41) others have indicated that hepatic Pparγ is pro-steatotic (27, 42–44). Pparγ is abundantly expressed in quiescent HSCs, with reduced expression and transcriptional activity during HSC activation, and several studies have shown that Pparγ agonists inhibit the activation and proliferation of HSCs (10, 20, 45, 46). These findings together suggest that Pparγ might be anti-fibrogenic in HSCs. This suggestion raises the intriguing question of whether the increased expression of Pparγ following Ad-L-Fabp transduction of passaged HSCs plays a role in attenuating the activation state observed in vivo.
A key question, unanswered by the current findings, is whether the loss of LDs is a cause or consequence of HSC activation in vivo. It was recently reported that the absence of retinoid-containing lipid droplets in HSCs in lecithin-retinol acyltransferase knockout (Lrat−/− mice) mice did not enhance HSC activation induced by bile duct ligation or by carbon tetrachloride administration (47). In this scenario, the loss of retinoid signaling was invoked as a consequence, but not a prerequisite, for HSC activation. The current findings place in context the importance of cell-specific events in lipid signaling as mediators of liver injury. It will be important, for example, to reconcile the role of these signaling events as implied from in vitro studies in isolated cell culture with their physiological functions in vivo. The current findings in germline L-Fabp−/− mice imply that there are distinctive roles for LD biology in hepatocytes and HSCs and it will be important to examine these implications using targeted cell-specific deletion strategies. These approaches will form the foundation of ongoing studies to explore some underlying mechanisms of liver injury and may allow us to place the current observations into proper perspective.
Supplementary Material
Acknowledgments
The work was supported by grants RO-1 DK 47995 from NIH/NIDDK and the President Research Fund of Saint Louis University to A. Chen, and RO-1 DK 56260, DDRCC P30 DK-52574, HL-38180 from NIH to N.O. Davidson. Mass Spectrometry facility (Washington University) was supported by NIH grants P41-RR00954, P0-DK20579, and P30-DK56341.
ABBREVIATIONS
- α-SMA
alpha-smooth muscle actin
- C/EBPα
CCAAT/enhancer-binding protein-alpha
- Cidec
cell death-inducing DFFA-like effector c
- CTGF
connective tissue growth factor
- DMEM
Dulbecco’s modified Eagle’s medium
- ECM
extracellular matrix
- FA
fatty acids
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- LDs
lipid droplets
- HSCs
hepatic stellate cells
- L-FABP
liver fatty acid binding protein (Fabp1)
- NASH
non-alcoholic steatohepatitis
- ESI-MS
electrospray ionization mass spectrometry
- PDGF-βR
platelet-derived growth factor-beta receptor
- Plin
perilipin
- PPARγ
peroxisome proliferator-activated receptor-gamma
- SREBP-1c
sterol regulatory element-binding protein-1c
- TG
triglyceride
- TFF
trans-fat fructose diet
- TGF-βR
transforming growth factor-beta receptor
References
- 1.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, et al. The Diagnosis and Management of Non-alcoholic Fatty Liver Disease: Practice Guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annu Rev Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brasaemle DL. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res. 2007;48:2547–2559. doi: 10.1194/jlr.R700014-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Okumura T. Role of lipid droplet proteins in liver steatosis. J Physiol Biochem. 2011;67:629–636. doi: 10.1007/s13105-011-0110-6. [DOI] [PubMed] [Google Scholar]
- 6.Yamada M, Blaner WS, Soprano DR, Dixon JL, Kjeldbye HM, Goodman DS. Biochemical characteristics of isolated rat liver stellate cells. Hepatology. 1987;7:1224–1229. doi: 10.1002/hep.1840070609. [DOI] [PubMed] [Google Scholar]
- 7.Hendriks HF, Brekelmans PJ, Buytenhek R, Brouwer A, de Leeuw AM, Knook DL. Liver parenchymal cells differ from the fat-storing cells in their lipid composition. Lipids. 1987;22:266–273. doi: 10.1007/BF02533990. [DOI] [PubMed] [Google Scholar]
- 8.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng JH, She H, Han YP, Wang J, Xiong S, Asahina K, Tsukamoto H. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008;294:G39–49. doi: 10.1152/ajpgi.00263.2007. [DOI] [PubMed] [Google Scholar]
- 10.Tsukamoto H, She H, Hazra S, Cheng J, Miyahara T. Anti-adipogenic regulation underlies hepatic stellate cell transdifferentiation. J Gastroenterol Hepatol. 2006;21 (Suppl 3):S102–105. doi: 10.1111/j.1440-1746.2006.04573.x. [DOI] [PubMed] [Google Scholar]
- 11.Tsukamoto H, She H, Hazra S, Cheng J, Wang J. Fat paradox of steatohepatitis. J Gastroenterol Hepatol. 2008;23 (Suppl 1):S104–107. doi: 10.1111/j.1440-1746.2007.05294.x. [DOI] [PubMed] [Google Scholar]
- 12.Storch J, Thumser AE. Tissue-specific functions in the fatty acid-binding protein family. J Biol Chem. 2010;285:32679–32683. doi: 10.1074/jbc.R110.135210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newberry EP, Xie Y, Kennedy S, Han X, Buhman KK, Luo J, Gross RW, et al. Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid-binding protein gene. J Biol Chem. 2003;278:51664–51672. doi: 10.1074/jbc.M309377200. [DOI] [PubMed] [Google Scholar]
- 14.Newberry EP, Xie Y, Kennedy SM, Luo J, Davidson NO. Protection against Western diet-induced obesity and hepatic steatosis in liver fatty acid-binding protein knockout mice. Hepatology. 2006;44:1191–1205. doi: 10.1002/hep.21369. [DOI] [PubMed] [Google Scholar]
- 15.Newberry EP, Kennedy SM, Xie Y, Luo J, Davidson NO. Diet-induced alterations in intestinal and extrahepatic lipid metabolism in liver fatty acid binding protein knockout mice. Mol Cell Biochem. 2009;326:79–86. doi: 10.1007/s11010-008-0002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charlton M, Viker K, Krishnan A, Sanderson S, Veldt B, Kaalsbeek AJ, Kendrick M, et al. Differential expression of lumican and fatty acid binding protein-1: new insights into the histologic spectrum of nonalcoholic fatty liver disease. Hepatology. 2009;49:1375–1384. doi: 10.1002/hep.22927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2011;301:G825–834. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol. 2008;295:G987–995. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newberry EP, Kennedy SM, Xie Y, Luo J, Crooke RM, Graham MJ, Fu J, et al. Decreased body weight and hepatic steatosis with altered fatty acid ethanolamide metabolism in aged L-Fabp −/− mice. J Lipid Res. 2012;53:744–754. doi: 10.1194/jlr.M020966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu J, Fu Y, Chen A. Activation of peroxisome proliferator-activated receptor-gamma contributes to the inhibitory effects of curcumin on rat hepatic stellate cell growth. Am J Physiol Gastrointest Liver Physiol. 2003;285:G20–30. doi: 10.1152/ajpgi.00474.2002. [DOI] [PubMed] [Google Scholar]
- 21.Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol. 2006;21 (Suppl 3):S84–87. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- 22.Newberry EP, Kennedy SM, Xie Y, Sternard BT, Luo J, Davidson NO. Diet-induced obesity and hepatic steatosis in L-Fabp/mice is abrogated with SF, but not PUFA, feeding and attenuated after cholesterol supplementation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G307–314. doi: 10.1152/ajpgi.00377.2007. [DOI] [PubMed] [Google Scholar]
- 23.Hellemans K, Rombouts K, Quartier E, Dittie AS, Knorr A, Michalik L, Rogiers V, et al. PPARbeta regulates vitamin A metabolism-related gene expression in hepatic stellate cells undergoing activation. J Lipid Res. 2003;44:280–295. doi: 10.1194/jlr.M200376-JLR200. [DOI] [PubMed] [Google Scholar]
- 24.Wolfrum C, Borrmann CM, Borchers T, Spener F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha - and gamma-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc Natl Acad Sci U S A. 2001;98:2323–2328. doi: 10.1073/pnas.051619898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Storch J, Zhou YX, Lagakos WS. Metabolism of apical versus basolateral sn-2-monoacylglycerol and fatty acids in rodent small intestine. J Lipid Res. 2008;49:1762–1769. doi: 10.1194/jlr.M800116-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamaguchi T, Matsushita S, Motojima K, Hirose F, Osumi T. MLDP, a novel PAT family protein localized to lipid droplets and enriched in the heart, is regulated by peroxisome proliferator-activated receptor alpha. J Biol Chem. 2006;281:14232–14240. doi: 10.1074/jbc.M601682200. [DOI] [PubMed] [Google Scholar]
- 27.Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, et al. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J Biol Chem. 2003;278:498–505. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 28.Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer B, Jr, et al. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest. 2003;111:737–747. doi: 10.1172/JCI17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsukamoto H. Fat paradox in liver disease. Keio J Med. 2005;54:190–192. doi: 10.2302/kjm.54.190. [DOI] [PubMed] [Google Scholar]
- 30.Motomura W, Inoue M, Ohtake T, Takahashi N, Nagamine M, Tanno S, Kohgo Y, et al. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem Biophys Res Commun. 2006;340:1111–1118. doi: 10.1016/j.bbrc.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 31.Steiner S, Wahl D, Mangold BL, Robison R, Raymackers J, Meheus L, Anderson NL, et al. Induction of the adipose differentiation-related protein in liver of etomoxir-treated rats. Biochem Biophys Res Commun. 1996;218:777–782. doi: 10.1006/bbrc.1996.0138. [DOI] [PubMed] [Google Scholar]
- 32.Chang BH, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, Chan L. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol. 2006;26:1063–1076. doi: 10.1128/MCB.26.3.1063-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imai Y, Varela GM, Jackson MB, Graham MJ, Crooke RM, Ahima RS. Reduction of hepatosteatosis and lipid levels by an adipose differentiation-related protein antisense oligonucleotide. Gastroenterology. 2007;132:1947–1954. doi: 10.1053/j.gastro.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 34.Chang BH, Li L, Saha P, Chan L. Absence of adipose differentiation related protein upregulates hepatic VLDL secretion, relieves hepatosteatosis, and improves whole body insulin resistance in leptin-deficient mice. J Lipid Res. 2010;51:2132–2142. doi: 10.1194/jlr.M004515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee TF, Mak KM, Rackovsky O, Lin YL, Kwong AJ, Loke JC, Friedman SL. Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by adipose differentiation-related protein (ADRP) J Cell Physiol. 2010;223:648–657. doi: 10.1002/jcp.22063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Ambrosio DN, Walewski JL, Clugston RD, Berk PD, Rippe RA, Blaner WS. Distinct populations of hepatic stellate cells in the mouse liver have different capacities for retinoid and lipid storage. PLoS One. 2011;6:e24993. doi: 10.1371/journal.pone.0024993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 38.Edvardsson U, Bergstrom M, Alexandersson M, Bamberg K, Ljung B, Dahllof B. Rosiglitazone (BRL49653), a PPARgamma-selective agonist, causes peroxisome proliferator-like liver effects in obese mice. J Lipid Res. 1999;40:1177–1184. [PubMed] [Google Scholar]
- 39.Westerbacka J, Kolak M, Kiviluoto T, Arkkila P, Siren J, Hamsten A, Fisher RM, et al. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes. 2007;56:2759–2765. doi: 10.2337/db07-0156. [DOI] [PubMed] [Google Scholar]
- 40.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 41.Nan YM, Han F, Kong LB, Zhao SX, Wang RQ, Wu WJ, Yu J. Adenovirus-mediated peroxisome proliferator activated receptor gamma overexpression prevents nutritional fibrotic steatohepatitis in mice. Scand J Gastroenterol. 2011;46:358–369. doi: 10.3109/00365521.2010.525717. [DOI] [PubMed] [Google Scholar]
- 42.Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- 43.Moran-Salvador E, Lopez-Parra M, Garcia-Alonso V, Titos E, Martinez-Clemente M, Gonzalez-Periz A, Lopez-Vicario C, et al. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011;25:2538–2550. doi: 10.1096/fj.10-173716. [DOI] [PubMed] [Google Scholar]
- 44.Panasyuk G, Espeillac C, Chauvin C, Pradelli LA, Horie Y, Suzuki A, Annicotte JS, et al. PPARgamma contributes to PKM2 and HK2 expression in fatty liver. Nat Commun. 2012;3:672. doi: 10.1038/ncomms1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF, Jr, Motomura K, Anania FA, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–35722. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 46.Zheng S, Chen A. Activation of PPARgamma is required for curcumin to induce apoptosis and to inhibit the expression of extracellular matrix genes in hepatic stellate cells in vitro. Biochem J. 2004;384:149–157. doi: 10.1042/BJ20040928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kluwe J, Wongsiriroj N, Troeger JS, Gwak GY, Dapito DH, Pradere JP, Jiang H, et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut. 2011;60:1260–1268. doi: 10.1136/gut.2010.209551. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.