

Abstract
Antigen-mediated mast cell (MC) degranulation is the critical early event in the induction of allergic reactions. Transient receptor potential channels (TRPC), particularly TRPC1, are thought to contribute to such MC activation. To explore the contribution of TRPC1 in MC-driven allergic reactions, we examined antigen-mediated anaphylaxis in Trpc1−/− and WT mice, and TRPC1 involvement in the activation of MCs derived from the bone marrow (BMMCs) of these mice. In vivo, we observed a similar induction of passive systemic anaphylaxis in the Trpc1−/− mice compared to WT controls. Nevertheless, there was delayed recovery from this response in Trpc1−/− mice. Furthermore, contrary to expectations, Trpc1−/− BMMCs responded to antigen with enhanced calcium signalling but with little defect in degranulation or associated signalling. In contrast, antigen-mediated production of TNF-α, and other cytokines, was enhanced in the Trpc1−/− BMMCs, as were calcium-dependent events required for these responses. Additionally, circulating levels of TNF-α in response to antigen were preferentially elevated in the Trpc1−/− mice, and administration of an anti-TNF-α antibody blocked the delay in recovery from anaphylaxis in these mice. These data thus provide evidence that, in this model, TRPC1 promotes recovery from the anaphylactic response by repressing antigen-mediated TNF-α release from MCs.
Keywords: anaphylaxis, calcium, degranulation, FcεRI, JUN, KIT, mast cells, NFAT, SCF, TNF-α, TRPC1
1. Introduction
The transient receptor potential (TRP) channel superfamily comprises a group of structurally and evolutionary-related cation channels which are largely non-selective in nature [1–3]. Six mammalian TRP channel sub-families are described: the canonical (TRPC1 to TRPC7); melastatin (TRPM1 to TRPM8); vallinoid (TRPV1 to TRPV6); ankyrin (TRPA); the polycistyns (TRPP), and mucolipins (TRPML) TRP subfamilies, the last three consisting of 1 to 4 members each [4]. These channels are widely expressed in most tissues and cells where they can be activated by receptor ligation or by various chemical and physical stimuli. Through the regulation of mast cell function, TRP channels have been reported to modify allergic reactions as seen in asthma [5, 6] and anaphylaxis [7].
The primary mechanism of mast cell activation in allergic reactions is antigen cross-linking of IgE bound to the high affinity IgE receptor (FcεRI) on the mast cell surface. This results in the release of a variety of preformed and de novo generated inflammatory mediators that act upon surrounding tissues, including airway smooth muscle, to induce the characteristic symptoms associated with the allergic response [8, 9]. Mast cells express a number of TRP channels, several of which have been described to differentially modulate antigen-mediated mast cell responses. Selective members of the TRPC family are proposed to function as positive regulators of antigen-mediated mast cell function by contributing to calcium influx following FcεRI aggregation. In this regard, we previously reported that TRPC5, in conjunction with the calcium channel Orai1 and the endoplasmic reticulum (ER) calcium sensor STIM1, is required for optimal influx of Ca2+ and degranulation in the RBL 2H3 rat mast cell line [10]. Typically, depletion of calcium stores in the ER, through activated inositol 1,4,5-trisphosphate (IP3) receptor channels, leads to the interaction of STIM1 in ER with Orai1 in the plasma membrane to allow calcium entry, a process referred to as store-operated calcium entry (SOCE) [11], although the participation of TRPC channels in this process is still debated. In contrast, studies conducted in mice deficient in the calcium-activated non-selective cation channel TRPM4, in conjunction with studies of mast cells derived from the bone marrow (BMMCs) of these mice, have demonstrated that TRPM4 functions as a negative regulator of mast cell-dependent anaphylaxis through membrane depolarization and suppressed influx of extracellular calcium into mast cells [7].
A role for TRPC1 in antigen-mediated mast cell activation has been proposed on the basis of reduced FcεRI-mediated calcium entry and reduced sensitivity to antigen in cultured rat RBL-2H3 cells and mouse BMMCs following TRPC1 knock down [12, 13]. Nevertheless, the role of TRPC1 in mast cell-dependent responses in situ is unknown. We have therefore explored the potential outcome of TRPC1 deletion on mast cell-dependent anaphylaxis in a Trpc1−/− mouse model. As reported here, we unexpectedly found that TRPC1 deficiency in this model resulted in a delayed recovery of antigen-induced anaphylaxis as monitored by the decrease in core body temperature. Furthermore, we observed an exaggerated antigen-induced calcium response in BMMCs derived from these mice, and a consequentially higher production of cytokines including TNF-α, in these cells; a response that appeared to account for the delayed recovery from anaphylaxis in the TRPC1-deficent mice.
2. Materials and Methods
2.1. Chemicals and reagents
Unless otherwise specified, all chemicals and reagents were purchased from Sigma Aldrich (St. Louis, MO).
2.2. Human mast cells
In initial experiments in which the expression of TRP channels was examined, we used human mast cells (HuMCs) derived from CD34+-peripheral blood progenitor cells [14] obtained from normal volunteers, following informed consent, under a protocol (NCT00001756) approved by the NIAID IRB.
2.3. Mice
Trpc1−/− and Trpc6−/− mice (129SvEv background), generated as reported earlier [15, 16], were housed in the animal facility within NIAID, NIH, Bethesda. Trpc6−/− mice, originally created on a mixed 129SvEv and C57Bl/6J background, were backcrossed to 129SvEv mice for at least 10 generations before they were used in the current experiments. Corresponding wild type (WT) mice were obtained from a colony at NIEHS. Subsequent generations and Trpc1−/−/Trpc6−/− double knockout mice were bred in the animal care facility within NIAID, NIH, under a protocol approved by the NIH/NIAID Institutional Animal Care and Use Committee.
2.4. Genotyping and PCR
The genotypes of these mice were confirmed using the following primers:
TRPC1 primers
A) C1 ex8F 5′ GGG ATG ATT TGG TCA GAC ATT AAG; B) C1 int8R 5′ GTG TAC CTA ACA TCA ACC ATG GTA C; C) PGKProm R1 5′ TGG ATG TGG AAT GTG TGC GAG GC. Reaction conditions: 95 °C for 2 min, 38 cycles of [95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s], then 72 °C for 7 min. To identify the WT allele we used A and B primers; with a fragment size of 368 bp. For the TRPC1 KO we used A and C primers, fragment size: 250 bp. Both alleles were amplified in the same reaction.
TRPC6 primers for KO
5′ ACG AGA CTA GTG AGA CGT GCT ACT TCC 3′ and 5′ GGG TTT AAT GTC TGT ATC ACT AAA GCC TCC 3′, and for WT type- reaction 5′ CAG ATC ATC TCT GAA GGT CTT TAT GC 3′ and 5′ TGT GAA TGC TTC ATT CTG TTT TGC GCC 3′. Reaction conditions: 94 °C for 4 min, 35 cycles of [94 °C 1 min, 60 °C for 1.3 min, 72 °C for 2.3 min], then 72 °C for 10 min, fragment sizes: 310 bp and 245 bp.
The expression of TRPC1 in cultured mast cells was confirmed by RT-PCR using the following primers: TRPC1 primers: 5′ ATG TAT ACA ACC AGC TCT ATT TTG 3′ and 5′ CGT CTT TGG AGA AGG AAT AAT G 3′, fragment size: 525 bp. The reaction conditions were as follows: 94 °C for 2 min, 40 cycles of [94 °C for 15 s, 56 °C for 30s, 72 °C for 1 min], then 72 °C for 10 min. Mouse brain total RNA (Clontech, Mountain View, CA) was used a positive control. Human TRPC1 primers were: 5′ATG TAT ACA ACC AGC TCT ATC TTG 3′ and 5′AGT CTT TGG TGA GGG AAT GAT G 3′. The fragment size was 525 bp.
2.5. In vivo studies, induction of anaphylaxis
Mice were sensitized intravenously via the retro-orbital plexus with 3 μg anti-DNP IgE mAb (DNP-specific IgE, clone H1-DNP-e-26.82; generous gift from Dr. Juan Rivera NIAMS, NIH), and an implantable electronic transponder was injected subcutaneously (IPTT-300; Bio Medic Data Systems, Seaford, DE). After 24 h, mice were injected retro-orbitally with 200 μg of DNP-HSA (Sigma-Aldrich) in PBS. Changes in core body temperature were measured every 5 min for 2 h using a temperature sensor (Bio Medic Data Systems)
2.6. In vivo Cytokine Capture Assay (IVCCA)
This method of measuring cytokine was adapted from Finkelman et al. [17]. Briefly, mice were sensitized with 3 μg anti-DNP IgE (clone H1-DNP-e-26.82)). After 24 h, the mice are injected with 10 μg of biotinylated anti-TNF-α antibody (eBioscience, San Diego, CA; clone TN3.19), then 1 h later, the mice were injected with 100 μg DNP-HSA (Sigma-Aldrich) to induce anaphylaxis. Changes in core body temperature were monitored as described above. Mice were bled at the end of experiment and levels of serum TNF-α-bound to the biotinylated anti-TNF-α antibody was measured by absorbance as described [17]
2.7. Culture of mouse BMMCs
Bone marrow mast cell progenitors were obtained by femur lavage and subsequently cultured in RPMI 1640 media supplemented with 10% FBS, 4 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 25 mM HEPES, 1 mM sodium pyruvate, 1% nonessential amino acids, 50 mM β-mercaptoethanol and 30 ng/ml mouse recombinant IL-3 (PeproTech, Rocky Hill, NJ) as described [18]. Unless indicated, all experiments were conducted between 6–7 weeks when >95% of mouse bone marrow-derived mast cells expressed high levels of FcεRI and CD117.
2.8. Toluidine blue staining
Mast cell morphology and granularity was assessed by toluidine blue staining. Approximately 1×105 BMMCs, attached to slides by cytospin, were fixed in Mota’s fixative (20% ethanol, 2.5% lead acetate, 2% acetic acid, in water solution), then stained (30% ethanol, 0.13% toluidine blue, 1% HCl, pH 4) as described [19].
2.9. Flow cytometry
Cells were fixed then stained with FITC-conjugated rat-anti mouse IgE, PE-conjugated rat-anti mouse KIT (CD117), as well as rat IgE, FITC IgE control and PE-conjugated rat IgG2bκ isotype control (BD Biosciences, San Jose, California) in PBS containing 0.1% BSA for 1h. The cells were then rinsed in the same buffer and surface FcεRI and KIT expression determined by FACSCalibur Analytic Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ).
2.10. Cell activation, degranulation, and cytokine production
BMMCs were sensitized by incubating overnight in cytokine-free media containing anti-mouse monoclonal dinitrophenyl (DNP)-IgE (100 ng/ml) (Sigma Aldrich) in RPMI medium. For degranulation studies, the cells were rinsed with HEPES buffer (10 mM HEPES pH 7.4, 137 mM NaCl, 2.7 mM KCl, 0.4 mM Na2HPO4 7H2O, 5.6 mM glucose, 1.8 mM CaCl2 2H2O) containing 0.04% BSA (Sigma Aldrich) then 2×104 cells were seeded into individual wells of a 96-well plate. The cells were activated by the addition of antigen (DNP-HSA) (0–100 ng/ml) contained in the same buffer for 30 min and degranulation monitored by the release of β-hexosaminidase into the supernatants [20]. The extent of degranulation was calculated as the percentage of the total content (cells and media) released into the supernatant. To determine cytokine release, BMMCs were similarly sensitized, rinsed with IL-3-free RPMI media, then aliquots (5×105 cells) were triggered for 6 h with antigen (10 ng/ml) in the presence and absence of stem cell factor (SCF, 10 ng/ml, PeproTech, Rocky Hill, NJ). Cytokines released into the supernatants were measured by mouse TNF-α, IL-6, and GM-CSF Quantikine ELISA kits (R&D Systems, Minneapolis, MN).
2.11. Measurement of intracellular calcium
BMMCs were incubated with 2 μM Fura-2 AM (Molecular Probes/Invitrogen, Eugene, OR) in HEPES buffer containing 0.4% BSA and 0.3 mM sulfinapyrazole at 37 °C for 30 min [21]. Following rinsing twice the cells were re-suspended in the HEPES/sulfinapyrazole buffer and placed in a 96-well black culture plate (Culture plate-96F, PerkinElmer Life Sciences, Waltham, MA) at 1×104 cells/100 μl/well. Fluorescence (excitation wavelengths: 340 and 380 nm; emission wavelength: 510 nm) was measured using a Wallac Victor plate reader (PerkinElmer Life Sciences) Changes in levels of cytosolic Ca2+ were monitored from the ratio of fluorescence readings after correction for fluorescence of the cells that had not been loaded with Fura-2 AM. In some experiments cells were stimulated in Ca2+-free medium that contained 20 μM EGTA. Calcium was then added 10 min later to adjust Ca2+ to normal levels (1.8 mM) where indicated.
2.12. Immunobloting
BMMCs were sensitized as for degranulation experiments and aliquoted (1X106 cells/100 μl) into 1.5 ml polyethylene screw cap tubes. The cells were then activated as described in the figure legends and the reactions terminated by adding boiling 100 μl of 2X boiling lysis buffer [22]. Proteins were resolved by electrophoresis using 4–12% NuPage BisTris gels (Life Sciences, Carlsbad, CA) under MES buffer running conditions according to the manufacturer’s directions. Following membrane transfer, immuno-reactive proteins were detected using the following Abs: anti-β actin (clone AC-15) (Sigma), anti-phospho-PLCγ1 (Tyr(P)-783) (Biosource, Camarillo, CA), anti-phospho-PLCγ2 (Tyr(P)-759), anti-phospho-AKT (Ser(P)-473), anti-phospho-ERK1/2 (Thr(P)-202, anti-phospho-LAT (Tyr(P)-171), anti-phospho-JNK (Thr(P)-183/Tyr(P)185), anti-phospho-p38 (Thr(P)-180/Tyr(P)182) (Cell Signaling Technology, Beverly, MA), and anti-phospho BTK (Tyr(P)-551) pAb (BD Pharmingen). The secondary antibodies were, depending or the species of the primary antibody, either anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (Sigma-Aldrich). Antibodies used for transcription factors were anti-phospho NFAT1 (pS54) (Biosource), anti-NFAT1, anti-phospho NF-κB (p65), anti-phospho c-jun (S63), anti c-jun, anti-phospho-ATF-2 (T71), and anti-phospho CREB (S133) (Cell Signalling). The anti-phospho antibodies used were chosen for their ability to detect proteins in their activated phosphorylated state such as anti-phospho-ATF-2 (T71). To normalize the protein loading, identically loaded samples are probed for β-actin. For selected proteins, phosphorylation was determined by scanning the ECL films using a Quantity One scanner (Bio-Rad, Hercules, CA).
2.13. Data presentation and statistics
Unless indicated all data are presented as the mean +/− S.E.M. where n = the number of individual cultures or animals. Significant differences between test groups was determined by either Student’s T test or ANOVA as indicated in the figure legends.
3. Results
3.1. TRP expression in human and mouse mast cells
As reported for RBL 2H3 cells [10], HuMCs and mouse BMMCs contained message for multiple TRP channel proteins including TRPC1 (Fig. 1A) along with TRPC2, TRPC3, TRPC5, TRPC6, TRPM4, TRPM5, TRPM8, and TRPA1 (data not shown). We were unable to detect mRNA for TRPC4. The present studies focused specifically on the role of TRPC1 in mast cell-dependent anaphylactic responses using Trpc1−/− mice.
FIGURE 1.

A: TRPC1 expression in HuMCs and mouse BMMCs. Expression of TRPC1 in HuMC (left panel: Human ) and BMMCs (right panel: Mouse) was determined by RT-PCR. Human brain total RNA (predicted size 525 bp) and mouse brain total RNA (predicted size 525 bp), were used as positive controls. B: Anaphylactic responses in WT and Trpc1−/− mice. Mice were sensitized using 3 μg anti-DNP IgE mAb. After 24 h, mice received an injection of 200 μg DNP-HSA and the change in core body temperature was monitored for 2 h. Data are presented as the mean ± SEM. (*p<0.05, **p<0.01; two way ANOVA). N=10. C: Mice were injected with a bolus of histamine (5 μMoles) retro-orbitally and the change in body temperature was measured every 5 min for 2 h. N=3.
3.2. In vivo induction of passive systemic anaphylaxis in Trpc1−/− and WT mice
Systemic anaphylaxis in WT and Trpc1−/− mice was induced through passive sensitization by i.v. injection with anti-DNP IgE followed by i.v. challenge with antigen (DNP-HSA) and then assessed by measurement of the decrease in core body temperature [22]. This regimen in WT mice produced a marked (~6 °C) drop in body temperature within 30 min of antigen challenge. This was followed by a slow recovery with core body temperatures approaching normal levels within 2 h after injection (Fig. 1B). An identical drop in core body temperature was observed in the Trpc1−/− mice during the initial 30 min. However, contrary to expectations, the recovery was substantially delayed when compared to WT mice (Fig. 1B).
3.3. Reversal of the delayed recovery from anaphylaxis in the Trpc1−/− mice by an anti-TNF-α antibody
Anaphylactic hypothermia is generally attributed to the release of histamine which is known to induce hypothermia by acting through the hypothalamus [23, 24]. As reported by others [25], a single injection of histamine elicited profound hypothermia similar to that produced by injection of antigen in WT mice (Fig. 1C). However, the extent and duration of the hypothermic response to histamine was unaffected by TRPC1-deficiency when compared to WT mice. These data indicate that the prolonged hypothermia in Trpc1−/− mice was not due to effects downstream of the actions of histamine during antigen-induced anaphylaxis.
TNF-α has been implicated as one factor responsible for symptoms of the late-phase anaphylactic response [26, 27] and as a cryogenic factor during mast cell-dependent hypothermia [28] or acute bacterial infection [29]. We therefore considered TNF-α as a possible candidate for the slow recovery of Trpc1−/− mice from anaphylactic hypothermia. The accumulation of circulating TNF-α was substantially higher during anaphylaxis in Trpc1−/− mice than WT mice (Fig. 2A). Moreover, in the Trpc1−/− mice receiving the anti-TNF-α antibody 1 h before antigen, the delay in recovery from hypothermia at later stages of anaphylaxis was not observed (Fig. 2B).
FIGURE 2. TNF-α release and anaphylaxis in WT and Trpc1−/− mice.

A: Mice were sensitized by injecting anti-DNP IgE (i.v.). The next day, anaphylaxis was induced by i.v. injection of DNP-HSA as described in Figure 1. After 2 h, blood was collected, and serum TNF-α levels were measured by ELISA. Data are presented as the mean ± SEM. (N=7 for WT and N=6 for Trpc1−/−; *p<0.05, two tailed Student’s t-test). B: Mice were sensitized using 3 μg anti-DNP IgE mAb. After 24 h the mice were injected with 10 μg anti-TNF-α antibody. After 1 h, mice were injected with antigen (DNP-HSA; 200 μg), and the change in core body temperature was monitored every 5 min for 2 h. The data are represented as the mean ± SEM. N=7 (*p<0.05, **p<0.01; two way ANOVA).
Taken together, these data provide evidence that the delayed recovery from anaphylaxis observed in the Trpc1−/− mice is mediated by the elevated generation of TNF-α. We thus examined the consequences of TRPC1 deficiency on the release of inflammatory mediators from activated mast cells.
3.4. TRPC1 deficiency results in enhanced mast cell cytokine generation but not degranulation
As TRPC1-deficiency is known to delay the cell cycle, at least in some cell types [30] [31], to define conditions for optimal release of inflammatory mediators from mast cells, we first determined the outcome of TRPC1 deficiency on mast cell development. As shown in Figure 3A and 3B, it was apparent on the basis of morphology and expression of KIT and FcεRI that cultured Trpc1−/− BMMCs had delayed maturation when compared to the WT BMMCs. Those derived from WT mice (Fig. 3A) after 4 weeks of culture consisted of a densely granulated homogeneous cell population with similar appearance to freshly isolated peritoneal mast cells (not shown) that expressed KIT and FcεRI (Fig. 3B). In contrast, Trpc1−/− BMMCs at 4 weeks were heterogeneous, hypo-granulated (Fig 3A) and had reduced FcεRI expression (Fig 3B), as well as markedly reduced degranulation in response to antigen both in the absence and presence of SCF (Fig. 3C and 3D). Nevertheless, after 6 weeks of culture, the degree of maturity, based on morphology and receptor expression, appeared to be identical for WT and Trpc1−/− BMMCs (Figs 4A and B). For this reason, all subsequent experiments were performed with 6–7 week old cultures when the expression of KIT and FcεRI became the same in both cell populations. Under these conditions, no differences were detected in FcεRI-mediated degranulation or in the ability of SCF to enhance this response [32] in WT and Trpc1−/− BMMCs (Fig. 4C and 4D).
FIGURE 3. Characteristics and activation of 4 wk old WT and Trpc1−/− BMMCs.

A: Toluidine blue staining revealed a difference in morphology and granularity between 4 wk old WT (left) and Trpc1−/− (right) BMMCs. B: Reduced expression of FcεRI and KIT receptor in Trpc1−/− as revealed by flow cytometry. C,D: β-hexosaminidase release assay in 4 wk old BMMCs prepared from WT and Trpc1−/− mice show reduced degranulation in Trpc1−/− BMMCs. Anti-DNP-IgE-sensitized cells were treated with increasing concentrations of antigen (C) or Ag and SCF (30 ng/ml) (D) for 30 min. The data are presented as the mean ± SEM (n=3) separate experiments conducted in duplicate (*p<0.05, **p<0.01; two tailed Student’s t-test).
FIGURE 4. Characteristics and release properties of 6 wk old WT and Trpc1−/− BMMCs.

A: Flow cytometry conducted on 7 wk old BMMCs prepared from WT and Trpc1−/− mice showed an identical expression of FcεRI and KIT. B: Toluidine blue staining of 6 wk old cultures of WT (left) and Trpc1−/− (right) BMMCS show identical granularity. C: β-hexosaminidase release in 6 wk old BMMC cultures prepared from WT and Trpc1−/− show identical degranulation. Anti-DNP-IgE-sensitized cells were treated with increasing concentrations of antigen (Ag) (C) or Ag and SCF (30 ng/ml) (D) for 30 min. The data are presented as the mean ± S.E. of (n=4) separate experiments conducted in duplicate. Production of TNF-α (E), GM-CSF (F), and IL-6 (G) were elevated in Trpc1−/− BMMCs. IgE sensitized cells were incubated with antigen (10 ng/ml) and SCF (10 ng/ml) individually or in combination for 6h. Values for TNF-α and IL-6 are presented as the mean ± SEM from 8–10 independent experiments. For GM-SCF, values are presented as the mean ± SEM from 3 independent experiments. (*p<0.05, **p<0.01,*** p<0.001, two tailed Student’s t-test).
We next examined the outcome of TRPC1 deficiency on FcεRI-mediated cytokine generation. WT and Trpc1−/− BMMCs were again challenged with Ag in the presence and absence of SCF which also optimizes cytokine production [21]. As shown in Figure 4E, 4F, and 4G, there was a marked enhancement of the generation of TNF-α in addition to other cytokines examined (IL-6, GM-CSF) in Trpc1−/− BMMCs in response to antigen, especially when administered in combination with SCF.
Together, the above data suggest that TRPC1 deficiency does not alter the balance of signaling pathways that regulate degranulation but does so in a manner that regulates cytokine production. The lack of effect on degranulation is consistent with the similar initial drop (<30 min) in body temperature observed in both WT and Trpc1−/− mice following antigen challenge when degranulation and the associated histamine release occurs. This is in contrast to the delayed recovery in body temperature in Trpc1−/− mice when circulating levels of TNF-α remain elevated (Fig. 2).
3.5. The calcium signal in activated BMMCs is enhanced by Trpc1 deficiency
To investigate the underlying cellular mechanisms that would account for the enhanced antigen induced TNF-α release from mast cells associated with TRPC1 deficiency, we examined the related signaling processes leading to both cytokine production and degranulation in both the WT and Trpc1−/− BMMCs. As TRPC1 channels are thought to contribute to calcium influx in mast cells [12, 13] thereby regulating degranulation and cytokine production, we first investigated the effect of TRPC1-deficiency on calcium signaling.
Contrary to previous studies employing knock down approaches, but consistent with the elevated cytokine production in the Trpc1−/− BMMCs, antigen-induced elevation of cytosolic Ca2+ was markedly enhanced in these cells as compared to WT BMMCs (Figure 5A). The ability of SCF to enhance this response was also potentiated in the Trpc1−/− BMMCs (data not shown).
FIGURE 5. Changes in cytosolic calcium in response to antigen and thapsigargin in WT and Trpc1−/− BMMCs.

Sensitized cells were loaded with Fura 2 then challenged with DNP-HSA (Ag; 10 ng/ml) (A, B), thapsigargin (Thaps; 800 nM) (C,D) or unchallenged (E) in the presence (A,C) or absence (B,D,E) of extracellular calcium. Calcium (Ca2+) was subsequently added to the cells in B,D,E after 10 min and intracellular calcium levels were monitored for a further 10 min. The data in A and C are the mean ± S.E from 10 and 7 separate experiments respectively conducted in duplicate and data in B,D,E are presented as the mean ± S.E from 3 separate experiments conducted in duplicate.
To further investigate this finding, BMMCs were exposed to antigen for 10 min in Ca2+-free media to determine the release from intracellular stores and, after the addition of Ca2+, the subsequent influx of Ca2+ as assessed from the rate of increase in cytosolic Ca2+. Under these conditions, we observed no differences in the antigen-induced increases in cytosolic calcium between the WT and Trpc1−/− BMMCs (Fig. 5B). Additional studies were then conducted with thapsigargin which depletes intracellular ER stores and thereby activates SOC channels independently of IP3 activity [33]. In the presence of Ca2+, the levels of cytosolic Ca2+ initially rose at similar rates in WT and Trpc1−/− BMMCs after addition of thapsigargin but, as in antigen stimulated cells, the levels continued to increase in Trpc1−/− BMMCs to much higher levels than WT BMMCs (Fig. 5C). Although reduced, a similar difference between WT and Trpc1−/− BMMCs was observed after delayed addition of Ca2+ following thapsigargin stimulation (Fig. 5D). These results suggest that although TRPC1-deficiency did not affect SOCE initially, the mechanism for feed-back inhibition of SOCE at high cytosolic Ca2+ [34] is impaired. A further indication that SOCE was unaffected by TRPC1-deficiency at low cytosolic Ca2+ was the similarity in rates of increase of cytosolic Ca2+ in WT and deficient BMMCs after re-provision of Ca2+ to Ca2+-deprived cells in the absence of any stimulant (Fig. 5E).
3.6. TRPC6 compensation does not account for observed phenotypes in Trpc1−/− mice and BMMCs
Knock-down of TRPC1 is reported to reciprocally upregulate TRPC6 expression in vascular smooth muscle cells [35, 36] although TRPC6, as well as STIM1, expression is not altered in TRPC1−/− mice [15] as used in the experiments described herein.
To examine therefore whether TRC6 could account for the enhanced calcium signal observed in the Trpc1−/− BMMCs, we generated BMMCs from both Trpc6−/− and Trpc1−/−/Trpc6−/− double knockout mice. As shown in Figure 6 A and B, respectively, neither the antigen-induced nor the thapsigargin-induced calcium response was affected by TRPC6 deficiency. Furthermore, the deficiency of both TRPC1 and TRPC6 had a similar hyperresponsive phenotype with respect to the calcium signal as observed in the Trpc1−/− BMMCs (Fig. 6C and 6D). Furthermore, the absence of effect on antigen-mediated degranulation, and the enhanced TNF-α production in Trpc1−/− BMMCs and the delayed recovery from antigen-induced systemic anaphylaxis in the Trpc1−/− mice were also respectively recapitulated in Trpc1−/−/Trpc6−/− double knockout BMMCs and mice (Fig, 6E, 6F and 6G). These data are consistent with the conclusion that TRPC6 plays no role in the mast cell-dependent phenotype associated with TRPC1 deficiency.
FIGURE 6. Calcium signaling and mediator release in Trpc6−/− and Trpc1−/−/Trpc6−/− BMMCs.

A–D: Changes in cytosolic calcium in response to DNP-HSA (Ag; 10 ng/ml)) (A,C) and thapsigargin (thaps: 800 nM) (B,D) in sensitized WT and Trpc1−/− (A,B) and WT and Trpc1−/−/Trpc6−/− BMMCs (C,D) E: DNP-HSA (Ag)-mediated degranulation in 6 week old sensitized BMMC cultures prepared from WT and Trpc1−/−/Trpc6−/− BMMCs. The data in A to E are presented as means ± S.E of (n=2–3) separate experiments conducted in duplicate. F: Release of TNF-α from sensitized Trpc1−/−/Trpc6−/− BMMCs. IgE-sensitized cells were incubated with antigen (10 ng/ml) and SCF (10 ng/ml) individually or in combination for 6h. The level of TNF-α was determined using ELISA. Values are presented as the mean ± SEM from 4 independent experiments. *p<0.05 for comparison with Ag/SCF G: Mice were sensitized using 3 μg anti-DNP IgE mAb. After 24 h mice received an injection of 200 μg DNP-HSA, and the change in core body temperature was monitored for 2 hours. Data is represented as the mean ±SEM. (*p<0.05, ** p<0.01). N=4.
3.7. TNF-α production correlates with intracellular calcium levels in antigen-stimulated mast cells
To further investigate the correlation between intracellular calcium levels and TNF-α production, we challenged BMMCs with antigen in the presence of increasing concentrations of extracellular calcium. As shown in Figure 7A, the antigen-dependent increase in intracellular calcium was highly dependent on the extracellular calcium concentration. Such manipulation of the calcium signal revealed that indeed TNF-α production is proportional to cytosolic calcium concentrations (Figure 7B). Furthermore these data reveal that antigen-mediated TNF-α production is absolutely dependent on calcium.
FIGURE 7. Dependency of antigen-mediated TNF-α production on intracellular calcium concentration.

The increases in intracellular calcium concentrations in response to antigen (Ag; DNP-HSA; 10 ng/ml) were modulated by varying the extracellular calcium concentrations (A) and then TNF-α release (B) was determined as described in Materials and Methods with the exception that the experiments were conducted in HEPES buffer containing the indicated concentrations of calcium. The data are means and SEM of 3 (A) or of 2 (B) separate experiments.
3.8. TRPC1 deficiency does not influence signaling events regulating the release of intracellular calcium
The characteristics of the calcium signal in Trpc1−/− BMMCs (Fig. 5D) suggested that Ca2+ release and initial Ca2+ influx were unaffected by TRPC1 deficiency and this appeared to be true for signaling pathways that lead to the release of calcium from intracellular stores. We examined whether there was differential activation of PLCγ1 and PLCγ2, along with other early activation events including phosphorylation of Akt (used as marker for the activation of PI3K), Btk, and LAT, required for generating and modulating the release of calcium from the ER. As shown in Figure 8a, there were no differences in these responses between the WT and Trpc1−/− BMMCs. It would thus appear that TRPC1-deficiency had minimal, if any, effect on signals leading to Ca2+-release from intracellular stores although the influx of Ca2+ was enhanced independently of the replenishment of these stores as indicated by the studies with thapsigargin.
FIGURE 8.
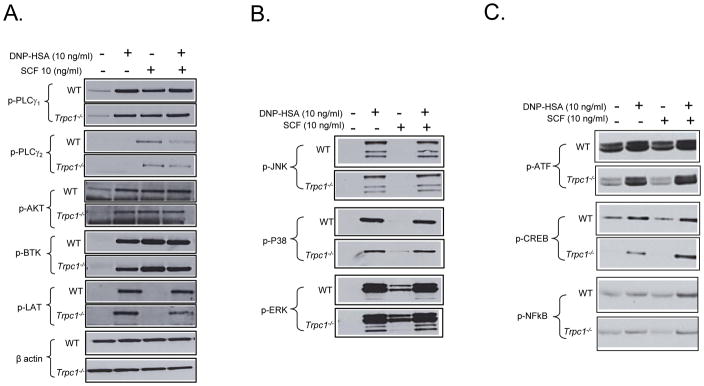
Effect of TRPC1 deficiency on phoshorylation of cytoplasmic signaling molecules regulating calcium signaling (A) or transcriptional regulation (B,C) important for mast cell activation.. Anti-DNP-IgE-sensitized BMMCs were stimulated with DNP-HSA (10 ng/ml) and/or SCF (10 ng/ml) for 10 min (A) or 30 min (B,C). Phosphorylation of different signaling molecules was determined by immune-blot analysis as described in Materials and Methods. The blots are representative of three independent experiments.
3.9. TRPC1 and regulation of transcriptional pathways
To verify that the increase in cytokine production was related to the enhanced calcium signal in the Trpc1−/− BMMCs, we compared the activation of the calcium-regulated transcription signaling pathways with that of calcium-independent pathways. As shown in Figure 8B, there was no increase in the phosphorylation of JNK, p38 and ERK MAP kinases (Fig. 8B), and ATF, NFκB, and CREB (Fig. 8C). If anything, the phosphorylation of some of these factors was slightly lower in the Trpc1−/− BMMCs in response to either SCF or antigen, or the combination thereof.
In contrast to these results, transcription factors that are described to be regulated specifically by calcium via calmodulin-dependent kinase-II or calcineurin, namely JUN [37] (Fig. 9A and 9B) and NFAT [38–40] (Fig. 9C and 9D) respectively, were markedly enhanced in Trpc1−/− BMMCs. From the above data we conclude that the elevation in cytokine production observed in the Trpc1−/− BMMCs is likely a consequence of the elevated calcium signal and subsequent calcium-dependent activation of AP1 and NFAT transcriptional pathways.
FIGURE 9.

Effect of TRPC1 deficiency on phoshorylation of the calcium-regulated transcription factors JUN (A,B) or NFAT (B,D). In C and D, the blots in A and B were respectively scanned to quantitate. These values were then normalized to the responses obtained with Ag alone in the WT cells. The blots in A and B are representative of 6–7 independent experiments. The data in B and D are presented as the mean +/− SEM of these data. **p<0.01, **P<0.001, 2 tailed Student’s t-test).
4. Discussion
To investigate the role of TRPC1 in mast cell function, we have used Trpc1−/− mice and TRPC1-deficient BMMC derived from these mice as experimental models. This allowed comparisons with previous studies where reduction of TRPC1 expression was achieved by knockdown of TRPC1 with siRNAs. Contrary to conclusions drawn from the knockdown studies, we find that TRPC1 deficiency augmented FcεRI-mediated signaling events (Fig. 9) leading to cytokine production in BMMCs (Fig. 4) and, in mice, a delayed recovery from antigen-induced hypothermia (Fig. 1), which was used as a proxy for systemic anaphylaxis. The notable observations in TRPC1-deficient BMMCs were delayed maturation (Fig 3), an enhanced calcium signal (Fig. 5) and an increase in Ca2+-dependent activation of NFAT and the AP1 component, JUN (Fig. 9). Other signalling events such as activation of the PI 3-kinase/Akt MAP kinases, and NFκB (Fig. 7) pathways were minimally, if at all, affected. Production of cytokines including IL-6, GM-CSF, and TNFα (Fig. 4) were substantially increased but paradoxically degranulation was not affected (Fig. 4). The accumulation of TNF-α in the circulation was also markedly elevated in Trpc1−/− mice (Fig. 2) and this appeared in concert with the delayed recovery from hypothermia (Fig. 1).
The delayed recovery in body temperature in antigen-challenged Trpc1 −/− mice was not apparent following injection of an equally effective concentration of histamine (Fig. 1). Histamine is thought to contribute the decline in body temperature during severe anaphylaxis [23] and, indeed, accelerated clearance of histamine is associated with an accelerated recovery from hypothermia during anaphylaxis in Sphk2−/− mice or after injection of sphingosine 1-phosphate [25]. However, in our studies, the delayed recovery could be effectively eliminated by injection of an anti-TNF-α antibody, providing evidence that TNF-α helped sustain the decline in temperature, especially at later stages of anaphylaxis. This observation is consistent with reports that TNF-α production is associated with late-phase anaphylactic reactions [27, 41] and mast cell-dependent hypothermia [28]; as well as our finding of higher circulating levels of TNF-α in anaphylactic Trpc1−/− mice. Collectively, the in vivo data indicates that TRPC1-deficiency enhances the anaphylactic response as a consequence of release of mast cell mediators, primarily TNF-α. Together with the observations in BMMC cultures, we conclude that, in mast cells, TRPC1-deficiency results in an amplified influx of Ca2+ and, as a consequence, enhancement of Ca2+-dependent signals and production of TNF-α and other cytokines. In this regard, TRPC1 appears to be a negative regulator of SOCE.
The participation of TRPCs in SOCE remains controversial. One view is that interaction of the endoplasmic reticulum (ER) Ca2+-sensor STIM1 with Orai proteins at the plasma membrane mediates SOCE [42, 43] and, indeed, these two proteins are absolutely essential for SOCE in various types of cells including mast cells [10]. This exclusive view has been challenged, however, on the basis that divalent cations such as Sr2+ and Mn2+ readily enter mast cells along with Ca2+ upon stimulation whereas conductance through the Orai/STIM complex (ICRAC, for calcium-release-activated current) is highly selective for Ca2+ [10, 33]. The alternate view is that TRPCs also interact with the STIM1/Orai1 complex to form channels with distinctive currents, typically referred to as ISOC (for store-operated calcium current), that are permeable to Sr2+ and Mn2+ as well as Ca2+ [44]. Several TRPCs have been considered as candidates for mediating ISOC via STIM/TRPC/Orai complexes of which TRPC1 has been the most widely studied in this regard [44].
TRPC1 is expressed in rodent mast cell lines along with other TRPCs [10, 13, 45] and another model is that TRPC1 is critical for localized Ca2+-entry and wave initiation within cell protrusions of stimulated mast cells. Knockdown of TRPC1 blocks these events [12] and degranulation at these sites [46]. It is postulated that localized Ca2+ entry through TRPC1 amplifies IP3-induced Ca2+-release from local ER Ca2+-stores within protrusions to promote the initial wave of Ca2+ which then propagates into the cell body to initiate IP3/SOCE-dependent Ca2+ oscillations throughout the cell [12] which leads to more global degranulation from the cell body [46]. Fyn is reported to be required for expression and function of TRPC1 as well as the ensuing Ca2+ influx and degranulation as these are diminished in Fyn −/− BMMC [13].
Other studies, as well as our study, suggest that TRPC1 can alternatively play a negative role in modulating SOC channel activity. For example, heterologous expression in HEK293 cells showed that TRPC1 by itself is unable to form functional tetrameric channels but is capable of forming heteromeric complexes with other individual TRPC subfamily members and by incorporating into such heteromeric complexes reduces Ca2+ permeability [47]. Moreover, knockdown of TRPC1 in neuronal Gn11 cells that normally express TRPC1 along with other TRPCs, enhanced Ca2+ influx in response to G protein-coupled receptor ligands. Indirect inhibitory mechanisms may also come into play and include effects on feed-back inhibition at high cytosolic levels of Ca2+ and on plasma membrane depolarization. TRPC1 is subject to Ca2+-dependent inactivation through a unique calmodulin-binding domain2 (CaMBD2) [48] and STIM/Orai-mediated ICRAC is inhibited at high cytosolic Ca2+ concentrations by several mechanisms [49] Therefore, the possibility exists that TRPC1 enhances Ca2+-dependent inactivation when complexed with STIM and Orai and thus negatively regulates channel activity when cytosolic Ca2+ is elevated. Ca2+-influx in mast cells is absolutely dependent on maintenance of the electrochemical polarity of the plasma membrane for which several mechanisms have been implicated to compensate for the depolarizing effect of influx of Ca2+ (reviewed in [33]) and, in the case of TRPC1, Na+ as well. Here, compensating repolarizing currents may be more effective in the absence of TRPC1 and might explain the differential effects of delayed addition of Ca2+ (Fig 5A versus 5B) where ion influx would be transient and oscillatory in antigen-stimulated cells and sustained in thapsigargin-stimulated cells. Electrophysiological studies are needed to clarify which, if any, of the above scenarios contribute to the amplification in Ca2+ influx in Trpc1−/− BMMCs.
The reason why amplification of the calcium signal augments cytokine production but not degranulation is also unclear but production of TNF-α and IL-6 are highly dependent on activation of NFAT and AP1 [39] as well as the strength and duration of signals in mast cells [50]. Our data (Figure 7) also reveals an absolute dependency of antigen-mediated TNF-α production on intracellular calcium concentrations. Optimal degranulation, in contrast, requires a relatively modest increase in cytosolic Ca2+ [51] and is dependent on the spatiotemporal characteristics of this increase [46] which may remain optimal either in the absence or presence of TRPC1.
In conclusion, our studies are consistent with a negative role for TRPC1 in regulating SOCE in mast cells and that TNF-α rather than histamine, is the likely mediator of delayed anaphylactic hypothermia. However, anaphylaxis and allergic diseases involve multiple cellular targets of mast cell mediators where TRPC1 may instead enhance responses of target cells and pathology as exemplified in a mouse model of asthma [52]. Our results are also at variance with the positive role for TRPC1 inferred from previous studies with BMMCs after knockdown of TRPC1 expression with siRNA technology. This discrepancy may relate to an incomplete and transient depletion of TRPC1 on knockdown as compared permanent deletion of TRPC1 in our studies which could permanently alter phenotypic characteristics in terms of expression of signaling molecules and other regulatory proteins. The variation and phenotypic plasticity in mast cells from different tissues [8, 53] and even mast cell lines [54–56] is well recognized, but the present studies also highlight caveats in the use of these different genetic approaches in studies of mast cell signaling and function. Even so, we believe that our results illustrate the key role of TNF-α in sustaining anaphylactic reactions, in particular hypothermia, and its potential as a therapeutic target.
Acknowledgments
Research in the authors’ laboratories was supported by funding from the Intramural Research programs within NIAID (N.M., A.D., A.M.G., D.D.M.), the NHLBI (M.A.B), the NIAMS (A.O) and NIEHS (project Z01-ES101864 to LB).
Abbreviations
- Ag
Antigen
- BMMCs
bone marrow-derived mast cells
- DNP
dinitrophenyl
- FcεRI
high affinity receptor for IgE
- Hu
human
- MC
mast cell
- SCF
stem cell factor
- TRPC
Transient receptor potential channel
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Birnbaumer L. The TRPC class of ion channels: A critical review of their roles in slow, sustained increases in intracellular Ca2+ concentrations. Annu Rev Pharmacol Toxicol. 2009;49:395–426. doi: 10.1146/annurev.pharmtox.48.113006.094928. [DOI] [PubMed] [Google Scholar]
- 2.Montell C. The history of TRP channels, a commentary and reflection. Pflugers Arch. 2011;461:499–506. doi: 10.1007/s00424-010-0920-3. [DOI] [PubMed] [Google Scholar]
- 3.Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev. 2010;62:381–404. doi: 10.1124/pr.110.002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nilius B, Voets T, Peters J. TRP channels in disease. Sci STKE. 2005;2005:re8. doi: 10.1126/stke.2952005re8. [DOI] [PubMed] [Google Scholar]
- 5.Colsoul B, Nilius B, Vennekens R. On the putative role of transient receptor potential cation channels in asthma. Clin Exp Allergy. 2009;39:1456–1466. doi: 10.1111/j.1365-2222.2009.03315.x. [DOI] [PubMed] [Google Scholar]
- 6.Banner KH, Igney F, Poll C. TRP channels: emerging targets for respiratory disease. Pharmacol Ther. 2011;130:371–384. doi: 10.1016/j.pharmthera.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Vennekens R, Olausson J, Meissner M, Bloch W, Mathar I, Philipp SE, Schmitz F, Weissgerber P, Nilius B, Flockerzi V, Freichel M. Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat Immunol. 2007;8:312–320. doi: 10.1038/ni1441. [DOI] [PubMed] [Google Scholar]
- 8.Gilfillan AM, Beaven MA. Regulation of mast cell responses in health and disease. Crit Rev Immunol. 2011;31:475–529. doi: 10.1615/critrevimmunol.v31.i6.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsai M, Grimbaldeston M, Galli SJ. Mast cells and immunoregulation/immunomodulation. Adv Exp Med Biol. 2011;716:186–211. doi: 10.1007/978-1-4419-9533-9_11. [DOI] [PubMed] [Google Scholar]
- 10.Ma HT, Peng Z, Hiragun T, Iwaki S, Gilfillan AM, Beaven MA. Canonical transient receptor potential 5 channel in conjunction with Orai1 and STIM1 allows Sr2+ entry, optimal influx of Ca2+, and degranulation in a rat mast cell line. J Immunol. 2008;180:2233–2239. doi: 10.4049/jimmunol.180.4.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins SR, Meyer T. Evolutionary origins of STIM1 and STIM2 within ancient Ca2+ signaling systems. Trends Cell Biol. 2011;21:202–211. doi: 10.1016/j.tcb.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen R, Torres A, Ma HT, Holowka D, Baird B. Ca2+ Waves initiate antigen-stimulated Ca2+ responses in mast cells. J Immunol. 2009;183:6478–6488. doi: 10.4049/jimmunol.0901615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki R, Liu X, Olivera A, Aguiniga L, Yamashita Y, Blank U, Ambudkar I, Rivera J. Loss of TRPC1-mediated Ca2+ influx contributes to impaired degranulation in Fyn-deficient mouse bone marrow-derived mast cells. J Leukoc Biol. 2010;88:863–875. doi: 10.1189/jlb.0510253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Radinger M, Jensen BM, Kuehn HS, Kirshenbaum A, Gilfillan AM. Generation, isolation, and maintenance of human mast cells and mast cell lines derived from peripheral blood or cord blood. Curr Protoc Immunol Chapter. 2010;7(Unit 7):37. doi: 10.1002/0471142735.im0737s90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dietrich A, Kalwa H, Storch U, Schnitzler M, Salanova B, Pinkenburg O, Dubrovska G, Essin K, Gollasch M, Birnbaumer L, Gudermann T. Pressure-induced and store-operated cation influx in vascular smooth muscle cells is independent of TRPC1. Pflugers Arch. 2007;455:465–477. doi: 10.1007/s00424-007-0314-3. [DOI] [PubMed] [Google Scholar]
- 16.Dietrich A, Mederos YSM, Gollasch M, Gross V, Storch U, Dubrovska G, Obst M, Yildirim E, Salanova B, Kalwa H, Essin K, Pinkenburg O, Luft FC, Gudermann T, Birnbaumer L. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finkelman F, Morris S, Orekhova T, Sehy D. The in vivo cytokine capture assay for measurement of cytokine production in the mouse. Curr Protoc Immunol Chapter. 2003;6(Unit 6):28. doi: 10.1002/0471142735.im0628s54. [DOI] [PubMed] [Google Scholar]
- 18.Jensen BM, Swindle EJ, Iwaki S, Gilfillan AM. Generation, isolation, and maintenance of rodent mast cells and mast cell lines. Curr Protoc Immunol Chapter. 2006;3:3.23.21–23.23.13. doi: 10.1002/0471142735.im0323s74. [DOI] [PubMed] [Google Scholar]
- 19.Kirshenbaum AS, Metcalfe DD. Growth of human mast cells from bone marrow and peripheral blood-derived CD34 + pluripotent progenitor cells. Methods Mol Biol. 2006;315:105–112. doi: 10.1385/1-59259-967-2:105. [DOI] [PubMed] [Google Scholar]
- 20.Choi OH, Lee JH, Kassessinoff T, Cunha-Melo JR, Jones SV, Beaven MA. Carbachol and antigen mobilize calcium by similar mechanisms in a transfected mast cell line (RBL-2H3 cells) that expresses m1 muscarinic receptors. J Immunol. 1993;151:5586–5595. [PubMed] [Google Scholar]
- 21.Hundley TR, Gilfillan AM, Tkaczyk C, Andrade MV, Metcalfe DD, Beaven MA. Kit and FcεRI mediate unique and convergent signals for release of inflammatory mediators from human mast cells. Blood. 2004;104:2410–2417. doi: 10.1182/blood-2004-02-0631. [DOI] [PubMed] [Google Scholar]
- 22.Tkaczyk C, Metcalfe DD, Gilfillan AM. Determination of protein phosphorylation in FcεRI-activated human mast cells by immunoblot analysis requires protein extraction under denaturing conditions. J Immunol Methods. 2002;268:239–243. doi: 10.1016/s0022-1759(02)00210-7. [DOI] [PubMed] [Google Scholar]
- 23.Makabe-Kobayashi Y, Hori Y, Adachi T, Ishigaki-Suzuki S, Kikuchi Y, Kagaya Y, Shirato K, Nagy A, Ujike A, Takai T, Watanabe T, Ohtsu H. The control effect of histamine on body temperature and respiratory function in IgE-dependent systemic anaphylaxis. J Allergy Clin Immunol. 2002;110:298–303. doi: 10.1067/mai.2002.125977. [DOI] [PubMed] [Google Scholar]
- 24.Herwig A, Ivanova EA, Lydon H, Barrett P, Steinlechner S, Loudon AS. Histamine H3 receptor and orexin A expression during daily torpor in the Djungarian hamster (Phodopus sungorus) J Neuroendocrinol. 2007;19:1001–1007. doi: 10.1111/j.1365-2826.2007.01620.x. [DOI] [PubMed] [Google Scholar]
- 25.Olivera A, Eisner C, Kitamura Y, Dillahunt S, Allende L, Tuymetova G, Watford W, Meylan F, Diesner SC, Li L, Schnermann J, Proia RL, Rivera J. Sphingosine kinase 1 and sphingosine-1-phosphate receptor 2 are vital to recovery from anaphylactic shock in mice. J Clin Invest. 2010;120:1429–1440. doi: 10.1172/JCI40659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon JR, Galli SJ. Release of both preformed and newly synthesized tumor necrosis factor alpha (TNF-α)/cachectin by mouse mast cells stimulated via the FcεRI. A mechanism for the sustained action of mast cell-derived TNF-α during IgE-dependent biological responses. J Exp Med. 1991;174:103–107. doi: 10.1084/jem.174.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang NI, Kim HK, Ko HM, Kim JH, You HJ, Choi IW, Im SY, Lee HK. Tumor necrosis factor-α develops late anaphylactic reaction through cytosolic phospholipase A2 activation. Int Arch Allergy Immunol. 2008;147:315–322. doi: 10.1159/000144039. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi T, Cottam HB, Chan M, Jin G, Tawatao RI, Crain B, Ronacher L, Messer K, Carson DA, Corr M. Mast cell-dependent anorexia and hypothermia induced by mucosal activation of Toll-like receptor 7. Am J Physiol Regul Integr Comp Physiol. 2008;295:R123–132. doi: 10.1152/ajpregu.00527.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leon LR, White AA, Kluger MJ. Role of IL-6 and TNF in thermoregulation and survival during sepsis in mice. Am J Physiol. 1998;275:R269–277. doi: 10.1152/ajpregu.1998.275.1.R269. [DOI] [PubMed] [Google Scholar]
- 30.Tajeddine N, Gailly P. TRPC1 protein channel is major regulator of epidermal growth factor receptor signaling. J Biol Chem. 2012;287:16146–16157. doi: 10.1074/jbc.M112.340034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madsen CP, Klausen TK, Fabian A, Hansen BJ, Pedersen S, Hoffmann EK. On the role of TRPC1 in control of Ca2+ influx, cell volume and cell cycle. Am J Physiol Cell Physiol. 2012 doi: 10.1152/ajpcell.00287.2011. [DOI] [PubMed] [Google Scholar]
- 32.Iwaki S, Tkaczyk C, Satterthwaite AB, Halcomb K, Beaven MA, Metcalfe DD, Gilfillan AM. Btk plays a crucial role in the amplification of FcεRI-mediated mast cell activation by Kit. J Biol Chem. 2005;280:40261–40270. doi: 10.1074/jbc.M506063200. [DOI] [PubMed] [Google Scholar]
- 33.Ma HT, Beaven MA. Regulators of Ca2+ signaling in mast cells: potential targets for treatment of mast cell-related diseases? Adv Exp Med Biol. 2011;716:62–90. doi: 10.1007/978-1-4419-9533-9_5. [DOI] [PubMed] [Google Scholar]
- 34.Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J Gen Physiol. 1995;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Selli C, Erac Y, Kosova B, Tosun M. Post-transcriptional silencing of TRPC1 ion channel gene by RNA interference upregulates TRPC6 expression and store-operated Ca2+ entry in A7r5 vascular smooth muscle cells. Vascul Pharmacol. 2009;51:96–100. doi: 10.1016/j.vph.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 36.Erac Y, Selli C, Kosova B, Akcali KC, Tosun M. Expression levels of TRPC1 and TRPC6 ion channels are reciprocally altered in aging rat aorta: implications for age-related vasospastic disorders. Age (Dordr) 2010;32:223–230. doi: 10.1007/s11357-009-9126-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saxena M, Busca A, Pandey S, Kryworuchko M, Kumar A. CpG protects human monocytic cells against HIV-Vpr-induced apoptosis by cellular inhibitor of apoptosis-2 through the calcium-activated JNK pathway in a TLR9-independent manner. J Immunol. 2011;187:5865–5878. doi: 10.4049/jimmunol.1100115. [DOI] [PubMed] [Google Scholar]
- 38.Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol. 2008;9:81–88. doi: 10.1038/ni1546. [DOI] [PubMed] [Google Scholar]
- 39.Andrade MV, Iwaki S, Ropert C, Gazzinelli RT, Cunha-Melo JR, Beaven MA. Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur J Immunol. 2011;41:760–772. doi: 10.1002/eji.201040718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gwack Y, Feske S, Srikanth S, Hogan PG, Rao A. Signalling to transcription: store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium. 2007;42:145–156. doi: 10.1016/j.ceca.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 41.Choi IW, Kim YS, Kim DK, Choi JH, Seo KH, Im SY, Kwon KS, Lee MS, Ha TY, Lee HK. Platelet-activating factor-mediated NF-κB dependency of a late anaphylactic reaction. J Exp Med. 2003;198:145–151. doi: 10.1084/jem.20022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shaw PJ, Feske S. Physiological and pathophysiological functions of SOCE in the immune system. Front Biosci (Elite Ed) 2012;4:2253–2268. doi: 10.2741/540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di Capite JL, Bates GJ, Parekh AB. Mast cell CRAC channel as a novel therapeutic target in allergy. Curr Opin Allergy Clin Immunol. 2011;11:33–38. doi: 10.1097/ACI.0b013e32834232b0. [DOI] [PubMed] [Google Scholar]
- 44.Cheng KT, Ong HL, Liu X, Ambudkar IS. Contribution of TRPC1 and Orai1 to Ca2+ entry activated by store depletion. Adv Exp Med Biol. 2011;704:435–449. doi: 10.1007/978-94-007-0265-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanchez-Miranda E, Ibarra-Sanchez A, Gonzalez-Espinosa C. Fyn kinase controls FcεRI receptor-operated calcium entry necessary for full degranulation in mast cells. Biochem Biophys Res Commun. 2010;391:1714–1720. doi: 10.1016/j.bbrc.2009.12.139. [DOI] [PubMed] [Google Scholar]
- 46.Cohen R, Corwith K, Holowka D, Baird B. Spatiotemporal resolution of mast cell granule exocytosis reveals correlation with Ca2+ wave initiation. J Cell Sci. 2012;125:2986–2994. doi: 10.1242/jcs.102632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Storch U, Forst AL, Philipp M, Gudermann T, Mederos y Schnitzler M. Transient receptor potential channel 1 (TRPC1) reduces calcium permeability in heteromeric channel complexes. J Biol Chem. 2012;287:3530–3540. doi: 10.1074/jbc.M111.283218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh BB, Liu X, Tang J, Zhu MX, Ambudkar IS. Calmodulin regulates Ca2+-dependent feedback inhibition of store-operated Ca2+ influx by interaction with a site in the C terminus of TrpC1. Mol Cell. 2002;9:739–750. doi: 10.1016/s1097-2765(02)00506-3. [DOI] [PubMed] [Google Scholar]
- 49.Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nature Rev Mol Cell Biol. 2012;13:549–565. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez-Espinosa C, Odom S, Olivera A, Hobson JP, Martinez ME, Oliveira-Dos-Santos A, Barra L, Spiegel S, Penninger JM, Rivera J. Preferential signaling and induction of allergy-promoting lymphokines upon weak stimulation of the high affinity IgE receptor on mast cells. J Exp Med. 2003;197:1453–1465. doi: 10.1084/jem.20021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beaven MA, Ozawa K. Role of calcium, protein kinase C, and MAP kinase in the activation of mast cells. Allergol Internat. 1996;45:73–84. [Google Scholar]
- 52.Yildirim E, Carey MA, Card JW, Dietrich A, Flake GP, Zhang Y, Bradbury JA, Rebolloso Y, Germolec DR, Morgan DL, Zeldin DC, Birnbaumer L. Severely Blunted Allergen-Induced Pulmonary Th2-cell Response and Lung Hyperresponsiveness in Type 1 Transient Receptor Potential Channel (TRPC1)-Deficient Mice. Am J Physiol Lung Cell Mol Physiol. 2012 doi: 10.1152/ajplung.00389.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moon TC, St Laurent CD, Morris KE, Marcet C, Yoshimura T, Sekar Y, Befus AD. Advances in mast cell biology: new understanding of heterogeneity and function. Mucosal Immunol. 2010;3:111–128. doi: 10.1038/mi.2009.136. [DOI] [PubMed] [Google Scholar]
- 54.Wong GW, Tang Y, Feyfant E, Sali A, Li L, Li Y, Huang C, Friend DS, Krilis SA, Stevens RL. Identification of a new member of the tryptase family of mouse and human mast cell proteases which possesses a novel COOH-terminal hydrophobic extension. J Biol Chem. 1999;274:30784–30793. doi: 10.1074/jbc.274.43.30784. [DOI] [PubMed] [Google Scholar]
- 55.Yamashita Y, Charles N, Furumoto Y, Odom S, Yamashita T, Gilfillan AM, Constant S, Bower MA, Ryan JJ, Rivera J. Cutting edge: genetic variation influences FcεRI-induced mast cell activation and allergic responses. J Immunol. 2007;179:740–743. doi: 10.4049/jimmunol.179.2.740. [DOI] [PubMed] [Google Scholar]
- 56.Tshori S, Razin E. Editorial: Mast cell degranulation and calcium entry--the Fyn-calcium store connection. J Leukoc Biol. 2010;88:837–838. doi: 10.1189/jlb.0610365. [DOI] [PubMed] [Google Scholar]