

Abstract
Toll-like receptors (TLRs) are type I transmembrane proteins that are key regulators of both innate and adaptive immune responses. To protect the host from viral and bacterial threats, TLRs trigger a pro-inflammatory immune response by detecting pathogen and danger associated molecular patterns. Considerable evidence has accumulated to show that the dysregulation of TLR signaling contributes to the development and progression of numerous diseases. Therefore, TLRs are emerging as important drug discovery targets. Currently, there is great interest in the development of TLR small molecule modulators for interrogating TLR signaling and treating diseases caused by TLR signaling malfunctions. In this Tutorial Review, we will outline methods for the discovery of TLR small molecule modulators and the up-to-date progress in this field. Small molecules targeting TLRs not only provide an opportunity to identify promising drug candidates, but also unveil knowledge regarding TLR signaling pathways.
1. Introduction
Toll-like receptors (TLRs) are evolutionarily conserved type I transmembrane proteins that detect pathogen associated molecular patterns (PAMPs) and danger associated molecular patterns (DAMPs).1 The ability to sense these molecular patterns make TLRs key regulators of both innate and adaptive immune responses. TLRs are comprised of an extracellular domain containing leucine-rich repeats (LxxLxLxxN, LRRs) and an intracellular Toll/interleukin-1 receptor (TIR) domain. All TLR ectodomains share a common horseshoe structure, but different amino acid composition allows for the recognition of a diverse set of PAMPs and DAMPs (Table 1). Upon ligand recognition, TLR homo-or heterodimerizes, which in turn triggers signaling, activates the intracellular TIR domain. TIR domain recruitment of MyD88 and other adaptor proteins activates signal transduction cascades, which culminate in the production of pro-inflammatory cytokines and chemokines. TLRs are named “Toll-like receptors” due to their structural and functional similarities to the protein encoded by the toll gene in Drosophila.2 The word “toll” means “great” in German, referring to an intriguing gene whose mutations make the Drosophila look unusual, and has nothing to do with road and bridge tolls.
Table 1.
Pathogen associated molecule patterns (PAMPs) and danger associated molecule patterns (DAMPs) that TLRs recognize
| TLRs | Localization | PAMPs/DAMPs |
|---|---|---|
| TLR1/TLR2 | Plasma membrane | Triacylated lipoprotein, peptidoglycan, saturated fatty acids |
| TLR2 | Plasma membrane | Peptidoglycan, polysaccharide krestin, β-defensin 3, phospholipomannan, serum amyloid A, haemagglutinin, biglycan, porins, lipoarabinomannan, Gp96, HMGB1, glucuronoxylomannan, anti-phospholipid antibodies, HSP 60/70, surfactant proteins A and D, eosinophil-derived neurotoxin |
| TLR3 | Endosome membrane | dsRNA |
| TLR4 | Plasma membrane/Endosome membrane | LPS, heme, saturated fatty acids, VSV glycoprotein G, RSV fusion protein, resistin, biglycan, lactoferrin, MMTV envelope protein, mannan, glucuronoxylomannan, Gp96, nickel, S100, glycosylinositolphospholipids, chitohexaose, FimH, HSP60/70/72/22, surfactant proteins A and D, serum amyloid A, fibrinogen, β-defensin 2, hyaluronan acid fragment, heparin sulfate fragment, HMGB1, neutrophil elatase, wheat amylase trypsin inhibitors |
| TLR4/TLR6 | Plasma membrane | OxLDL, amyloid-β fibril |
| TLR5 | Plasma membrane | flagellin |
| TLR2/TLR6 | Plasma membrane | Diacylated lipopeptides, lipoteichoic acid, zymosan, saturated fatty acids |
| TLR7 | Endosome membrane | ssRNA, Anti-phospholipids antibodies, RNA associated auto-antigens |
| TLR8 | Endosome membrane | ssRNA, Anti-phospholipids antibodies |
| TLR9 | Endosome membrane | Unmethylated CpG DNA, IgG-chromatin complexes |
| TLR10a | Plasma membrane | unknown |
| TLR11b | Endosome membrane | profilin-like protein |
| TLR12b | Endosome membrane | unknown |
| TLR13b | Endosome membrane | 23s rRNA |
| TLR14c | Plasma membrane | unknown |
| TLR15d | Plasma membrane | unknown |
| TLR16e | Endosome membrane | unknown |
| TLR21-23f | unknown | unknown |
Note:
Mice lack TLR10;
TLRs 11–13 are represented in human by a pseudogene and are not functional;
TLR14 is present in fish, not in human or mice;
TLR15 is present in chickens, not in human or mice;
TLR16 is only found in Xenopus;
TLR21-23 are present in fish and frogs.
TLRs are highly conserved in vertebrates and invertebrates. TLRs 1, 2, 4,5, 6, and 10 are expressed on the plasma membrane, whereas TLRs 3, 7, 8, 9, 11, 12, and 13 are present in intracellular vesicles. This variety in localization allows TLRs to protect the host against threats present in the extracellular environment as well as those internalized. In vertebrates, TLRs are comprised of 6 major families, TLR1 (TLR1, 2, 6, 10, 14 and 15), TLR3, TLR4, TLR5, TLR7 (TLR7, 8, 9) and TLR11 (TLR11-13 in mice).3 Humans have 10 functional TLRs (TLR1-10), whereas twelve functional TLRs (TLR1-9 and TLR11-13) have been identified in mice. Native ligands have been identified for TLR1-9 as well as TLR13.
Human and animal genetic studies have shown that the dysregulation of innate immune TLR signaling contributes to the development and progression of various diseases including sepsis, autoimmune disease, and neuropathic pain (Table 2), a topic that has been extensively reviewed in the literature.4, 5 Due to their significant biomedical relevance, TLRs have emerged as important drug targets.6 Currently, there is great interest in the development of TLR small molecule modulators (Table 3) for interrogating TLR signaling and treating diseases caused by TLR signaling malfunctions. In this Tutorial Review, we will summarize methods for the discovery of small molecule TLR modulators by highlighting representative small molecule, peptide, and oligonucleotide agents that target various TLRs. To keep a focused scope, small molecules targeting downstream mediators (e.g. TAK-1, TBK-1, IRF3, NF-κB, Akt and MAPKs) will not be covered.
Table 2.
TLR family proteins as targets in drug development
| Disease | TLR involved | Therapeutic approach |
|---|---|---|
| Sepsis | TLR2/4/9 | Antagonist |
| Asthma | TLR7/8/9 | Agonist |
| Acute/chronic inflammation | TLR2/4 | Antagonist |
| Chronic obstructive pulmonary disease | TLR2/4 | Antagonist |
| Cardiovascular disease | TLR2/4 | Antagonist |
| Neuropathic pain, chronic pain | TLR4 | Antagonist |
| Autoimmune diseases (including systemic lupus erythematosus, rheumatoid arthritis, systemic sclerosis, Sjögren’s syndrome) | TLR1-9 | Antagonist |
| Cancer (including colon cancer, gastric cancer, breast cancer, melanoma, hepatocellular carcinoma, lung cancer, glioma, prostate cancer, ovarian cancer, cervical squamous cell carcinomas, chronic lymphocytic leukemia) | TLR1-9 | Agonist |
Table 3.
Small molecule TLR modulators
| TLRs | Small molecule agonist | Small molecule antagonist |
|---|---|---|
| TLR1/TLR2 | Pam3CSK4 | Compounds 1–5 |
| TLR3 | Poly (I:C), Poly (A:U), IPH-3102, Rintatolimod | Compound 6 |
| TLR4 | LPS, Lipid A, CRX-547, CRX-527, E6020, morphine-3-glucuronide, saturated fatty acids, compounds 11, 19 | Lipid A mimetic (D1), SPA4, STM28, Eritoran, E5531, xanthohumol, JTT-705, auranofin, sulforaphane, cinnamaldehyde, taxanes, 6-shogaol, isoliquiritigenin, OSL07, glycyrrhizin, isoliquiritigenin, caffeic acid phenethyl ester, compounds 10, 12–18, 20–33 |
| TLR5 | Flagellin-derived peptides | None |
| TLR2/TLR6 | FSL1, compounds 34–37 | None |
| TLR7 | Isatoribine, Loxoribine, Imiquimod, gardiquimod, AZD8848, IMO-8400, ANA773, IMO-3100, SM360320, 852A | 2′-O-methyl-modified RNAs, IRS-954 (DV-1079) |
| TLR8 | VTX-1463, VTX-2337, IMO-8400, 2,3-Diamino-furo[2,3-c] pyridine | VTX-763 |
| TLR9 | IMO-8400, IMO-3100, SAR-21609, AZD1419, SD-101, IMO-2055, IMO-2125, QAX-935, AVE0675, DIMS0150, MGN-1703, MGN-1706, ISS1018, Agatolimod | IRS-954 (DV-1079) |
| TLR13 | ACGGAAAGACCCC | None |
Note: For the chemical structures of compounds 1–37, see Supplementary Table S1.
2. Discovery of small molecule modulators of TLRs
2.1 TLR ligand derivatives
TLRs recognize evolutionarily conserved PAMPs and DAMPs. Native ligands for all human TLRs except for TLR10 have been identified (Table 1), which has provided excellent templates for the development of small molecule modulators.
2.1.1 TLR3
TLR3 recognizes double stranded RNA (dsRNA), providing an important defense against viral infections. Polyinosinic-polycytidylic acid (poly(I:C)) and polyadenylic-polyuridylic acid (poly(A:U)) are synthetic analogues of dsRNA. Due to their specific TLR3 activity, these molecules are widely used as TLR3 agonists in research.7
Innate Pharma has developed a synthetic dsRNA agent, IPH-3102, as a specific TLR3 agonist.8 IPH-3102 does not have activity against the RNA helicase MDA-5/RIG-1 for which poly(I:C) shows stimulation activity. MDA-5/RIG-1 stimulation contributes to the in vivo toxicity of poly(I:C) due to overproduction of type I interferons. Further studies showed that IPH-3102 induces apoptosis in TLR3-overexpressing tumor cell lines and demonstrated significant anti-tumor efficacy in vivo.8 Since TLR3 is over-expressed by a large subset of patients in multiple cancers, IPH-3102 has been suggested as an immune-stimulating agent for potential cancer treatment.
2.1.2 TLR4
Lipopolysaccharides (LPS) are a group of natural ligands for TLR4. LPS presents on the cell membrane of Gram-negative bacteria. The generic structure of LPS consists of an outer core, inner core, and a lipid A motif. Lipid A activates TLR4 signaling by binding to the TLR4 accessory protein myeloid differentiation-2 (MD-2). Structural differences in lipid A, such as changes in the number and length of acyl chains and the functional groups at the bioisosteric position of the 1-phosphate, can affect immune response. These types of modifications can alter the activities of lipid A mimetics from canonical agonism to antagonism.9 Peri and co-workers have synthesized novel lipid A mimetics such as IAXO-101 (Supplementary Table S1, compound 10).10 These mimetics have been shown to inhibit LPS-stimulated TLR4 activation by preventing LPS from binding to the accessory proteins (e.g. CD14, MD-2) thereby inhibiting TLR4 signaling.10
Eritoran (E5564) is a lipid A analogue developed by Eisaias an investigational drug for the treatment of sepsis.11 Eritoran prevents septic over-inflammation by competing with LPS for binding to MD-2 (Fig. 1). However, a Phase III clinical trial of Eritoran in patients with sepsis failed due to the lack of efficacy. Nonetheless, the high-resolution structure of TLR4/MD-2/Eritoran reported by Lee and co-workers (Fig. 1) laid crucial groundwork for future development of TLR4 inhibitors.12
Fig. 1.
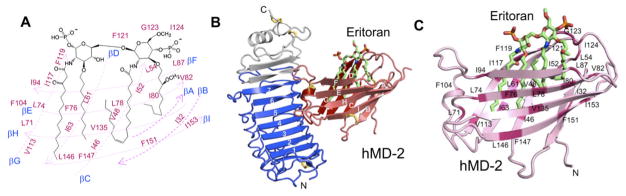
High-resolution X-ray crystal structure of the human TLR4/MD-2/Eritoran complex. (A) Chemical structure of Eritoran. (B) Overall structure of the TLR4/hMD-2/Eritoran complex (PDB ID 2Z65, resolution 2.70 Å). (C) A close-up view of the binding site of Eritoran on the MD-2 surface. This figure is reprinted with permission.12
Monophosphoryl lipid A (MPLA) (Supplementary Table S1, compound 11) is a low-toxicity derivative of LPS with beneficial immune-stimulatory activity.13 MPLA is an adapter-inducing, interferon-β (TRIF)-biased TLR4 agonist with lower toxicity due to a lesser production of pro-inflammatory factors.13, 14 As such, the US Food and Drug Administration (FDA) has approved MPLA for use as a human vaccine adjuvant. Over 33,000 doses of MPLA have been administered. Vaccine adjuvants combine antigens with drugs in order to increase the adaptive immune response. In fact, TLR agonists generally make excellent vaccine adjuvants because they activate both the innate and adaptive immune responses as well as escalate B and T cell immunity.
2.1.3 TLR2/TLR6
TLR2 recognizes lipoteichoic acid and diacylated lipopeptides from Gram-positive bacteria and yeast, in concert with TLR6 as a heterodimer. Pam2CSK4 and MALP-2 (Supplementary Table S1, compounds 34 and 35) are synthetic diacylated lipopeptides and potent TLR2/TLR6 agonists. For Pam2CSK4 agonism, the Cys-Ser dipeptide unit and at least one C16 acyl group with an appropriately oriented ester carbonyl group are essential.15, 16 While not required for agonist activity, four lysines are included in the structure of Pam2CSK4 to increase solubility. Further structure-activity relationship (SAR) studies have led to structurally simpler and water-soluble analogues (Supplementary Table S1, compounds 36 and 37) possessing strong TLR2-agonistic activities in human, but not in murine cells.15, 17
2.2 High throughput screening
2.2.1 In silico screening
In the last decade, significant progress has been made in TLR structural analysis.18 The structures of TLR/ligand complexes including TLR1/TLR2/Pam3CSK4, TLR3/dsRNA, TLR4/MD-2/LPS, TLR5/flagellin and TLR2/TLR6/Pam2CSK4 have been solved.19 These structures allow for the development of TLR small molecule modulators using in silico screening as an economical alternative.
MD-2 is an accessory protein of TLR4, with an indispensable role in LPS activation of TLR signaling.1 Among all TLRs, TLR4 is the only one that has been confirmed to form an active heterodimer with MD-2, although there is still speculation about whether TLR2 also binds to MD-2.20 Therefore, targeting the TLR4/MD-2 interaction is a promising strategy for the development TLR4-specific modulators. Yin and co-workers have developed a novel in silico screening methodology incorporating Molecular Mechanics (MM)/implicit solvent methods to evaluate binding free energies. This new method has been applied to the identification of disruptors of the TLR4/MD-2 complex.21 As a brief outline of this methodology, fast molecular docking is used to generate binding poses and subsequent molecular dynamics (MD) simulation is used to rank the ligand poses according to their binding affinities as implemented in QUANTUM. The selected hits were then counter-screened against approximately 500 representative human proteins to eliminate the non-specific inhibitors. T5342126 (Supplementary Table S1, compound 12) has been identified as a highly specific TLR4/MD-2 interaction disruptor (Fig. 2).21 Follow-up studies showed that T5342126 suppresses LPS-induced activation of the serine-threonine kinase, Akt-1,21 and pro-inflammatory factors induced by E. coli.22 Furthermore, T5342126 potentiates morphine-induced analgesia in vivo.23
Fig. 2.

The small-molecule inhibitor, T5342126, identified through virtual screening. (A) The chemical structure of T5342126. (B) The TLR4/MD-2 complex as the target for T5342126. (C) T5342126 binds to the same site on the TLR4 surface that the MD-2 protein recognizes. This figure is reprinted with permission.21
In search of TLR3 small-molecule antagonists, Yin and co-workers have performed in silico screening with the Enamine database (~1.2 million compounds) against the extracellular domain of TLR3.24 Analysis of the database using the Glide program identified nine initial hits. The majority of these identified hits shared a D-amino acid scaffold. This motif was suggested to represent a novel pharmacophore for targeting the RNA-binding site of TLR3.24 Further SAR studies lead to CU-CPT4a (Supplementary Table S1, compound 9) as a potent TLR3-specific antagonist. CU-CPT4a prevents dsRNA binding to TLR3, thereby blocking dsRNA-induced inflammation in murine macrophage RAW 264.7 cells.24
2.2.2 Target-based screening
In order to identify peptides directly targeting TLR4, Yang and co-workers used a yeast two-hybrid system to screen for TLR4 antagonists. The screen of a random 16-mer randomized peptide library of was conducted using full-length TLR4 as bait.25 Peptide No.9 (Supplementary Table S1, compound 13) has been found to suppress LPS-induced NF-κB activation, Iκ-Bα phosphorylation and the release of inflammatory factors IL-1, IL-6, and TNF-α.25 Further analysis showed peptide No.9 functions through specifically binding to the TLR4 extracellular domain.25
Peptides that reproduce the interactions between TLR4 and LPS are also potential TLR4 modulators. To identify peptidomimetics that target the TLR4/LPS interface, Tiwari and co-workers have screened a 7-mer phage-display peptide library.26 Experimental data showed that the LPS mimics are capable of inducing inflammatory cytokine secretion. Additionally, these mimics can act as TLR4 agonists and functional adjuvants for vaccine development.26
MyD88 mediates all TLR signaling pathways except for TLR3.1 Therefore, the TLR/MyD88 interaction is a general target for regulating TLR signaling. Tobias and co-workers have constructed a β-lactamase (Bla) fragment complementation system for identifying small molecules that disrupt the TLR4/MyD88 interaction.27 In this study, the authors developed a stable HeLa reporter cell line, expressing MyD88/Bla(a) and TLR4/Bla(b). When TLR signaling is activated, the two β-lactamase fragments complement each other by virtue of spontaneous TLR4/MyD88 binding via their TIR domains.27 Inhibition of MyD88/TLR4 binding leads to the disruption of enzyme complementation and a loss of lactamase activity. After screening approximately 16,000 compounds using this reporter system, 45 hits were identified.27 Five MyD88/TLR4 interaction disruptors (Supplementary Table S1, compounds 14–18) were selected after eliminating compounds that directly inhibit β-lactamase. These inhibitors specifically suppress LPS-induced pro-inflammatory factors, while showing no apparent cytotoxicity.27
The HEK 293 NF-κB reporter cells offer a superior system for TLR target-based screening because of its readiness for high throughput assay.1 All TLR pathways shares NF-κB as a common downstream signalling factor.1 NF-κB activity can be measured in HEK-Blue hTLR4 cells, which are stably transfected with human TLR4, CD14, MD-2 and a secreted embryonic alkaline phosphatase (SEAP) reporter gene. Watkins, Yin, and co-workers successfully employed this method to identify morphine (Supplementary Table S1, compound 19) as a TLR4 agonist.28 Further studies also showed that morphine binds to the TLR4 accessory protein MD-2 and induces neuro-inflammation.23 Additionally, naloxone and naltrexone (Supplementary Table S1, compounds 20 and 21) have been found to be TLR4 antagonists, which block LPS binding to MD-2.29 Interestingly, (+)-naloxone and (+)-naltrexone can reverse neuropathic pain and reduce opioid side effects such as addiction, tolerance, reinforcement, and constipation.29, 30 Another example of using the NF-κB reporter assay comes from Chow’s group who has run a high throughput screening assay in TLR2-transfected HEK-293 cells.31 They have established compounds 1–4 (Supplementary Table S1) as novel TLR1/TLR2 antagonists.31 However, compounds 1–4 also non-specifically inhibit TLR4 signaling.31
2.2.3 Phenotypic screening
Nitric oxide (NO) is an important pro-inflammatory factor in TLR signaling. Yin and co-workers have screened the National Cancer Institute (NCI) Diversity library (1,364 compounds) using a phenotypic assay that monitors the NO production in macrophage cells. CU-CPT22 (Supplementary Table S1, compound 5) has been identified as a TLR1/TLR2-specific antagonist that blocks Pam3CSK4 binding to the TLR1/TLR2 complex.32 In a similar fashion, NCI 126224 (Supplementary Table S1, compound 24) was identified as a potent TLR4 signaling inhibitor that suppresses LPS-induced NO over-production.50
Yamada and co-workers have screened the Takeda chemical library by testing the ability of compounds to inhibit TLR4 signaling in LPS-treated macrophages.33 The screen identified a novel cyclohexene derivative (Supplementary Table S1, compound 22) as a TLR4 inhibitor.33 Further optimization lead to TAK-242 (Supplementary Table S1, compound 23), which suppresses LPS-induced NO, TNF-α and IL-6 over-production with a sub-micromolar IC50 value. TAK-242 does not inhibit TLR1/TLR2, TLR2/TLR6, TLR3, TLR5, TLR7, TLR9 and IL-1 receptor, showing high selectivity.33–35 Further biochemical analysis showed that TAK-242 suppresses TLR4 signaling by directly binding Cys747 in the intracellular domain of TLR4, preventing its interactions with adaptor molecules.36, 37 Intravenous administration of TAK-242 (0.1 mg/kg) inhibits the over-production of pro-inflammatory factors in a dose dependent fashion when tested in a mouse endotoxin shock model.33 Even though its clinical trial was discontinued in Phase III due to lack of efficacy,38 TAK-242 has been widely used as a research tool to probe TLR4.
Phytochemicals and natural products are abundant with TLR modulators.39 The mitotic spindle inhibitor paclitaxel (Supplementary Table S1, compound 25) is derived from the bark of the Pacific yew.40 Marketed as Taxol, this drug is used to treat multiple forms of cancer and restenosis. Paclitaxel binds to MD-2 and activates TLR signaling, promoting apoptosis of cancer cells.40 Despite their structural similarities, the taxane family can inhibit TLR4 responses,41 demonstrating the difficulty of predicting the effect of small molecules on signaling. Heme (Supplementary Table S1, compound 26) groups found in blood hemoglobin can agonize TLR4 signaling.42 This ability is of biological relevance because blood clotting can result in the release of free heme into the bloodstream, stimulating the innate immune response to cell damage. Hwang and co-workers have found that curcumin (Supplementary Table S1, compound 27) inhibits LPS-induced pro-inflammation factors.43 Curcumin inhibits TLR4 activation by interfering with cysteine residue-mediated TLR4 receptor dimerization.43 Ravindran and co-workers have identified chitohexaose (Supplementary Table S1, compound 28), a small molecule polysaccharide, as a TLR4 agonist.44 Unlike LPS, Chitohexaose directly binds to TLR4 instead of its accessory protein MD-2. Chitohexaose activates TLR4 signaling through an alternate pathway that blocks LPS-induced overproduction of pro-inflammatory factors in vitro, and endotoxemia in vivo.44
2.3 Structure-based rational design
The X-ray crystal structure of the TLR4/MD-2 complex reveals critical molecular recognition sites on the TLR4/MD-2 interface. Kuroki and co-workers have developed a synthetic peptide that corresponds to the Glu(24)-Lys(47) binding site on TLR4. This peptide effectively suppresses the association of MD-2 and TLR4, attenuating LPS-induced NF-κB activation and IL-8 secretion.45 Likewise, Yin and co-workers designed a truncated 17-mer peptide (MD-2-I) (Supplementary Table S1, compound 29) that maintains TLR4-binding affinity of the full-length MD-2.46 This rationally designed peptide disrupts the TLR4/MD-2 interactions and blocks the TLR4-mediated inflammatory response both in vitro and in vivo.46, 47
TLRs and TLR adaptor proteins (e.g. MyD88, MAL, TRIF, TRAM, and SARM) contain TIR domains.18 TIR/TIR interactions mediate TLR dimerization and the recruitment of adaptor proteins, as well as initiating intracellular TLR signaling activation. The BB loop, a 14 amino acid region within the TIR domain, enables interactions between TLRs or between TLRs and their adaptor proteins. Vogel and co-workers have synthesized BB loop-derived decoy peptides based on the MyD88, TIRAP, TIRF and TRAM TIR domains (Supplementary Table S1, compounds 30–33).48 These BB loop peptides all inhibited TLR4 signaling with remarkable specificity.48 The effectiveness of decoy peptides as TLR inhibitors has been demonstrated by additional work on the TLR4 and MAL TIR domains. 49
3. Perspectives and conclusions
Mimicking the natural ligands of TLRs has proven to be a successful approach for the creation of TLR modulators. Ligand mimics have generated drug candidates currently under clinical trials. More recently, rational design has emerged as an attractive alternative approach. TLRs are very difficult to prepare in large quantities or to crystallize, making structural and biophysical experiments taxing. However, ground-breaking progress in TLR structural biology has been made in the past decade. The structures of TLRs 1–6 have all been determined in complex with their cognate ligands, which provides the basis for rational design or in silico docking. More sophisticated structures will provide opportunities for the further development of TLR signaling modulators. Currently, as the structures of TLRs 7–10 are not available, phenotypic screening and modification of natural ligand are still the most viable methods for these TLRs. It should be noted that each method for small molecule discovery has its advantages and disadvantages. Combinations of different approaches (e.g. phenotypic screening and structure-based rational design) may provide more desirable outcomes. The small molecules targeting TLRs not only provide the opportunity to identify promising drug candidates, but also enable novel biology to be unveiled.
Supplementary Material
Fig. 3.
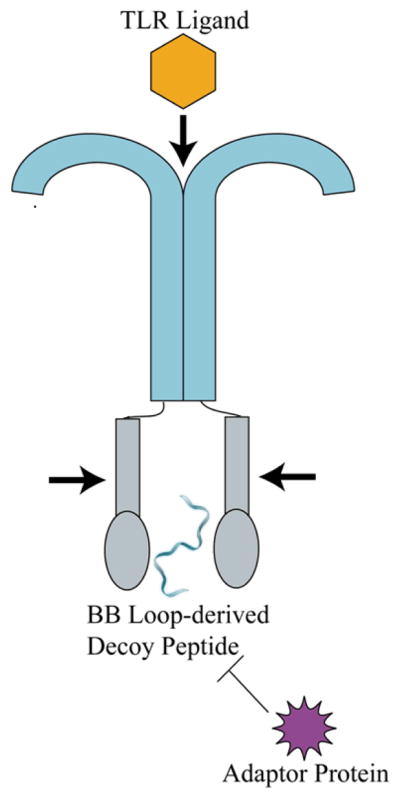
Schematic of BB loop-derived peptides interacting with TLRs. BB loop-derived decoy peptides bind to the TLR TIR domain, preventing the TLR dimerization and its downstream signaling.
Acknowledgments
We acknowledge the financial support from the National Institutes of Health (NIH GM101279). We thank James I. Godfroy III and Shuting Zhang for proofreading the manuscript.
Biographies

Hang Hubert Yin received his doctorate in chemistry from Yale University in 2004 under the supervision of Professor Andrew Hamilton. He joined the faculty of the University of Colorado at Boulder in 2007 from the University of Pennsylvania, where he was a postdoctoral fellow with Professor William DeGrado. His current research interests lie at the interface of chemistry, biology, and engineering with particular focuses on structure-based drug design, cell signaling biochemistry, and biotechnology development. Yin has received the NSF Career award, CAPA Distinguished Junior Faculty Award, AACR Elion Award, NIH CEBRA Award, and Early Career Award in Chemistry of Drug Abuse and Addiction.

Xiaohui Wang received his Bachelor of Engineering in 2004 from Beijing Technology and Business University, his PhD in Chemical Biology in 2010 from Changchun Institute of Applied Chemistry, Chinese Academy of Science under the supervision of Prof. Xiaogang Qu. He has since joined the research group of Prof. Hang Hubert Yin at the University of Colorado at Boulder. His research involves the discovery of small molecule modulators of innate immune receptors and integral membrane proteins.

Christina Smith earned her bachelorin molecular biology from the University of Coloradoat Boulder in 2009. After two years working at Amgen, Christina returned to the University of Colorado at Boulderfor graduate studies. Christina is currently pursuing a PhD in biochemistry in Prof Hang Hubert Yin’s lab. Her primary research interests are Toll-like receptor signaling and drug development.
Notes and references
- 1.Takeuchi O, Akira S. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 2.Anderson KV, Bokla L, Nusslein-Volhard C. Cell. 1985;42:791–798. doi: 10.1016/0092-8674(85)90275-2. [DOI] [PubMed] [Google Scholar]
- 3.Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, Hood LE, Aderem A. Proc Natl Acad Sci USA. 2005;102:9577–9582. doi: 10.1073/pnas.0502272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Netea MG, Wijmenga C, O’Neill LA. Nat Immunol. 2012;13:535–542. doi: 10.1038/ni.2284. [DOI] [PubMed] [Google Scholar]
- 5.Buchanan MM, Hutchinson M, Watkins LR, Yin H. J Neurochem. 2010;114:13–27. doi: 10.1111/j.1471-4159.2010.06736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Connolly DJ, O’Neill LA. Curr Opin Pharmacol. 2012;12:510–518. doi: 10.1016/j.coph.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Hennessy EJ, Parker AE, O’Neill LA. Nat Rev Drug Discov. 2010;9:293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 8.Basith S, Manavalan B, Lee G, Kim SG, Choi S. Expert Opin Ther Pat. 2011;21:927–944. doi: 10.1517/13543776.2011.569494. [DOI] [PubMed] [Google Scholar]
- 9.Stover AG, Da Silva Correia J, Evans JT, Cluff CW, Elliott MW, Jeffery EW, Johnson DA, Lacy MJ, Baldridge JR, Probst P, Ulevitch RJ, Persing DH, Hershberg RM. J Biol Chem. 2004;279:4440–4449. doi: 10.1074/jbc.M310760200. [DOI] [PubMed] [Google Scholar]
- 10.Peri F, Piazza M. Biotechnol Adv. 2012;30:251–260. doi: 10.1016/j.biotechadv.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 11.Barochia A, Solomon S, Cui X, Natanson C, Eichacker PQ. Expert Opin Drug Metab Toxicol. 2011;7:479–494. doi: 10.1517/17425255.2011.558190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Persing DH, Coler RN, Lacy MJ, Johnson DA, Baldridge JR, Hershberg RM, Reed SG. Trends Microbiol. 2002;10:S32–37. doi: 10.1016/s0966-842x(02)02426-5. [DOI] [PubMed] [Google Scholar]
- 14.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. Science. 2007;316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 15.Agnihotri G, Crall BM, Lewis TC, Day TP, Balakrishna R, Warshakoon HJ, Malladi SS, David SA. J Med Chem. 2011;54:8148–8160. doi: 10.1021/jm201071e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu W, Li R, Malladi SS, Warshakoon HJ, Kimbrell MR, Amolins MW, Ukani R, Datta A, David SA. J Med Chem. 2010;53:3198–3213. doi: 10.1021/jm901839g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salunke DB, Shukla NM, Yoo E, Crall BM, Balakrishna R, Malladi SS, David SA. J Med Chem. 2012;55:3353–3363. doi: 10.1021/jm3000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song DH, Lee JO. Immunol Rev. 2012;250:216–229. doi: 10.1111/j.1600-065X.2012.01167.x. [DOI] [PubMed] [Google Scholar]
- 19.Kang JY, Lee JO. Annu Rev Biochem. 2011;80:917–941. doi: 10.1146/annurev-biochem-052909-141507. [DOI] [PubMed] [Google Scholar]
- 20.Dziarski R, Wang Q, Miyake K, Kirschning CJ, Gupta D. J Immunol. 2001;166:1938–1944. doi: 10.4049/jimmunol.166.3.1938. [DOI] [PubMed] [Google Scholar]
- 21.Joce C, Stahl JA, Shridhar M, Hutchinson MR, Watkins LR, Fedichev PO, Yin H. Bioorg Med Chem Lett. 2010;20:5411–5413. doi: 10.1016/j.bmcl.2010.07.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chavez SA, Martinko AJ, Lau C, Pham MN, Cheng K, Bevan DE, Mollnes TE, Yin H. J Med Chem. 2011;54:4659–4669. doi: 10.1021/jm2003365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, Reddy A, Somogyi AA, Hutchinson MR, Watkins LR, Yin H. Proc Natl Acad Sci USA. 2012;109:6325–6330. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng K, Wang X, Yin H. J Am Chem Soc. 2011;133:3764–3767. doi: 10.1021/ja111312h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang QW, Mou L, Lv FL, Zhu PF, Wang ZG, Jiang JX, Wang JZ. Biochem Biophys Res Commun. 2005;329:846–854. doi: 10.1016/j.bbrc.2005.01.162. [DOI] [PubMed] [Google Scholar]
- 26.Shanmugam A, Rajoria S, George AL, Mittelman A, Suriano R, Tiwari RK. Plos One. 2012;7:e30839. doi: 10.1371/journal.pone.0030839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee HK, Brown SJ, Rosen H, Tobias PS. Mol Pharmacol. 2007;72:868–875. doi: 10.1124/mol.107.038349. [DOI] [PubMed] [Google Scholar]
- 28.Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Scientific World Journal. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J, van Steeg K, Kopajtic TA, Loram LC, Sfregola C, Galer E, Miles NE, Bland ST, Amat J, Rozeske RR, Maslanik T, Chapman TR, Strand KA, Fleshner M, Bachtell RK, Somogyi AA, Yin H, Katz JL, Rice KC, Maier SF, Watkins LR. J Neurosci. 2012;32:11187–11200. doi: 10.1523/JNEUROSCI.0684-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hutchinson MR, Lewis SS, Coats BD, Rezvani N, Zhang Y, Wieseler JL, Somogyi AA, Yin H, Maier SF, Rice KC, Watkins LR. Neuroscience. 2010;167:880–893. doi: 10.1016/j.neuroscience.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spyvee MR, Zhang H, Hawkins LD, Chow JC. Bioorg Med Chem Lett. 2005;15:5494–5498. doi: 10.1016/j.bmcl.2005.08.080. [DOI] [PubMed] [Google Scholar]
- 32.Cheng K, Wang X, Zhang S, Yin H. Angew Chem Int Ed Engl. 2012;51:12246–12249. doi: 10.1002/anie.201204910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada M, Ichikawa T, Ii M, Sunamoto M, Itoh K, Tamura N, Kitazaki T. J Med Chem. 2005;48:7457–7467. doi: 10.1021/jm050623t. [DOI] [PubMed] [Google Scholar]
- 34.Ii M, Matsunaga N, Hazeki K, Nakamura K, Takashima K, Seya T, Hazeki O, Kitazaki T, Iizawa Y. Mol Pharmacol. 2006;69:1288–1295. doi: 10.1124/mol.105.019695. [DOI] [PubMed] [Google Scholar]
- 35.Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. Eur J Pharmacol. 2008;584:40–48. doi: 10.1016/j.ejphar.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 36.Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y, Ii M. Br J Pharmacol. 2009;157:1250–1262. doi: 10.1111/j.1476-5381.2009.00297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. Mol Pharmacol. 2011;79:34–41. doi: 10.1124/mol.110.068064. [DOI] [PubMed] [Google Scholar]
- 38.Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J. Crit Care Med. 2010;38:1685–1694. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- 39.Chahal DS, Sivamani RK, Rivkah Isseroff R, Dasu MR. Phytother Res. 2012 doi: 10.1002/ptr.4886. [DOI] [PubMed] [Google Scholar]
- 40.Zimmer SM, Liu J, Clayton JL, Stephens DS, Snyder JP. J Biol Chem. 2008;283:27916–27926. doi: 10.1074/jbc.M802826200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Resman N, Gradisar H, Vasl J, Keber MM, Pristovsek P, Jerala R. FEBS Lett. 2008;582:3929–3934. doi: 10.1016/j.febslet.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 42.Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, Bozza MT. J Biol Chem. 2007;282:20221–20229. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- 43.Zhao L, Lee JY, Hwang DH. Nutr Rev. 2011;69:310–320. doi: 10.1111/j.1753-4887.2011.00394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Panda SK, Kumar S, Tupperwar NC, Vaidya T, George A, Rath S, Bal V, Ravindran B. PLoS Pathog. 2012;8:e1002717. doi: 10.1371/journal.ppat.1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishitani C, Mitsuzawa H, Sano H, Shimizu T, Matsushima N, Kuroki Y. J Biol Chem. 2006;281:38322–38329. doi: 10.1074/jbc.M606904200. [DOI] [PubMed] [Google Scholar]
- 46.Slivka PF, Shridhar M, Lee GI, Sammond DW, Hutchinson MR, Martinko AJ, Buchanan MM, Sholar PW, Kearney JJ, Harrison JA, Watkins LR, Yin H. Chembiochem. 2009;10:645–649. doi: 10.1002/cbic.200800769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu L, Ghosh N, Slivka PF, Fiorini Z, Hutchinson MR, Watkins LR, Yin H. Chembiochem. 2011;12:1827–1831. doi: 10.1002/cbic.201100211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toshchakov VY, Vogel SN. Expert Opin Biol Ther. 2007;7:1035–1050. doi: 10.1517/14712598.7.7.1035. [DOI] [PubMed] [Google Scholar]
- 49.Toshchakov VY, Szmacinski H, Couture LA, Lakowicz JR, Vogel SN. J Immunol. 2011;186:4819–4827. doi: 10.4049/jimmunol.1002424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang S, Cheng K, Wang X, Yin H. Bioorg Med Chem. 2012;20:6073–6079. doi: 10.1016/j.bmc.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.