

Abstract
Most age-associated neurodegenerative diseases involve the aggregation of specific proteins within the nervous system. In Alzheimer’s disease, the insidious pathogenic process begins many years before the symptoms emerge, and the lesions that characterize the disease – senile plaques and neurofibrillary tangles – ramify systematically through the brain. We review evidence that the β-amyloid and tau proteins, which aggregate to form senile plaques and neurofibrillary tangles, respectively, are induced to misfold and self-assemble by a process of templated conformational change that amplifies a toxic species. Recent data also indicate that the spread of these lesions from one site to another is mediated by the cellular uptake, transport and release of endogenous seeds formed by the cognate proteins. This simple pathogenic principle suggests that the formation, trafficking and metabolism of pathogenic protein seeds are promising therapeutic targets for Alzheimer’s disease and other neurodegenerative disorders.
Many age-related neurodegenerative diseases involve the anomalous aggregation of specific proteins within or among the cells of the nervous system. These diseases include Alzheimer’s disease (AD), Parkinson’s disease, amyotrophic lateral sclerosis, frontotemporal dementia, and many others. Because these are chronic disorders, the signs and symptoms generally become evident only after the degenerative trajectory is well underway. Furthermore, the regenerative capacity of post-mitotic cells such as neurons is negligible. For these reasons, an effective disease-modifying therapy will probably require that treatment be initiated early, which in turn demands a fuller understanding of the seminal events in the pathogenic cascade. Recent evidence indicates that, in a variety of neurodegenerative diseases, protein aggregation is initiated, and the aggregates continue to proliferate, by a prion-like process of templated protein corruption, or seeding. In this brief review, we will describe the potential role of protein seeding in the instigation and spread of the cardinal lesions that characterize AD. We believe that this disease concept has the potential to unify our approach to a broad class of clinically and pathologically dissimilar disorders.
Protein aggregation and the prion paradigm
Most proteins fold into their functional three-dimensional architecture shortly after they are generated within the cell; others, called intrinsically unfolded proteins, retain a degree of structural ambiguity. Under pathogenic conditions, some proteins misfold into a configuration that is rich in β-sheet secondary structure, which is often quite stable and capable of binding to, and conformationally corrupting, cognate molecules. The resulting aggregates include the intracellular inclusions and/or extracellular deposits that are typically associated with each disease. In addition to the larger lesions that can be detected with the light microscope, small, soluble aggregates known as oligomers have been increasingly implicated in the cytotoxic effects of some pathogenic proteins.
The ability of misfolded proteins to propagate by the corruption of like molecules underlies the unusual nature of the prion diseases, which can be infectious, genetic, or idiopathic in origin1. The infectivity of prions is made possible in part by their remarkable stability; it enables them to survive the transfer from one organism to another, where they initiate a new nexus of prion formation. In genetic and idiopathic variants of prion disease, which account for the great majority of cases in humans, the prions probably originate endogenously. In this instance, it is thought that an intrinsic prion protein misfolds, evades cellular clearance, and then initiates the sequential corruption of other prion protein molecules.
Presently there is no evidence that the non-prion proteopathies are infectious in the same manner as prion diseases. We contend, however, that the pathogenesis of multiple disorders is governed by a prion-like mechanism of endogenous misfolding and templated corruption of disease-specific proteins. This principle may help to illuminate both the earliest events that give rise to neurodegenerative diseases, and the means by which they ramify through the nervous system.
Alzheimer disease (AD)
The 2 proteins that, in their aggregated state, histopathologically define AD are β-amyloid (Aβ) and tau. The Aβ protein is an approximately 40- to 42 amino acid cleavage product of the Aβ precursor protein (APP) that misfolds and self-assembles to form senile plaques and cerebral Aβ angiopathy. The tau protein normally promotes the stability of microtubules, but in the disease condition, tau polymerizes into filaments that constitute neurofibrillary tangles (NFTs). Although these lesions are not exclusive to AD, when plaques and NFTs are sufficiently numerous in patients with clinical dementia, they are pathognomonic for the disease.
In the past several decades, a number of groups have analyzed plaques and NFTs in postmortem brains of humans at various ages. The findings, broadly considered, suggest that the lesions spread systematically, possibly along neuronal pathways that interconnect different regions2 (see also the following discussion of tauopathy). Magnetic resonance imaging studies in living subjects support this view—the brain regions most prominently affected in AD are tightly linked components of neuronal networks, suggesting that pathogenic factor(s) travel via neuronal transport and transsynaptic diffusion.3– 4 Evidence that the agents involved in the instigation and spread of AD-like lesions are specific proteinaceous seeds has emerged from experiments with genetically modified rodents.
The seeded initiation and spread of aggregated Aβ
In a paradigm similar to that used to study the transmission of prion disease, the intracerebral injection of dilute brain extracts from patients with AD induces the formation of senile plaques and cerebral amyloid angiopathy in mice expressing mutant human APP transgenes.2 Present data indicate that the essential inductive agent in this model is aggregated Aβ: (1) the brain extract is active only if it contains misfolded Aβ (control brain extracts are ineffective); (2) seeding is prevented when Aβ is neutralized by adding formic acid or anti-Aβ antibodies to the extract; (3) seeding does not occur in nontransgenic rodent hosts, which generate a variant Aβ sequence that does not readily aggregate; (4) Aβ-rich brain extracts from APP-transgenic mice seed as potently as do extracts from AD brains, ruling out factors that are specific to the human brain; and (5) the immunoreactivity in the brains of the host mice is not the injectate itself because the induced deposits appear only after a lag period, the duration of which depends on the characteristics of both the seed and the host.2 Finally, a recent study has shown that cerebral Aβ amyloidosis can be seeded by pure, aggregated, synthetic Aβ, albeit with less potency than Aβ that has aggregated within the living brain.5
When viewed from the perspective of in vitro and in vivo experiments, we can begin to draw some tentative conclusions about the nature of Aβ seeds and the seeding paradigm. It seems likely that Aβ, like prions, can misfold and aggregate into structurally and functionally variant strains.2,6 The Aβ seeds also vary considerably in size, ranging from small, soluble multimers to large, insoluble aggregates.7 At least some Aβ seeds are able to induce Aβ deposition in the brain following injection into the peritoneal cavity.8 Finally, following the injection of Aβ-laden extracts into 1 brain region, Aβ deposits appear in noncontiguous but axonally interconnected areas; this suggests that the pathogenic seeds can expand along defined pathways, possibly via neuronal transport mechanisms.2
Whereas the aforementioned studies used an exogenous seeding paradigm, endogenously generated Aβ aggregates have been found to traffic preferentially among interconnected brain regions in APP -transgenic mice.9 In the context of a growing literature on the spatiotemporal manifestations of Aβ pathology, these findings in experimental models implicate proteopathic Aβ seeds as an essential element in the initiation and expansion of aggregated Aβ in AD. As we will now discuss, endogenously generated aggregates of tau also can mediate the development of tauopathy in the brain.
The seeded initiation and spread of tauopathy in vivo
In addition to its key role in AD, the tau protein polymerizes into characteristic NFTs in a variety of other neurological conditions.10 One of the most intriguing aspects of AD is the temporal and spatial spread of disease pathology through the brain from regions of initial vulnerability.11 This is especially well defined for tauopathy, which is highly correlated with cognitive decline.12 Early studies of the distribution of NFTs in AD led to the conclusion that superficial cells of the entorhinal cortex are among the first neurons to be affected and thereafter a hierarchical pattern of progression occurs, ultimately affecting anatomically closely connected limbic and association cortices.13 These observations did not distinguish between 2 possibilities: the selective vulnerability of this group of neurons might be due to an intrinsic property of these relatively large projection neurons, or the vulnerability may be due to their connectivity to one another.
In the last year, de Calignon et al14 in the Hyman laboratory and Liu et al15 in the Duff laboratory (see the discussion later) approached this problem by directly testing the latter possibility. These laboratories independently generated similar lines of genetically engineered mice that express a pathogenic human tau transgene16– 17 under a promoter restricted to the entorhinal cortex.18 By immunostaining as well as by in situ hybridization, the tau transgene was shown to be expressed uniquely in entorhinal projection neurons, including their axons and terminals. Importantly, the models have recapitulated the temporal and spatial aspects of NFT distribution, demonstrating that NFTs emanating from the entorhinal cortex can propagate pathology in neuroanatomically connected areas.
The entorhinal cortex projects massively to the dentate gyrus via the perforant path, with the projections terminating in the middle third of the molecular layer (MML) of the dentate gyrus. Thus, anatomically, it was possible to address the question of whether deafferentation of downstream neurons (i.e., in the dentate gyrus) led to their vulnerability independent of intrinsic factors that would lead to tau accumulation and to NFTs because the neurons to which the entorhinal cells project were normal, wild-type neurons that did not express the transgene. As the mice aged, the granule cells of the dentate gyrus did indeed develop NFTs. As shown in Figure 1, at age 12 months the dentate gyrus does not contain immunoreactivity for human tau, whereas by 24 months, numerous granule cells are immunopositive. Surprisingly, these lesions included human tau, despite the fact that human tau messenger RNA was not detectable within the cell bodies.
Figure 1.
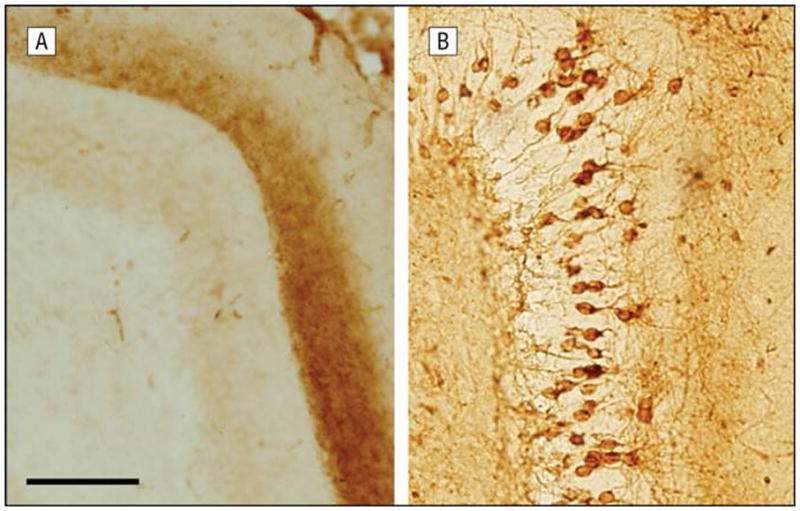
Age-associated development of neurofibrillary lesions in the dentate gyrus of mice expressing human tau selectively in the entorhinal cortex. The dentate gyrus lacks Alz50 immunoreactivity at 12 months of age (A), but by the age of 24 months, numerous granule cells are immunopositive (B). These lesions include human tau, despite the fact that human tau messenger RNA was not detectable within the cells (scale bar = 100μm).
Further evidence that there was little if any human tau messenger RNA in the dentate gyrus granule cell bodies came from a laser-capture microscopy experiment in which human tau–immunopositive, in situ–negative neurons were collected and tested by quantitative reverse transcription–polymerase chain reaction.14 Again, no human tau message was detected. Moreover, reporter lines expressing 2 different fluorescent markers of gene expression were examined, and neither showed expression in the dentate gyrus.15 Taken together, these experiments indicate that human tau is released from the terminals of the entorhinal cortex neurons and propagated to the dentate gyrus granule cells. Additional evidence supporting this idea was the observation that human tau could be (rarely) detected in both microglia and astrocytes in the terminal zone in the molecular layer of the dentate gyrus. This argues in favor of tau being released and taken up into other cells.
The second intriguing observation was made when the lesions were examined using antibodies specific for human or mouse tau. The aggregates contained human tau, presumably misfolded on the basis of the P301L mutation. However, endogenous mouse tau was found to coaggregate with the human tau core.14 In fact, in many cells, the mouse tau aggregates were morphologically more robust than the human tau nidus. This finding suggested that the human tau acted as a misfolded template for endogenous wild-type mouse tau and that the latter was then able to aggregate and form a stable tau aggregate.
Collectively, these results suggest that there may well be 2 critical factors that determine the hierarchical march of NFTs across association and limbic areas: their connectivity does matter, and there is an important role played by the release of misfolded tau and uptake from axon terminals that results in downstream neurons being exposed to misfolded, mislocalized tau. We postulate that this leads to the propagation of NFTs across anatomically connected brain regions in AD. These results also suggest the possibility that characteristics of the downstream neurons are critical for whether another NFT will form.
Among the uncertainties are the following: the mechanism of release and uptake of misfolded tau, the ways in which that misfolded tau evades degradation programs, and how misfolded tau interacts with the (normally axonal) endogenous tau to form mislocalized aggregates in the cytoplasm. Individual cells may also initially differ in their ability to deal with misfolded proteins, based on proteasomal degradation capabilities, or other misfolded protein response pathways. Many of these issues can be addressed effectively in cell culture models (see the following discussion). Finally, the relationship between the release and uptake of misfolded tau and neurodegenerative phenotypes—synaptic loss and ultimately neuronal loss remains to be explored, although recent observations using a synaptoneurosomal preparation of human AD brain material are consistent with the accumulation of proteasome-resistant tau in both presynaptic and postsynaptic compartments in the AD brain.19
The medial perforant path, which originates in layer II of the entorhinal cortex and terminates in the MML of the dentate gyrus, is progressively affected by NFT formation prior to the appearance of substantial cognitive decline, and it is devastated in AD.20 The Duff laboratory has undertaken a detailed histopathological investigation of anatomically selective tau-transgenic mice to define the age-related evolution of tauopathy originating in the entorhinal cortex (Figure 2). In young mice, human tau accumulates at the end zones of axonal terminals in the perforant pathway, within the MML of the dentate gyrus, and in cells and neurites of the subiculum and entorhinal cortex (both medial and lateral). As the mice age more than 18 months, human tau accumulates as thioflavin S–positive, silver-positive NFTs in the somatodendritic compartment of the entorhinal cortex, and far less tau staining is seen in the MML. Significantly, human tau now appears in granule cells of the dentate gyrus, including in cells with no endogenously expressed human tau.14 Postmortem studies of patients with AD show little (or late) pathology in granule cells relative to CA1 pyramidal cells. High levels of tau in the granule cells of the mouse probably reflect the high levels of the protein expressed in layer II neurons compared with layer III. However, progressively denser immunostaining in CA1 is observed in very old mice and with pathology-related antibodies such as MC1 compared with antibodies that recognize all tau such as CP27. Full characterization of these mice is reported by Liu et al15 and de Calignon et al.14
Figure 2.
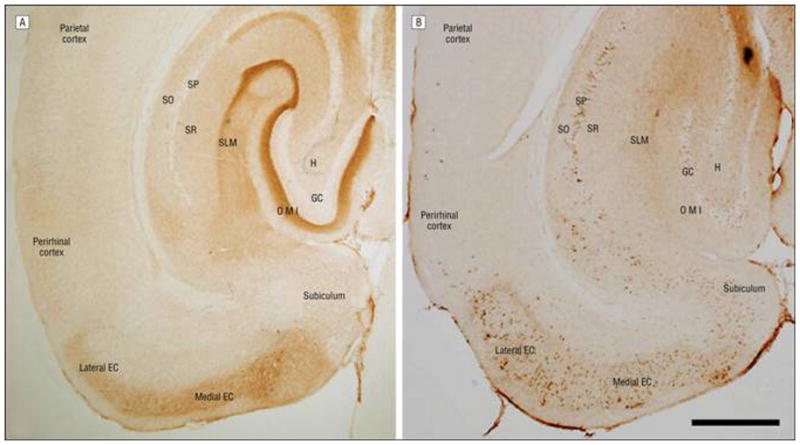
Monosynaptic and trans-synaptic cortico-hippocampal and cortico-cortico connections radiating from the entorhinal cortex in young and old neuropsin-tTA-tau transgenic mice. Young mice show accumulation of MC1-immunoreactive human tau in cell bodies and axonal tracts of the EC, the presubiculum, the parasubiculum, and projection areas (A), whereas old mice show relocation of tau to cell bodies and the appearance of human tau in synaptically connected areas in the hippocampus and neocortex (B) (scale bar = 500 μm). GC indicates granule cell layer of the dentate gyrus; H, hilus; I, inner molecular layer of the dentate gyrus; M, middle molecular layer of the dentate gyrus; O, outer molecular layer of the dentate gyrus; SLM, stratum lacunosum moleculare; SO, stratum oriens; SP, stratum pyramidale; and SR, stratum radiatum.
The observation of human tau protein in the granule cells—cells that are physically separated by a synapse from where the tau is produced in entorhinal cortex neurons21—strongly supports the idea that human tau can transfer between cells, and it appears most likely that this occurs as free tau conformers are released and then internalized via the extracellular space. Although tau usually exists as a cytoplasmic protein closely associated with microtubules, it has been found outside cells in the cerebrospinal and interstitial fluids, even in the absence of overt neurodegeneration.22 It has also been found in secreted vesicles termed exosomes,23– 24 although exosome-mediated secretion may only be a response to overabundant or abnormal tau as normal tau present at endogenous levels is not seen in exosomes.25 It is known that tau can be internalized from outside cells where it can promote templating to endogenous tau, both from in vitro (see later discussion) and in vivo26 studies.
Several models can be proposed for the propagation of pathological tau (reviewed by Frost and Diamond27). To better understand the mechanisms and requirements for tau uptake, the Duff laboratory has examined whether tau conformers can be taken up by primary neurons using neurons grown in Campenot (microfluidic) chambers to allow for separate analysis of somatodendritic and axonal compartments. Primary neurons grown in microfluidic chambers were incubated with fluorescently labeled low-molecular-weight conformers either in the somatodendritic chamber or in the axonal chamber. Conformers were internalized; in experiments where dextran was added, the conformers colocalized with dextran, indicating that the mechanism of uptake was bulk endocytosis. Conformers were transported down axons, which emerge in the opposing chamber. By adding tau to different chambers, it was possible to demonstrate that uptake and transport can be both anterograde and retrograde. This is of significance for spread because although the entorhinal cortex to granule cell pathway is anterograde, spread to other regions could be through retrograde or anterograde movement. Interestingly, monomeric tau did not bind to cells, which would make it unlikely that normal tau (presumed to be soluble and monomeric) in the interstitial fluid would be internalized by cells in vivo. Based on both published and presented data from researchers in the field, it is possible that intracellular tau aggregates, released via secretion as a means of clearance or on degeneration of axons or somatodendritic compartments, could be internalized by anatomically connected cells and then anterogradely and retrogradely transported to remote brain regions. Aggregates could accumulate, clogging cellular degradation machinery such as the ubiquitin-proteasome system and autophagy. Subsequently, aggregated proteins that failed to be sequestered or degraded may cause local membrane rupture of degradative organelles, leading to their release into the cytosol, where they could physically interact with intracellular soluble protein and trigger the endogenous misfolding of tau. Thus, the temporally and spatially distinct distribution of tau pathology that defines the early stages of AD could be explained by the uptake, templating, and release of aggregated tau between neurons in neuroanatomically connected circuits. The cellular mechanisms involved in the propagation of aggregated assemblies of tau protein will be considered next.
Transcellular propagation of tau aggregation in vitro
As discussed earlier, in vivo studies implicate prion-like seeding mechanisms in the initiation and spread of both extracellular (Aβ) and intracellular (tau) protein aggregates. In parallel, in vitro cell biology studies are shedding light on how these phenomena might occur through the transcellular propagation of protein aggregates. The Diamond laboratory has begun to address this problem by using biochemistry and tissue culture to test the possibility of transcellular propagation of aggregated tau. These experiments began with an analysis of the structure of tau fibrils formed by recombinant protein with or without disease-associated mutations. Single amino acid substitutions in monomeric tau were sufficient to drive the resultant fibrils into distinct conformations. Surprisingly, however, fibrils derived from mutant tau drove wild-type tau monomers into a stably self-propagating fibrillar conformation distinct from that which would form if wild-type tau monomer was allowed to fibrillize alone.28 This phenomenon, termed templated conformational change, had been recognized as a defining feature of prion protein strains.29– 30 It had also previously been demonstrated for Aβ,6 but it was now clear that templated conformational change is likely to be a feature of many protein misfolding diseases, including tauopathies, and might underlie their phenotypic diversity. In other words, different aggregate conformations might produce different disease phenotypes.
To determine whether basic cellular mechanisms mediate the propagation of aggregates among cells, the first question was whether protein fibrils derived from recombinant tau would be preferentially taken up by cells in culture. The fibrils were covalently labeled with a fluorescent dye, and any extracellular protein bound to the external cell membrane was destroyed by protease treatment. Under these conditions, tau aggregates (but not monomers) were clearly taken up into cells. Electron microscopic imaging demonstrated that this process occurs through the formation of massive vesicles, termed macropinosomes (Brandon B. Holmes, BS, unpublished data, 2012).
The next question to be addressed was whether exposure of cells to exogenous tau aggregates can influence the intracellular aggregation of tau. Indeed, extracellular aggregates consisting of recombinant tau are taken into cells, where they directly contact the intracellular protein and convert it to a fibrillar form31 (Figure 3). Thus, the aggregation state can be directly propagated from outside to inside the cell. Subsequent work demonstrated that newly formed aggregates are released into the extracellular space, where they are taken up by recipient cells and can repeat the seeding phenomenon.32 These observations were supported by the finding that an antitau antibody prevents the intercellular propagation of aggregation by binding directly to tau aggregates and blocking cell uptake.32 This antibody can be used to purify tau fibrils from the conditioned medium of cells that express aggregates.32
Figure 3.
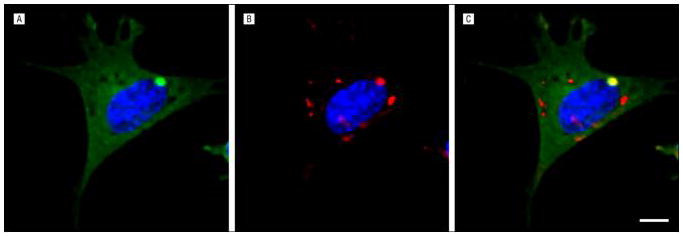
Induction of intracellular aggregation by extracellular fibrils. The C17.2 neural cells expressing a tau–yellow fluorescent protein fusion (green) (A) were exposed to recombinant fibrils labeled with AlexaFluor 546 (red) (B); in the merged image, note the colocalization of externally derived fibrils with an intracellular tau–yellow fluorescent protein inclusion, representing the induction of intracellular aggregation (C) (scale bar = 10 μm) (image courtesy of Brandon B. Holmes, BS).
Data from the Diamond laboratory and others support a model in which protein aggregates that form in a vulnerable cell are released into the extracellular space. These aggregates are free (not encapsulated by a membrane) and are in a fibrillar form. Currently, there is no evidence to suggest that this process is limited to neurons. How, then, can these basic cellular mechanisms be put in the context of emerging human and animal studies that suggest an important role for neural networks in the progression of pathology?
These data suggest the possibility that transsynaptic movement of aggregates among neurons is not the only means of transcellular propagation within the nervous system. Rather, a basic cellular mechanism involves the flux of aggregates into and out of many different types of cells. This could represent a protein quality control or protection mechanism. Meanwhile, the uptake of macromolecules from the extracellular space is a well-described aspect of cellular physiology. Only in the context of fibril-forming proteins, which have the ability to self-propagate aggregates by seeded polymerization, would this process facilitate the spread of pathology. According to this model, neural networks are involved mainly because neurons themselves are the most vulnerable to the accumulation of aggregates; they are capable of specific and long-distance aggregate movement by anterograde and/or retrograde transport, and synapses are the sites at which neurons are in close proximity to one another. This could allow aggregates to move across long anatomical distances simply by traversing short synapses in series. Synaptic activity itself may or may not promote transcellular propagation. Regardless, the main concept is that extracellular protein aggregates play a fundamental role in pathogenesis. The observation that tau protein generated in entorhinal neurons can transfer to other neurons within the entorhinal cortex and can cross a synapse to other cells is of significant interest as it suggests that tau can be trapped in the extracellular space before it is taken up into recipient cells, for example, through the use of antibodies (immunotherapy). It is likely, however, that such an intervention would be most effective at the earliest stages of the disease, when pathology is localized and there is limited cognitive impairment.
Conclusions
A growing body of data indicates that the propagation of pathogenic protein aggregates across neural systems, and hence the disruption of function of those neural systems, might be mediated by misfolded protein seeds that are released and taken up by anatomically connected neurons. If so, blocking this process may help arrest the progression of disease. In light of the growing spectrum of disorders involving the accumulation and spread of misfolded proteins, efforts to detect pathogenic protein aggregates and impede their movement between cells could change how we diagnose and treat neurodegenerative diseases.
Acknowledgments
Funding/Support: This work was supported by the following grants from the National Institutes of Health: R21AG040589, P51RR165, P51OD11132 and P50AG025688 (Dr Walker); R01NS071835 (Dr Diamond); 1F31NS079039; and R01NS074874 (Dr Duff); and R21AG038835-01A1 and R21NS067127 (Dr Hyman). This work also was supported by grants from the CART Foundation (Dr Walker), the Muscular Dystrophy Association (Dr Diamond), the Tau Consortium (Dr Diamond), and the American Health Assistance Foundation (Drs Diamond and Hyman), and by a postdoctoral fellowship from the American Health Assistance Foundation (Dr Duff’s laboratory).
Footnotes
Author Contributions: Study concept and design: Walker, Diamond, Duff, and Hyman. Acquisition of data: Walker, Diamond, Duff, and Hyman. Analysis and interpretation of data: Walker, Diamond, Duff, and Hyman. Drafting of the manuscript: Walker, Diamond, Duff, and Hyman. Critical revision of the manuscript for important intellectual content: Walker, Diamond, Duff, and Hyman. Obtained funding: Walker, Diamond, Duff, and Hyman. Administrative, technical, and material support: Walker, Diamond, Duff, and Hyman. Study supervision: Walker, Diamond, Duff, and Hyman.
Conflict of Interest Disclosures: Dr Hyman has stock options with Novartis and consults or has sponsored research agreements for Pfizer, Neurophage, Neotope, Biogen, GlaxoSmithKline, Siemens, Elan, Acumen, and Takeda, none of which were involved in these projects.
Previous Presentation: This work was presented at the Alzheimer’s Disease Centers Meeting; April 21, 2012; New Orleans, Louisiana.
Additional Contributions: We gratefully acknowledge the invaluable contributions of our collaborators and coworkers, in particular Mathias Jucker, Ph.D. Harry LeVine, Ph.D., Rebecca Rosen, Ph.D., Amarallys Cintron, B.S., Li Liu, Ph.D., Jessica Wu, Ph.D., Alix de Calignon, M.Sc., Manuela Polydoro, Ph.D., Tara Spires, D.Phil., George Carlson, Ph.D., Karen Ashe, Ph.D., and Mark Mayford, Ph.D. In addition, Manuela Polydoro, Ph.D. assisted with figure preparation and Brandon B. Holmes, B.S. provided cell images and editorial comments to Dr Diamond.
Contributor Information
Dr. Lary C. Walker, Email: lary.walker@emory.edu.
Dr. Marc I. Diamond, Email: diamondm@neuro.wustl.edu.
Dr. Karen E. Duff, Email: ked2115@columbia.edu.
Dr. Bradley T. Hyman, Email: bhyman@PARTNERS.ORG.
References
- 1.Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011 Jan;3(1):a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011 Oct;70(4):532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raj A, Kuceyeski A, Weiner M. A network diffusion model of disease progression in dementia. Neuron. 2012 Mar 22;73(6):1204–1215. doi: 10.1016/j.neuron.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron. 2012 Mar 22;73(6):1216–1227. doi: 10.1016/j.neuron.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stohr J, Watts JC, Mensinger ZL, et al. Purified and synthetic Alzheimer’s amyloid beta (Abeta) prions. Proc Natl Acad Sci U S A. 2012 Jul 3;109(27):11025–11030. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005 Jan 14;307(5707):262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 7.Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. J Neurosci. 2011 Oct 12;31(41):14488–14495. doi: 10.1523/JNEUROSCI.3088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisele YS, Obermuller U, Heilbronner G, et al. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010 Nov 12;330(6006):980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronnback A, Sagelius H, Bergstedt KD, et al. Amyloid neuropathology in the single Arctic APP transgenic model affects interconnected brain regions. Neurobiol Aging. 2012 Apr;33(4):831, e811–839. doi: 10.1016/j.neurobiolaging.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 10.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 11.Price JL, Davis PB, Morris JC, White DL. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol Aging. 1991 Jul-Aug;12(4):295–312. doi: 10.1016/0197-4580(91)90006-6. [DOI] [PubMed] [Google Scholar]
- 12.Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003 May 13;60(9):1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 13.Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984 Sep 14;225(4667):1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 14.de Calignon A, Polydoro M, Suarez-Calvet M, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012 Feb 23;73(4):685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu L, Drouet V, Wu JW, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santacruz K, Lewis J, Spires T, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005 Jul 15;309(5733):476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spires TL, Orne JD, SantaCruz K, et al. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006 May;168(5):1598–1607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yasuda M, Mayford MR. CaMKII activation in the entorhinal cortex disrupts previously encoded spatial memory. Neuron. 2006 Apr 20;50(2):309–318. doi: 10.1016/j.neuron.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 19.Tai HC, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT. The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated With Dysfunction of the Ubiquitin-Proteasome System. Am J Pathol. 2012 Aug 3; doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996 Jul 15;16(14):4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witter MP. The perforant path: projections from the entorhinal cortex to the dentate gyrus. Prog Brain Res. 2007;163:43–61. doi: 10.1016/S0079-6123(07)63003-9. [DOI] [PubMed] [Google Scholar]
- 22.Yamada K, Cirrito JR, Stewart FR, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011 Sep 14;31(37):13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saman S, Kim W, Raya M, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012 Feb 3;287(6):3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simon D, Garcia-Garcia E, Gomez-Ramos A, et al. Tau overexpression results in its secretion via membrane vesicles. Neurodegener Dis. 2012;10(1–4):73–75. doi: 10.1159/000334915. [DOI] [PubMed] [Google Scholar]
- 25.Faure J, Lachenal G, Court M, et al. Exosomes are released by cultured cortical neurones. Mol Cell Neurosci. 2006 Apr;31(4):642–648. doi: 10.1016/j.mcn.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 26.Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009 Jul;11(7):909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 2010 Mar;11(3):155–159. doi: 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J Biol Chem. 2009 Feb 6;284(6):3546–3551. doi: 10.1074/jbc.M805627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994 Dec;68(12):7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Legname G, Nguyen HO, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci U S A. 2006 Dec 12;103(50):19105–19110. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009 May 8;284(19):12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012 Jun 1;287(23):19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]