

Abstract
Twelve alkyl analogues (1 - 12) of the high-affinity serotonin transporter (SERT) inhibitor 6-nitroquipazine (6-NQ) were synthesised and studied using in vitro radioligand competition binding assays to determine their binding affinity (Ki). The putative antidepressant activity of five of the binders with the highest SERT binding affinities was studied by the forced swim and locomotor activity mouse tests. The three dimensional (3D) structures of 8 and 9 were determined using NOE NMR technique. Flexible docking of the compounds was undertaken to illustrate the binding of the compounds in the SERT model. Our results showed that several of the 6-NQ analogues are high-affinity SERT inhibitors and indicated that the octyl (8), decyl (10) and dodecyl (12) 6-NQ analogues exhibit moderate antidepressant activity.
Keywords: Serotonin transporter, 6-nitroquipazine alkyl analogues, Radioligand competition assay, NMR spectroscopy, Flexible docking, Porsolt forced swim test
INTRODUCTION
The serotonin (5-hydroxytryptamine, 5-HT) transporter (SERT) is a putative twelve transmembrane α-helical protein that belongs to the neurotransmitter:sodium symporter (NSS) transporter family (1). Located in the membrane of presynaptic neurons, the transporter plays an important role in the termination of serotonergic neurotransmission by removing 5-HT from the synaptic cleft. In addition to being targeted by psychostimulants such as cocaine and amphetamines, SERT is targeted by the two main classes of antidepressant drugs, the tricyclic antidepressants (TCAs) and selective serotonin reuptake inhibitors (SSRIs), which inhibit the transport of 5-HT into the presynaptic neuron.
6-nitroquipazine (6-NQ) is a highly potent and selective SERT inhibitor with higher affinity for SERT than most TCAs and SSRIs (2-4). Multiple studies have described the synthesis and in vitro SERT binding affinities of 6-NQ analogues (5-10) and several radiolabelled 6-NQ analogues, as well as radiolabelled 6-NQ itself, have also been developed for use as imaging agents (11-21). Furthermore, dual-acting 6-NQ analogues that inhibit SERT as well as antagonise the presynaptic autoinhibitory 5-HT1A receptors have been developed (22). These receptors are activated by the increases in synaptic 5-HT and upon activation decrease the serotonergic neurotransmission, hence causing a delay in the onset of antidepressant action (23). This delay typically lasts 2 to 4 weeks until the presynaptic autoinhibitory receptors have become desensitised and the firing of the 5-HT neurons is normalised (23). Dual-acting compounds that inhibit SERT and antagonise the 5-HT1A autoinhibitory receptors may thus reduce the time of onset of action of antidepressant drugs.
In the present study, twelve 6-NQ alkyl analogues (2-(4-alkyl-piperazin-1-yl)-6-nitroquinolines, 1 - 12) were synthesised and their affinities (Ki) to SERT and the 5-HT1A receptor were determined. The three dimensional (3D) structure of 8 and 9 were determined by NMR spectroscopy (NOE technique). To illustrate the interaction of the compounds with SERT, a SERT homology model (24) based on a homologous bacterial leucine transporter (LeuT) x-ray crystal structure (25), was used in a flexible docking study of the compounds. Furthermore, the alkyl analogues with the highest SERT affinities were screened in vivo using forced swim and locomotor activity tests.
MATERIALS AND METHODS
Materials
All reagents were purchased from commercial sources: Iodides were acquired from Aldrich (St. Louis, MO (methyl-octyl)) and from Alfa Aesar (Johnson Matthey Company, Karlsruhe, Germany (nonyl-dodecyl)). 6-nitroquipazine was obtained from 2-chloroquinoline (ABCR, Karlsruhe, Germany), piperazine (Aldrich, St. Louis, MO), nitric and sulfuric acid (POCh, Gliwice, Poland). NaH was purchased from Lancaster Synthesis (Alfa Aesar, Johnson Matthey Company, Karlsruhe, Germany). Solvents (except THF) were purchased from POCh (Gliwice, Poland). All air and water-sensitive reactions were carried out under argon. THF, purchased from ChemPur (Piekary Śląskie, Poland), was dried with sodium and distilled under argon from sodium benzophenone ketyl. Thin-layer chromatography (TLC) was performed on 0.2 mm Merck silica gel 60 F254 silica plates (Merck, Darmstadt, Germany) and compounds were visualised under 245 nm ultraviolet irradiation and/or phosphomolybdic acid. Column chromatography was performed using Baker gel 60 (230e400 mesh) (Baker, Deventer, The Netherlands) with the indicated solvents.
1H NMR, 13C NMR, IR and elemental analysis data
The 1H and 13C NMR spectra were obtained on a Bruker AVANCE DMX 400WB or at Bruker AVANCE DPX 200 MHz instrument (Bruker BioSpin, Fällanden, Switzerland) in CDCl3 with TMS as an internal reference. Chemical shifts were expressed in δ units and coupling constants (J) were expressed in hertz (Hz). The following abbreviations were used to describe peak patterns when appropriate: s (singlet), d (doublet), t (triplet), q (quartet), qt (quintet), m (multiplet), p (pseudo-) and b (broad-). For the two-dimensional experiments, the pulse sequences, acquisition, and the processing parameters were taken from the standard Bruker software library. Infrared spectra (KBr) were recorded on a Shimadzu FTIR-8300 spectrometer (Duisburg, Germany). Elemental analyses were performed on a Perkin-Elmer 2400 analyser (Perkin Elmer, Norwalk, CT, USA) and were within ± 0.4% of the theoretical values. The infrared spectra were recorded on a Nicolet Magna 550 IR spectrophotometer (Nicolet, Madison, WI, USA).
General preparation procedure of 6-nitro-4’-alkylquipazine analogues
A 1.1 mol-equivalent of sodium hydride (NaH) was added to an ice cooled (0°C) solution of 6-nitroquipazine (0.2 g, 0.77 mmol) in anhydrous tetrahydrofurane (THF). After 30 minutes, 0.77 mmol of alkyl iodide (RI) was added and the mixture was heated under reflux for 70 hours. The reaction mixture was cooled to room temperature and the excess of sodium hydride was decomposed with a small amount of water. Solvents were evaporated under reduced pressure and the crude products were purified with the aid of column chromatography on silica gel with 0-4% AcOEt/hexane (1-8) or 10-30% AcOEt/cyclohexane (9-12). Figure 1 shows the general synthesis procedure of 1-12.
Figure 1.
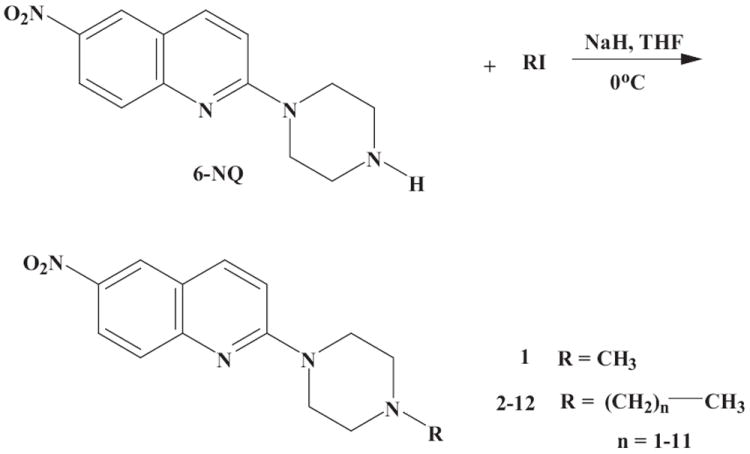
General synthesis scheme of 6-NQ aryl analogues 1 - 12. RI, alkyl iodide; THF, tetrahydrofurane.
2-(4-Methyl-piperazin-1-yl)-6-nitroquinoline (1)
C14H16N4O2. M = 272.30 g/mol. Yield 72%. Rf: 0.44 (10% MeOH/DCM); 1H NMR (200 MHz, CDCl3): δ 8.53 (d, 1H, J = 2.5 Hz), 8.29 (dd, 1H, J1 = 2.7 Hz, J2 = 9.3 Hz), 7.96 (d, 1H, J = 8.6 Hz), 7.66 (d, 1H, J = 9.3), 7.06 (d, 1H, J = 9.3), 3.87 (t, 4H, J = 5.3), 2.55 (t, 4H, J = 5.3), 2.37 (s, 3H); 13C NMR (50.3 MHz, CDCl3): δ 158.4, 151.5, 141.8, 138.5, 127.0, 124.2, 123.6, 121.0, 110.8, 60.5, 53.0, 44.3; IR (CHCl3): ν = 2944.6, 2852.8, 1616.7, 1496.4, 1324.9, 1229.2 cm-1; Anal. Calcd. for C14H16N4O2 · HCl · 0.25 H2O: C, 53.68; H, 5.63; N, 17.88; Found: C, 53.80; H, 5.56; N, 17.65%.
2-(4-Ethyl-piperazin-1-yl)-6-nitroquinoline (2)
C15H18N4O2. M = 286.33 g/mol. Yield 87 %. Rf: 0.52 (10% MeOH/DCM); 1H NMR (200 MHz, CDCl3): δ 8.53 (d, 1H, J = 2.54 Hz), 8.31 (dd, 1H, J1 = 2.7 Hz, J2 = 9.3 Hz), 7.96 (d, 1H, J = 9.3 Hz), 7.65 (d, 1H, J = 9.3 Hz), 7.06 (d, 1H, J = 9.3 Hz), 3.88 (t, 4H, J = 5.3 Hz), 2.59 (t, 4H, J = 5.3 Hz), 2.52 (q, 2H, J = 7.3 Hz), 1.16 (t, 3H, J = 7.3 Hz); 13C NMR (50.3 MHz, CDCl3): δ 158.4, 151.5, 141.8, 138.5, 127.0, 124.2, 123.5, 120.9, 110.8, 58.7, 53.0, 44.6, 29.7; IR (CHCl3): ν = 2937.0, 2820.1, 1616.0, 1496.4, 1324.9, 1233.5 cm-1; Anal. Calcd. for C15H18N4O2 · HCl : C, 55.81; H, 5.93; N, 17.36; Found: C, 55.87; H, 5.98; N, 17.10%.
2-(4-Propyl-piperazin-1-yl)-6-nitroquinoline (3)
C16H20N4O2. M = 300.36 g/mol. Yield 72 %. Rf: 0.53 (10% MeOH/DCM); 1H NMR (200 MHz, CDCl3): δ 8.53 (d, 1H, J = 2.5 Hz), 8.29 (dd, 1H, J1 = 2.7 Hz, J2 = 9.3 Hz), 7.96 (d, 1H, J = 8.6 Hz), 7.66 (d, 1H, J = 9.3 Hz), 7.06 (d, 1H, J = 9.3 Hz), 3.87 (t, 4H, J = 5.3 Hz), 2.58 (t, 4H, J = 5.3 Hz), 2.38 (t, 2H, J = 7.45 Hz), 1.63-1.52 (m, 2H), 0.95 (t, 3H, J = 7,31 Hz); 13C NMR (50.3 MHz, CDCl3): δ 158.4, 151.5, 141.8, 138.5, 127.0, 124.2, 123.6, 121.0, 110.8, 60.5, 53.0, 44.6,19.9, 11.8; IR (CHCl3): ν = 2930.1, 2856.3, 1616.0, 1496.9, 1324.6, 1232.7 cm-1; Anal. Calcd. for C16H20N4O2 · 2HCl · 0.5 H2O: C, 50.27; H, 6.06; N, 14.66; Found: C, 50.32; H, 6.34; N, 14.63%.
2-(4-Butyl-piperazin-1-yl)-6-nitroquinoline (4)
C17H22N4O2. M = 314.17 g/mol. Yield 69 %. Rf: 0.41 (10% MeOH/DCM); 1H NMR (200 MHz, CDCl3): δ 8.52 (d, 1H, J = 2.6 Hz), 8.29 (dd, 1H, J1 = 3,0 Hz, J2 = 9.3 Hz), 7.95 (d, 1H, J = 9.3 Hz), 7.64 (d, 1H, J = 9.3 Hz), 7.06 (d, 1H, J = 9.3 Hz), 3.87 (t, 4H, J = 5.0 Hz), 2.58 (t, 4H, J = 5.0 Hz), 2.41 (t, 2H, J = 5.5 Hz), 1.54-1.3 (m, 4H), 0.95 (t, 3H, J = 7.05 Hz); 13C NMR (50.3 MHz, CDCl3): δ 158.4, 151.5, 141.8, 138.5, 127.0, 124.2, 123.4, 121.0, 110.8, 58.4, 53.0, 44.7, 28.9, 20.7, 14.0; IR (CHCl3): ν = 2930.1, 2856.3, 1616.0, 1496.9, 1324.6, 1232.7 cm-1; Anal. Calcd. for C17H22N4O2 · HCl : C, 58.20; H, 6.61; N, 15.97; Found: C, 57.99; H, 6.78; N, 15.68%.
2-(4-Pentyl-piperazin-1-yl)-6-nitroquinoline (5)
C18H24N4O2. M = 328.19 g/mol. Yield 79 %. Rf: 0.57 (10% MeOH/DCM); 1H NMR (200 MHz, CDCl3): δ 8.49 (d, 1H, J = 2.6 Hz), 8.27 (dd, 1H, J1 = 2.6 Hz, J2 = 9.3 Hz), 7.93 (d, 1H, J = 9.3 Hz), 7.63 (d, 1H, J = 9.3 Hz), 7.04 (d, 1H, J = 9.3 Hz), 3.86 (t, 4H, J = 5.2 Hz), 2.57 (t, 4H, J = 5.2 Hz), 2.39 (t, 2H, J = 7.3 Hz), 1.65-1.50 (m, 2H), 1.40-1.25 (m, 4H), 0.91 (t, 3H, J = 6.7 Hz); 13C NMR (50.3 MHz, CDCl3): δ 158.4, 151.5, 141.8, 138.5, 127.0, 124.2, 123.5, 120.9, 110.8, 58.7, 53.0, 44.6, 29.7, 26.48, 22.6, 14.0; IR (CHCl3): ν = 2933.6, 2861.1, 1616.2, 1496.9, 1324.6, 1233.0 cm-1; Anal. Calcd. for C18H25N4O2 · HCl : C, 59.25; H, 6.91; N, 15.36; Found: C, 59.42; H, 7.05; N, 15.34%.
2-(4-Hexyl-piperazin-1-yl)-6-nitroquinoline (6)
C19H26N4O2. M = 342.44 g/mol. Yield 85 %. Rf: 0.55 (10% MeOH/DCM); 1H NMR (200 MHz, CDCl3): δ 8.53 (d, 1H, J = 2.6 Hz), 8.29 (dd, 1H, J1 = 2.6 Hz, J2 = 9.3 Hz), 7.96 (d, 1H, J = 9.3 Hz), 7.65 (d, 1H, J = 9.3 Hz), 7.06 (d, 1H, J = 9.3 Hz), 3.87 (t, 4H, J = 5.0 Hz), 2.58 (t, 4H, J = 5.0 Hz), 2.40 (t, 2H, J = 7.2 Hz), 1.60-1.50 (m, 2H), 1.40-1.20 (m, 6H), 0.90 (t, 3H, J = 3.9 Hz); 13C NMR (50.3 MHz, CDCl3): δ 158.4, 151.5, 141.8, 138.5, 127.0, 124.2, 123.6, 120.9, 110.8, 58.7, 53.0, 44.7, 31.7, 27.2, 26.8, 22.6, 14.0; IR (CHCl3): ν = 2932.4, 2859.0, 1616.1, 1497.0, 1324.3, 1232.4 cm-1; Anal. Calcd. for C19H26N4O2 · HCl: C, 60.27; H, 7.19; N, 14.79; Found: C, 60.27; H, 7.15; N, 14.81%.
2-(4-Heptyl-piperazin-1-yl)-6-nitroquinoline (7)
C20H28N4O2. M = 356.46 g/mol. Yield 84 %. Rf: 0.48 (10% MeOH/DCM); 1H NMR (200 MHz, CDCl3): δ 8.51 (d, 1H, J = 2.6 Hz), 8.28 (dd, 1H, J1 = 2.6 Hz, J2 = 9.3 Hz), 7.94 (d, 1H, J = 9.3 Hz), 7.64 (d, 1H, J = 9.3 Hz), 7.05 (d, 1H, J = 9.3 Hz), 3.86 (t, 4H, J = 5.2 Hz), 2.58 (t, 4H, J = 5.2 Hz), 2.40 (t, 2H, J = 7.3 Hz), 1.58-1.50 (m, 2H), 1.40-1.20 (m, 8H), 0.89 (t, 3H, J = 6.7 Hz); 13C NMR (50.3 MHz, CDCl3): δ 158.3, 151.4, 141.6, 138.4, 126.9, 124.1, 123.4, 120.9, 110.7, 58.6, 53.0, 44.6, 31.7, 29.1, 27.4, 26.7, 22.5, 14.0; IR (CHCl3): ν = 2931.1, 2857.4, 1616.2, 1496.9, 1324.4, 1233.0 cm-1; Anal. Calcd. for C20H28N4O2 · HCl : C, 61.14; H, 7.44; N, 14.26; Found: C, 61.22; H, 7.50; N, 14.32%.
2-(4-Octyl-piperazin-1-yl)-6-nitroquinoline (8)
C21H30N4O2. M = 370.49 g/mol. Yield 82 %. Rf: 0.42 (10% MeOH/DCM); 1H NMR (200 MHz, CDCl3): δ 8.52 (d, 1H, J = 2.6 Hz), 8.29 (dd, 1H, J1 = 2.6 Hz, J2 = 9.3 Hz), 7.95 (d, 1H, J = 9.3 Hz), 7.65 (d, 1H, J = 9.3 Hz), 7.06 (d, 1H, J = 9.3 Hz), 3.87 (t, 4H, J = 5.0 Hz), 2.58 (t, 4H, J = 5.0 Hz), 2.40(t, 2H, J = 7.3 Hz), 1.58-1.50 (m, 2H), 1.40-1.20 (m, 10H), 0.89 (t, 3H, J = 6.3 Hz); 13C NMR δc(50.3 MHz, CDCl3):158.3, 151.4, 141.6, 138.4, 126.9, 124.1, 123.4, 120.8, 110.7, 58.6, 53.0, 44.6, 31.7, 29.4, 29.1, 27.4, 26.7, 22.5, 14.0; IR (CHCl3): ν = 2930.1, 2856.3, 1616.0, 1496.9, 1324.6, 1232.7 cm-1; Anal. Calcd. for C21H30N4O2 · 2HCl: C, 56.88; H, 7.27; N, 12.64; Found: C, 57.03; H, 7.19; N, 12.08%.
2-(4-Nonyl-piperazin-1-yl)-6-nitroquinoline (9)
C22H32N4O2. M = 384.51 g/mol. Yield 63%. M.p.: 79-81 °C. 1H NMR (400 MHz, CDCl3): δ 8.51 (s, 1H, Harom), 8.28 (d, 1H, J = 8.5 Hz, Harom), 7.95 (d, 1H, J = 9.0 Hz, Harom), 7.64 (d, 1H, J = 9.0 Hz, Harom), 7.06 (d, 1H, J = 9.0 Hz, Harom), 3.90 (s, 4H, N(CH2)2(CH2)2NC9H19), 2.63 (s, 4H, N(CH2)2(CH2)2NC9H19), 2.44 (s, 2H, CH2), 1.57 (s, 2H, CH2), 1.28 (m, 12H, 6 × CH2), 0.88 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 158.58, 151.70, 142.09, 138.89, 127.33, 124.47, 123.85, 121.26, 111.04, 58.90, 53.17, 44.69, 32.07, 29.73, 29.62, 29.47, 27.70, 26.80, 22.87, 14.32. Anal. Calcd. For C22H32N4O2: C, 68.72; H, 8.39; N, 14.57%. Found C, 68.36; H, 8.15; N, 13.92%. ESI-MS: m/z [%]: 385.2 [M + H]+ 100.
2-(4-Decylo-piperazin-1-yl)-6-nitroquinoline (10)
C23H34N4O2. M = 398.54 g/mol. Yield 58%. M.p.: 98-99 °C. 1H NMR (400 MHz, CDCl3): δ 8.50 (s, 1H, Harom), 8.27 (d, 1H, J = 9.2 Hz, Harom), 7.93 (d, 1H, J = 9.2 Hz, Harom), 7.63 (d, 1H, J = 9.2 Hz, Harom), 7.04 (d, 1H, J = 9.2 Hz, Harom), 3.88 (s, 4H, N(CH2)2(CH2)2NC10H21), 2.60 (s, 4H, N(CH2)2(CH2)2NC10H21), 2.41 (br.t, 2H, J = 7.2 Hz, CH2), 1.55 (br.s, 2H, CH2), 1.28 (m, 14H, 7 × CH2), 0.87 (t, 3H, J = 6.0 Hz, CH3). 13C NMR (100 MHz, CDCl3): δ 158.83, 151.95, 142.28, 139.08, 127.54, 124.70, 124.07, 121.47, 111.27, 59.16, 53.45, 44.99, 32.33, 30.01, 30.01, 29.85, 29.75, 27.95, 27.13, 23.12, 14.56. Anal. calcd. for C23H34N4O2: C, 69.31; H, 8.60; N, 14.06%. Found C, 70.24; H, 8.56; N, 14.39%. ESI-MS: m/z [%]: 399.2 [M + H]+ 100.
2-(4-Undecyl-piperazin-1-yl)-6-nitroquinoline (11)
C24H36N4O2. M = 412.51 g/mol. Yield 55%. M.p.: 89-90 °C. 1H NMR (400 MHz, CDCl3): δ 8.51 (d, 1H, J = 2.8 Hz, Harom), 8.28 (dd, 1H, J1= 2.4 Hz, J2 = 9.2 Hz, Harom), 7.95 (d, 1H, J = 9.2 Hz, Harom), 7.65 (d, 1H, J = 9.2 Hz, Harom), 7.06 (d, 1H, J = 9.6 Hz, Harom), 3.91 (s, 4H, N(CH2)2(CH2)2NC11H23), 2.63 (s, 4H, N(CH2)2(CH2)2NC11H23), 2.44 (br.t, 2H, J = 7.2 Hz, CH2), 1.57 (s, 2H, CH2), 1.31 (m, 16H, 8 × CH2), 0.88 (t, 3H, J = 6.4 Hz, CH3). 13C NMR (100 MHz, CDCl3): δ 158.59, 151.70, 142.15, 138.89, 127.37, 124.45, 123.85, 121.29, 111.04, 58.89, 53.16, 44.68, 32.11, 29.81, 29.77, 29.73, 29.64, 29.54, 27.70, 26.76, 22.88, 14.31. Anal. calcd. for C24H36N4O2: C, 69.87; H, 8.79; N, 13.58%. Found C, 68.73; H, 8.73; N, 13.11%. ESI-MS: m/z [%]: 413.2 [M + H]+ 100.
2-(4-Dodecyl-piperazin-1-yl)-6-nitroquinoline (12)
C25H38N4O2. M = 426.59 g/mol. Yield 53%. M.p.: 95-96 °C. 1H NMR (400 MHz, CDCl3): δ 8.51 (d, 1H, J = 2.0 Hz, Harom), 8.28 (dd, 1H, J1= 2.0 Hz, J2 = 9.2 Hz, Harom), 7.95 (d, 1H, J = 9.2 Hz, Harom), 7.65 (d, 1H, J = 9.2 Hz, Harom), 7.05 (d, 1H, J = 9.2 Hz, Harom), 3.91 (s, 4H, N(CH2)2(CH2)2NC12H25), 2.64 (s, 4H, N(CH2)2(CH2)2NC12H25), 2.45 (br.t, 2H, J = 7.2 Hz, CH2), 1.57 (br.s, 2H, CH2), 1.28 (m, 18H, 9 × CH2), 0.88 (t, 3H, J = 6.0 Hz, CH3). 13C NMR (100 MHz, CDCl3): δ 158.58, 151.70, 142.18, 138.92, 127.39, 124.47, 123.88, 121.31, 111.05, 58.87, 53.14, 44.65, 32.13, 29.87, 29.85, 29.82, 29.78, 29.73, 29.56, 27.69, 26.71, 22.90, 14.33. Anal. calcd. for C25H38N4O2: C, 70.39; H, 8.98; N, 13.13%. Found C, 69.82; H, 8.55; N, 12.75%. ESI-MS: m/z [%]: 427.2 [M + H]+ 100.
Nuclear magnetic resonance (NMR) spectroscopy of 8 and 9
The NMR spectra were recorded at 303 K on Varian INOVA 500 spectrometer (Varian, Palo Alto, USA) operated at 499.8, 125.7 and 50.51 MHz for 1H, 13C and 15N nuclei, respectively. Eight mg of the compounds (8 and 9) were dissolved in 0.7 ml DMSO-d6 (Dr. Glaser AB, Basel, Switzerland) and transferred to a 5 mm NMR tube. Chemical shifts (δ, ppm) were referenced against the internal reference 3-trimethylsilyl-propionic acid (TSPA; Dr. Glaser AB, Basel, Switzerland). The spectrometer was equipped with an inverse 1H{13C} 5 mm Nalorac probe with an actively shielded z-gradient coil (ID-PFG) (Varian, Palo Alto, USA).
1H NMR was run using the standard program implemented in the Varian software (Varian, Palo Alto, USA). Double pulsed field gradient spin echo nuclear overhauser effect (DPFGSE-NOE) experiments were run using a pulse sequence published by Stott et al. (26) using shaped selective π soft pulses generated by a standard Varian program (Varian, Palo Alto, USA). The rsnob π pulse was calibrated for each individual multiplet. The Echo-antiecho phase sensitive gradient-selected 1H-13C HSQCAD (heteronuclear single quantum coherence adiabatic version) NMR spectra (27) were obtained with a spectral width of 5000 Hz, 2048 points in the 1H dimension and 8000 Hz, 800 × 2 increments in the 13C dimension; 128 transients per t1 increment, with a relaxation delay of 1 s and 1J(C,H) = 135 Hz. The data were linearly predicted to 1600 points and zero-filled to 4096 points in F1 before Fourier transformation. The gradient selected 1H-13C HMBC (heteronuclear multiple bond coherence) spectra were performed with an acquisition time of 0.2 s, 1H - 90° pulse width of 7.8 μs, 13C - 90° pulse width of 11.5 μs, a spectral width of 5000 Hz, 2048 data points in the 1H dimension and 25000 Hz, 1024 increments in the 13C dimension, relaxation delay of 1.2 s. The data were acquired as absolute value mode, with 64 transients per t1 increment. The experiment was optimized for nJ(C,H) = 8 Hz and low pass filter for 1J(C,H) = 140 Hz was used. The data were linear predicted to 2048 points and zero filled to 4096 points in F1 prior to Fourier transformation. The 1H/15N-HSQC (heteronuclear single quantum coherence) NMR spectra were obtained using external CH3NO2 (POCh, Warszawa, Poland) as the 15N reference (using the internally locked substitution method) (28). The spectral parameters were as follows: π/2 pulse lengths 7.2 μs (1H) and 21.5 μs (15N); acquisition time 0.2 s; spectral windows 8000 Hz (F2) and 10000 Hz (F1); 2048 data points in the 1H dimension and 256 increments in that of 15N; 1J(N,H) = 80 Hz; relaxation delay 1.0 s; 4 transients per increment. The spectra were measured in DMSO-d6 (Dr. Glaser AB, Basel, Switzerland) at 298 K or in DMF-d7 (Dr. Glaser AB, Basel, Switzerland) at 253 K using a Varian INOVA 500-MHz NMR spectrometer (Varian, Palo Alto, USA) equipped with an inverse 1H{31P/15N} 5 mm Z-SPEC Nalorac probe with an actively shielded z-gradient coil (Varian, Palo Alto, USA). The coherence selection with two pulsed field gradient (PFG) pulses of relative amplitudes of +(γ I+γ S): −(γ I−γ S) and −(γ I−γ S): +(γ I+γ S), for heteronuclear echo and anti-echo, respectively, was applied. Such implementation enabled us to obtain nearly pure absorption lineshapes along F1, combined with absolute value mode in F2 dimension. The spectral parameters were as follows: π/2 pulse lengths 7.2 μs (1H) and 21.5 μs (15N); acquisition time 0.2 s; spectral windows 8000 Hz (F2) and 23000 Hz (F1); 2048 data points in the 1H dimension and 256 increments in that of 15N; nJ(N,H) = 3 Hz; relaxation delay 1.0 s; 8 transients per increment.
In vitro binding affinity assay
SERT
The assay was performed in accordance with the method described by Owens et al. (2) with slight modifications. Rat cerebral cortex was homogenized in 30 volumes of ice-cold 50 mM Tris-HCl containing 150 mM NaCl and 5 mM KCl, pH = 7.7 at 25 °C and centrifuged at 20.000 × g for 20 minutes. The supernatant was decanted and pellet was resuspended in 30 volumes of buffer and centrifuged again. The resulting pellet was resuspended in the same quantity of the buffer and centrifuged third time in the same conditions. [3H]-Citalopram (spec. act. 50 Ci/mmol, NEN Chemicals, USA) was used for labelling 5-HT-transporter. 240 μl of the tissue suspension, 30 μl of 1 μM imipramine (Sigma, USA) (displacer), 30 μl of 1nM [3H]-citalopram and 100 μl of the analysed compound were incubated at 22°C for 1 h. The concentrations of analysed compounds ranged from 10-10 to 10-4 M. Incubations were terminated by vacuum filtration over Whatman GF/B filters (Whatman GE, USA) and washed two times with 100 μl of ice-cold buffer. The radioactivity was measured using a WALLAC 1409 DSA – liquid scintillation counter (Wallac, USA). The assay was done in duplicate. Radioligand binding data were analysed using iterative curve fitting routines (GraphPAD/Prism, Version 3.0, San Diego, CA). Ki values were calculated from the Cheng-Prusoff equation (29):
where LO is the labelled ligand concentration and KD is the dissociation constant of labelled ligand.
5-HT1A receptor
[3H]8-Hydroxy-2-(di-n-propylamino)tetraline (8-OH-DPAT, spec. act. 106 Ci/mmol, NEN Chemicals) was used for labelling of the 5-HT1A receptors. The membrane preparation and assay procedure were carried out according to a previously described procedure (30) with slight modifications. In brief, the hippocampus tissue was homogenised in 20 volumes of 50 mM Tris-HCl buffer (pH 7.7 at 25°C) using Ultra-Turrax® T 25 and then centrifuged at 32000g for 10 min. The supernatant fraction was discarded, the pellet resuspended in the same volume of Tris-HCl buffer and the solution centrifuged again. Before the third centrifugation, the samples were incubated at 37°C for 10 min. The final pellet was resuspended in Tris-HCl buffer containing 10 μM pargyline, 4 mM CaCl2 and 0.1 % ascorbic acid. Samples containing 1 ml of the tissue suspension (5 mg wet weight), 100 μl of 10 μM serotonin for non-specific binding, 100 μl of [3H]8-OH-DPAT, and 100 μl of analysed compound were incubated at 37°C for 15 minutes. The incubation was followed by rapid vacuum filtration through Whatman GF/B glass filters and the remainder was washed three times with 5 ml cold buffer (50 mM Tris-HCl, pH 7.7) using a Brandel cell harvester. The final [3H]8-OH-DPAT concentration was 1 nM and the concentrations of the analysed compounds ranged from 10-10 to 10-4 M.
Homology modelling and docking
SERT
The construction of the homology model of SERT based on the LeuT in an outward-facing conformation (PDB id 3F3A) (25) has been described elsewhere (24) and will only shortly be described here. The LeuT and SERT (UniProt accession number P31645) sequences were aligned and adjusted to fit a comprehensive alignment of NSS members (31). The homology models were generated using the BuildModel macro available in ICM version 3.5, and were energy refined using the refineModel macro of ICM (32).
Starting geometries of the ligands were built using ICM (32) based on the information obtained from the NMR spectroscopy data of 8, which showed that the nitroquinoline and alkyl substituents occupied diequatorial positions in the piperazine ring and that the alkyl substituent was in extended conformation and that the piperazine N4’ nitrogen atom formed the primary protonation site of the ligand molecule. The N4’ atom of the ligands was hence charged using default ECEPP/3 partial charges (33).
A flexible docking protocol (24) was used to dock 6-NQ and the twelve alkyl analogues. It consisted of (1) detection of a ligand binding pocket using ICM PocketFinder (34), (2) torsional sampling of the side chains of the amino acids constituting the binding pocket using biased probability Monte Carlo (BPMC) (35) and (3) 4D flexible ligand docking (36). Using this protocol, the side chains of amino acids Y95, D98, L99, W103, R104, Y107, Y175, Y176, I179, F335, S336, L337, F341, V343, K399, D400, P403, L405, L406, F407, S438, T439, E493 and T497 were sampled. The side chain sampling resulted in the generation of 47 SERT binding pocket conformations (24) into which 6-NQ and the twelve analogues were docked using the 4D docking approach (36). Three parallel docking runs of 6-NQ and the analogues were performed. Following the 4D docking, refinement of the best scored complex of each ligand was performed, in which the side-chain torsional angles of the amino acids in the vicinity of the ligands were sampled using the BPMC procedure (35).
The refined SERT-ligand complexes were scored using the ICM scanScoreExternal macro (32). The scoring function includes terms for steric, entropic, hydrogen bonding, hydrophobic and electrostatic interactions, together with a correction term that takes into account the number of atoms in the ligand to avoid the bias towards larger ligands (37).
5-HT1A receptor
A homology model of the 5-HT1A receptor (UniProt accession number P08908) was constructed using the crystal structure of the human β2-adrenergic receptor in complex with alprenolol, a beta blocker and 5-HT1A receptor antagonist (38), as template (PDB id 3NYA (39). The final model consisted of amino acids S34-K418 except amino acids 222-339 of IL3. A disulphide bridge between C109 and C187 was included. Following the model construction, the ICM refineModel macro (32) was used to remove possible close contacts between amino acids in the model and to relax the structure. A stereochemical quality analysis of the model and 3NYA template structure was then performed using the PROCHECK, ERRAT and VERIFY 3D programmes available via the SAVES server (http://nihserver.mbi.ucla.edu/SAVES/). The results show that the model structure was of similar stereochemical quality as the template structure.
ICM PocketFinder (tolerance level 3) (34) detected that the following amino acids constituted the orthosteric binding site of the 5-HT1A receptor: A93, Y96, Q97, F112, I113, D116, V117, C120, C187, I189, K191, Y195, T196, S199, T200, A203, W358, F361, F362, V364, A365, L368, T379, L381, I385, W387 and Y390. Three parallel docking runs of 6-NQ, 1-12 and (R)- and (S)-alprenolol into the orthosteric binding site using ICM were performed.
In vivo tests
Based on the results from the in vitro binding studies, 8, 9, 10, 11 and 12 were selected for in vivo tests.
Animals and housing
The in vivo experiments were performed on male Albino Swiss mice (24 - 28 g). The animals were kept at a room temperature (20 ± 1°C) on a natural day-night cycle (April - May) and housed under standard laboratory conditions. They had free access to food and tap water before the experiments. Each experimental group consisted of 6 - 8 animals/dose, and all the animals were used only once.
Drug treatments and analysis of results
8-OH-DPAT (Research Biochemical Inc.) and N-{2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl}-N-(2-pyridinyl)cyclohexane-carboxamidetrihydrochloride (WAY-100635, synthesised by Dr. J. Boksa, Institute of Pharmacology, Polish Academy of Sciences, Kraków, Poland), were used as aqueous solutions. 9, 10, 11 and 12 were suspended in a 1% aqueous solution of Tween 80 whereas 8 was suspended in dimethyl sulfoxide (DMSO). 8-OH-DPAT and WAY-100635 were injected subcutaneously (sc), whereas 8, 9, 10, 11 and 12 were given intraperitoneally (ip) in volumes of 10 ml/kg. The comparisons of the compounds were carried out by a one-way analysis of variance (ANOVA) followed by intergroup comparisons using the Dunnett’s test (when one drug was administered) or by the Newman-Keuls test (when two drugs were administered).
Forced swim test
The experiment was carried out according to the method of Porsolt et al. (40). The mice were individually placed in a glass cylinder (25 cm high, 10 cm in diameter) containing 6 cm of water maintained at 23 - 25°C, and were left therein for 6 minutes. A mouse was regarded as immobile when it remained floating on water, making only small movements to keep its head above water. The total duration of immobility was measured by an experimenter during the final 4 minutes of a 6 minutes test session after a 2 minutes habituation period.
Locomotor activity test
The spontaneous locomotor activity of mice was recorded in photoresistor actometers (24 cm in diameter and illuminated by two light beams), which were connected to a counter for the recording of light-beam interruptions. The mice were placed individually in the actometers, and the number of light beam crossings was counted twice: during the first 6 minutes, i.e. at the time equal to the observation period in the forced swimming test, and during a 30-minutes experimental session.
RESULTS AND DISCUSSION
Synthesis of 6-NQ alkyl analogues and in vitro binding studies
The general synthesis scheme of the twelve 6-NQ analogues is shown in Figure 1. The results of the in vitro studies showed that the long-chain analogues (8 - 12) have the highest binding affinity (Ki) in SERT, ranging from 1.8 ± 0.2 nM to 20.8 ± 3.8 nM, respectively (Table 1). The binding affinities of the shorter analogues varied more and the methyl analogue (1, quipazine) had the lowest affinity of the analogues with a Ki of 1300 ± 300 nM (Table 1).
Table 1.
6-nitroquipazine (6-NQ) analogue affinities (Ki, nM) in SERT.
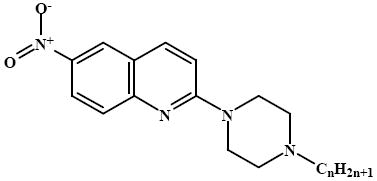
| ||
|---|---|---|
| Compound | n | SERT |
| 6-NQ | 0 | 0.17 ± 0.03a |
| 1 | 1 | 1300 ± 300 |
| 2 | 2 | 86.1 ± 8.5 |
| 3 | 3 | 495.7 ± 34.3 |
| 4 | 4 | 24.0 ± 2.6 |
| 5 | 5 | 80.5 ± 7.4 |
| 6 | 6 | 72.9 ± 8.1 |
| 7 | 7 | 54.1 ± 4.7 |
| 8 | 8 | 20.8 ± 3.8 |
| 9 | 9 | 1.8 ± 0.2 |
| 10 | 10 | 4.5 ± 0.7 |
| 11 | 11 | 12.6 ± 1.3 |
| 12 | 12 | 16.1 ± 1.2 |
From (6)
As 6-NQ analogues may be dual-acting compounds that inhibit SERT and antagonise the presynaptic 5-HT1A receptor and hence reduce the onset time of antidepressant action (22), the affinities of the analogues to this receptor were also determined. The results, however, showed that they had low 5-HT1A receptor affinity (in the μM range). The butyl analogue (3) had the highest 5-HT1A receptor affinity with a Ki = 842.3 ± 12.6 nM (supporting information).
NMR studies
The DPFG NOE results for compound 8 are shown in Figure 2, while the corresponding results for compound 9 are shown in the supplementary information (Figure S1). The NMR studies suggested that the piperazine ring of 8 and 9 were in a fixed chair conformation with substituents on nitrogen atoms occupying the equatorial positions. The high frequency position of NH signal (11.02 ppm in 1H NMR) and 1J (N, H) = 75 Hz showed that NH did not exchange and was involved in hydrogen bonding, most probably with DMSO. The protonation site was confirmed by DPGSE-NOE experiment, which showed that irradiation of NH resonance produced nOe enhancements at 3.06 ppm, i.e., the H3’a H3’e and H1” signals overlapped (Figure 2). Both signals were in close vicinity of piperazine N4’protonation site. Tautomerisation of a proton between the piperazine N1’ and N4’ nitrogen atoms was excluded. The NMR experiment also suggested that quinoline N1 and piperazine N1’ atoms could form a secondary protonation site. The attempts to locate another hydrogen atom were, however, unsuccessful even at -20°C in DMF-d7. Apparently the exchange of a proton, possibly between quinoline N1 and piperazine N1’, was very fast. This also may be a reason that it was not possible to observe resonance position of these atoms neither in HSQC nor in HMBC experiments.
Figure 2.
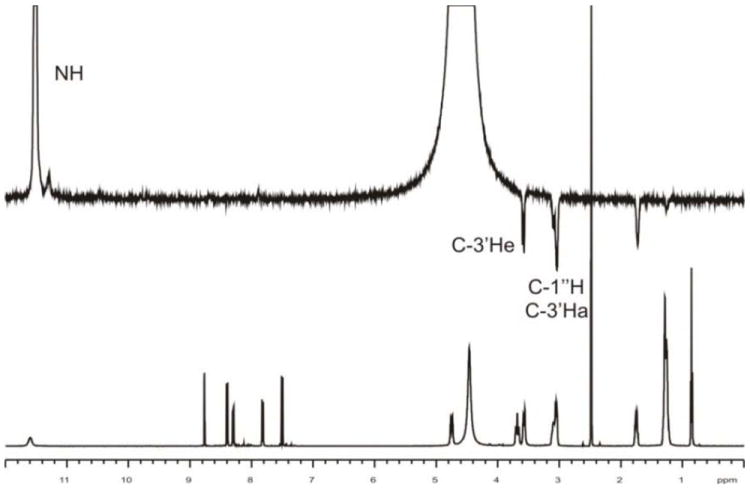
DPFG NOE results of 8 × HCl in DMSO-d6 solution. Upper spectrum shows nOe effects and bottom spectrum shows a trace of 1H NMR in studied solvent. Irradiation of NH signal results in NOE enhancements at marked positions.
Docking
SERT
The compounds were docked into the ligand binding pocket detected by ICM PocketFinder (34) in an outward-facing SERT homology model that had previously been generated using the x-ray crystal structure of LeuT (PDB id 3F3A (25)) as template (24).
LeuT, a prokaryotic homologue of SERT, was the first, and is so far the only, NSS family member to be crystallised. The LeuT crystal structures published represent three major conformations of the transport cycle: the outward-facing conformation (e.g. PDB id 3F3A (25)), in which the central substrate binding site is accessible from the extracellular environment but inaccessible to the intracellular environment, the occluded conformation (e.g. PDB id 2A65 (41)), where a thin extracellular gate and a thick intracellular gate separate the central substrate binding site from either environments, and the inward-facing conformation (PDB id 3TT3 (42)), where the central substrate binding site is inaccessible from the extracellular environment but accessible from the cytoplasm. In the present study, a SERT homology model (24) based on the outward-facing LeuT conformation (25) was chosen for docking as studies have indicated that inhibitors of SERT stabilise outward-facing conformations of the transporter (38, 39).
The binding pocket detected by ICM PocketFinder (34) in the model extended from the putative central substrate binding site up towards the extracellular environment (Figure 3). The lower parts of this pocket corresponded to the putative substrate binding site of SERT detected through experimental studies (43-48). This region also corresponds to the central high-affinity leucine binding site of LeuT (41). In addition to the central substrate binding site, the vestibular region extending from the extracellular environment towards the putative substrate binding site was also included in the binding pocket. The vestibular region contained several charged amino acids (R104, K399, D400 and E493) and EL4 was oriented like a lid over the region (Figure 3).
Figure 3.
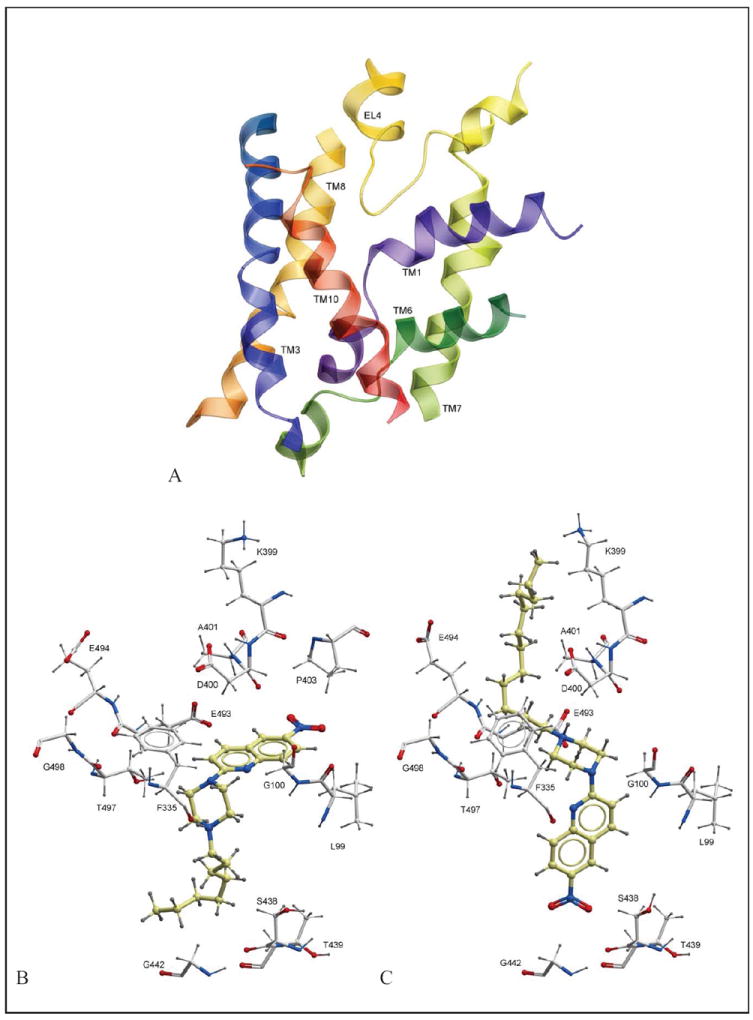
Docking results. (A) The binding pocket (ribbon representation) of the outward-facing SERT homology model, (B) Orientation of 8 in the binding pocket, (C) Orientation of 12 in the binding pocket. Amino acids surrounding 8 and 12 in (B) and (C) are shown in xstick representation.
The docking scores of the SERT-ligand complexes are listed in the supporting information and show that docking scores did not correlate to the binding affinities measured experimentally (Table 1). The main reason for the lack of correlation is the structural flexibility of the compounds and putative structural limitations in our SERT models. Structural flexibility is challenging for the sampling the conformational space of the ligand during docking. The entropic term of the scoring function is proportional to the number of rotatable bonds in the ligand. Scoring of structurally flexible compounds during docking is therefore challenging.
Two main orientations of the ligands were observed during docking. Illustrations of these orientations, 8 and 12 in the binding pocket are shown in Figure 3. The nitroquinolone moiety of 8 was pointing towards EL4 amino acids D400 (backbone) and P403. The octyl side chain of 8 was located in the central region of the pocket, surrounded by the side chains of A169 (TM3), I172 (TM3), A173 (TM3) and F341 (TM6) (Figure 3). The protonated nitrogen of the piperazine ring was in the vicinity of the backbone oxygen of F335 (TM6) (Figure 3). A similar orientation has been proposed by our group by docking of another series of long-chain arylpiperazine nitroquipazine analogues (49). In comparison, 12 had an opposite orientation to that of 8 in the binding pocket. Its −NO2 moiety was located in the lower parts of the central binding pocket, pointing towards S438 in TM8, whereas the protonated piperazine moiety was located near EL4, possibly forming ionic interactions with D400 (EL4) and/or E493 (TM10) (Figure 3). Its dodecyl side chain was located in the extracellular regions of the binding pocket, juxtaposed between EL4 and TMs 6 and 10 (Figure 3). Of the thirteen compounds docked, five preferred a binding mode similar to that of 12, whereas the remaining eight were oriented in a similar manner as 8 (supporting information). When oriented similar to 8, the longer alkyl analogues may form favourable hydrophobic interactions with amino acids located in TM3 and TM8 (A169, A172, A173, F341) of SERT. These hydrophobic interactions may contribute to more favourable affinity values of the longer alkyl analogues than the shorter.
5-HT1A receptor
To study the possible interactions of the analogues in the 5-HT1A receptor, a model of the receptor was constructed using the crystal structure of the human β2-adrenergic receptor in complex with alprenolol (39) as template. Alprenolol is a beta blocker but also a 5-HT1A receptor antagonist (38). Docking of the compounds into the orthosteric binding site of the 5-HT1A receptor model indicated that the compounds are weak 5-HT1A receptor binders, which is in agreement with the results of the 5-HT1A receptor in vitro binding studies results (Table S1, supporting information). The experimental 5-HT1A receptor binding affinities indicated that compound 3 is the strongest 5-HT1A receptor binder. The best docking score was likewise obtained with compound 3 (-4.56 kcal/mol, Table S1). In comparison, the alprenolol docking scores were -14.42 kcal/mol for the R enantiomer and -19.08 kcal/mol for the S enantiomer. This is also in agreement with experimental results showing that the S-enantiomer binds stronger than the R enantiomer (38). The orientations of both alprenolol enantiomers were furthermore very similar to that of alprenolol in the β2-adrenergic receptor (rmsd approx. 2 Å). This suggests that the model was suitable for docking of 5-HT1A receptor binders.
In vivo tests
The in vitro binding affinities of the 6-NQ analogues with the longest alkyl side chains were within the range of known antidepressant drugs (2, 4) and 8 - 12 were hence selected for in vivo testing in mice using the forced swim test developed by Porsolt et al. (40). The forced swim test, also known as the behavioural despair test, is a common way to evaluate the antidepressant effect of compounds in animals (rats or mice). During the test, the animals are forced to swim in a glass cylinder from which they cannot escape. After a short time of vigorous activity, the animals adopt an immobile posture. Administration of antidepressants and psychostimulants reduces the immobility time; however, psychostimulants additionally cause marked motor stimulation (40). The forced swim test is thus often followed by a locomotor activity test.
The results of the forced swim test showed that 8 (20 mg/kg) and 10 (5 mg/kg) caused a reduction in mice immobility time by 15 % and 12 %, respectively, whereas 12 reduced the immobility time by 10 % and 8 % at doses of 5 and 10 mg/kg, respectively (Figure 4). 9 and 11 were ineffective (Figure 4). The results of the locomotor activity test indicated that the reduced immobility time observed in the forced swim test after administration of 8, 10 and 12 was not the result of increased spontaneous locomotor activity (Table 2).
Figure 4.
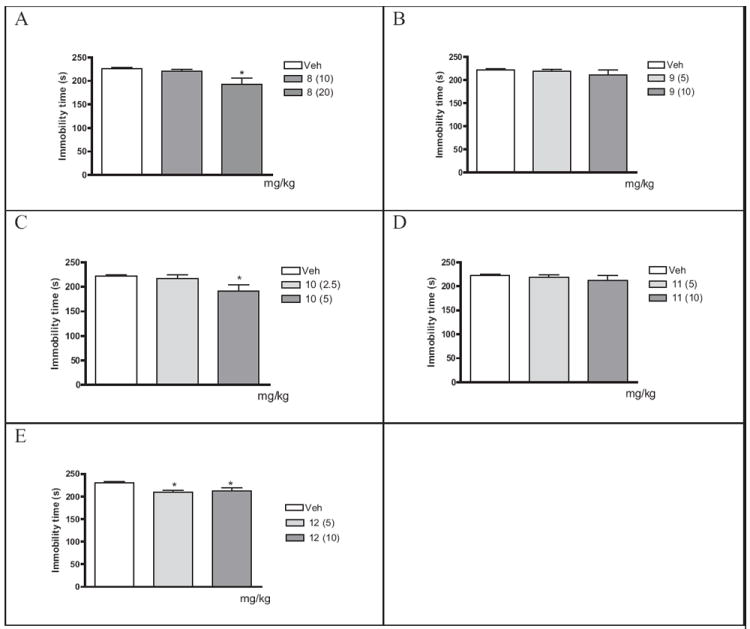
Effects of 8 (A), 9 (B), 10 (C), 11 (D) and 12 (E) in the forced swim test in Albino Swiss mice. Each bar represents the mean ± SEM of 9-10 mice. All analogues were injected 30 min before the test. *p<0.05 vs. respective vehicle group (Dunnett’s test).
Table 2.
Effect of 8 (20 mg/kg), 10 (5 mg/kg) and 12 (10 mg/kg) on mice locomotor activity.
| Treatment | Locomotor activity | |
|---|---|---|
|
| ||
| 6 minutes | 30 minutes | |
| Vehicle | 554 ± 46 | 1514 ± 173 |
| 8 | 453 ± 41 | 1409 ± 199 |
| 10 | 239 ± 41*** | 897 ± 120** |
| 12 | 628 ± 55 | 1774 ± 187 |
Number of light beam crossings ± S.E.M. 8, 10 and 12 were injected ip 30 min before the test.
p<0.001,
p<0.01 vs. vehicle group, (n=10). 6 minutes: F(2,27)=18.47, P<0.001; 30 minutes: F(2,27)=6.505, P<0.01.
CONCLUSION
In this study, a combination of in vitro, in silico and in vivo approaches has been used to evaluate twelve 6-nitroquipazine analogues (1 - 12). The in vitro binding studies showed that the analogues with the longest side chains were high-affinity SERT inhibitors, though their affinity in the 5-HT1A receptor was low. Three analogues were found to have moderate antidepressant activity in the in vivo Porsolt forced swim test (8, 10 and 12). A reason for the moderate antidepressant activity may the polar head group of the compounds that may contribute to low bioavailability and low concentration of the compounds at the site of action.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Nevronor program of the Research Council of Norway (project 176956/V40), the Polish-Norwegian Research Fund (grant PNRF-103-AI-1/07) and the University of Tromsø, Norway. The work was also supported by grants from National Institutes of Health USA (grant numbers R01 GM071872, U01 GM094612, U54 GM094618, and RC2 LM 010994). Mari Gabrielsen gratefully acknowledges support and training from BioStruct, the Norwegian national graduate school in Structural biology.
References
- 1.Saier MH., Jr A functional-phylogenetic classification system for transmembrane solute transporters. Microbiol Mol Biol Rev. 2000;64:354–411. doi: 10.1128/mmbr.64.2.354-411.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther. 1997;283:1305–22. [PubMed] [Google Scholar]
- 3.Vaatstra WJ, Deiman-Van Aalst WM, Eigeman L. Du 24565, a quipazine derivative, a potent selective serotonin uptake inhibitor. Eur J Pharmacol. 1981;70:195–202. doi: 10.1016/0014-2999(81)90214-4. [DOI] [PubMed] [Google Scholar]
- 4.Owens MJ, Knight DL, Nemeroff CB. Second-generation SSRIs: human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol Psychiatry. 2001;50:345–50. doi: 10.1016/s0006-3223(01)01145-3. [DOI] [PubMed] [Google Scholar]
- 5.Gerdes JM, DeFina SC, Wilson PA, Taylor SE. Serotonin transporter inhibitors: synthesis and binding potency of 2’-methyl- and 3’-methyl-6-nitroquipazine. Bioorg Med Chem Lett. 2000;10:2643–6. doi: 10.1016/s0960-894x(00)00546-1. [DOI] [PubMed] [Google Scholar]
- 6.Lee BS, Chu S, Lee BC, Chi DY, Choe YS, Jeong KJ, et al. Syntheses and binding affinities of 6-nitroquipazine analogues for serotonin transporter. Part 1. Bioorg Med Chem Lett. 2000;10:1559–62. doi: 10.1016/s0960-894x(00)00290-0. [DOI] [PubMed] [Google Scholar]
- 7.Lee BS, Chu S, Lee KC, Chi DY, Choe YS, Kim SE, et al. Syntheses and binding affinities of 6-nitroquipazine analogues for serotonin transporter: Part 3. A potential 5-HT transporter imaging agent, 3-(3-[18F]fluoropropyl)-6-nitroquipazine. Bioorg Med Chem. 2003;11:4949–58. doi: 10.1016/j.bmc.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 8.Lee JH, Choi YH, Lim YJ, Lee BS, Chi DY, Jin C. Syntheses and binding affinities of 6-nitroquipazine analogues for serotonin transporter. Part 5: 2’-Substituted 6-nitroquipazines. Bioorg Med Chem. 2007;15:3499–504. doi: 10.1016/j.bmc.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Moon BS, Lee BS, Chi DY. Syntheses and binding affinities of 6-nitroquipazine analogues for serotonin transporter. Part 4: 3-Alkyl-4-halo-6-nitroquipazines. Bioorg Med Chem. 2005;13:4952–9. doi: 10.1016/j.bmc.2005.05.031. [DOI] [PubMed] [Google Scholar]
- 10.Se Lee B, Chu S, Lee BS, Yoon Chi D, Song YS, Jin C. Syntheses and binding affinities of 6-nitroquipazine analogues for serotonin transporter. Part 2: 4-substituted 6-nitroquipazines. Bioorg Med Chem Lett. 2002;12:811–5. doi: 10.1016/s0960-894x(02)00028-8. [DOI] [PubMed] [Google Scholar]
- 11.Biegon A, Mathis CA, Hanrahan SM, Jagust WJ. [125I]5-iodo-6-nitroquipazine: a potent and selective ligand for the 5-hydroxytryptamine uptake complex. II. In vivo studies in rats. Brain Res. 1993;619:236–46. doi: 10.1016/0006-8993(93)91617-2. [DOI] [PubMed] [Google Scholar]
- 12.Hashimoto K, Goromaru T. Preparation of [3H]6-nitroquipazine, a potent and selective 5-hydroxytryptamine uptake inhibitor. Radioisotopes. 1990;39:168–9. doi: 10.3769/radioisotopes.39.4_168. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto K, Goromaru T. 4-Bromo-6-nitroquipazine: a new ligand for studying 5-hydroxytryptamine uptake sites in vivo. Neuropharmacology. 1992;31:869–74. doi: 10.1016/0028-3908(92)90124-8. [DOI] [PubMed] [Google Scholar]
- 14.Jagust WJ, Eberling JL, Biegon A, Taylor SE, VanBrocklin HF, Jordan S, et al. Iodine-123-5-iodo-6-nitroquipazine: SPECT radiotracer to image the serotonin transporter. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 1996;37:1207–14. [PubMed] [Google Scholar]
- 15.Jagust WJ, Eberling JL, Roberts JA, Brennan KM, Hanrahan SM, VanBrocklin H, et al. In vivo imaging of the 5-hydroxytryptamine reuptake site in primate brain using single photon emission computed tomography and [123I]5-iodo-6-nitroquipazine. Eur J Pharmacol. 1993;242:189–93. doi: 10.1016/0014-2999(93)90079-w. [DOI] [PubMed] [Google Scholar]
- 16.Karramkam M, Dolle F, Valette H, Besret L, Bramoulle Y, Hinnen F, et al. Synthesis of a fluorine-18-labelled derivative of 6-nitroquipazine, as a radioligand for the in vivo serotonin transporter imaging with PET. Bioorg Med Chem. 2002;10:2611–23. doi: 10.1016/s0968-0896(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 17.Lundkvist C, Loc’h C, Halldin C, Bottlaender M, Ottaviani M, Coulon C, et al. Characterization of bromine-76-labelled 5-bromo-6-nitroquipazine for PET studies of the serotonin transporter. Nucl Med Biol. 1999;26:501–7. doi: 10.1016/s0969-8051(99)00019-0. [DOI] [PubMed] [Google Scholar]
- 18.Mathis CA, Taylor SE, Biegon A, Enas JD. [125I]5-iodo-6-nitroquipazine: a potent and selective ligand for the 5-hydroxytryptamine uptake complex. I. In vitro studies. Brain Res. 1993;619:229–35. doi: 10.1016/0006-8993(93)91616-z. [DOI] [PubMed] [Google Scholar]
- 19.Mathis CA, Taylor SE, Enas JD, Akgun E. Binding potency of 6-nitroquipazine analogues for the 5-hydroxytryptamine reuptake complex. J Pharm Pharmacol. 1994;46:751–4. doi: 10.1111/j.2042-7158.1994.tb03896.x. [DOI] [PubMed] [Google Scholar]
- 20.Sandell J, Yu M, Emond P, Garreau L, Chalon S, Nagren K, et al. Synthesis, radiolabeling and preliminary biological evaluation of radiolabeled 5-methyl-6-nitroquipazine, a potential radioligand for the serotonin transporter. Bioorg Med Chem Lett. 2002;12:3611–3. doi: 10.1016/s0960-894x(02)00787-4. [DOI] [PubMed] [Google Scholar]
- 21.Guo Y, Chen X, Jia H, Ji X, Liu B. Synthesis and biological evaluation of one novel technetium-99m-labeled nitroquipazine derivative as an imaging agent for serotonin transporter. Appl Radiat Isot. 2008;66:1804–9. doi: 10.1016/j.apradiso.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 22.Perrone R, Berardi F, Colabufo NA, Lacivita E, Larizza C, Leopoldo M, et al. Design and synthesis of long-chain arylpiperazines with mixed affinity for serotonin transporter (SERT) and 5-HT(1A) receptor. J Pharm Pharmacol. 2005;57:1319–27. doi: 10.1211/jpp.57.10.0011. [DOI] [PubMed] [Google Scholar]
- 23.Blier P. Pharmacology of rapid-onset antidepressant treatment strategies. The Journal of clinical psychiatry. 2001;62(Suppl 15):12–7. [PubMed] [Google Scholar]
- 24.Gabrielsen M, Kurczab R, Ravna AW, Kufareva I, Abagyan R, Chilmonczyk Z, et al. Molecular mechanism of serotonin transporter inhibition elucidated by a new flexible docking protocol. Eur J Med Chem. 2011 doi: 10.1016/j.ejmech.2011.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–61. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stott K, Stonehouse J, Keeler J, Hwang T-L, Shaka AJ. Excitation Sculpting in High-Resolution Nuclear Magnetic Resonance Spectroscopy: Application to Selective NOE Experiments. J Am Chem Soc. 1995;117:4199–200. [Google Scholar]
- 27.Summers MF, Marzilli LG, Bax Ad. Complete proton and carbon-13 assignments of coenzyme B12 through the use of new two-dimensional NMR experiments. J Am Chem Soc. 1986;108:4285–94. [Google Scholar]
- 28.Harris RK, Becker ED, De Menezes SM, Granger P, Hoffman RE, Zilm KW. Further conventions for NMR shielding and chemical shifts (IUPAC Recommendations 2008) Magn Reson Chem. 2008;46:582–98. doi: 10.1002/mrc.2225. [DOI] [PubMed] [Google Scholar]
- 29.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 30.Middlemiss DN, Fozard JR. 8-Hydroxy-2-(di-n-propylamino)-tetralin discriminates between subtypes of the 5-HT1 recognition site. European journal of pharmacology. 1983;90:151–3. doi: 10.1016/0014-2999(83)90230-3. [DOI] [PubMed] [Google Scholar]
- 31.Beuming T, Shi L, Javitch JA, Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol Pharmacol. 2006;70:1630–42. doi: 10.1124/mol.106.026120. [DOI] [PubMed] [Google Scholar]
- 32.Abagyan R, Totrov M, Kuznetsov D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. Journal of Computational Chemistry. 1994;15:488–506. [Google Scholar]
- 33.Nemethy G, Gibson KD, Palmer KA, Yoon CN, Paterlini G, Zagari A, Rumsey S, Scheraga H. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. Journal of Physical Chemistry. 1992;96:6472–84. [Google Scholar]
- 34.An J, Totrov M, Abagyan R. Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol Cell Proteomics. 2005;4:752–61. doi: 10.1074/mcp.M400159-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 36.Bottegoni G, Kufareva I, Totrov M, Abagyan R. Four-dimensional docking: a fast and accurate account of discrete receptor flexibility in ligand docking. J Med Chem. 2009;52:397–406. doi: 10.1021/jm8009958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schapira M, Abagyan R, Totrov M. Nuclear hormone receptor targeted virtual screening. J Med Chem. 2003;46:3045–59. doi: 10.1021/jm0300173. [DOI] [PubMed] [Google Scholar]
- 38.Kuipers W, Link R, Standaar PJ, Stoit AR, Van Wijngaarden I, Leurs R, et al. Study of the interaction between aryloxypropanolamines and Asn386 in helix VII of the human 5-hydroxytryptamine1A receptor. Mol Pharmacol. 1997;51:889–96. doi: 10.1124/mol.51.5.889. [DOI] [PubMed] [Google Scholar]
- 39.Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, et al. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc. 2010;132:11443–5. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977;229:327–36. [PubMed] [Google Scholar]
- 41.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–23. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 42.Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481:469–74. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adkins EM, Barker EL, Blakely RD. Interactions of tryptamine derivatives with serotonin transporter species variants implicate transmembrane domain I in substrate recognition. Mol Pharmacol. 2001;59:514–23. doi: 10.1124/mol.59.3.514. [DOI] [PubMed] [Google Scholar]
- 44.Barker EL, Moore KR, Rakhshan F, Blakely RD. Transmembrane domain I contributes to the permeation pathway for serotonin and ions in the serotonin transporter. J Neurosci. 1999;19:4705–17. doi: 10.1523/JNEUROSCI.19-12-04705.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Celik L, Sinning S, Severinsen K, Hansen CG, Moller MS, Bols M, et al. Binding of serotonin to the human serotonin transporter. Molecular modeling and experimental validation. J Am Chem Soc. 2008;130:3853–65. doi: 10.1021/ja076403h. [DOI] [PubMed] [Google Scholar]
- 46.Field JR, Henry LK, Blakely RD. Transmembrane domain 6 of the human serotonin transporter contributes to an aqueously accessible binding pocket for serotonin and the psychostimulant 3,4-methylene dioxymethamphetamine. J Biol Chem. 2010;285:11270–80. doi: 10.1074/jbc.M109.093658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaufmann KW, Dawson ES, Henry LK, Field JR, Blakely RD, Meiler J. Structural determinants of species-selective substrate recognition in human and Drosophila serotonin transporters revealed through computational docking studies. Proteins. 2009;74:630–42. doi: 10.1002/prot.22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walline CC, Nichols DE, Carroll FI, Barker EL. Comparative molecular field analysis using selectivity fields reveals residues in the third transmembrane helix of the serotonin transporter associated with substrate and antagonist recognition. J Pharmacol Exp Ther. 2008;325:791–800. doi: 10.1124/jpet.108.136200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaronczyk M, Wolosewicz K, Gabrielsen M, Nowak G, Kufareva I, Mazurek AP, et al. Synthesis, in vitro binding studies and docking of long-chain arylpiperazine nitroquipazine analogues, as potential serotonin transporter inhibitors. Eur J Med Chem. 2012;49:200–10. doi: 10.1016/j.ejmech.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.