

Abstract
Pancreatic ductal adenocarcinoma (PDA) remains a lethal human malignancy with historically limited success in treatment. The role of aberrant Notch signaling, which requires the constitutive activation of γ-secretase, in the initiation and progression of PDA is well defined and inhibitors of this pathway are currently in clinical trials. Here we investigated the in vivo therapeutic effect of PF-03084014, a selective γ-secretase inhibitor, alone and in combination with gemcitabine in pancreatic cancer xenografts. PF-03084014 treatment inhibited the cleavage of nuclear Notch 1 intracellular domain and Notch targets Hes-1 and Hey-1. Gemcitabine treatment showed good response but not capable of inducing tumor regressions and targeting the tumor-resident cancer stem cells (CD24+CD44+ and ALDH+ tumor cells). A combination of PF-03084014 and gemcitabine treatment resulted tumor regression in 3 of 4 subcutaneously implanted xenograft models. PF-03084014, and in combination with gemcitabine reduced putative cancer stem cells, indicating that PF-03084014 target the especially dangerous and resilient cancer stem cells within pancreatic tumors. Tumor re-growth curves plotted after drug treatments demonstrated that the effect of the combination therapy was sustainable than that of gemcitabine. Notably, in a highly aggressive orthotopic model, PF-03084014 and gemcitabine combination was effective in inducing apoptosis, inhibition of tumor cell proliferation and angiogenesis, resulting in the attenuation of primary tumor growth as well as controlling metastatic dissemination, compared to gemcitabine treatment. In summary, our preclinical data suggest that PF-03084014 has greater anti-tumor activity in combination with gemcitabine in PDA and provides rationale for the further investigation of this combination in PDA.
Keywords: Notch, gamma-secretase inhibitor, PF-03084014, pancreatic cancer
1. Introduction
Pancreatic ductal adenocarcinoma (PDA) affects more than quarter a million people worldwide and most of the patients die within the first year of diagnosis [1]. This poor prognosis is largely due to intense therapeutic resistance and tumor recurrence after surgical removal [2]. Although, novel therapeutic approaches are actively sought to improve outcomes of patients with PDA, there is no therapy that can offer significant survival advantage to patients with this deadly disease [3]. Therefore, there is an urgent need to develop novel and effective treatment strategies that can be rapidly introduced into the clinic.
Activating mutations in the KRAS proto-oncogene occur almost ubiquitously in PDA and in its putative precursor lesions, pancreatic intraepithelial neoplasia (PanIN) [4]. Notch-1 is a key downstream mediator of Kras and sustained Notch1 signaling was shown to be essential in maintaining the transformed phenotype of Ras mutant cells. Moreover, Notch signaling has been reported to synergize with Kras, promoting PanIN initiation and progression [5]. Recent studies have suggested that the ability of Kras to transform cells also depends on cooperation with the Notch signaling pathway [6]. To date, attempts to develop Kras targeted agents to specifically kill cancer cells have been disappointing [7]. Notch signaling inhibition in the setting of PDA treatment would be particularly attractive because this pathway does not appear to play a major role in the normal adult pancreas homeostasis. Notch signaling is implicated in tumor angiogenesis and metastasis [8]. Indeed, down-regulation of Notch receptors using specific siRNA or treatment with γ-secretase inhibitor is correlated with reduced proliferative rates, increased apoptosis, decreased anchorage-dependent growth and deceased invasion properties of pancreatic cancer cells [9; 10]. Hypoxic conditions, which is prevalent in PDA, has been known to stimulate the Notch signaling pathway, and it has been recently shown that pancreatic cancer cells can be sensitized by Notch inhibition [11].
Proteolytic release of the Notch intracellular domain (NICD) by γ-secretase plays a key role in Notch-dependent nuclear signaling. NICD translocates to the nucleus where it activates the transcription of Notch target genes. The strategy of blocking Notch signaling using γ-secretase inhibition has been reported to be effective in targeting tumor cells, cancer stem cells (CSCs), and tumor endothelial cells [12; 13]. PF-03084014 is a non-competitive, and selective small molecule inhibitor of γ-secretase developed by Pfizer and is currently in phase I/II clinical trials against different malignancies [14]. PF-03084014 binds to γ-secretase, blocking proteolytic activation of Notch receptors; as a consequence, Notch signaling is inhibited, leading to apoptosis induction in tumor cells [15; 16].
Here, we investigated the in vivo efficacy of PF-03084014 alone and in combination with GEM in patient derived pancreatic cancer xenografts. Mice bearing engrafted tumors were treated with vehicle, PF-03084014, GEM or a combination of both agents. By impairing Notch signaling, a combination of PF-03084014 and GEM effectively suppressed tumor cell proliferation, induces apoptosis, curb CSCs, inhibited tumor growth, delayed tumor recurrence and prevents metastatic spread in clinically relevant models established from the tumors resected from PDA patients. These results suggest that GEM plus γ-secretase inhibitor combination may be a promising strategy worth to investigate further in pancreatic cancer.
2. Materials and methods
2.1. Drugs
PF-03084014 ((S)-s-((S)-5,7-difluoro-1,2,3,4,--tetrahydro-naphthalen-3-ylamino)-N-(1-(2-methyl-1-1(neopentylamino) propan-2-yl)-1H-imidazol-4-yl) pentanamide) was synthesized at Pfizer, Groton, CT. GEM (Eli Lilly) was purchased from the Johns Hopkins Hospital pharmacy. Chemical structures of PF-03084014 and GEM were reported elsewhere [17; 18].
2.2. Efficacy of PF-03084014, GEM and the combination of PF-03084014 and GEM in subcutaneously (s.c) implanted PDA xenografts
All experiments using mice were approved by the Johns Hopkins University Animal Care and Use Committee. Mice were maintained in accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International. Pancreatic cancer xenografts from the Johns Hopkins PancXenoBank collection were used for the study. These xenografts were generated from freshly resected pancreatic tumor specimens from patients at the time of surgery, and are propagated subcutaneously (s.c.) on the flanks of athymic mice. We used 4 different pancreatic cancer xenografts (Panc215, Panc266, Panc354 and Panc265) for the experiments. Freshly collected tumors from these xenografts were cut into cubes of 1–2 mm3 pieces, and the pieces were s.c implanted on both flanks of 6-week-old female nu/nu athymic mice (Harlan). When cohorts of tumors reached ~200 mm3, mice (5 mice/group) were randomly assigned to control and three treatment groups, with 8–10 tumors in each arm: 1) control; 2) PF-03084014 (150 mg/kg, twice daily, p.o, 7-days-on/7-days-off schedule for 4 weeks); 3) GEM (25 mg/kg, i.p, twice a week for 4 weeks) and 4) GEM + PF-03084014 in the above mentioned dose and frequency for 4 weeks. PF-03084014 and GEM dose were selected based on previously published reports [14; 19]. Tumors were measured twice per week using a digital caliper allowing to discriminate size modifications >0.1 mm. Individual tumor volumes were calculated using the formula: tumor volume V = a × b2/2, a being the largest dimension of the tumor, b the smallest. In order to check the effect of the treatment on tumor re-growth, animals from Panc266 was kept for another 2 months post 4 weeks drug treatment and tumor size was monitored.
2.3. Efficacy of PF-03084014, GEM and the combination of PF-03084014 with GEM in orthotopically implanted PDA xenograft model
Panc265 xenograft, a highly aggressive tumor in orthotopic setting, was selected for the study. Briefly, s.c. grown tumors were harvested and cut into cubes of 1–2 mm3. Mice were anesthetized using volatile gas anesthesia. The abdomen was sterilized and opened via a subcostal left incision of 1 cm. After visualization of the spleen and adherent pancreas, a small pocket was prepared inside the pancreas using microscissors, into which one piece of the tumor was implanted. Orthotopic tumor growth was accessed by ultrasound. When tumors reached a volume of ~200 mm3, mice were randomized (9–10 mice in each group) into 1) control; 2) PF-03084014 (2 cycles of 150 mg/kg, twice daily, p.o, 7-days-on/7-days-off schedule); 3) GEM (25 mg/kg, i.p., twice a week for 3 weeks) and 4) GEM + PF-03084014 in the above mentioned dose and frequency for 3 weeks. Animal body weights were taken twice weekly. On day 21, mice were euthanized; spleen, liver, kidneys, lungs and lymph nodes were inspected using a 10x lens for gross visible metastases and the tissues were preserved in 10% formalin solution. In addition to macroscopic inspection, suspected lesions and random histological sections of the liver, spleen, kidneys, lungs and lymph nodes were examined microscopically for the presence or absence of micro-metastases in each animal.
2.4. Measurement putative pancreatic cancer stem cells (CD44+CD24+ and ALDH+ cancer cells)
In PDA, both CD44+CD24+ and ALDH+ tumor cells display functional properties of CSCs [20; 21]. Upon implantation, these phenotypically distinct cells were equally tumorigenic in NOD/SCID and NSG mice [20; 22]. We assessed the presence of CSCs in the four individual patient-derived xenografts used in the present study. Fresh tumors (s.c. grown) harvested from untreated mice of Panc 215, Panc265, Panc354 and Panc265 xenografts were used for the study. CD44+CD24+ and ALDH+ cancer cells were determined by FACS analysis as previously reported procedures [23]. Briefly, single-cell suspensions were generated from the fresh tumors by mincing tumors using sterile razors, followed by incubation in dispase and collagenase type IV at 37°C for 2 hours with agitation. The cell suspension was filtered using a 70-μm filter (BD Biosciences) and further purified by density centrifugation using Ficoll-Paque Plus (GE Healthcare). Both mouse-derived cells and non-viable cells were excluded based on anti-mouse PerCP and propidium iodide (PI) staining. Viable tumor cells were washed and stained with ALDEFLUOR reagent and antibodies against human CD44, CD24 and analyzed using a FACSAria flow cytometer (BD Biosciences), as previously described [23]. Gates were created on the basis of cellular staining with isotype control antibodies.
Treatment effect of PF-03084014 on CD44+CD24+ in Panc215 xenograft was assed by FACS analysis. Tumor samples harvested after 4 weeks of treatments were used for this study. ALDH expression was analyzed by immuno-histochemistry as previously reported procedure [20]. Paraffin embedded tumor sections prepared from Panc215 treatment arms were used for the ALDH immuno-histochemical analysis. Two separate tumors each from treatment arms were analyzed. Slides were deparaffinized and rehydrated by standard procedures. Antigen retrieval was performed by incubating the slides in boiling sodium citrate buffer (10 mM, pH 6.0) for 30 minutes. Endogenous peroxidases were quenched by incubating the slides in 3% hydrogen peroxide in methanol for 10 minutes at room temperature. The slides were incubated with a mouse monoclonal antibody against human ALDH1 (clone 44; BD Biosciences) diluted 1:50 at room temperature for 1 hour followed by antibody detection with the use of a Vectastain Elite developer kit (Vector Labs) and 3,3′-diamniobenzidine, as recommended by the manufacturer. Positive staining was defined as intense 3,3′-diamniobenzidine signal in tumor cells. For quantitation, average number of cells stained positive for ALDH was counted in 5 random fields each from the tumors of 2 separate animals under at 40x magnification.
2.5. Protein extraction and Immuno-blotting
In order to investigate the pharmacodynamic effect of PF-03084014, we conducted immunobloting using post-treatment primary tumors harvested from orthotopically implanted Panc265 model. Protein extracts were prepared from tumors in cell lysis buffer as previously described [24]. Protein lysates (25 μg) were boiled in Laemmli sample buffer, resolved by electrophoresis on 4% to 12% Bis-Tris precast gels (Bio-Rad Laboratories), and transferred to Immobilon-P membranes (Millipore). Membranes were blocked at room temperature with 5% nonfat milk (Pierce) for 1 hour. Membranes were incubated overnight with primary antibodies against rabbit N1ICD (Val1744), p MEK1/2 Ser217/221, total MEK, c-PARP (all from Cell Signaling Technology, Inc.), Hes-1 and Hey-1 (both from Abcam). Membranes were probed with secondary HRP-conjugated antibody (GE Healthcare) and bound antibodies were detected by SuperSignal West Pico/Femto chemiluminescent substrate (Thermo Scientific). Equal loading was verified with β-actin antibody.
2.6. Hematoxylin and eosin (H&E), Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) and Ki-67 staining
Excised tumors or organs from the vehicle and treatment groups were fixed in 10% neutral buffered formalin and processed into paraffin blocks. Tumors harvested from three mice each per treatment group were analyzed. Sections were deparaffinized in xylene and rehydrated in graded-alcohol washes. H&E staining was performed using standard procedure. Ki-67 staining was performed using an anti- MIB-1 (Ki-67) antibody (Ventana Medical Systems; clone K2, 1:100 dilutions) as previously described [18]. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining was done by a commercial apoptosis detection kit (DeadEndTM Fluorometric TUNEL System, Promega), according to the recommendations of the manufacturer [18]. Sections were examined microscopically and representative field were photographed under 10 or 40x magnifications. Average number of cells stained positive for Ki-67 and TUNEL were counted in 5 random fields each from the tumors of 3 separate animals under at 40x magnification.
2.7. N1ICD and CD-31 Immuno-histochemistry
Immuno-histochemical staining of nuclear anti-activated Notch1 (Abcam; ab8925, 1:250 dilutions) and anti-murine CD-31 (Dianova GmbH, Germany; Clone SZ31, 1:30 dilutions) were conducted on formalin-fixed paraffin sections as per the manufactures protocol and previously published procedures [18; 25]. Tumors harvested from three mice each per treatment group of Panc215 and Panc354 were analyzed. All sections were treated with horseradish peroxidase (HRP)-conjugate for one hour and then developed with 3,3′ diaminobenzidine. Finally, the slides were counterstained with haematoxylin and mounted. Sections were examined in Nikon Eclipse microscope for CD-31 positive cells and N1ICD stained nuclei. Representative fields were photographed under 20 or 40x magnifications.
2.8. Statistical analysis
The statistical significance between groups was analyzed by one-way ANOVA and Bonferroni multiple comparison tests using GraphPad Prism. The difference between experimental groups was considered significant at P value <0.05. Line graphs show mean ± SEM.
3. Results
3.1. Combination treatment of PF-03084014 and GEM inhibits the tumor growth in s.c implanted PDA xenografts
When tested as a single agent, PF-03084014 monotherapy failed to impart antitumor activity in pancreatic cancer xenografts. Tumor growth curves of Panc215, Panc265, Panc354 and Panc266 were not statistically significant when compared to vehicle treated animals (Fig. 1). Although, GEM treatment alone caused significant tumor growth inhibition, the combined treatment of GEM with PF-03084014 produced further significant inhibition of tumor growth when compared to GEM in 3 of 4 xenografts (Fig. 1). Additionally, while GEM treatment failed to induce tumor regression, the combination treatment resulted in tumor regression in 3 of 4 xenografts (Fig. 1). In Panc266 xenografts, which were maintained without therapy for follow-up, tumor growth resumed within 2 weeks after the cessation of treatment, indicating that GEM treatment imparts only transient growth inhibitory effects (Fig. 1) as observed in the clinic. In contrast, the GEM with PF-03084014 combination significantly suppressed the tumor growth and the effect was maintained till 92 days, the last day of follow-up (Fig. 1). This suggests that the antitumor effects were sustained after dosing was completed and indicates that combining PF-03084014 to GEM might be efficacious in preventing tumor recurrence.
Figure 1.

A combination of PF-03084014 and gemcitabine (GEM) produces durable tumor growth inhibition in subcutaneously implanted pancreatic cancer xenografts. Tumors from four individual patient-derived pancreatic cancer xenografts were implanted in athymic mice. Animals with established tumors were dosed with vehicle, PF-03084014, GEM or a combination of GEM with PF-03084014 as mentioned in the materials and methods. Tumor size was evaluated twice per week by caliper measurements. After 4 weeks drug treatment, Panc266 xenograft animals were maintained without further therapy. Tumor re-growth curves plotted after drug treatments demonstrate that the effect of the combination therapy was sustainable than that of GEM. Points, mean of tumor volume; bars, SEM (N = 8–10 tumors; *p<0.05, **p<0.01 compared to Control; #p<0.05, # #p<0.01 compared to GEM).
3.2. PF-03084014 targets putative cancer stem cells
We assessed the baseline CD44+CD24+ and ALDH+ tumor cells in the four individual patient-derived xenografts used in this study by flow cytometry. In agreement with our previous data in seven individual patient-derived xenografts [23], the results of the present study showed that percentage of CSCs varied widely from xenografts to xenografts (Table 1).
Table 1.
Cancer stem cell quantification in xenografts by Fluorescence Activated Cell Sorting (FACS)
| Pancreatic cancer xenografts | % ALDH+ cells | % CD44+CD24+ cells |
|---|---|---|
| Panc215 | 9.41 | 3.70 |
| Panc265 | 0.82 | 8.47 |
| Panc354 | 0.75 | 16.60 |
| Panc266 | 4.67 | 0.64 |
NOTE: Four individual patient-derived pancreatic tumors grown subcutaneously (s.c.) in nude mice were harvested in sterile conditions. Single cell suspensions were generated by mincing tumors, followed by incubation in Dispase and Collagenase IV (Sigma) at 37°C for 2h.
The cell suspension was filtered and further purified by density centrifugation using Ficoll-Paque Plus (GE Healthcare). Cells were labeled using the ALDEFLUOR reagent (Stem Cell Technologies) and then stained with antibodies against mouse CD31 (BD Biosciences), lineage cocktail (Miltenyi Biotec), and H-2Kd (BD Biosciences). Non-mouse cells were separated and stained with antibodies against human CD44, CD24 (BD Biosciences). Cells were analyzed using FACSAria Flow Cytometer (BD Biosciences).
We examined the treatment effect of PF-03084014 on CD44+CD24+ and ALDH+ tumor cells, Flow cytometry of tumor cells sorted from Panc215 (4 weeks treatment) showed that GEM treatment was not capable of diminishing the CD44+CD24+ tumor cells and in fact resulted in the enrichment of CD44+CD24+ tumor cells compared to vehicle treated mice (Fig. 2A). However, PF-03084014 and combination of GEM with PF-03084014 resulted in a 3.72 and 3.04-fold decrease in CD24+CD44+ tumor cells compared with GEM treatment, respectively (Fig. 2A). We have recently reported that ALDH expression in PDA was associated with worse overall survival in patients [20]. Both PF-03084014 and combination of PF-03084014 and GEM are effective in reducing ALDH+ tumor cells (Fig. 2B). PF-03084014 alone, and combination of PF-03084014 and GEM decreased in ALDH+ tumor cells compared with GEM treatment (3.4 and 3.7-fold, respectively).
Figure 2.

PF-03084014 monotherapy and combination of PF-03084014 with GEM is highly effective in reducing putative pancreatic cancer stem cells. A) Flow cytometry of tumor cells sorted from Panc215 (after 4 weeks of treatment). GEM treatment was not capable of diminishing the CSC pool as compared to the vehicle treated mice. However, PF-03084014 alone, and combination of PF-03084014 with GEM decreased in CD24+CD44+ tumor cells compared with GEM treatment (3.72 and 3.04-fold, respectively). B) Representative photomicrographs of ALDH staining, indicating that PF-03084014 and combination of PF-03084014 with GEM is highly effective in reducing ALDH+ cells. Both ALDH+ and ALDH- tumor cells were counted in 5 random high power fields of 2 tumors each from treatment arms and expressed as % ALDH+ cells. Arrowheads point towards ALDH+ cells. PF-03084014 alone, and combination of PF-03084014 with GEM decreased in ALDH+ tumor cells compared with GEM treatment (3.4 and 3.7-fold, respectively). Numbers inside the figures shows the average (%) of positively stained cells.
3.3. Efficacy of PF-03084014, GEM and combination of GEM and PF-03084014 in highly aggressive orthotopic PDA model
We next examined the effects of PF-03084014 in a spontaneously metastatic orthotopic xenografts model. We used fresh tumors harvested from Panc265 which were surgically implanted into the pancreas of athymic nude mice. In this model absence of treatment leads to metastatic spread of pancreatic cancels to visceral organs and lymph nodes (Fig. 2 and Table 2). Body weights at the end of the experiments were not significantly different compared to vehicle treated animals (Fig. 3A). PF-03084014 alone (p<0.05), GEM (p<0.01) and the combination of PF-03084014 and GEM treatment (p<0.01) significantly reduced the primary tumor volume compared to the tumors in the vehicle treated mice (Fig. 3B). However, combination of PF-03084014 and GEM treatment significantly reduced the primary tumor volume compared to GEM treatment alone (p<0.05; Fig. 3B).
Table 2.
Incidence of metastasis in treatment groups.
| Group | Number (percentage) of mice affected with metastasis | ||||
|---|---|---|---|---|---|
|
| |||||
| Spleen | Liver | Lungs | Kidneys | Lymph nodes | |
| Vehicle | 9/10 (90) | 3/10 (30) | 2/10 (20) | 3/10 (30) | 7/10 (70) |
| PF-03084014 | 7/9 (78) | 2/9 (22) | 1/9 (11) | 0/9 (0) | 4/9 (44) |
| GEM | 10/10 (100) | 2/10 (20) | 2/10 (20) | 2/10 (20) | 3/10 (30) |
| GEM + PF-03084014 | 6/10 (60) | 0/10 (0) | 0/10 (0) | 1/10 (10) | 2/10 (20) |
Figure 3.
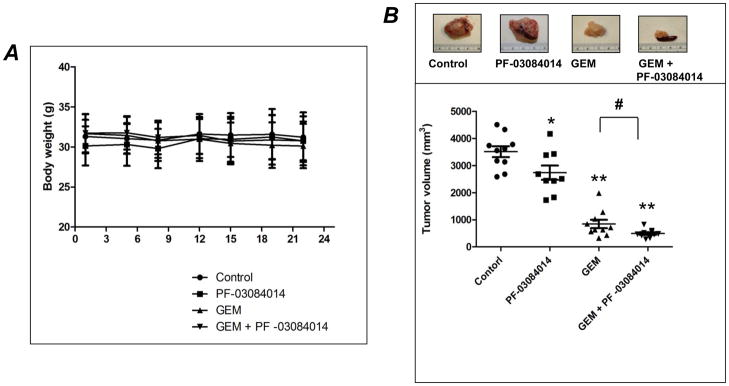
Combination of PF-03084014 with GEM blocks primary tumor progression in orthotopic pancreatic cancer xenograft (Panc265) model. A) Body weights of mice remained relatively stable during treatment period showing that therapies were well-tolerated and did not alter body weight compared to vehicle treated animals. B) Representative photomicrographs of primary tumors resected from control, PF-03084014, GEM and GEM plus PF-03084014 treated mice (upper panel). Primary tumor volumes of mice demonstrating that GEM + PF-03084014 combination is remarkably effective in suppressing primary tumor growth (lower panel). Photographs of excised primary tumors from representative mouse are shown in inset. Points, mean of tumor volume, bars SEM, N = 9–10 mice/group. *p<0.05 and **p<0.01 vs. control; # p<0.05 vs. GEM.
3.4. Combination of PF-03084014 and GEM prevented the development of metastasis in the lungs and liver
In order to examine the effect of treatment on metastasis formation in Panc265 xenografts, we carefully examined the organs using 10x lens and H&E stained sections microscopically. Both vehicle and GEM treated animals showed metastatic spread in spleen, liver, lungs, kidneys and lymph nodes (Table 2). While 20% of mice in the GEM treatment showed metastasis in the lungs and liver, combination of PF-03084014 and GEM effectively prevented the development of metastasis in the lungs and liver (Fig. 4 and Table 2), which are the common metastatic site of PDA patients. While 100% mice in the GEM treatment arm developed splenic metastasis, 40% mice in the combination of PF-03084014 and GEM group were free of spleen metastasis (Table 2).
Figure 4.

GEM + PF-03084014 combination therapy prevents the metastasis in liver and lungs. Representative H&E stained sections showing that GEM + PF-03084014 combination therapy prevents the metastasis in liver and lungs (10x magnifications). Arrowheads point towards metastatic area.
3.5. Effect of therapy on Notch targets
We investigated the effect of treatment on Notch targets, cell proliferation, apoptosis, tumor angiogenesis and intratumoral necrosis using post-treatment tumors harvested from xenografts. Formation of N1ICD protein following Notch receptor cleavage by γ-secretase is a crucial step in Notch signaling. N1ICD moves to the nucleus, becoming part of a larger transcriptional complex regulating the expression of well-known Notch-regulated transcription factors Hes-1 and Hey-1. Immunohistochemical staining of tumors from Panc215 and Panc354 clearly demonstrated that PF-03084014 is highly effective in blocking the cleavage of N1ICD (Fig. 5A).
Figure 5.

PF-03084014 treatment blocks the activation of N1ICD and inhibits the expression of Notch targets. A) Representative photo micrographs of Panc215 and Panc354 showing that PF-03084014 and combination of GEM plus PF-03084014 strongly inhibit the nuclear staining of N1ICD. B) Immunobloting of lysates from Panc265 orthotopic experiment demonstrating that PF-03084014 and combination of GEM plus PF-03084014 strongly inhibit N1ICD and the expression of Notch targets Hes-1 and Hey-1. Cleaved-PARP (c-PARP) levels were elevated in GEM and GEM plus PF-03084014 treatment, indicating higher apoptosis compared to Control or PF-03084014 treatment groups. While the treatments could not modulate MEK1/2 protein expression, PF-03084014 and combination of GEM plus PF-03084014 treatment inhibit MEK1/2 phosphorylation. Numbers on the left side of the blots indicate the molecular weight of the proteins in kilodaltons.
Westernblot analysis of lysates from Panc265 orthotopic experiment showed that PF-03084014 could suppress the clevage of N1ICD as well as the protein expression of Notch targets Hes-1 and Hey-1 (Fig. 5B). Kras mediates its effect in part through activation of downstream pathways such as MEK [26]. PF-03084014 treatment could inhibit the phosphorylation of MEK. Our results are in agreement with a recent report that γ-secretase inhibitor treatment resulted in inhibition of MEK signaling in cancer cells [27].
3.6. Effect of therapy on cell proliferation, apoptosis, tumor angiogenesis and intratumoral necrosis
When compared to vehicle treated tumors, apoptosis induction was not evident in PF-03084014 alone treatment (Fig. 6). However, the treatment was effective in suppressing tumor cell proliferation compared to the vehicle treated tumors (Fig. 6). Combination of PF-03084014 and GEM was significantly effective in suppressing tumor cell proliferation and apoptosis induction compared to GEM (Fig. 6).
Figure 6.

Combination PF-03084014 with GEM induces apoptosis, suppresses cell proliferation. Representative photomicrographs of TUNEL and Ki-67 stained tumor sections (40x magnifications). Quantitation of TUNEL+ and Ki-67+ tumor nuclei (bottom). Data represent the mean ± SD of TUNEL or Ki-67 positive nuclei in five different high-power fields per xenograft sections from 3 independent tumours of treatment arms. *p<0.05 and ***p<0.0001 vs. control; ## p<0.001 vs. GEM.
It has been reported that tumor endothelial cells use Notch signaling in building the tumor vasculature [8]. Tumor sections were stained for CD-31, an endothelial cell-specific surface marker and the vessel areas were counted. Treatment with PF-03084014 and PF-03084014 plus GEM combination showed significant reduction in endothelial-specific antigen CD-31, when compared with vehicle-treated tumors of Panc215 (Fig. 7). In both Panc215 and Panc354, the combination treatment could greatly reduce the vessel density compared to GEM treatment (p<0.001; Fig. 7). Although this class of inhibitors has been shown to induce intratumoral necrosis [11; 18], we failed to observe any preferential exacerbation of this phenomenon with PF-03084014 compared to GEM alone in xenografts used for the study (Only Panc354 is shown, Suppl. Fig. 1).
Figure 7.

Representative photo micrograph of CD31 immunostaining (20x) in Panc215 and Panc354. In Panc215, treatment with PF-03084014 or combination of GEM plus PF-03084014 leads to a significant reduction in endothelial-specific antigen CD-31, when compared to vehicle-treated tumors. Arrowheads point towards CD31 stained endothelial cells. *p<05, ***p<0.0001 vs. control; ##p<0.001 vs. GEM.
4. Discussion
PDA is a devastating disease with an extremely poor life expectancy and no effective treatment [3]. GEM is widely used as first-line chemotherapy for patients with PDA, but the treatment offers modest advantage in survival [28]. Our current study showed that a combination of PF-03084014 and GEM is highly effective in producing durable tumor regression compared to GEM in pancreatic cancer xenografts (Fig. 1). The efficacy of the combination therapy was sustained in the absence of further dosing and was not associated with body weight loss in mice (Fig 1 and 3). PF-03084014 alone and in combination with GEM was effective in reducing putative CSCs (Fig. 2). Further, in a highly aggressive orthotopic model, PF-03084014 and GEM combination was significantly effective in controlling the primary tumor growth as well as preventing metastatic dissemination to the lungs and liver compared to GEM treatment (Fig. 3 and 5). A simplified schematic diagram illustrating the mechanisms responsible for the enhanced anti-tumor and anti-metastatic properties of PF-03084014 plus GEM combination in pancreatic cancer xenografts is shown in Fig. 8.
Figure 8.

A simplified schematic diagram illustrating the mechanisms responsible for the enhanced anti-tumor and anti-metastatic properties of PF-03084014 plus GEM combination in pancreatic cancer xenografts.
Notch promotes cell survival and treatment resistance in numerous human malignancies [29; 30], while pharmacological inhibition of Notch signaling suppresses tumor growth [31]. Furthermore, in many tumor models including orthotopic and genetically modified models, Notch inhibition demonstrates combinatorial efficacy with standard cytotoxic agents [11; 32; 33].
The strong evidence linking aberrant Notch signaling to tumorigenesis and the maintenance of CSCs makes Notch an appealing therapeutic target [34]. Notch signaling is known to be a survival pathway for CSCs and is the most intensively studied putative therapeutic targets in CSC. Notch may be a particularly powerful target for the tumor CSC populations, which is resistant to standard treatments such as chemotherapy and radiation, but has been demonstrated to be sensitive to inhibition of stem cell maintenance pathways such as Notch [35]. Notch pathway components are enriched in PDA patients resistant to GEM treatment and other molecular targeted agents [36; 37]. Recent reports indicated that GEM treatment enriches the pancreatic CSC population. Challenging the pancreatic cancer cell lines to increasing concentrations of GEM resulted in epithelial-mesenchymal transition (EMT) and increased expression of the stem cell markers CD24, CD44 and epithelial-specific antigen [38]. It has been recently demonstrated that acquisition of EMT phenotype of GEM-resistant pancreatic cancer cells is linked with activation of the Notch signaling pathway, suggesting that the blockade of the Notch signaling by novel strategies could be a potential targeted therapeutic approach for overcoming chemo-resistance in PDA [39].
Recent studies underscore the emergence of γ-secretase class of inhibitors as tractable inhibitors of Notch signaling in solid tumors [14–18]. For example, the γ-secretase inhibitor PF-03084014 used in the current study was also shown to significantly enhance the antitumor activity of docetaxel in multiple xenograft models of triple-negative breast cancer [40]. Similarly, Arcaroli and colleagues recently reported that a combination of PF-03084014 and irinotecan reduced CSCs and slowed tumor recurrence in patient-derived colorectal adenocarcinoma model [41]. Another γ-secretase inhibitor, MRK-003, showed preferential efficacy in pancreatic cancer cell lines when tested in a panel of >400 human solid tumor-derived cell lines [33]. MRK-003 treatment completely inhibited tumor development in a genetically engineered model of invasive pancreatic cancer. Moreover, in the same model, a combination of MRK-003 and GEM resulted in the killing of tumor endothelial cells, causing extensive hypoxic necrosis and prolonging survival [11]. Our recent finding confirms that MRK-003 down-regulates Notch target genes resulting in the inhibition of tumor growth, induction of apoptosis and intra-tumoral necrosis as well as potentiation of GEM sensitivity in patient-derived PDA xenografts [18]. In addition, MRK-003 exposure to pancreatic cancer cell lines leads to the reduction of CSCs [18]. A recent clinical investigation of MK-0752 (clinical version of MK-003) demonstrated good tolerability, evidence of Notch pathway inhibition and clinical benefit in adult patients with advanced solid tumors [42]. Recent reports demonstrated that in vitro treatment with PF-03084014 resulted in G0-G1 cell cycle arrest, apoptosis and strong activity against tumor cell migration and endothelial cell tube formation [16; 17]. A recent clinical investigation of PF-03084014 revealed that the drug was well tolerated in patients with advanced solid tumors, with early evidence of clinical activity and favorable PK characteristics [43].
We have previously reported that GEM treatment was initially effective in blocking PDA progression, but the tumors re-grew following cessation of GEM treatment [23; 44]. Analysis of post-GEM treated tumors revealed that GEM treatment was not capable of diminishing putative CSCs [23]. These findings in clinically relevant human pancreatic cancer xenograft model, which was established from freshly resected tumors from PDA patients, may explain the reason for the high frequency of tumor recurrence in PDA patients administered with GEM as a first-line therapy or in clinical trials that use GEM as an anchor drug together with other agents. Improvement in the clinical outcome of PDA patients is likely to be achieved by using specific agents capable of controlling CSCs.
Our results showed that upon on completion of treatment on day 28, PF-03084014 alone treatment failed to show significant anti-tumor effect when compared to vehicle treated mice in Panc215, Panc354 and Panc265 s.c. implanted models (Fig 1). However, the treatment was significantly effective in delaying tumor growth in Panc266 s.c implanted model as well as in Panc265 orthotopic models. While PF-03084014 alone was equally or more effective in suppressing CSCs as compared to the PF-03084014 plus GEM combination in the Panc215 xenograft model, the treatment showed insignificant anti-tumor effect compared to vehicle treated mice when the experiment was terminated on day 28 (Fig. 2).
We have recently reported that CSCs isolated from patient-derived pancreatic cancer xenografts have been found to confer greater tumor-forming capability when transplanted into mice than other types of cancer cells from the same tumor [22]. CSCs are particularly aggressive type of tumor cells, resistant to conventional cancer therapy, and thus implicated an underlying cause of tumor recurrence and metastasis. Therapies that target rapidly dividing non-stem cell bulk populations (NSCs) in the tumor may produce rapid tumor shrinkage, but they are unlikely to produce long-term remissions if the CSCs responsible for maintaining the disease are also not targeted [45; 46]. It is postulated that by developing therapeutics that target CSCs can address the problem of cancer recurrence and metastasis. It is important to note that agents that eliminate the CSCs within a tumor may bring little or no immediate reduction in tumor size. However, tumor growth is not sustainable without CSCs to replenish the bulk population and the tumor will eventually degenerate as bulk tumor cells are depleted. Therefore it is paramount importance to develop targeted therapies, which also eliminate CSC, thereby destroying the disease’s sole source for new tumor cells.
The Notch pathway has tremendous potential as a new target in cancer therapy [47]. One major side effect that emerged from the clinical trials of a first-line Notch-inhibiting drug was gastrointestinal toxicity [48]. Intermittent dosing schedules of Notch inhibitor as well as corticosteroid administration has been recently reported to largely spare the gut while maintaining anti-tumor efficacy [49]. Early clinical trial reports indicated manageable toxicity and preliminary evidence of efficacy in breast cancer patients undergoing a combination of γ-secretase inhibitor with chemotherapy [50]. Moreover, a decrease in CSCs was observed in tumors of patients undergoing serial biopsies [50]. The intermittent dosing (7 days on/7 days off) of PF-03084014 used for the present study has been recently reported to reduce its gastrointestinal toxicity while efficacious in tumor growth inhibition [14]. We examined distal ileum sections from mice treated with PF-03084014, GEM and GEM + PF-03084014. Our results reveal that there is no evidence of goblet cell metaplasia upon PF-03084014 treatment (Only Panc215 is shown, Suppl. Fig 1B).
In conclusion, the preclinical findings presented here demonstrate that GEM antitumor activity was significantly enhanced when combined with γ-secretase inhibitor, PF-03084014. Our preclinical results justify further evaluation γ-secretase inhibitor and GEM combination in PDA.
Supplementary Material
Acknowledgments
N.V. Rajeshkumar, A. Maitra and M. Hidalgo are supported by a Stand Up To Cancer Dream Team Translational Cancer Research Grant, a Program of the Entertainment Industry Foundation (SU2C-AACR-DT0509). A. Maitra is supported by NIH CA113669
Footnotes
Note: Contents of this manuscript was partially presented as a poster (Abstract # 3262) at the 103rd annual meeting of AACR, Chicago, IL, March 31- April 04, 2012
Conflict of Interest Statement
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–188. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 4.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, Murtaugh LC. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105:18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, Osborne BA, Gottipati S, Aster JC, Hahn WC, Rudolf M, Siziopikou K, Kast WM, Miele L. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med. 2002;8:979–986. doi: 10.1038/nm754. [DOI] [PubMed] [Google Scholar]
- 7.Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, Schoffski P, Post S, Verslype C, Neumann H, Safran H, Humblet Y, Perez Ruixo J, Ma Y, Von Hoff D. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–1438. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 8.Garcia A, Kandel JJ. Notch: a key regulator of tumor angiogenesis and metastasis. Histol Histopathol. 2012;27:151–156. doi: 10.14670/hh-27.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Zhang Y, Li Y, Banerjee S, Liao J, Sarkar FH. Down-regulation of Notch-1 contributes to cell growth inhibition and apoptosis in pancreatic cancer cells. Mol Cancer Ther. 2006;5:483–493. doi: 10.1158/1535-7163.MCT-05-0299. [DOI] [PubMed] [Google Scholar]
- 10.Wang Z, Banerjee S, Li Y, Rahman KM, Zhang Y, Sarkar FH. Down-regulation of notch-1 inhibits invasion by inactivation of nuclear factor-kappaB, vascular endothelial growth factor, and matrix metalloproteinase-9 in pancreatic cancer cells. Cancer Res. 2006;66:2778–2784. doi: 10.1158/0008-5472.CAN-05-4281. [DOI] [PubMed] [Google Scholar]
- 11.Cook N, Frese KK, Bapiro TE, Jacobetz MA, Gopinathan A, Miller JL, Rao SS, Demuth T, Howat WJ, Jodrell DI, Tuveson DA. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med. 2012;209:437–444. doi: 10.1084/jem.20111923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Souza B, Miyamoto A, Weinmaster G. The many facets of Notch ligands. Oncogene. 2008;27:5148–5167. doi: 10.1038/onc.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dufraine J, Funahashi Y, Kitajewski J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene. 2008;27:5132–5137. doi: 10.1038/onc.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A, Tsaparikos K, Jani JP, Hosea N, Sands M, Randolph S, Smeal T. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. 2010;9:1618–1628. doi: 10.1158/1535-7163.MCT-10-0034. [DOI] [PubMed] [Google Scholar]
- 15.Lanz TA, Wood KM, Richter KE, Nolan CE, Becker SL, Pozdnyakov N, Martin BA, Du P, Oborski CE, Wood DE, Brown TM, Finley JE, Sokolowski SA, Hicks CD, Coffman KJ, Geoghegan KF, Brodney MA, Liston D, Tate B. Pharmacodynamics and pharmacokinetics of the gamma-secretase inhibitor PF-3084014. J Pharmacol Exp Ther. 2010;334:269–277. doi: 10.1124/jpet.110.167379. [DOI] [PubMed] [Google Scholar]
- 16.Zhang CC, Pavlicek A, Zhang Q, Lira ME, Painter CL, Yan Z, Zheng X, Lee NV, Ozeck M, Qiu M, Zong Q, Lappin PB, Wong A, Rejto PA, Smeal T, Christensen JG. Biomarker and Pharmacologic Evaluation of the gamma-Secretase Inhibitor PF-03084014 in Breast Cancer Models. Clin Cancer Res. 2012;18:5008–5019. doi: 10.1158/1078-0432.CCR-12-1379. [DOI] [PubMed] [Google Scholar]
- 17.Samon JB, Castillo-Martin M, Hadler M, Ambesi-Impiobato A, Paietta E, Racevskis J, Wiernik PH, Rowe JM, Jakubczak J, Randolph S, Cordon-Cardo C, Ferrando AA. Preclinical analysis of the gamma-secretase inhibitor PF-03084014 in combination with glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther. 2012;11:1565–1575. doi: 10.1158/1535-7163.MCT-11-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizuma M, Rasheed ZA, Yabuuchi S, Omura N, Campbell NR, de Wilde RF, De Oliveira E, Zhang Q, Puig O, Matsui W, Hidalgo M, Maitra A, Rajeshkumar NV. The Gamma Secretase Inhibitor MRK-003 Attenuates Pancreatic Cancer Growth in Preclinical Models. Mol Cancer Ther. 2012;11:1999–2009. doi: 10.1158/1535-7163.MCT-12-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donadelli M, Dando I, Zaniboni T, Costanzo C, Dalla Pozza E, Scupoli MT, Scarpa A, Zappavigna S, Marra M, Abbruzzese A, Bifulco M, Caraglia M, Palmieri M. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis. 2011;2:e152. doi: 10.1038/cddis.2011.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rasheed ZA, Yang J, Wang Q, Kowalski J, Freed I, Murter C, Hong SM, Koorstra JB, Rajeshkumar NV, He X, Goggins M, Iacobuzio-Donahue C, Berman DM, Laheru D, Jimeno A, Hidalgo M, Maitra A, Matsui W. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst. 2010;102:340–351. doi: 10.1093/jnci/djp535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 22.Ishizawa K, Rasheed ZA, Karisch R, Wang Q, Kowalski J, Susky E, Pereira K, Karamboulas C, Moghal N, Rajeshkumar NV, Hidalgo M, Tsao M, Ailles L, Waddell TK, Maitra A, Neel BG, Matsui W. Tumor-initiating cells are rare in many human tumors. Cell Stem Cell. 2010;7:279–282. doi: 10.1016/j.stem.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajeshkumar NV, Rasheed ZA, Garcia-Garcia E, Lopez-Rios F, Fujiwara K, Matsui WH, Hidalgo M. A combination of DR5 agonistic monoclonal antibody with gemcitabine targets pancreatic cancer stem cells and results in long-term disease control in human pancreatic cancer model. Mol Cancer Ther. 2010;9:2582–2592. doi: 10.1158/1535-7163.MCT-10-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajeshkumar NV, Tan AC, De Oliveira E, Womack C, Wombwell H, Morgan S, Warren MV, Walker J, Green TP, Jimeno A, Messersmith WA, Hidalgo M. Antitumor effects and biomarkers of activity of AZD0530, a Src inhibitor, in pancreatic cancer. Clin Cancer Res. 2009;15:4138–4146. doi: 10.1158/1078-0432.CCR-08-3021. [DOI] [PubMed] [Google Scholar]
- 25.Jiang J, Thyagarajan-Sahu A, Krchnak V, Jedinak A, Sandusky GE, Sliva D. NAHA, a novel hydroxamic acid-derivative, inhibits growth and angiogenesis of breast cancer in vitro and in vivo. PLoS One. 2012;7:e34283. doi: 10.1371/journal.pone.0034283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanrahan AJ, Solit DB. RAF/MEK Dependence of KRAS-Mutant Pancreatic Ductal Adenocarcinomas. Cancer Discov. 2012;2:666–669. doi: 10.1158/2159-8290.CD-12-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De La OJ, Murtaugh LC. Notch and Kras in pancreatic cancer: at the crossroads of mutation, differentiation and signaling. Cell Cycle. 2009;8:1860–1864. doi: 10.4161/cc.8.12.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 29.Al-Ejeh F, Smart CE, Morrison BJ, Chenevix-Trench G, Lopez JA, Lakhani SR, Brown MP, Khanna KK. Breast cancer stem cells: treatment resistance and therapeutic opportunities. Carcinogenesis. 2011;32:650–658. doi: 10.1093/carcin/bgr028. [DOI] [PubMed] [Google Scholar]
- 30.Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB, Schwartz GK. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009;69:573–582. doi: 10.1158/0008-5472.CAN-08-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groth C, Fortini ME. Therapeutic approaches to modulating Notch signaling: current challenges and future prospects. Semin Cell Dev Biol. 2012;23:465–472. doi: 10.1016/j.semcdb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z, Li Y, Ahmad A, Azmi AS, Banerjee S, Kong D, Sarkar FH. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim Biophys Acta. 2010;1806:258–267. doi: 10.1016/j.bbcan.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plentz R, Park JS, Rhim AD, Abravanel D, Hezel AF, Sharma SV, Gurumurthy S, Deshpande V, Kenific C, Settleman J, Majumder PK, Stanger BZ, Bardeesy N. Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2009;136:1741–1749. e1746. doi: 10.1053/j.gastro.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Ahmad A, Li Y, Azmi AS, Miele L, Sarkar FH. Targeting notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Res. 2011;31:1105–1113. [PubMed] [Google Scholar]
- 35.Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B, Miele L. Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010;16:3141–3152. doi: 10.1158/1078-0432.CCR-09-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feldmann G, Mishra A, Bisht S, Karikari C, Garrido-Laguna I, Rasheed Z, Ottenhof NA, Dadon T, Alvarez H, Fendrich V, Rajeshkumar NV, Matsui W, Brossart P, Hidalgo M, Bannerji R, Maitra A, Nelkin BD. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol Ther. 2011;12:598–609. doi: 10.4161/cbt.12.7.16475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garrido-Laguna I, Uson M, Rajeshkumar NV, Tan AC, de Oliveira E, Karikari C, Villaroel MC, Salomon A, Taylor G, Sharma R, Hruban RH, Maitra A, Laheru D, Rubio-Viqueira B, Jimeno A, Hidalgo M. Tumor engraftment in nude mice and enrichment in stroma- related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin Cancer Res. 2011;17:5793–5800. doi: 10.1158/1078-0432.CCR-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol. 2007;14:3629–3637. doi: 10.1245/s10434-007-9583-5. [DOI] [PubMed] [Google Scholar]
- 39.Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang C, Yan Y, Zong Q, Fang D, Painter C, Zhang Q, Pavlicek A, Lira M, John-Baptiste A, Christensen J. Gamma -secretase inhibitor PF-03084014 diminishes the tumor-initiating cells and demonstrates synergy with docetaxel in breast cancer xenograft models. Cancer Res; Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; 2012 Mar 31–Apr 4; Chicago, IL: Philadelphia (PA). 2012. p. Abstract nr 3492. [Google Scholar]
- 41.Arcaroli JJ, Powell RW, Varella-Garcia M, McManus M, Tan AC, Quackenbush KS, Pitts TM, Gao D, Spreafico A, Dasari A, Touban BM, Messersmith WA. ALDH(+) tumor-initiating cells exhibiting gain in NOTCH1 gene copy number have enhanced regrowth sensitivity to a gamma-secretase inhibitor and irinotecan in colorectal cancer. Mol Oncol. 2012;6:370–381. doi: 10.1016/j.molonc.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ, Blackman S, Watters J, Loboda A, Podtelezhnikov A, Lunceford J, Chen C, Giannotti M, Hing J, Beckman R, Lorusso P. Phase I Pharmacologic and Pharmacodynamic Study of the Gamma Secretase (Notch) Inhibitor MK-0752 in Adult Patients With Advanced Solid Tumors. J Clin Oncol. 2012;30:2307–2313. doi: 10.1200/JCO.2011.39.1540. [DOI] [PubMed] [Google Scholar]
- 43.Messersmith WA, Lorusso P, Cleary JM, Dasari A, Zhang X, Shaik MN, Courtney RD, Randolph S, Shapiro G. A phase I dose-escalation study of the novel gamma secretase inhibitor PF-03084014 in patients (pts) with advanced solid tumors. J Clin Oncol. 2011;29(suppl):abstr 3100. [Google Scholar]
- 44.Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, Shumway SD, Mizuarai S, Hirai H, Maitra A, Hidalgo M. MK-1775, a Potent Wee1 Inhibitor, Synergizes with Gemcitabine to Achieve Tumor Regressions, Selectively in p53-Deficient Pancreatic Cancer Xenografts. Clin Cancer Res. 2011;17:2799–2806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones RJ, Matsui WH, Smith BD. Cancer stem cells: are we missing the target? J Natl Cancer Inst. 2004;96:583–585. doi: 10.1093/jnci/djh095. [DOI] [PubMed] [Google Scholar]
- 46.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Purow B. Notch inhibition as a promising new approach to cancer therapy. Adv Exp Med Biol. 2012;727:305–319. doi: 10.1007/978-1-4614-0899-4_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garber K. Notch emerges as new cancer drug target. J Natl Cancer Inst. 2007;99:1284–1285. doi: 10.1093/jnci/djm148. [DOI] [PubMed] [Google Scholar]
- 49.Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E, Sulis ML, Barnes K, Sawai C, Homminga I, Meijerink J, Aifantis I, Basso G, Cordon-Cardo C, Ai W, Ferrando A. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15:50–58. doi: 10.1038/nm.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schott AF, Landis M, Dontu G, Griffith KA, Layman RM, Krop I, Paskett LA, Wong H, Drobolecki LE, Froehlich AM, Paranilam J, Hayes DF, Wicha MS, Chang JC. Preclinical and Clinical Studies of Gamma Secretase Inhibitors with Docetaxel on Human Breast Tumors. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-11-3326. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.