

Abstract
Antiangiogenesis is a promising antitumor strategy that inhibits tumor vascular formation to suppress tumor growth. Specifically, targeting VEGF has shown therapeutic benefits in many cancer types, leading to its approval as the first antiangiogenic drug by the Food and Drug Administration in the United States. It is known, however, that patients will experience unfavorable side effects as the VEGF and/or VEGF receptor signaling pathway is also required for homeostasis in normal tissues. Moreover, due to the cytostatic nature of antiangiogenic, cancer cells that are not killed by these drugs later develop an even more malignant phenotype, resulting in tumor invasion and metastasis. Although there have been many attempts to reduce drug resistance and increase therapeutic efficacy by combining antiangiogenic drugs with chemotherapy, the cumulative toxicity of antiangiogenic combinations limits their feasibility as treatments, as chronic angiogenesis inhibition typically reduces the antitumor effect of the co-administered chemotherapeutics. To overcome these problems, it is critical to explore new strategies that limit tumor resistance and side effects and also increase the exposure of chemotherapy drugs at the tumor site. Here, we review current understanding of antiangiogenic drugs and introduce a new combination strategy that links direct antiangiogenic protein and enzyme prodrug system with dual-targeting antiangiogenic and antiproliferative therapeutic effect in tumor microenvironment. This strategy has the potential to overcome these clinical hindrances and may serve as a paradigm for the next generation of antiangiogenic drugs.
Keywords: Antiangiogenesis, bevacizumab, chemotherapy, 5-fluorouracil, endothelial cell-targeting
Introduction
Tumors initially grow as avascular nodules by absorbing nutrients and removing waste through simple diffusion. When they grow beyond approximately 1 mm in diameter, however, tumors require a delicate network of blood vessels to supply nutrients and oxygen and remove waste products [1]. The neovasculation process in tumors is called “tumor angiogenesis” and begins with the “angiogenic switch” [2]. Tumors can produce several angiogenic activators to attract and activate endothelial cells, which is a critical step in mediating angiogenesis. Activation of endothelial cells initiates their proliferation, which in turn induces sprouting from existing vessels and migration and adhesion of the endothelial cells to form a lumen. New formation of vessels under angiogenesis continues to provide the necessary nutrients for cancer cells to grow and survive [2].
The angiogenic process involves interaction between angiogenic factors (inducers) and endothelial cells (responders) and can be inhibited directly or indirectly. Direct angiogenic inhibition occurs through direct suppression of endothelial cell proliferation and migration via endogenous proteins such as endostatin, angiostatin, and tumstatin [3]. In contrast, indirect angiogenic inhibition occurs through neutralizing ligands, such as VEGF and platelet-derived growth factor (PDGF), and blocking the activities of receptor tyrosine kinases, such as VEGF receptor (VEGFR) and PDGF receptor (PDGFR).
Many antiangiogenic targeting molecules have been tested and are now used for cancer treatment. VEGF is a well-recognized angiogenic factor that plays a crucial role in regulating tumor angiogenesis [4] and normal vascular development [5,6]. It is secreted by starving cancer cells and binds to the receptors in endothelial cells to elicit several responses, including microvascular permeability [7]; matrix-degrading enzyme secretion; and endothelial cell proliferation, migration, and survival [8]. Therefore, antiangiogenesis via inhibition of the VEGF signaling pathway is considered a good antitumor strategy antitumor.
The United States Food and Drug Administration (FDA) has approved several antiangiogenic drugs that have shown promising antitumor effects in patients [9]. One of these drugs is bevacizumab (Avastin; Roche), a monoclonal antibody that neutralizes VEGF to prevent new vascular formation. Bevacizumab was the first approved angiogenesis inhibitor and was approved for use in combination treatment with cytotoxic chemotherapy for metastatic colorectal cancer in 2004 [10]. It was subsequently approved for patients with non-small cell lung cancer [11], glioblastoma [12], and renal cell cancer [13]. Its use was also approved for metastatic breast cancer when combined with a conventional chemotherapeutic agent, paclitaxel [14]. Other angiogenesis inhibitors now used clinically are sorafenib and sunitinib, small-molecule inhibitors that can block the activity of both VEGFR tyrosine kinase and PDGFR [15]. The FDA approved sorafenib for treatment of renal cancer in 2005 [16] and hepatocellular carcinoma in 2007 [17]. In 2007, sunitinib became the first antiangiogenic drug approved for two cancers at the same time, renal cell carcinoma and imatinib-resistant gastrointestinal stromal tumor [18].
These three FDA-approved antiangiogenic drugs (bevacizumab, sorafenib, and sunitinib) all belong to the same category of antiangiogenic drugs that inhibit vascular growth indirectly. Other antiangiogenic agents are in the late stages of clinical development. In this review, however, we will focus primarily on the clinical hindrances to using VEGF as a therapeutic approach and evaluate preclinical evidence that may provide alternative option of antiangiogenic treatments.
Clinical limitations of current FDA-approved antiangiogenic agents
Ideally, antitumor drugs have high therapeutic efficacy and low toxicity. Conventional cytotoxic agents provide good antitumor activity [19] but have relatively high toxicity because they lack cancer-specific targeting, resulting in severe side effects [20]. In contrast to conventional cytotoxic agents, endogenously direct antiangiogenic proteins work by blocking oxygen and nutrient supplies to tumors, thereby suppressing their growth. This approach has several theoretical advantages. First, a direct antiangiogenic protein (e.g., endostatin) is less likely than a cytotoxic agent to induce drug resistance because it targets genetically stable endothelial cells instead of genetically unstable tumor cells [3]. Second, an antiangiogenic approach would have fewer off-target side effects because only tumor-associated endothelial cells proliferate, unlike quiescent normal endothelial cells [3]. In vitro preclinical data and in vivo animal models have indeed provided experimental results to support the prediction that antiangiogenic therapy is an effective therapeutic strategy, with a low incidence of drug resistance and virtually no toxicity.
However, because most FDA-approved antiangiogenic drugs work through an indirect antiangiogenic approach focusing on VEGF signaling pathway—that is, they neutralize proangiogenic factors or block receptor tyrosine kinase activity to inhibit vascular growth—the absence of tumor specificity by these agents frequently does lead to off-target side effects [21]. The VEGF pathway functions not only in normal growth and development but also in homeostasis in many organs [22]. Most of the adverse effects of VEGF inhibitors are modest and manageable, but some have been associated with serious, life-threatening complications, such as gastrointestinal perforation during short-term treatment and cardiac function failure during long-term treatment [23,24]. Both monoclonal antibodies and tyrosine kinase inhibitors share similar side effects, including hypertension, arterial thromboembolic events, proteinuria, wound healing complications, hemorrhaging, gastrointestinal perforation, and reversible posterior leukoencephalopathy syndrome [25].
Michael Klagsbrun, once stated, “The angiogenesis world is redundant—you knock out one angiogenic factor and another one pops up to take its place” [26]. Indeed, accumulating evidence has demonstrated that use of a single antiangiogenic agent, the strategy most often used now, is unable to sufficiently inhibit tumor angiogenesis, leading to tumor recurrence and drug resistance [27,34,35]. In addition to the VEGF pathway, several other signaling pathways regulate tumor angiogenesis and serve as important alternative sources for tumor growth stimuli [28].
Adaptive drug resistance has been predicted to be a major issue with indirect angiogenesis inhibitors as tumor endothelial cells can still be stimulated by alternative angiogenic factors that are not blocked. For example, blocking the VEGF signaling pathway may induce placental growth factor to take over tumor angiogenesis [27]. By themselves, tumor cells can also produce angiogenic factors to escape external inhibition and switch their signaling output to induce drug resistance [29], as evidenced by the disease progression that occurs when patients’ tumors become resistant to anti-VEGF therapy [30]. Clinical reports have also shown that anti-VEGF therapy often prolongs patients’ overall survival by only months and that it has no curative potential [31]. For instance, intrinsic non-responsiveness has been observed in which a tumor initially shrinks for months but then increases in invasiveness and metastatic potential after a short drug holiday, i.e., in the time between drug treatment cycles [32-34].
Several mechanisms related to redundant angiogenic factor receptor regulation, the hypoxic tumor microenvironment, tumor-associated stromal cells, and tumor endothelial properties have been proposed to explain anti-VEGF resistance [35]; however, the mechanisms underlying it are still not clearly understood because patients’ clinical outcomes vary [36]. Interestingly, several recent studies found that glioblastoma stem cells can themselves differentiate into endothelial phenotype and thereby cause vascularization that promotes tumor progression and metastasis [37,38]. Together, these studies suggest that antiangiogenic drug development may need to focus on stopping tumor angiogenesis through two distinct mechanisms: i) blocking endothelial cells from forming vessels under the classical angiogenesis process and ii) preventing cancer stem cells from differentiating into functional endothelial cells [37,38].
Strategies to overcome resistance to antiangiogenic agents
Researchers and clinicians have proposed the use of combination therapy to reduce tumors’ resistance to antiangiogenic treatments and increase the drugs’ therapeutic efficacy; however, overlapping adverse effects that can cause shorter progression-free survival in patients are barriers to the combinations’ use [39]. There are two major combination strategies to overcome resistance.
Combining antiangiogenic treatments and conventional cytotoxic chemotherapy
The dose of a chemotherapeutic drug is determined based on the well-established concept of maximum tolerated dose in order to provide the best antitumor efficacy; however, it is not yet clearly defined the dose and treatment schedule of cytotoxic drug. ‘The more frequent is better’ or ‘less is more’ are controversial. A higher dose of a cytotoxic drug is expected to have more antitumor effects but not to improve survival time due to adverse side effects [40], whereas a low dose of chemotherapy (metronomic therapy) may limit adverse side effects and inhibit angiogenesis by inhibiting endothelial cell proliferation and thus formation of new vasculature in the tumor microenvironment, even if the dose is not sufficient to kill tumor cells [41]. Therefore, the strategy proposed to overcome resistance to antiangiogenic therapy is using combination therapy consisting of cytostatic antiangiogenic agents and conventional cytotoxic chemotherapy for high antitumor efficacy. The preclinical rationale for the combination of an antiangiogenic agent and chemotherapy is based on antiangiogenic therapy’s ability to normalize vascular flow, which can result in increased oxygenation for the delivery of chemotherapeutic agents [42] and can be useful for maximizing therapeutic activity [43]; however, the interaction between antiangiogenic drugs and conventional chemotherapeutic agents needs to be further examined because chronic angiogenesis inhibition typically reduces tumor uptake of coadministered chemotherapeutics.
In order to maximize the benefit of vascular normalization-enhanced chemotherapy drug delivery, intermittent treatment schedules and provascular strategies have been proposed and practiced to increase chemotherapeutic drug exposure in preclinical and clinic studies [44]. Several studies have shown that some antiangiogenic drugs can induce normalization of the tumor vasculature, resulting in a transient increase in chemotherapeutic drug penetration; however, continuous treatment with angiogenesis inhibitors may decrease blood flow to tumors, thereby also decreasing the tumors’ uptake of coadministered cytotoxic drugs. By scheduling intermittent antiangiogenic treatments, it is possible to allow tumor vasculature to recover between each treatment cycle of drug administration and minimize the impact of angiogenesis inhibitors on the delivery of cytotoxic agents to the tumors [45]. The potential benefits of facilitating tumor cell recovery from cytotoxic drug treatment for each cycle of renormalization of the tumor vasculature should be further investigated [44]. Of note, many antiangiogenic drugs can be administered safely over extended periods with manageable toxicity compared with standard maximum tolerated dose of chemotherapies, which are often accompanied by severe adverse effects.
Combining multiple antiangiogenic agents
Several angiogenic factors can redundantly regulate tumor angiogenesis. For example, when patients are treated with anti-VEGF therapy, hypoxia will likely be induced in the tumor microenvironment. Tumor cells and tumor-associated fibroblasts as well as microphages are stimulated by hypoxia to secrete angiogenic factors other than VEGF to rescue hypoxic condition in the tumor microenvironment [15]; At different stages of breast cancer cell development, up to six angiogenic factors and several intracellular factors in addition to VEGF modulate angiogenesis [46]. Monotherapy is not sufficient to inhibit tumor angiogenesis. Therefore, next-generation antiangiogenic agents, including sunitinib and sorafenib, can inhibit the activities of multiple tyrosine kinases, such as PDGFR and VEGFR. The ability of these agents to block multiple molecular targets increases their antitumor activity and decreases drug resistance potential [47].
In addition to developing multi-targeted tyrosine kinases inhibitors, current clinical investigators have pursued the drug resistance rationale of preclinical and clinical experience by combining multiple antiangiogenic inhibitors to overcome some of the known mechanisms of tumor resistance. Combining direct (targeting endothelial cells) and indirect (targeting growth factors/receptors) inhibitors [48] or two indirect inhibitors [49] are now emerging in early clinical trials. Despite positive data from preclinical studies, simultaneous inhibition of several angiogenic pathways has not yet been established in clinical practice. Recent reports from early clinical trials showed that overlapping toxicities from antiangiogenic combinations might limit long-term feasibility of this approach due to cumulative toxicity [48]. It will be more important to determine their tolerability and validate whether sequential treatment with these agents can provide similar benefits in terms of reducing the toxic effects of the combinations. As more antiangiogenic agents targeting pathways other than VEGF are tested in clinical trials, opportunities for using novel antiangiogenic inhibitors in combination may arise. However, a major concern of these combination strategies is the lack of interpretative biomarkers to monitor treatment efficacy, which means that it would need to be tested in large late phase trials to confirm clinical benefits.
A novel antiangiogenic approach: linking direct angiogenic inhibitors and enzyme prodrug system
Direct angiogenic inhibitor: endostatin
Following Folkman’s original principle, antiangiogenic treatment should come directly from endogenous antiangiogenic proteins [50]. Endostatin, which is a 20-kDa cleavage product of collagen XVIII from its C-terminus, represents the most studied endogenous antiangiogenic protein. O’Reilly and colleagues discovered endostatin in 1997 [51] and determined that it plays a role in inhibiting endothelial cell proliferation and migration by binding to the α5β1, αvβ3, and αvβ5 integrins [52]. Endostatin confers the broadest spectrum of antiangiogenic activities through its downregulation of several angiogenesis pathways [53]. It has no detectable toxicity even after long-term delivery [54], does not cause acquired drug resistance, and confers no additional toxic effects when co-administered with conventional chemotherapy or other angiogenesis inhibitors [55].
Gene expression profile analysis showed that endostatin downregulates a number of angiogenic factors, including VEGF-A and FGF-2, and upregulates other known endogenous angiogenesis inhibitors, such as thrombospondin 1 [53]. Among all proposed mechanisms, integrins αvβ3 and α5β1 have specifically been linked to endostatin’s antiangiogenic activity [56]. Because these integrins are selectively expressed in growing endothelial cells, endostatin’s targeting of angiogenic endothelial cells is highly specific. The broad molecular targets of endostatin suggest that its role in mediating antiangiogenesis involves multiple signaling systems.
Over 100 reports from preclinical studies and clinical trials have demonstrated significant antitumor activity of endostatin, which had virtually no toxicity and did not elicit any drug resistance in patients [55]. However, due to the short half-life of endostatin [57], no significant outcome was observed in clinical trials, and the best response in most cases was disease stabilization [58]. In China, Wang and colleagues succeeded in overcoming the short half-life of endostatin by integrating the metal-chelating sequence (MGGSHHHHH) at endostatin’s N-terminus to provide additional zinc binding sites reduced thermally induced endostatin degradation [59]. This modified endostatin, named Endostar, has also been approved for non-small cell lung cancer patients in China [60]. Although endostatin is not the only factor that has a broach spectrum of antiangiogenic activities, it has been tested in clinical trials more than any other antiangiogenic protein [61].
In order to overcome the weaknesses of endostatin, researchers have attempted to modify this protein to increase its protein stability and/or antitumor activity. For example, linking the Fc domain of IgG to the N-terminus of endostatin prolonged endostatin’s protein stability and antitumor effect relative to unmodified endostatin [62]. In addition, endostatin fused to either the HER2 monoclonal antibody, angiostatin, or antagonist integrin receptor RGD peptide demonstrated increased antitumor effect and antiangiogenic activities in multiple cancer types, including colon, ovarian, and pancreatic cancers [63-65]. These studies show that protein engineering modification method can compensate for the protein’s weak antitumor activities. Moreover, chemotherapy remains as the primary cancer treatment because of its strongest curative potential. Therefore, targeted antiangiogenic agents that carry targeted chemotherapeutic agents may provide an extremity of therapeutic effect and have less chance of inducing drug resistance.
Enzyme prodrug system: cytosine deaminase/5-fluorocytosine
Enzyme prodrug therapy is an antitumor strategy that metabolizes an inactive prodrug into an active drug and preferentially kills proliferating cells by blocking cell DNA/RNA synthesis and replication so as to inhibit cell proliferation. When used in short treatment cycles, enzyme prodrug therapies can kill large numbers of tumor cells [66]. Cytosine deaminase is an enzyme that can catalyze conversion of the prodrug 5-fluorocytosine (5-FC) into the chemotherapeutic agent 5-fluorouracil (5-FU). 5-FU, which has been used against cancer for about 40 years, acts as a thymidylate synthase inhibitor. Interrupting the action of thymidylate synthase blocks synthesis of the pyrimidine thymidine, which is a nucleoside required for DNA replication. Interestingly, the cytosine deaminase-uracil phosphoribosyltransferase fusion protein (encoded by a fusion gene we will refer to as CD) is able to enhance the enzymatic conversion of 5-FC to 5-FU to a greater extent than cytosine deaminase alone [67] and has demonstrated enhanced therapeutic effects in cancer cells [68,69]; however, systemic treatment with either 5-FU or enzyme prodrug therapy induces off-target effects that can cause unwanted side effects. Moreover, liver enzymes can metabolize and inactivate 5-FU [70], lowering the overall effectiveness of systemic treatment. Furthermore, chemodrug concentrations at the tumor sites following systemic treatment with enzyme prodrugs is quite low, particularly in proliferating tissues like bone marrow [71]. Therefore, the development of tumor-specific targeting strategies for prodrug therapy would be beneficial.
To enhance the efficacy and reduce the normal-tissue toxicity of anticancer drugs, investigators have developed numerous selective tumor therapies, including the highly promising antibody-directed enzyme prodrug therapy (ADEPT), gene-directed enzyme prodrug therapy (GDEPT), and virus-directed enzyme prodrug therapy [72]. Each of these new therapies provides a targeted chemotherapeutic effect for the selective release of cytotoxic agents from non-toxic prodrugs at the tumor site.
Dual-targeting by antiangiogenesis and prodrug chemotherapy linkage
A novel antiangiogenic drug is urgently needed to provide high therapeutic efficacy and decrease the incidence of tumor recurrence and risk of both tumor invasion and metastasis. We deeply believe a novel strategy that targets antiangiogenic agents carrying targeted chemotherapeutic agents would provide an extremity of therapeutic effect and have less chance of inducing drug resistance. One such drug is on the horizon; the recently developed antitumor therapeutic EndoCD fusion protein (endostatin linked to CD) has demonstrated high efficacy and specificity in vitro and in vivo. The EndoCD approach aims to not only restrict the action of a strongly cytotoxic drug to cancer sites but also limit tumor endothelial cells from undergoing neovascularization. Endostatin targets unique tumor endothelial cells to provide tumor-specific antiangiogenic activity. It also carries CD to the local tumor area, where it converts the nontoxic prodrug 5-FC to the anti-metabolite chemotherapy drug 5-FU. Using this approach, a concentration of 5-FU 70 times higher than the standard clinical 5-FU dose was detected at the tumor sites, resulting in a highly cytotoxic effect on tumor cells surrounded by endothelial cells [73,74] (Figure 1). Furthermore, EndoCD/5-FC therapy also decreased tumor growth and colorectal liver metastasis incidence compared with bevacizumab/5-FU treatment in orthotopic animal models of human breast cancer and colorectal metastases from liver cancer, respectively.
Figure 1.
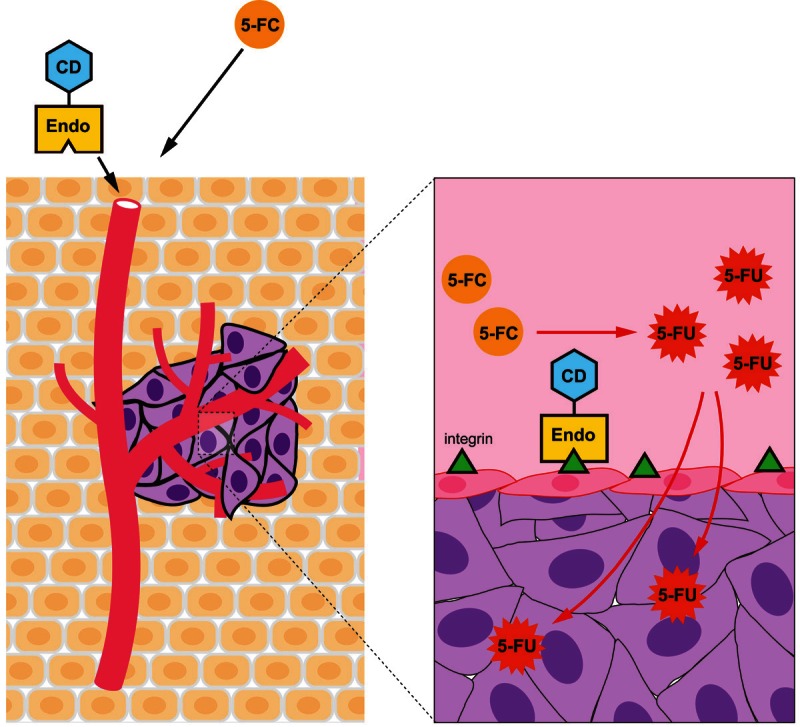
Model of dual targeting of antiangiogenic and antiproliferative therapy. EndoCD and 5-FC are sequentially administered. EndoCD targets integrin receptor on tumor endothelial cells first and then converts pro-drug 5-FC in tumor microenvironment. Pro-drug 5-FC is continuously converted to chemo-drug 5-FU by EndoCD. By using this strategy, high concentrations of 5-FU will accumulate specifically in tumor microenvironment to kill proliferating endothelial cells and tumor cells. EndoCD, endostatin-cytosine deaminase-uracil phosphoribosyl transferase; 5-FC, 5-fluorocytosine; 5-FU, 5-fluorouracil.
Life-threatening cardiotoxicity is a potential side effect in patients receiving antiangiogenic treatment. Those who received bevacizumab had an incidence of left ventricular dysfunction ranging from 1.7% to 3%. Moreover, 5-FU has also been shown to induce ischemic complications in cancer patients [75]. Carmeliet and colleagues used small animal magnetic resonance imaging to directly analyze end-diastolic volume (EDV) and end-systolic volume (ESV) in order to calculate the left ventricular ejection fraction (LVEF; LVEF=(ΣEDV-ΣESV)/ΣEDV) of mice before (pretreatment basal level) and after treatment [76]. Using this technique, we determined that the LVEF was significantly decreased in bevacizumab/5-FU-treated mice 3 months after treatment. In contrast, the LVEF was only slightly changed in EndoCD/5FC-treated mice even after 6 months of treatment [74]. One of the biological functions of VEGF is to maintain myocardial angiogenesis, and a loss of VEGF in mice has been shown to induce ischemic cardiomyopathy [76]. Our study also showed that circulating VEGF levels were significantly reduced relative to baseline in bevacizumab/5-FU- but not EndoCD/5-FC-treated mice. Moreover, coronal vessel density was also decreased relative to baseline in the heart tissues of mice treated with bevacizumab/5-FU- but not EndoCD/5-FC.
Bevacizumab/5-FU treatment can potentially induce cardiomyopathy and/or cardiac function failure in cancer patients. Therefore, EndoCD/5-FC protein therapy may provide a greater advantage than bevacizumab/5-FU because it has minimal cardiac impact. Moreover, EndoCD/5-FC demonstrated greater therapeutic efficacy, had a better safety profile, and provided stronger inhibition of tumor invasion or metastasis than did bevacizumab/5-FU [74]. Together, the EndoCD fusion protein and 5-FC function in both tumor targeted chemotherapy and antiangiogenesis and could serve as an alternative option for antiangiogenic therapy.
Conclusion and future opportunities
Antiangiogenesis, the novel concept of antitumorigenesis developed by Folkman, has become a reality over the last three decades and has been applied in the clinic. Inhibition of tumor growth by targeting the VEGF or VEGFR pathway became the first approved clinical antiangiogenic modality. Although antitumor efficacy was fully tested in preclinical studies, accumulating clinical reports have shown that these drugs have a cytostatic function but not curative potential and that they can further induce tumor recurrence, tumor invasion, and metastasis [33]. It has also become apparent that systemic treatment interrupts normal organ homeostasis to induce side effects, whereas targeted therapy is intended to provide a predictable safety profile [77]. Even though the mechanisms of resistance to antiangiogenic therapies are not yet clearly defined, efforts are under way to address the optimal scheduling and combinations of angiogenesis inhibitors. Early clinical trials have raised concerns that overlapping and cumulative toxicities from antiangiogenic combinations may limit the feasibility of such combinations in long-term treatment.
In our opinion, investigators must move beyond thinking of VEGF and its receptors as the major vascular targets and focus on the tumor endothelial cells that line the tumor blood vessels, supplying and draining the tumor microenvironment. As an endogenous angiogenesis inhibitor, endostatin triggers a signaling network and induces a broad spectrum of changes in the genetic profiles of endothelial cells [78]. Furthermore, we believe it is critical to increase antitumor efficacy by exposing chemotherapeutic agents only in the tumor microenvironment; this is the principle behind the EndoCD fusion protein—direct targeting of uniquely proliferating endothelial cells at tumor sites to decrease the potential for off-target side effects [79]. Moreover, by carrying and limiting chemotherapeutic drugs to the tumor area, EndoCD simultaneously enhances the cytostatic effect. Our studies demonstrated that EndoCD/5-FC strengthens both antiangiogenic and antiproliferative agents and revealed a novel possibility for antiangiogenic modalities [74]. We anticipate that targeting the vasculature and enhancing the chemotherapeutic effect in the tumor microenvironment will become the next new wave of cancer therapy.
Finally, we have preliminary data to show that EndoCD/5-FC is cytotoxic to breast cancer stem cell-like cells (unpublished data). Cancer stem cells (CSCs), also known as tumor-initiating cells, are known for their resistance to chemotherapy and antiangiogenic therapy. CSCs harbor the capability of not only self-renewal but also differentiation into multiple lineages of tumor cells growing in various types of organs [80]. Currently, there are no effective ways to target CSCs. For example, glioblastoma cancer stem cells can, by themselves, differentiate into neo-formed vessels contributing to tumor progression and metastasis [37,38]. Counter to their intent, antiangiogenic therapies may lead to tumor progression by increasing invasion and metastasis, likely due to the activation of the CSC population [32,34]. It is hoped that results from those clinical studies as well as others still ongoing will be able to provide yet another strategy to target CSCs and have important implications for future treatment options for cancer patients.
Acknowledgements
We thank Dr. Amelia Scholtz and Kathryn Carnes from the Scientific Publications Department at MD Anderson for editing the review. This work was supported by the Breast Cancer Research Foundation, National Breast Cancer Research Foundation, Patel Memorial Breast Cancer Research Fund, The University of Texas MD Anderson-China Medical University and Hospital Sister Institution Fund, Cancer Research Center of Excellence (D0H102-TD-C-111-005, Taiwan), and NSC99-2632-B-039-001-MY3 (Taiwan).
References
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 3.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2:727–739. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 4.Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- 5.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 6.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 7.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 8.Terman BI, Stoletov KV. VEGF and Tumor Angiogenesis. Einstein Quart J Biol and Med. 2001;18:59–66. [Google Scholar]
- 9.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 10.Ratner M. Genentech discloses safety concerns over Avastin. Nat Biotechnol. 2004;22:1198. doi: 10.1038/nbt1004-1198. [DOI] [PubMed] [Google Scholar]
- 11.Cohen MH, Gootenberg J, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) plus Carboplatin and Paclitaxel as first-line treatment of advanced/metastatic recurrent nonsquamous non-small cell lung cancer. Oncologist. 2007;12:713–718. doi: 10.1634/theoncologist.12-6-713. [DOI] [PubMed] [Google Scholar]
- 12.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14:1131–1138. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 13.Summers J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus interferon for advanced renal cell carcinoma. Oncologist. 2010;15:104–111. doi: 10.1634/theoncologist.2009-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 15.Ivy SP, Wick JY, Kaufman BM. An overview of small-molecule inhibitors of VEGFR signaling. Nat Rev Clin Oncol. 2009;6:569–579. doi: 10.1038/nrclinonc.2009.130. [DOI] [PubMed] [Google Scholar]
- 16.Eto M, Naito S. Molecular targeting therapy for renal cell carcinoma. Int J Clin Oncol. 2006;11:209–213. doi: 10.1007/s10147-006-0577-2. [DOI] [PubMed] [Google Scholar]
- 17.Flaherty KT. Sorafenib: delivering a targeted drug to the right targets. Expert Rev Anticancer Ther. 2007;7:617–626. doi: 10.1586/14737140.7.5.617. [DOI] [PubMed] [Google Scholar]
- 18.Rock EP, Goodman V, Jiang JX, Mahjoob K, Verbois SL, Morse D, Dagher R, Justice R, Pazdur R. Food and Drug Administration drug approval summary: Sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced renal cell carcinoma. Oncologist. 2007;12:107–113. doi: 10.1634/theoncologist.12-1-107. [DOI] [PubMed] [Google Scholar]
- 19.Morgan G, Ward R, Barton M. The contribution of cytotoxic chemotherapy to 5-year survival in adult malignancies. Clin Oncol (R Coll Radiol) 2004;16:549–560. doi: 10.1016/j.clon.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Orditura M, Quaglia F, Morgillo F, Martinelli E, Lieto E, De Rosa G, Comunale D, Diadema MR, Ciardiello F, Catalano G, De Vita F. Pegylated liposomal doxorubicin: pharmacologic and clinical evidence of potent antitumor activity with reduced anthracycline-induced cardiotoxicity (review) Oncol Rep. 2004;12:549–556. [PubMed] [Google Scholar]
- 21.Hasani A, Leighl N. Classification and toxicities of vascular disrupting agents. Clin Lung Cancer. 2011;12:18–25. doi: 10.3816/CLC.2011.n.002. [DOI] [PubMed] [Google Scholar]
- 22.Ferrara N. Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: therapeutic implications. Semin Oncol. 2002;29:10–14. doi: 10.1053/sonc.2002.37264. [DOI] [PubMed] [Google Scholar]
- 23.Kramer I, Lipp HP. Bevacizumab, a humanized anti-angiogenic monoclonal antibody for the treatment of colorectal cancer. J Clin Pharm Ther. 2007;32:1–14. doi: 10.1111/j.1365-2710.2007.00800.x. [DOI] [PubMed] [Google Scholar]
- 24.Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7:332–344. doi: 10.1038/nrc2106. [DOI] [PubMed] [Google Scholar]
- 25.Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. 2009;6:465–477. doi: 10.1038/nrclinonc.2009.94. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt C. Why do tumors become resistant to antiangiogenesis drugs? J Natl Cancer Inst. 2009;101:1530–1532. doi: 10.1093/jnci/djp425. [DOI] [PubMed] [Google Scholar]
- 27.Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De Mol M, Autiero M, Wyns S, Plaisance S, Moons L, van Rooijen N, Giacca M, Stassen JM, Dewerchin M, Collen D, Carmeliet P. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 28.Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010;21:21–26. doi: 10.1016/j.cytogfr.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Viloria-Petit A, Crombet T, Jothy S, Hicklin D, Bohlen P, Schlaeppi JM, Rak J, Kerbel RS. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61:5090–5101. [PubMed] [Google Scholar]
- 30.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 31.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loges S, Mazzone M, Hohensinner P, Carmeliet P. Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell. 2009;15:167–170. doi: 10.1016/j.ccr.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 34.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Azam F, Mehta S, Harris AL. Mechanisms of resistance to antiangiogenesis therapy. Eur J Cancer. 2010;46:1323–1332. doi: 10.1016/j.ejca.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 36.Rini BI. Vascular endothelial growth factor-targeted therapy in metastatic renal cell carcinoma. Cancer. 2009;115:2306–2312. doi: 10.1002/cncr.24227. [DOI] [PubMed] [Google Scholar]
- 37.Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- 38.Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, De Maria R. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 39.Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, Richel DJ, Voest EE, Dijkstra JR, Vink-Borger ME, Antonini NF, Mol L, van Krieken JH, Dalesio O, Punt CJ. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 40.Nieto Y. The verdict is not in yet. Analysis of the randomized trials of high-dose chemotherapy for breast cancer. Haematologica. 2003;88:201–211. [PubMed] [Google Scholar]
- 41.Tuma RS. Dosing study seen as victory for clinical trials, mathematical models. J Natl Cancer Inst. 2003;95:254–255. doi: 10.1093/jnci/95.4.254. [DOI] [PubMed] [Google Scholar]
- 42.Sweeney CJ, Miller KD, Sissons SE, Nozaki S, Heilman DK, Shen J, Sledge GW Jr. The antiangiogenic property of docetaxel is synergistic with a recombinant humanized monoclonal antibody against vascular endothelial growth factor or 2-methoxyestradiol but antagonized by endothelial growth factors. Cancer Res. 2001;61:3369–3372. [PubMed] [Google Scholar]
- 43.Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408–1416. [PubMed] [Google Scholar]
- 44.Ma J, Waxman DJ. Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol Cancer Ther. 2008;7:3670–3684. doi: 10.1158/1535-7163.MCT-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 46.Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R, Moghaddam A, Whitehouse R, Bicknell R, Harris AL. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res. 1997;57:963–969. [PubMed] [Google Scholar]
- 47.Teicher BA. Antiangiogenic agents and targets: A perspective. Biochem Pharmacol. 2010;81:6–12. doi: 10.1016/j.bcp.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 48.Moreno Garcia V, Basu B, Molife LR, Kaye SB. Combining antiangiogenics to overcome resistance: rationale and clinical experience. Clin Cancer Res. 2012;18:3750–3761. doi: 10.1158/1078-0432.CCR-11-1275. [DOI] [PubMed] [Google Scholar]
- 49.Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer. 2010;10:505–514. doi: 10.1038/nrc2868. [DOI] [PubMed] [Google Scholar]
- 50.Boehm T, Folkman J, Browder T, O’Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 51.O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 52.Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci U S A. 2003;100:4766–4771. doi: 10.1073/pnas.0730882100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 53.Abdollahi A, Hahnfeldt P, Maercker C, Grone HJ, Debus J, Ansorge W, Folkman J, Hlatky L, Huber PE. Endostatin’s antiangiogenic signaling network. Mol Cell. 2004;13:649–663. doi: 10.1016/s1097-2765(04)00102-9. [DOI] [PubMed] [Google Scholar]
- 54.Zhang DW, Li HL, Yao Q, Yang WL, Wang HL, Zhai DX, Zhou ZH. The synergistic effect of recombinant human endostatin (YH-16) combined with oxaliplatin on human colorectal carcinoma. J Int Med Res. 2010;38:111–126. doi: 10.1177/147323001003800113. [DOI] [PubMed] [Google Scholar]
- 55.Folkman J. Antiangiogenesis in cancer therapy--endostatin and its mechanisms of action. Exp Cell Res. 2006;312:594–607. doi: 10.1016/j.yexcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 56.Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer Res. 2005;65:3967–3979. doi: 10.1158/0008-5472.CAN-04-2427. [DOI] [PubMed] [Google Scholar]
- 57.Kulke MH, Bergsland EK, Ryan DP, Enzinger PC, Lynch TJ, Zhu AX, Meyerhardt JA, Heymach JV, Fogler WE, Sidor C, Michelini A, Kinsella K, Venook AP, Fuchs CS. Phase II study of recombinant human endostatin in patients with advanced neuroendocrine tumors. J. Clin. Oncol. 2006;24:3555–3561. doi: 10.1200/JCO.2006.05.6762. [DOI] [PubMed] [Google Scholar]
- 58.Herbst RS, Hess KR, Tran HT, Tseng JE, Mullani NA, Charnsangavej C, Madden T, Davis DW, McConkey DJ, O’Reilly MS, Ellis LM, Pluda J, Hong WK, Abbruzzese JL. Phase I study of recombinant human endostatin in patients with advanced solid tumors. J. Clin. Oncol. 2002;20:3792–3803. doi: 10.1200/JCO.2002.11.061. [DOI] [PubMed] [Google Scholar]
- 59.Jiang LP, Zou C, Yuan X, Luo W, Wen Y, Chen Y. N-terminal modification increases the stability of the recombinant human endostatin in vitro. Biotechnol Appl Biochem. 2009;54:113–120. doi: 10.1042/BA20090063. [DOI] [PubMed] [Google Scholar]
- 60.Wang J, Sun Y, Liu Y, Yu Q, Zhang Y, Li K, Zhu Y, Zhou Q, Hou M, Guan Z, Li W, Zhuang W, Wang D, Liang H, Qin F, Lu H, Liu X, Sun H, Luo S, Yang R, Tu Y, Wang X, Song S, Zhou J, You L, Yao C. [Results of randomized, multicenter, double-blind phase III trial of rh-endostatin (YH-16) in treatment of advanced non-small cell lung cancer patients] . Zhongguo Fei Ai Za Zhi. 2005;8:283–290. doi: 10.3779/j.issn.1009-3419.2005.04.07. [DOI] [PubMed] [Google Scholar]
- 61.Cao Y. Endogenous angiogenesis inhibitors and their therapeutic implications. Int J Biochem Cell Biol. 2001;33:357–369. doi: 10.1016/s1357-2725(01)00023-1. [DOI] [PubMed] [Google Scholar]
- 62.Lee TY, Tjin Tham Sjin RM, Movahedi S, Ahmed B, Pravda EA, Lo KM, Gillies SD, Folkman J, Javaherian K. Linking antibody Fc domain to endostatin significantly improves endostatin half-life and efficacy. Clin Cancer Res. 2008;14:1487–1493. doi: 10.1158/1078-0432.CCR-07-1530. [DOI] [PubMed] [Google Scholar]
- 63.Tysome JR, Wang P, Alusi G, Briat A, Gangeswaran R, Wang J, Bhakta V, Fodor I, Lemoine NR, Wang Y. Lister Vaccine Strain of Vaccinia Virus Armed with the Endostatin-Angiostatin Fusion Gene: An Oncolytic Virus Superior to dl1520 (ONYX-015) for Human Head and Neck Cancer. Hum Gene Ther. 2011 Sep;22:1101–8. doi: 10.1089/hum.2010.172. [DOI] [PubMed] [Google Scholar]
- 64.Belur LR, Podetz-Pedersen KM, Sorenson BS, Hsu AH, Parker JB, Carlson CS, Saltzman DA, Ramakrishnan S, McIvor RS. Inhibition of angiogenesis and suppression of colorectal cancer metastatic to the liver using the Sleeping Beauty Transposon System. Mol Cancer. 2011;10:14. doi: 10.1186/1476-4598-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jing Y, Lu H, Wu K, Subramanian IV, Ramakrishnan S. Inhibition of ovarian cancer by RGD-P125A-endostatin-Fc fusion proteins. Int J Cancer. 2011 Aug 1;129:751–61. doi: 10.1002/ijc.25932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frei E 3rd, Teicher BA, Holden SA, Cathcart KN, Wang YY. Preclinical studies and clinical correlation of the effect of alkylating dose. Cancer Res. 1988;48:6417–6423. [PubMed] [Google Scholar]
- 67.Chung-Faye GA, Chen MJ, Green NK, Burton A, Anderson D, Mautner V, Searle PF, Kerr DJ. In vivo gene therapy for colon cancer using adenovirus-mediated, transfer of the fusion gene cytosine deaminase and uracil phosphoribosyltransferase. Gene Ther. 2001;8:1547–1554. doi: 10.1038/sj.gt.3301557. [DOI] [PubMed] [Google Scholar]
- 68.Ramnaraine M, Pan W, Goblirsch M, Lynch C, Lewis V, Orchard P, Mantyh P, Clohisy DR. Direct and bystander killing of sarcomas by novel cytosine deaminase fusion gene. Cancer Res. 2003;63:6847–6854. [PubMed] [Google Scholar]
- 69.Erbs P, Regulier E, Kintz J, Leroy P, Poitevin Y, Exinger F, Jund R, Mehtali M. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 2000;60:3813–3822. [PubMed] [Google Scholar]
- 70.Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet. 1989;16:215–237. doi: 10.2165/00003088-198916040-00002. [DOI] [PubMed] [Google Scholar]
- 71.Springer CJ, Niculescu-Duvaz I. Prodrug-activating systems in suicide gene therapy. J Clin Invest. 2000;105:1161–1167. doi: 10.1172/JCI10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu G, McLeod HL. Strategies for enzyme/prodrug cancer therapy. Clin Cancer Res. 2001;7:3314–3324. [PubMed] [Google Scholar]
- 73.Ou-Yang F, Lan KL, Chen CT, Liu JC, Weng CL, Chou CK, Xie X, Hung JY, Wei Y, Hortobagyi GN, Hung MC. Endostatin-cytosine deaminase fusion protein suppresses tumor growth by targeting neovascular endothelial cells. Cancer Res. 2006;66:378–384. doi: 10.1158/0008-5472.CAN-05-1578. [DOI] [PubMed] [Google Scholar]
- 74.Chen CT, Yamaguchi H, Lee HJ, Du Y, Lee HH, Xia W, Yu WH, Hsu JL, Yen CJ, Sun HL, Wang Y, Yeh ET, Hortobagyi GN, Hung MC. Dual targeting of tumor angiogenesis and chemotherapy by endostatin-Cytosine deaminase-uracil phosphoribosyltransferase. Mol Cancer Ther. 2011;10:1327–1336. doi: 10.1158/1535-7163.MCT-10-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yeh ET, Bickford CL. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol. 2009;53:2231–2247. doi: 10.1016/j.jacc.2009.02.050. [DOI] [PubMed] [Google Scholar]
- 76.Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, Ehler E, Kakkar VV, Stalmans I, Mattot V, Perriard JC, Dewerchin M, Flameng W, Nagy A, Lupu F, Moons L, Collen D, D’Amore PA, Shima DT. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med. 1999;5:495–502. doi: 10.1038/8379. [DOI] [PubMed] [Google Scholar]
- 77.Verheul HM, Pinedo HM. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat Rev Cancer. 2007;7:475–485. doi: 10.1038/nrc2152. [DOI] [PubMed] [Google Scholar]
- 78.Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604–617. doi: 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kerbel RS. Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. Bioessays. 1991;13:31–36. doi: 10.1002/bies.950130106. [DOI] [PubMed] [Google Scholar]
- 80.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]