

Abstract
A LC-ESI-MS/MS method for the determination of crenolanib (CP-868,596) in human serum was developed and validated employing d4-CP-868,596 as an internal standard (ISTD). In addition to human serum, the method was also partially validated for crenolanib determination in human cerebrospinal fluid (CSF) samples. Sample aliquots (50μl of serum or CSF) were prepared for analysis using liquid-liquid extraction (LLE) with tert-butyl methyl ether. Chromatography was performed using a phenomenex Gemini C18 column (3 μm, 100 mm × 4.6 mm I.D.) in a column heater set at 50 °C and an isocratic mobile phase (methanol/water/formic acid at a volume ratio of 25/25/0.15, v/v/v). The flow rate was 0.45 mL/min, and the retention time for both analyte and ISTD was less than 3.5 minutes. Samples were analyzed with an API-5500 LC-MS/MS system (ESI) in positive ionization mode coupled to a Shimadzu HPLC system. The ion transitions monitored were m/z 444.4→373.1 and m/z 448.2 →374.2 for crenolanib and ISTD, respectively. The method was linear over the range of 5 ng/mL to 1000 ng/mL for serum and 0.5 ng/mL to 1000 ng/mL for CSF. For human serum, both intra-day and inter-day precision were <4%, while intra-day and inter-day accuracy were within 8% of nominal values. Recovery was greater than 50% for both the analyte and ISTD. For CSF samples, both intra-day and inter-day precision were <9% except at the lower limit of quantification (LLOQ) which was <17%. The intra-day and inter-day accuracy were within 11% of the nominal CSF concentrations. After validation, this method was successfully applied to the analysis of serial pharmacokinetic samples obtained from a child treated with oral crenolanib.
Keywords: Crenolanib (CP-868, 596); human serum; liquid-liquid extraction (LLE); liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS); pharmacokinetic studies
1. Introduction
Platelet-derived growth factors (PDGFs) comprise a class of growth factors thought to support malignant cellular proliferation and survival through several mechanisms, including upregulation of angiogenesis[1]. Recent studies have linked elevated expression of PDGF receptors (PDGFRs) to the pathogenesis of pediatric gliomas [2–5], suggesting that selective inactivation of PDGF/PDGFR signaling could have a clinically significant impact in pediatric glioma therapy [6]. The investigational drug, crenolanib (CP-868,596; AROG Pharmaceuticals, Dallas, TX), is an orally bioavailable tyrosine kinase inhibitor that has been shown to preferentially antagonize PDGFR relative to other receptor tyrosine kinases. Crenolanib has demonstrated therapeutic potential in a human glioma xenograft model (unpublished results) and proven tolerable in early phase clinical trials in adults as well [7]. Patients are currently being enrolled on a phase I clinical trial in children (ClinicalTrials.gov number NCT01393912) with either newly diagnosed diffuse intrinsic pontine glioma (DIPG) or recurrent high grade glioma (HGG).
The pharmacokinetics of crenolanib are presently unknown in children, so the characterization of crenolanib disposition in this study population requires a method for determination of serum crenolanib concentrations. Crenolanib is a novel agent, and no bioanalytical methods are published in the literature. Hence, in the present paper, we describe a simple and robust method using standard LC-MS/MS that was developed and validated to analyze crenolanib concentrations in serum or cerebrospinal (CSF) samples from pediatric patients enrolled in a phase I trial of single-agent crenolanib. Concentration-time data generated with this analytical method will help define the general pharmacokinetic disposition of crenolanib and contribute to the understanding of the dose-exposure-response relationship for crenolanib in pediatric populations, and, in doing so, provide information needed to optimize dosing strategies for future clinical trials.
2. Experimental
2.1. Chemicals
Crenolanib (CP-868,596 freebase, >95.6%,) and its isotope, d4-CP-868,596 were provided by AROG Pharmaceuticals (Dallas, Texas, USA). Methanol was obtained from Fisher Scientific (Fairlawn, NJ, USA), and tert-Butyl methyl ether anhydrous (TBME; 99.8% purity) was purchased from Sigma-Aldrich. Formic acid (FA, 98% purity) was purchased from Fluka BioChemika (Buchs, Switzerland). Blank human serum was obtained from Innovative Research (Novi, MI, USA). All water was purified using a Millipore Milli-Q UV plus and Ultra-Pure Water System (Tokyo, Japan). Other chemicals were purchased from standard sources and were of the highest quality available.
2.2. Apparatus and conditions
2.2.1 Chromatographic conditions
The HPLC system consisted of a Shimadzu (Kyoto, Japan) system controller (CBM-20A), pump (LC-20ADXR), autoinjector (SIL-20AC), online degasser (DGU-20A3), and column heater (CTO-20AC). Chromatographic separation was performed at 50 °C using a Gemini C18 analytical column (3.0 μm, 100 mm × 4.6 mm I.D.); (Phenomenex, USA) preceded by a KrudKatcher Ultra (Phenomenex, USA). The analyte and ISTD were eluted using a mobile phase that consisted of methanol/water/formic acid in a volume ratio of 25/25/0.15 v/v/v at a flow rate of 0.45mL/min. The total sample acquisition time was 7.0 minutes.
2.2.2. Mass spectrometric conditions
Mass spectra were obtained using an AB SCIEX API 5500Qtrap (Toronto, Canada) with an ESI source. The mass spectrometer was operated using Analyst software (Version 1.5.1, Applied Biosystems, Foster City, CA). Multiple reaction monitoring (MRM) and positive ion mode with unit resolution for both Q1 and Q3 were used for detection. The optimized MS/MS conditions were as follows: ion spray source temperature at 650 °C, curtain (CUR) gas pressure at 25 psi, both gas 1 (GS1) and gas 2 (GS2) pressure at 70.0 psi, ionspray voltage (IS) at 5000 V, collision-activated dissociation (CAD) set at medium, declustering potential (DP) at 88 V, entrance potential (EP) at 11.0 V, collision energy (CE) at 37.5 V for crenolanib and at 39 V for ISTD, and collision exit potential (CXP) at 40 V. The transitions monitored were m/z 444.4>373.1 for crenolanib and m/z 448.2>374.2 for the ISTD.
2.3. Patient Sample Collection and Storage
Whole blood samples (3 mL) were collected in silicone-coated vacutainer tubes for serum processing. After collection, the tubes were placed upright for 30 minutes at room temperature to clot and then centrifuged for 10 minutes at 4 °C and 1500 g to separate the serum. All collected serum samples were transferred into 2.0 mL screw-top tubes and immediately frozen and stored at −80 °C until analysis.
2.4. Sample preparation
2.4.1. Stock solutions
Stock solutions were prepared by dissolving crenolanib and the internal standard in 100% methanol to concentrations of 0.5 mg/mL and 1 mg/mL, respectively. The crenolanib stock solution was diluted to 5000 ng/mL in 50% methanol/water (v/v). Working solutions were made for each calibrator and quality control concentration by diluting the 5000 ng/mL working solution to the appropriate concentration with 50% methanol/water (v/v). The internal standard was diluted in the same solvent solution as crenolanib to a working solution of 100 ng/mL. The stock solutions were stored at −80 °C and the working solutions were stored at 4°C.
2.4.2. Calibration curve and quality controls
The calibration samples were prepared by spiking 50μl of blank matrix to concentrations of 5, 10, 50, 100, 300, 600, and 1000 ng/mL, and 0.5, 1, 10, 50, 100, 300, 600, and 1000 ng/mL for human serum and CSF respectively. Both curves were designed to reflect the expected range of sample concentrations. Three quality control concentrations were prepared at 30, 200, and 800 ng/mL for human serum and 2, 200, and 800 ng/mL for CSF in the same manner as the calibration samples. Artificial cerebrospinal fluid (aCSF) was used for preparing calibrators and quality controls for the CSF samples. aCSF was prepared with NaCl (148mM), KCL (4mM), MgCl2 (0.8mM), CaCl2 (1.4mM), Na2HPO4 (1.2mM), NaH2PO4 (0.3mM), and filled to volume using sterile distilled water.
2.4.3. Human serum sample preparation
Liquid-liquid extraction (LLE) was used to extract 50μl aliquots of either serum or CSF samples spiked with 5 μl of ISTD working solution. After addition of 1.5mL of TBME, samples were vortexed for 5 minutes then centrifuged at 3000 × g for 5 minutes. The upper organic layer was transferred to a new tube and dried under house nitrogen gas for approximately 20 minutes. Samples were reconstituted in 150μl of mobile phase and 10μl was injected on the LC-MS/MS system for analysis.
2.5. Method validation
2.5.1 Linearity and lower limit of quantitation
Calibration curves were evaluated using least square linear regression weighted with 1/x2 The coefficient of determination (r2) was used to evaluate the linearity of each calibration curve. The LLOQ was defined as the lowest concentration in the calibration curve that had accuracy within 20% of the accepted true value and a signal/noise (S/N) ratio greater than 5. The lower limit of detection (LOD) was defined as the lowest concentration that will result in an instrument response so that S/N will be greater than 3.
2.5.2 Accuracy, precision, and recovery
The accuracy and precision were evaluated both within and between day over three days. A four concentration accuracy/precision study was performed at the LLOQ (5 ng/mL for human serum, 0.5 ng/mL for CSF), low (30 ng/mL for human serum, 2 ng/mL for CSF), medium (200 ng/mL), and high (800 ng/mL) concentrations (n≥5 at each level). The acceptance criterion for accuracy was ±15% of nominal concentration at all concentration levels. Acceptable precision was ±15% at the low, medium, and high concentrations and ±20% at the assay LLOQ. Recovery was assessed by calculating the ratio of the instrument response of extracted low (30ng/mL) and high (800ng/mL) concentration samples to that of samples prepared by spiking blank extracted matrix at the same concentrations.
2.5.3 Selectivity, carryover, and matrix effects
Selectivity was assessed by extraction of LLOQ and blank samples in six different sources of human serum. Sample carryover was assessed by monitoring wash samples after injections of crenolanib at 1000 ng/mL. Matrix effects were evaluated by comparing post extraction spiked samples to mobile phase spiked samples. The matrix factor (MF) was calculated by dividing the instrument response of the post extracted samples by instrument response of the mobile phase spiked samples. Both low (30ng/mL) and high (800ng/mL) contentrations were evaluated in pooled human serum.
2.5.4 Stability
The stability of crenolanib in human serum (30 ng/ml and 800 ng/mL) was evaluated at ambient temperature or 4 °C for up to 24 hours and at −80 °C for up to 90 days. Pools of blank human serum at low and high concentrations of 30 ng/mL and 800 ng/mL were prepared and aliquotted. Baseline values were established by assaying sample aliquots the same day of preparation, and all other aliquots were stored until analysis under conditions appropriate to the corresponding stability test. Freeze-thaw stability was assessed by subjecting unextracted spiked serum samples to a total of three freeze-thaw cycles. Stability of crenolanib in CSF was evaluated at ambient temperature and at 4°C at both low (2 ng/mL) and high concentrations (800 ng/mL) in the same manner as the human serum samples.
2.6. Application of method to patient pharmacokinetic samples
Serial samples for pharmacokinetic studies of crenolanib in serum were obtained as part of a clinical Phase I study of pediatric patients with diffuse intrinsic pontine glioma or high grade glioma (ClinicalTrials.gov number NCT01393912). Whole blood was collected at the following times: pre-dose, and 1, 2, 4, 8, 24, and 48 h after an oral dosage of 130 mg/m2. Blood samples (3.0 mL) were collected in silicone-coated BD Vacutainer tubes (BD, Franklin Lakes, NJ, USA) and processed and quantitated according to the method described above.
3. Results and discussion
3.1. Chromatography
A Phenomenx Gemini c18 (3 μ, 100 mm × 4.6 mm) column was selected to provide the best peak shape. During method development, several other column chemistries and c18 columns were systematically explored using a consistent range of conditions (sample preparation and mobile phase) and most columns were deemed unacceptable, primarily due to significant peak tailing. Acetonitrile was initially tested as the organic component of the mobile phase, but peak splitting was observed regardless of the injection volume or organic content of the injection solvent. This problem was corrected by changing the organic component to methanol. Increasing the formic acid content of the mobile phase from the initial concentration of 0.1% to 0.3% and jacketing the column at 50°C also helped improve the peak shape and reduce the overall retention time. Examples of typical chromatograms are shown in Figure 1.
Figure 1.

Chromatograms of a blank plasma sample (top), LLOQ sample in CSF (middle), and a LLOQ sample in human serum (bottom).
3.2. Mass spectrometry
Analysis was performed on an AB Sciex API-5500QTrap LC/MS/MS system at unit (Q1) and unit (Q3) resolution in positive MRM mode. All mass spectrometry parameters were optimized through direct infusion. Q1 scans were used to select the parent ion and product ion scans were used to select the transition ions. The source of the fragments were confirmed with precursor scans shown in Figure 2.
Figure 2.
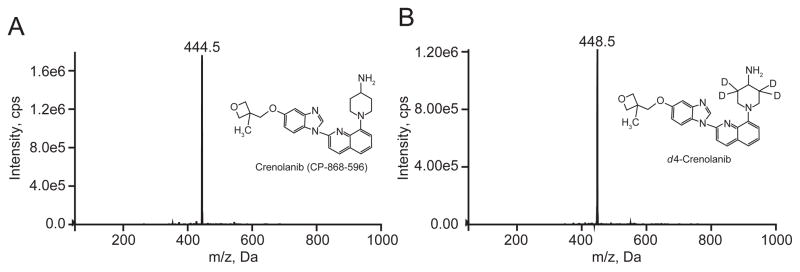
A.Precursor ion scan of 373.1 m/z for crenolanib. B. Precursor ion scan of 374.2 m/z for d4-CP-868,596.
3.3. Precision, accuracy and recovery
The intra-day and inter-day precision and accuracy results are presented in Table 1. Precision is presented in terms of %CV and the accuracy as %error. These results show that the method is both accurate and precise. The intra-day precision was <4% and the accuracy within 8% of nominal concentrations for all levels in human serum. For the inter-day studies in human serum, precision was <4% and the accuracy within 7% for all levels. For CSF, the intra-day accuracy was within 11% and the precision <9% while the inter-day study demonstrated similar accuracy (within 10%) and precision (<6%) for all levels except the LLOQ which was <17%. The calculated recovery for both analyte and ISTD was >50% in human serum and is listed in Table 2.
Table 1.
Accuracy and Precision Results
Inter-day and Intra-day accuracy and precision results for quantitation of crenolanib in serum (n=6) and CSF (n=5)
| Concentratio n (ng/ml) | Inter-day | Intra-day | ||
|---|---|---|---|---|
|
| ||||
| Accurac | Accurac | |||
| CV (%) | y (%) | CV (%) | y (%) | |
| Serum | ||||
| 5 | 3.6 | 97.7 | 3.6 | 98.6 |
| 30 | 3.6 | 103.0 | 2.3 | 104.4 |
| 200 | 2.6 | 101.0 | 1.3 | 100.6 |
| 800 | 3.5 | 93.8 | 3.7 | 92.6 |
| CSF | ||||
| 0.5 | 16.2 | 108.6 | 8.1 | 104.9 |
| 2 | 5.0 | 97.1 | 4.5 | 94.3 |
| 200 | 2.6 | 108.6 | 1.5 | 110.7 |
| 800 | 2.1 | 109.4 | 2.0 | 110.8 |
Table 2.
Recovery and Matrix Factor
Recovery and matrix factor for crenolanib and ISTD in human serum (n=3)
| Recovery | MF | |||
|---|---|---|---|---|
|
| ||||
| CV (%) | Avg. (%) | CV (%) | Avg. (%) | |
| Crenolanib | ||||
| Low QC* | 31.6 | 56.8 | 15.8 | 1.25 |
| High QC | 15.9 | 66.6 | 1.5 | 1.03 |
| Combined | 23.0 | 61.7 | 15.1 | 1.14 |
| ISTD | ||||
| Low QC* | 32.3 | 50.7 | 18.2 | 1.23 |
| High QCƚ | 16.6 | 54.5 | 7.2 | 1.07 |
| Combined | 22.8 | 52.6 | 16.0 | 1.14 |
Crenolanib conc. 30 ng/mL ; ISTD 10 ng/mL
Crenolanib conc. 800 ng/mL ; ISTD 10 ng/mL
3.4. Linearity and lower limit of quantitation
The crenolanib calibration curves were linear from 5 to 1,000 ng/mL with correlation coefficients (r2) greater than 0.998 for both curves in human serum. In CSF, the curves were linear from 0.5 to 1000 ng/mL with correlation coefficients (r2) greater than 0.996. For human serum, all LLOQ samples from both intra-day and inter-day deviated by less than 20% from the expected concentration with an overall average accuracy of 98.6% and 97.7% repectively. The S/N was typically ≥100 at the LLOQ and the LOD was 0.075 ng/mL. In CSF, all intra-day LLOQ samples were within 20% with an overall average accuracy of 104.9%. There were 2 out of 15 LLOQ samples in the interday study that did not meet the 20% criteria but the overall accuracy was 108.6% with a %CV of 16.2%. The signal to noise in CSF was typically ≥7 at the LLOQ and the LOD was 0.15 ng/mL. Initially, working solutions were prepared in 25% methanol/water (v/v), but curve and QC concentrations were inconsistent even when prepared in mobile phase. Once the organic in the working solution was increased to 50% methanol/water (v/v), no further quantitation inconsistencies were observed. This was likely due to insolubility of the analyte in such a highly aqueous solution.
3.5. Selectivity,carryover,and matrix effects
Selectivity was assessed by monitoring both extracted blank human serum and LLOQ samples for any coeluting peaks in six different lots of serum. No coeluting peaks were observed for crenolanib or the ISTD. Minimal matrix effect (MF=1.14, Table 2) was observed in human serum with a slight ion enhancement. This was not a concern as both crenolanib and the ISTD behaved similarily as would be expected with a deuterated ISTD. Carryover was assessed by monitoring blank samples after the injection of the high standard. Insignificant carryover was observed (<10% of the LLOQ) but did not interfere with quantitation. Selectivity in aCSF was assessed by monitoring two different preparations of aCSF, and no coeluting peaks were observed in either.
3.6. Stability
Pools of spiked human serum and aCSF samples were prepared at low and high concentrations and were aliquotted for single use and stored separately at 4 °C, ambient temperature, and −80 °C. These samples were then extracted and analyzed at the appropriate time points against a concurrently prepared calibration curve with QC’s. Crenolanib was stable in both matrices at 4 °C and ambient temperature for up to 24 hours with ≤5% change from time 0. Crenolanib serum samples were also stable at least 90 days at −80 °C and through three freeze thaw cycles agreeing within 8% of time 0 samples. Extract stability when reconstituted in mobile phase was confirmed for at least 48 hours with ≤4% change from original values.
3.7. Application of method to clinical pharmacokinetic study
To demonstrate the applicability of the method, we analyzed serum samples collected from a pediatric patient enrolled in a Phase I clinical trial of crenolanib. After administration of an oral dose of crenolanib (130 mg/m2), serial whole blood samples were collected over 48 hours. The samples were processed and analyzed by the method described in this report. A representative serum concentration-time profile for the pediatric patient is depicted in Figure 3. Because the serum elimination of crenolanib followed a biphasic decay (Figure 3), the concentration-time data was fit with a two-compartment, first-order elimination, first-order absorption pharmacokinetic structural model using ADAPT V with maximum likelihood estimation method (MLEM) parameter estimation.
Figure 3.

Crenolanib serum concentration–time profile in a single pediatric patient after one oral dose of crenolanib (130 mg/m2) and representative chromatogram from patient at 24 hour time point (inset).
4. Conclusion
A rapid LC-MS/MS method for quantitation of crenolanib in human serum samples was developed and validated. The method was also partially validated for use with human CSF. The method was simple and rugged and demonstrated excellent precision, accuracy, and reproducibility. The lower limit of quantitation of 5 ng/mL for human serum and 0.5 ng/mL for CSF provided an excellent signal to noise ratio indicating the possibility of measuring even lower concentrations. We have successfully applied this LC–MS/MS method by measuring crenolanib in human serum from a clinical pharmacokinetic study of children treated with crenolanib.
Highlights.
A novel method for the determination of crenolanib by lc/ms/ms is described.
The method was fully validated in human serum and partially validated in CSF.
The curve ranges were 5–1000 ng/ml and 0.5–1000 ng/ml for human serum and CSF respectively.
The method was applied to a clinical pharmacokinetic study of children treated with crenolanib.
Acknowledgments
This research was supported by a Cancer Center Support (CORE) Grant CA 21765, AROG Pharmaceuticals LLC, and by the American Lebanese Syrian Association (ALSAC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Westermark B, Heldin CH, Nister M. Glia. 1995;15:257–263. doi: 10.1002/glia.440150307. [DOI] [PubMed] [Google Scholar]
- 2.Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, Lowe J, Gajjar A, Zhao W, Broniscer A, Ellison DW, Grundy RG, Baker SJ. J Clin Oncol. 2010;28:3061–3068. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouffet E, Hawkins C. J Clin Oncol. 2010;28:1337–1344. doi: 10.1200/JCO.2009.25.5463. [DOI] [PubMed] [Google Scholar]
- 4.Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. Genes Dev. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kesari S, Stiles CD. Neuron. 2006;51:151–153. doi: 10.1016/j.neuron.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Ahluwalia MS, Patel M, Peereboom DM. Expert Rev Anticancer Ther. 2011;11:1739–1748. doi: 10.1586/era.11.166. [DOI] [PubMed] [Google Scholar]
- 7.Lewis NL, Lewis LD, Eder JP, Reddy NJ, Guo F, Pierce KJ, Olszanski AJ, Cohen RB. J Clin Oncol. 2009;27:5262–5269. doi: 10.1200/JCO.2009.21.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]