

Abstract
Objective
To evaluate the use of array comparative genomic hybridization (aCGH) for prenatal diagnosis, including assessment of variants of uncertain significance, and the ability to detect abnormalities not detected by karyotype, and vice versa.
Methods
Women undergoing amniocentesis or chorionic villus sampling (CVS) for karyotype were offered aCGH analysis using a targeted microarray. Parental samples were obtained concurrently to exclude maternal cell contamination and determine if copy number variants (CNVs) were de novo, or inherited prior to issuing a report.
Results
We analyzed 300 samples, most were amniotic fluid (82%) and CVS (17%). The most common indications were advanced maternal age (N = 123) and abnormal ultrasound findings (N = 84). We detected 58 CNVs (19.3%). Of these, 40 (13.3%) were interpreted as likely benign, 15 (5.0%) were of defined pathological significance, while 3 (1.0%) were of uncertain clinical significance. For seven (~2.3% or 1/43), aCGH contributed important new information. For two of these (1% or ~1/150), the abnormality would not have been detected without aCGH analysis.
Conclusion
Although aCGH-detected benign inherited variants in 13.3% of cases, these did not present major counseling difficulties, and the procedure is an improved diagnostic tool for prenatal detection of chromosomal abnormalities.
Keywords: aCGH, chromosomal abnormality, chromosomal microarray analysis, prenatal, copy number variants, CVS, amniotic fluid
INTRODUCTION
Prenatal diagnosis of cytogenetic abnormalities has focused primarily on detection of the most common aneuploidy in humans, trisomy 21. The currently accepted approach is to perform a combination of screening procedures in the first and second-trimester based on maternal age, levels of serum analytes, and ultrasound-detected markers or abnormalities in the fetus (Wapner et al., 2003; Malone et al., 2005; ACOG, 2007a; Reddy, 2007). Integrated first- and second-trimester screening detects approximately 95% of fetuses with Down syndrome with a 5% rate for an invasive procedure (Reddy, 2007). Such screening also provides a risk evaluation for trisomy 18, neural tube defects, and more recently Smith–Lemli–Opitz syndrome (Craig et al., 2006; ACOG, 2007a). This strategy is limited by lack of detection of many unbalanced structural chromosomal abnormalities (i.e. deletions and duplications) from submicroscopic chromosomal aberrations below the resolution of a standard karyotype analysis (Stankiewicz and Beaudet, 2007). Such disorders can be diagnosed by locus-specific fluorescence in situ hybridization (FISH), but it is not feasible to perform FISH for all possible deletion and duplication syndromes, or to identify which pregnancies are at increased risk with any regularity. In general, neither family history nor advanced maternal age can help guide the clinical indication for testing.
Array-based comparative genomic hybridization (aCGH) allows for fast and accurate detection of unbalanced structural and numerical chromosomal abnormalities (Pinkel et al., 1998; Mohammed et al., 2001; Vissers et al., 2003; Barrett et al., 2004). While experience with diagnostic aCGH in the pediatric population is extensive (Bejjani et al., 2005; Cheung et al., 2005; Shaffer et al., 2006; Lu et al., 2007; Stankiewicz and Beaudet, 2007), experience with its use for clinical prenatal diagnosis is still limited. Rickman and colleagues hybridized DNA extracted from amniotic fluid (AF) and chorionic villus sampling (CVS) cultures with known cytogenetic abnormalities and confirmed the abnormality in 22/30 cases using a whole-genome bacterial artificial chromosome (BAC) array and in 29/30 cases using a clinical ‘targeted’ array (Rickman et al., 2005). The first prospective validation study on 98 AF and CVS samples demonstrated a 5% detection rate of clinically significant abnormalities (Sahoo et al., 2006a). Other smaller studies have investigated the use of aCGH for analysis of cell-free DNA in amniotic fluid (Larrabee et al., 2004; Miura et al., 2006; Lapaire et al., 2007).
Here we present our experience with prenatal diagnosis on DNA extracted from CVS or AF using a targeted clinical CGH array on 300 cases. Our data indicate that cytogenetic abnormalities are readily detected. We confirm the value of aCGH for characterization of small supernumerary marker chromosomes (sSMCs) of unknown origin and for the detection of submicroscopic copy number changes in cases with apparently balanced structural chromosomal abnormalities or with a normal karyotype.
METHODS
Patients and samples
Samples included in this dataset were received between September 2005 and February 2008 for clinical prenatal aCGH testing by the Baylor College of Medicine Medical Genetics Laboratories. Patients underwent pretest counseling as described (Darilek et al., 2008) and provided informed consent for adjunct aCGH testing. We received direct CVS, uncultured AF, or established cultures of such samples, as well as one fibroblast culture from a fetal skin biopsy and one cystic hygroma fluid sample. Blood samples from both parents were requested with the fetal sample to test for possible maternal cell contamination (MCC) and for expedited characterization of potential familial copy number variants (CNVs) where necessary. Blood samples were received from both parents in 293 (94.8%) of cases, from the mother only in 15 (4.9%) cases, and from neither parent in 1 (0.3%) case. This report does not include data on 98 samples from our validation study (Sahoo et al., 2006a), but includes 8 samples described in a report on a novel DNA extraction method from uncultured AF (Bi et al., 2008).
Cell culture and DNA extraction
G-banded karyotypes and, if requested, FISH, to detect aneuploidy for chromosomes 13, 18, 21, X, and Y were performed on all samples using standard protocols, either in the referring laboratory or in our laboratory. Typically, 25–30 mg of CVS tissue or 25 mL of AF (at a gestational age of at least 16 weeks), were required if both karyotype analysis and aCGH were performed in our laboratory. When karyotype analysis was performed in the referring laboratory, 15 mg of CVS and 15 mL of AF were required. Direct analysis was performed on uncultured CVS (5 mg) or AF (5–7 mL) if a sufficient amount of sample was submitted. Backup cultures were established with the remainder of the samples to perform confirmatory karyotype, FISH or aCGH studies if needed. High molecular weight DNA was extracted from amniotic fluid cell pellets, chorionic villi or cultured cells using the QIAamp DNA Blood Midi Kit (Qiagen, Valencia, CA) with minor modifications. For samples with limiting amounts of DNA, 10–30 ng each of the fetal DNA and reference DNA were subjected to whole-genome amplification (WGA) using the GenomePlex WGA kit (Sigma-Aldrich Corporation, St. Louis, MO). All fetal DNA samples were tested for MCC using the PCR-based Identifiler system (AmpFLSTR, Applied Biosystem, Foster City, CA). Prior to labeling and hybridization, fetal gender was determined by amelogenin gene polymerase chain reaction (PCR) amplification. Test DNA and reference DNA of the same gender were then cohybridized to the array. Reference DNAs were derived from peripheral blood leukocytes of a phenotypically normal male and female.
Array platforms
Targeted clinical BAC arrays were used for 190 samples, including the Baylor College of Medicine (BCM) BAC Chromosomal Microarray V5 or 6; the others were hybridized to V6 of the BCM oligonucleotide Chromosomal Microarray. Arrays were designed to provide redundancy with high sensitivity and specificity for detection of clinically significant unbalanced chromosomal abnormalities, while minimizing detection of benign CNVs or CNVs of uncertain clinical significance. Manufacturing, composition and clinical application of these arrays have been previously described (Cheung et al., 2005; Roa et al., 2005; Sahoo et al., 2006b; Lu et al., 2007). The BCM V5 BAC array contained 853 distinct clones, targeted to cover 75 genomic disorders, 41 relevant subtelomeric regions with an average of 10 clones per region, and 43 unique pericentromeric regions. The BCM V6 BAC arrays contained 1476 distinct clones, covering over 140 genomic disorders, 41 relevant subtelomeric regions, 43 unique pericentromeric regions, and selected backbone clones distributed at an equivalent of a 650 band-interval. The V6 oligonucleotide arrays are custom designed emulations of the V6 BAC array manufactured by Agilent Technologies, Inc. (Santa Clara, CA) (Ou et al., 2008). Upon transition from a BAC to an oligo array platform, the oligonucleotide BAC-emulation design was preferred over more randomly spaced oligonucleotides to assure optimal coverage of clinically relevant regions.
Hybridization and data analysis
DNA labeling and hybridization were performed as described for BAC arrays (Cheung et al., 2005; Sahoo et al., 2006b; Lu et al., 2007). Briefly, two dye-label reversal hybridizations were done for each sample. Microarray image files were quantified using GenePix Pro 5 software, and signal intensities from both hybridizations were normalized and combined to determine a single-fold change value for each clone. Inferences were made for all clones using these final combined data values. All analyses were performed on log2 ratios using a code for the normalization and inference implemented in the R statistical programing language (Shaw et al., 2004; Cheung et al., 2005; Sahoo et al., 2006b; Lu et al., 2007).
For the oligonucleotide array, DNA labeling and hybridization were performed according to the manufacturer’s protocol (Agilent Technologies). Microarrays were scanned on an Agilent G2565 scanner and image files were quantified using Agilent’s Feature extraction software (V9.0). Text file outputs were converted to BAC-level emulation data by combining the oligo aCGH data corresponding to regions encompassed by BAC clones, as described (Ou et al., 2008).
Data interpretation and confirmatory analysis
Detected copy number gains or losses were compared to known CNVs in publicly available databases and in our own database of results from more than 12 000 samples. Parental samples were analyzed by aCGH, specifically to examine the presence or absence of CNVs detected in the fetus. If copy number changes were de novo and/or encompassed known genomic disorders, confirmatory FISH studies were also performed on metaphase spreads or interphase nuclei prepared from AF or CVS cultures using standard procedures.
RESULTS
Clinical indications and sample types
A total of 309 samples were submitted (Table 1), 254 (82.2%) AF and 53 (17.2%) CVS. For AF, analysis was performed on cultured amniocytes in 166 cases (65 of which were submitted as established cultures), on DNA directly extracted from AF cell pellets without (n = 38) or with (n = 50) prior to WGA. For CVS, analysis was performed on cultured villi in 27 (20 of which were submitted as established cultures), on directly extracted DNA in 21, and DNA obtained after WGA in 5 cases. Hence, analyses were completed with uncultured material for 88/189 (46.6%) of AF and for 26/33 (78.8%) of CVS. There was one fetal skin fibroblast culture and one cystic hygroma fluid sample. For two cases, MCC in the submitted sample was detected prior to aCGH analysis. The first was a direct AF for which results were reported from the amniocyte culture after it was found to be free of MCC. The second was an established CVS culture and follow-up amniocentesis was required to complete both the fetal aCGH and karyotype analysis.
Table 1.
Analyzed samples
| Sample type (N = 309) | DNA preparation | ||
|---|---|---|---|
| Amniotic fluid | 254 (82.2%) | ||
| Uncultured amniotic fluid | 189/254 (74.4%) | Direct | 38/189 (20.1%) |
| WGA | 50/189 (26.5%) | ||
| Culture | 101/189 (53.4%) | ||
| Amniocyte culture | 65/254 (25.6%) | Culture | 65/65 (100%) |
| Chorionic villus samples | 53 (17.2%) | ||
| Uncultured CVS | 33/53 (35.2%) | Direct | 21/33 (63.6%) |
| WGA | 5/33 (15.2%) | ||
| Culture | 7/33 (21.2%) | ||
| CVS culture | 20/53 (64.8%) | Culture | 20/20 (100%) |
| Fibroblast culture | 1 (0.3%) | Culture | 1 (100%) |
| Cystic hygroma fluid | 1 (0.3%) | Culture | 1 (100%) |
WGA, whole-genome amplification performed prior to DNA labeling.
The indications that were recorded (N = 367; Table 2) included one (N = 249) or up to three (N = 115) of the following: advanced maternal age of over 35 years at time of delivery (N = 123; 33.5%), abnormalities detected on prenatal ultrasound (N = 84; 22.9%), a previous child with or another family history of a genetic disorder or chromosomal abnormality (N = 87; 23.7%), further work-up for a chromosomal abnormality detected on the fetal karyotype (N = 28; 7.6%), parental concern (N = 33; 9.0%), abnormal maternal serum screening results (N = 9; 2.5%); unspecified (N = 3; 0.8%). For 17% of patients, more than one indication was recorded. The ultrasound abnormalities included both major and minor abnormalities, ranging from severe congenital heart defects to such anomalies as an isolated choroid plexus cyst (Table 2).
Table 2.
Indications for invasive prenatal diagnostic procedure
| Indication | N w/o other indicationsa | N with other indicationsb | Total Nc (%#) | % of all 367 indicationsd |
|---|---|---|---|---|
| Advanced maternal age (≥ 35 years at delivery) | 87 | 36 | 123 | 33.5 |
| Abnormal US | 67 | 17 | 84 | 22.9 |
| > 2 abnormalities detected | 32 (38.1) | |||
| 1 anomaly detected | 21 (25.0) | |||
| IUGR only | 3 (3.6) | |||
| Increased NT only | 4 (4.7) | |||
| CPC only | 1 (1.2) | |||
| Single umbilical artery only | 3 (3.6) | |||
| Unspecified | 20 (23.8) | |||
| Family history of genetic or other abnormality | 46 | 41 | 87 | 23.7 |
| Chromosomal abnormality detected on prenatal karyotype in current pregnancy | 22 | 6 | 28 | 7.6 |
| Parental concern | 24 | 9 | 33 | 9.0 |
| Abnormal maternal serum screening result | 3 | 6 | 9 | 2.5 |
| Unspecified | 3 | — | 3 | 0.8 |
| Total of 367 indications● | 249 | 115 | 367 | |
| Total of 300 analyses | 249 (83%) | 51 (17%) | 300 |
Some patients had more than one indication, hence 4 columns are shown:
listed indication was the only reason for the invasive procedure;
listed indication was one of two or more indications;
total for each indication;
% of total for each indication;
% refers to total cases with abnormal ultrasound.
Likely benign copy number changes
Of the total 309 submitted samples, 9 were not analyzed for reasons unrelated to the aCGH procedure (insufficient quality of the submitted cultured samples, MCC detected in the submitted sample, or test canceled by the submitter, parental samples unavailable). No deletions or duplications were seen in 242 (80.7%) of the remaining 300 samples. Copy number changes were detected in 58 (19.3%). Of these, 40 (13.3%) were CNVs that were interpreted as likely benign and of no clinical significance (Table 1, see Supporting Information). Of these, 39 were inherited from a phenotypically normal parent and 1 was de novo, but had been previously seen multiple times in our database of over 12 000 aCGH hybridization results from all previously analyzed clinical cases and phenotypically normal individuals.
Copy number changes of established or uncertain clinical significance
Eighteen (6.0%) copy number changes (Table 3) were either of established clinical significance, although of varying severity and penetrance (n = 15; 5.0%) or of uncertain clinical significance (n = 3; 1%). Two of the 18 were de novo CNVs, detected in the fetal DNA but not in parental DNAs, and not found in public CNV databases or in the laboratory’s aCGH database. The laboratory did not perform independent assessments of paternity in these two cases, but assumed paternity as described. The first de novo case (Table 3, case no. 7; Figure 1(A)) was referred because of micrognathia and micropenis detected on prenatal ultrasound in a male fetus with a 46,XY karyotype. A de novo copy number loss in the fetal DNA, not present in the mother’s DNA, of one clone in Xq27.3 was identified by aCGH, but not by FISH, as can be the case for smaller CNVs. The clinical significance of this CNV is uncertain, but the deleted region does not contain any genes, and it likely was not the cause of the phenotype. This result did not influence the pregnancy management. The infant died 1 day after birth and had a diaphragmatic hernia, ambiguous genitalia, scalp edema, a two-vessel cord, and a suspected clinical diagnosis of Fryns syndrome. The second de novo case (Table 3 case no. 11; Figure 1(B)) was analyzed because of fetal polydactyly detected on prenatal ultrasound, and showed a deletion containing known genes in 15q26.3 of at least 800 kb. The result was interpreted to possibly be the cause of the polydactyly, bearing an increased risk for other disabilities not detectable on prenatal ultrasound. It contributed to the parents’ decision to terminate the pregnancy; additional fetal samples for follow-up studies were not available. Although it can be argued that this aCGH finding is likely of clinical significance, we more cautiously classified it as ‘uncertain’.
Table 3.
Abnormalities of defined or uncertain clinical significance detected by aCGH
| Case No. | Array | Indication | aCGH result | Other result | Interpretation/Outcome |
|---|---|---|---|---|---|
| 1 | V5 | Abnormal fetal karyotype, unknown sSMC: 47,XX, +mar[12]/46,XX[3] |
arr cgh 12q12(RP11-79013,RP11-242B24)x3 | ish r(12)(p11q11)(D12Z3+)[16]/12(p11q11)(D12Z3x2)[4] | Female fetus with mosaic ring chromosome 12. Pregnancy continued. |
| 2 | V5 | Advanced maternal age | arr cgh 21(14 BAC)x3 | 47,XX, +21 | Female fetus with Down syndrome due to trisomy 21. Pregnancy terminated. |
| 3 | V5 | Apparently balanced de novo translocation on fetal karyotype: 46,XY,t(2;9)(q11.2;q34) |
arr cgh 9q34.3(RP11-432J22 > RP11-974F22)x1dn | ish der(9)t(2;9)(q11.2;q34) (D9S325-) | Male fetus with 9q34 deletion syndrome. Pregnancy terminated. |
| 4 | V6 | Abnormal fetal karyotype, unknown sSMC: 47,XX, +mar[11]/46,XX[9] |
arr cgh 12p11.22(RP11-847A19 > RP11-8P13)x3 | nuc ish 12cen(D12Z3x3), 12p11.2(RP11-8P13x3)[5]/12cen(D12Z3x2), 12p11.2(RP11-8P13x2)[195] | Female fetus with mosaic ring chromosome 12. Pregnancy continued. Development appropriate at 6 months of age. Karyotype on blood after birth confirmed r(12) in 7/10 cells. |
| 5 | V5 | Abnormal fetal karyotype, unknown sSMC: 47,XY, +mar |
arr cgh 21q21.1(RP11-625C23, RP11-840D8, RP11-143A3)x3 | nuc ish 21q11.2q21.1(RP11-625C23x3,RP11-840D8x3) | Male fetus with ring chromosome 21 confirmed at birth. Pregnancy continued. |
| 6 | V5 | Maternal carrier of MECP2 region duplication (Xq28) | arr cgh Xq28(RP11-157E12- > RP4-671D9)x3mat | arr cgh Xq28(RP11-157E12- > RP4-671D9)x3 in mother |
Female fetus with maternally inherited heterozygous duplication in MECP2 region. Pregnancy continued |
| 7 | V5 | Micrognathia, micropenis, 46,XY | arr cgh Xq27.3(RP11-387H19)x0dn (Figure 1(A)) | ish Xq27.3(RP11-387H19)x1; maternal aCGH normal; region does not contain any known genes | Male fetus with de novo deletion Xq27.3. Pregnancy continued. Neonatal demise with diaphragmatic hernia, 2-vessel cord, scalp edema, suspected Fryns syndrome (Xq27 deletion likely not the cause of the phenotype). |
| 8 | V6 Oligo | Maternal carrier of PMD gene duplication | arr cgh Xq22.2(RP11-1123D8- > RP11-832L2)x2mat | nuc ish Xq22(PLPx2) | Male fetus affected with Pelizaeus Merzbacher disease. Pregnancy terminated. |
| 9 | V6 Oligo | Maternal balanced translocation (10;17) and mosaic unbalanced chromosomal abnormality found in CVS: 46,XY,add(1)(p22), add(11)(p15)[8]/46,XY[2] |
arr cgh 1q21.1(RP11-315I20- > RP11-102F23)x1dn, 11p15.5(RP5-998N23- > RP11-38L8)x1 on CVS | ish del(1)(q21.1q21.1)(RP11-769J20-),del(11)(p15.5p15.5) (RP11-38L8)dn on CVS; ish del(1)(q21.1q21.1)(RP11-769J20-)on AF | Male fetus with TAR syndrome possibility on aCGH results, followed by ultrasound confirmation of TAR (del 11p 15.5 on CVS was culture artifact). Pregnancy continued, diagnosis confirmed after birth. |
| 10 | V6 Oligo | Maternal carrier of STS gene deletion (Xp22.31) | arr cgh Xp22.31(RP11-483M24- > RP11-143E20)x0mat | 46,XY.ish Xp22.3(STS-) | Male fetus with steroid sulfatase deficiency. Pregnancy continued, diagnosis confirmed after birth. |
| 11 | V6 Oligo | Abnormal ultrasound: Polydactyly | arr cgh 15q26.3(RP11-168G16- > RP11-505E24)x1dn (Figure 1(B)) | 46,XY.ish del(15)(q26.3q26.3) (RP11-168G16-,RP11-505E24-)dn | Male fetus with 15q26.3 de novo deletion of at least 800 Kb, confirmed by FISH. Pregnancy termination, no sample available for confirmatory testing. |
| 12 | V6 Oligo | Advanced maternal age; abnormal fetal karyotype: 46,XY,?dup(8)(p21.3p23.1) | arr cgh 8p22(RP11-520F7, RP11-90I3)x3 | nuc ish 8p22(RP11-90I3x3) | Male fetus with duplication 8p22. Pregnancy termination for chromosomal abnormality. |
| 13 | V6 Oligo | Abnormal fetal karyotype, unknown sSMC: 47,XX, +mar/46,XX |
arr cgh 22q11.1q11.2(RP11-48E19- > RP11-466H9)x3 | nuc ish 22q11.21(RP11-9106x3)[66]/22q11.21(RP11-9106x2)[134] | Female fetus with marker chromosome containing three clones from 22q11, in cat-eye syndrome critical region. Pregnancy terminated. |
| 14 | V6 Oligo | Cryptic translocation in mother 46,XX,t(8;12)(p23.1;p13.31).ish t(8;12)(D8S504-,VIJTYAC 14+; VIJTYAC 14-,D8S504+) | arr cgh del(8)(p23.1p23.2) (RP11-16G12- > RP11-91J19)x1,dup(12) (p13.31p13.33),(RP11-69M1- > 598F7)x3 | Retrospective evaluation showed 46,XY,der(8)t(8;12)(p23.1;p13.31)mat on fetal karyotype, confirmed by arr cgh | Male fetus with 8;12 unbalanced translocation. Fetal demise. Cotwin had same cryptic translocation as mother. Pregnancy continued. |
| 15 | V6 Oligo | Abnormal ultrasound: Twin pregnancy, fetal hydrops with cystic hygroma in this twin | arr cgh X(123 BAC)x1, 8p23.1(RP11-241I4)x1 | 45,X but found XYY by aneuploidy FISH in cystic hygroma fluid (16/19 cells). Mother normal in 8p23.1, father was not available | Female fetus with 45,X. Subsequent IUFD. Pregnancy continued (see case 16). |
| 16 | V6 Oligo | Abnormal ultrasound(see #15): Twin pregnancy, normal appearing ultrasound for this twin | arr cgh Y(12 BAC)x2 | 47,XYY on amniotic fluid karyotype. Follow-up FISH on uncultured whole blood after birth: nuc ish Xcen(DXZ1x1), Ycen(DYZ3x2)[141]/Xcen (DXZ1x1)[30]/Xcen(DXZ1x1), Ycen(DYZ3x1)[29] and 47,XYY[20] karyotype | Male fetus with 47,XYY/45,X/46,XY mosaic karyotype. Pregnancy continued, preterm delivery after premature rupture of membranes. |
| 17 | V6 Oligo | Abnormal ultrasound: hypoplastic left heart | arr cgh X(123 BAC)x1 (arr cgh was performed against male reference DNA). | Y chromosome present by PCR to determine fetal gender prior to array; 45,X on fetal karyotype; FISH 45,X[94]/46,XY[6] | Fetus with 45,X/46,XY mosaicism. Pregnancy continued. Neonate with mild Turner syndrome phenotype (webbed neck, low hairline, wide-spaced nipples) and hypoplastic left heart. |
| 18 | V6 Oligo | Abnormal fetal karyotype: 46,XX,del(3)(p26).ish del(3)(pter-)mat |
arr cgh 3p26.3p26.2(RP11-385A18- > RP11-129J10)x1mat, Xq12(RP11-349K4)x3pat | ~5.1 Mb deletion in 3p26.2-3p26.3; also present in the apparently normal mother. FISH previously performed by referring lab. | Female fetus with maternally Inherited ~5.1 Mb deletion. Mother appears phenotypically normal. Fetus also with likely benign paternally inherited duplication in Xq12. Normal targeted prenatal ultrasound at 22 weeks gestation. Pregnancy continued. |
Figure 1.
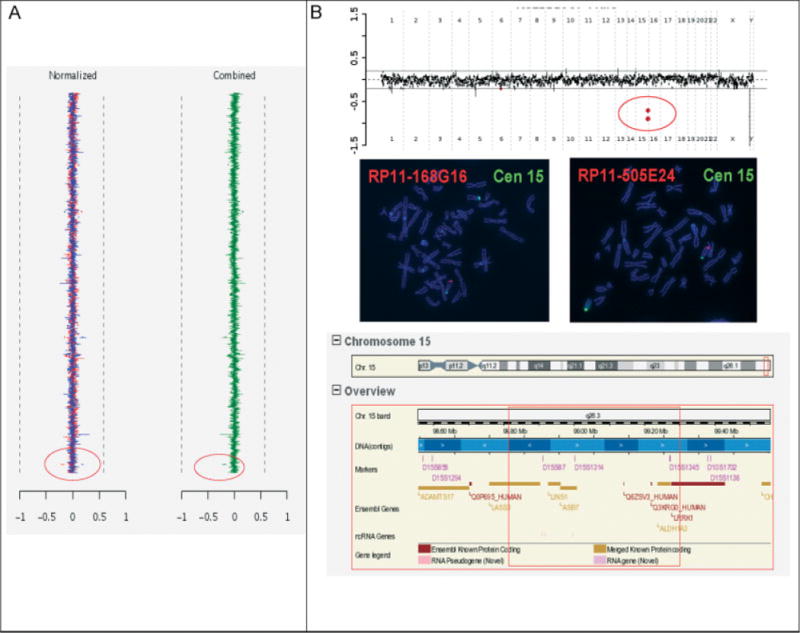
Copy number losses of uncertain clinical significance. (A) BAC aCGH result on a fetal sample, showing de novo copy number loss of one BAC clone in Xq27.3 (Table 3 case no. 7). Normalized data from both dye-reversal hybridizations are shown on the left in red and blue and combined data are shown on the right in green. The red circles indicate the clone affected by copy number loss. (B) Top panel: Oligo aCGH result showing two deleted clones in 15q26 (Table 3 case no. 11) (indicated by the red circle); middle panel: FISH results on metaphase spreads with the deleted clones; bottom panel: schematic of the genomic region from the Ensembl genome browser showing gene content of the deletion (red square)
The third case with a result of uncertain clinical significance (Table 3 case no. 18) showed a large (~5.1 Mb) deletion in 3p26.2–p26.3, detected both by karyotype and aCGH, that was also present in the mother who has no reported medical problems. The 15 remaining cases were interpreted as ‘of established clinical significance’, although the phenotypic consequences were not always fully predictable (e.g. for sSMCs).
Copy number abnormalities were further detected in eight samples that were submitted for work-up of a cytogenetic abnormality found on prior fetal karyotype analysis (Table 3 case nos. 1, 3, 4, 5, 9, 12, 13, 18). There were four cases (Table 3 case nos. 2, 15, 16, 17) with whole-chromosome aneuploidy (one 47,XX, +21, two 45,X, and one 47,XYY). Those were equally well detected by aCGH and karyotype analysis, but the aCGH data were available before the karyotype result for the fetus with trisomy 21. We are not aware of any aCGH failure to detect abnormalities known to be present based on karyotype or FISH, except for cases where aCGH analysis was requested to investigate the origin of a de novo sSMC found on the fetal karyotype. Out of nine samples that were submitted for this indication, there were four (Table 3 case nos. 1, 4, 5, 13) with a gain of one or several BAC clones on specific chromosomes, resulting in the diagnosis of two ring chromosomes 12, one ring chromosome 21 and a bisatellited marker containing material from the 22q11 cat-eye syndrome critical region. When aCGH did not detect genomic imbalance in the presence of a marker chromosome, we reported that counseling could be more reassuring, whereas for those with copy number gain, we reported that the presence of the de novo sSMC could result in an abnormal phenotype.
In one case, aCGH confirmed a duplication in 8p22 initially detected on the fetal karyotype (Table 3 case no. 12); this pregnancy was terminated after counseling. There were four deletions or duplications for which the pregnancy was known to be at elevated risk by virtue of maternal carrier status (MECP2 duplication, PMD duplication, STS deletion, or balanced translocation; Table 3 case nos. 6, 8, 10, and 14, respectively). There were two cases where aCGH provided a diagnosis that would have otherwise remained unascertained. In one (Table 3 case no. 3) previously reported case (Simovich et al., 2007), aCGH was performed to determine copy number balance at the breakpoints of a de novo apparently balanced (2;9) translocation and revealed a deletion at the 9q34 breakpoint, indicating that the fetus was affected with the 9q34 deletion syndrome (Yatsenko et al., 2005). A second (Table 3 case no. 9) was submitted to investigate a mosaic add(1)(p22), add(11)(p15) chromosomal abnormality, detected in a cultured CVS sample. While this finding was ultimately shown to be a culture artifact, the aCGH identified an unsuspected deletion in 1q21. This deletion was de novo and overlapped with the deleted region associated with thrombocytopenia-absent radius (TAR) syndrome (Klopocki et al., 2007; Uhrig et al., 2007). A targeted ultrasound was recommended and revealed bilaterally absent radii in the fetus, consistent with this diagnosis.
DISCUSSION
We found that aCGH yielded new clinically relevant results in 7 (2.3%) of 300 (1/43) analyses, that provided additional information for prenatal genetic counseling and risk assessment. These cases included four marker chromosomes that were rapidly identified, the discovery of a 9q34 deletion in an apparently balanced translocation, a case of TAR syndrome and a deletion of at least 800 kb in 15q26.3. It is virtually certain that the 9q34 deletion (Yatsenko et al., 2005) and the 1q21 (TAR) deletion (Klopocki et al., 2007) are associated with a clinical disorder. Although not proven, and therefore classified as ‘uncertain’, it is also likely that the de novo 15q26.3 deletion is associated with a clinical phenotype.
In two of these seven cases (0.7% or 1/150; 9q34 and 1q21 TAR syndrome deletion), aCGH detected a known disorder that would not have been found if only a karyotype or aneuploidy FISH had been performed. This risk of 1/150 is equivalent to the term pregnancy risk for Down syndrome of a 38-year-old woman, and to the term risk for all common aneuploidies of a 36-year-old woman (ACOG, 2007b). This risk is also higher than the currently quoted risk of a procedure-related loss after amniocentesis of 1/300–1/500, and the similar risk of loss after CVS (ACOG, 2007b).
We identified the origin of four out of nine sSMC. The quoted risk for an abnormal phenotype is 14.7% for nonsatellited sSMCs, and 10.9% for satellited sSMCs (Warburton, 1991). Determining the origin of sSMCs is important for accurate genetic counseling, but can be laborious and difficult by conventional cytogenetic methods and FISH. If the genetic material contained in the marker is represented on the array, i.e. the marker contains euchromatic material, it should show a copy number gain for this region. Although a high percentage of SMCs (59%) are mosaic (Crolla et al., 2005), levels of mosaicism as low as 15%, inclusive of SMCs, can be detected by aCGH (Ballif et al., 2006; Ballif et al., 2007; Cheung et al., 2007). In our study, in the four cases in which the sSMCs were detected by aCGH, the percent of cells in the cultures that contained the sSMC ranged from 33 to 100%. In the five cases in which the sSMCs were not detected, mosaicism ranged from 25 to 85% in the initial culture. Of note, three of these (67, 83, and 85% mosaicism) were karyotypically determined to be bisatellited markers and confirmed by FISH analysis to originate from chromosomes 14/22 and 13/21. Markers derived from these acrocentric chromosomes are occasionally seen in normal individuals and have historically been predicted to contain only heterochromatin. Since our array was targeted to unique (euchromatin-containing) pericentromeric sequences, SMCs that contain only heterochromatin material would not be expected to be detectable using this methodology. Therefore, a normal result by aCGH provides reassurance that the marker is likely benign.
An abnormal ultrasound finding was the sole indication for the aCGH analysis in only four of the cases that proved to be abnormal (Table 3 case nos. 7, 11, 15, 17). Furthermore, the limb abnormalities in the TAR case were detected after the aCGH result became available. This suggests that reserving prenatal aCGH analysis for pregnancies with an abnormal prenatal ultrasound may not be the optimal diagnostic strategy. This is not surprising because many of the genomic disorders represented on the targeted array are not associated with clinical features that are detectable on prenatal ultrasound. For some, such features may be present, but are atypical or variable (e.g. congenital heart defect or intra-uterine growth restriction). Moreover, it has been shown that genome-wide arrays detect pathological abnormalities in up to 17% of patients with mental retardation (Shaw-Smith et al., 2004; de Vries et al., 2005; Schoumans et al., 2005; Friedman et al., 2006; Krepischi-Santos et al., 2006; Lugtenberg et al., 2006; Menten et al., 2006; Rosenberg et al., 2006) and in 10–27% of children with autism (Jacquemont et al., 2006; Sebat et al., 2007). These conditions are not typically associated with birth defects detectable by prenatal ultrasound.
There is understandable concern that CNVs of uncertain significance might lead to termination of normal pregnancies. Thus, an important question is how often such a finding will be detected by aCGH used for prenatal diagnosis. Our arrays were focused on disease regions and likely to give a lower rate of abnormalities of uncertain significance than would be expected with more complex tiling arrays. We had three such findings (1%) with variable uncertainty. In addition to the 15q26.3 deletion, there was the Xq27.3 de novo deletion, which was likely coincidental with the diagnosis of Fryns syndrome, but a causative role cannot be excluded with certainty. The 3p26 deletion, although inherited, was classified as uncertain based on its larger size and possibly variable penetrance of a phenotype. The deleted region contains 11 known genes, and as reviewed by Barber (Barber, 2008), copy number loss of this region has been observed with (Dijkhuizen et al., 2006; Malmgren et al., 2007) and without a clinical phenotype, particularly in a case assessed through prenatal diagnosis (Knight et al., 1995). However, the majority of detected CNVs were smaller, present in a healthy parent and relatively common (13.3%). Most were also prevalent in the normal population, and hence, can be assigned a low risk with relative confidence. This group included ten deletions, four of regions containing known genes, but with no other evidence for benign CNVs, that could be considered of slightly elevated risk. Six were of regions without genes or with genes, but frequently deleted in the normal population. There were 30 duplications, 6 of regions containing known genes, but with no other evidence for benign CNVs, and 24 of regions without genes or with genes, but frequently duplicated in the normal population (Supporting Information Table 1). The experience with over 12 000 pediatric blood samples from patients with disabilities and normal controls is extremely helpful in interpreting common CNVs. However, the possibility that some have reduced penetrance for a disability must be considered. For example, one of the presumed benign variants, a duplication in the region of the Kallmann gene on Xp22.3 found in a male fetus and as a heterozygous change in his mother, was slightly problematic the first time it was detected. It was then interpreted to be of relatively low risk, but with some uncertainty. Subsequently, this duplication was observed in phenotypically normal males, indicating that it was a benign CNV.
Finally, results were obtained from uncultured amniocytes in 46.6% of submitted AF, and from uncultured chorionic villi in 78.8% of submitted CVS.
CONCLUSION
We found that aCGH reliably detected clinically significant copy number changes in 5.0% of fetal samples, confirming previous data (5.1%) from a smaller validation study (Sahoo et al., 2006a), while detection of CNVs of uncertain clinical significance remained at an acceptable low rate (1%). We believe that genetic counseling that informs prospective parents of the additional benefit of aCGH and also of the potential detection of a CNV of uncertain significance should precede all aCGH testing (Darilek et al., 2008). Larger studies will be needed to determine if aCGH will become the first-line test to detect chromosomal abnormalities in fetal samples and to establish whether the improved overall detection rates of clinically significant chromosomal abnormalities will justify offering aCGH more universally to all pregnant women.
Supplementary Material
Footnotes
Presented at the 14th Meeting of the International Society of Prenatal Diagnosis, Vancouver, June1–4, 2008.
CONFLICT OF INTEREST STATEMENT
The Medical Genetics Laboratories of the Department of Molecular and Human Genetics at Baylor College of Medicine (BCM), where the authors are employed as trainees, staff or faculty members, offers extensive genetic laboratory testing and derives revenue from this activity. BCM currently uses oligonucleotide arrays manufactured by Agilent Technologies.
References
- ACOG. ACOG Practice Bulletin No. 77: screening for fetal chromosomal abnormalities. Obstet Gynecol. 2007a;109:217–227. doi: 10.1097/00006250-200701000-00054. [DOI] [PubMed] [Google Scholar]
- ACOG. ACOG Practice Bulletin No. 88, December 2007. Invasive prenatal testing for aneuploidy. Obstet Gynecol. 2007b;110:1459–1467. doi: 10.1097/01.AOG.0000291570.63450.44. [DOI] [PubMed] [Google Scholar]
- Ballif BC, Hornor SA, Sulpizio SG, et al. Development of a high-density pericentromeric region BAC clone set for the detection and characterization of small supernumerary marker chromosomes by array CGH. Genet Med. 2007;9:150–162. doi: 10.1097/gim.0b013e3180312087. [DOI] [PubMed] [Google Scholar]
- Ballif BC, Rorem EA, Sundin K, et al. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am J Med Genet A. 2006;140:2757–2767. doi: 10.1002/ajmg.a.31539. [DOI] [PubMed] [Google Scholar]
- Barber JC. Terminal 3p deletions: phenotypic variability, chromosomal non-penetrance, or gene modification? Am J Med Genet A. 2008;146A:1899–1901. doi: 10.1002/ajmg.a.32387. [DOI] [PubMed] [Google Scholar]
- Barrett MT, Scheffer A, Ben-Dor A, et al. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc Natl Acad Sci USA. 2004;101:17765–17770. doi: 10.1073/pnas.0407979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejjani BA, Saleki R, Ballif BC, et al. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: is less more? Am J Med Genet A. 2005;134:259–267. doi: 10.1002/ajmg.a.30621. [DOI] [PubMed] [Google Scholar]
- Bi W, Breman AM, Venable SF, et al. Rapid prenatal diagnosis using uncultured amniocytes and oligonucleotide array CGH. Prenat Diagn. 2008 doi: 10.1002/pd.2087. In Press. [DOI] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Scott DA, et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am J Med Genet A. 2007;143:1679–1686. doi: 10.1002/ajmg.a.31740. [DOI] [PubMed] [Google Scholar]
- Cheung SW, Shaw CA, Yu W, et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med. 2005;7:422–432. doi: 10.1097/01.gim.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- Craig WY, Haddow JE, Palomaki GE, et al. Identifying Smith-Lemli-Opitz syndrome in conjunction with prenatal screening for Down syndrome. Prenat Diagn. 2006;26:842–849. doi: 10.1002/pd.1518. [DOI] [PubMed] [Google Scholar]
- Crolla JA, Youings SA, Ennis S, Jacobs PA. Supernumerary marker chromosomes in man: parental origin, mosaicism and maternal age revisited. Eur J Hum Genet. 2005;13:154–160. doi: 10.1038/sj.ejhg.5201311. [DOI] [PubMed] [Google Scholar]
- Darilek S, Ward P, Pursley A, et al. Pre- and postnatal genetic testing by array-comparative genomic hybridization: genetic counseling perspectives. Genet Med. 2008;10:13–18. doi: 10.1097/GIM.0b013e31815f1ddb. [DOI] [PubMed] [Google Scholar]
- de Vries BB, Pfundt R, Leisink M, et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen T, van Essen T, van der Vlies P, et al. FISH and array-CGH analysis of a complex chromosome 3 aberration suggests that loss of CNTN4 and CRBN contributes to mental retardation in 3pter deletions. Am J Med Genet A. 2006;140:2482–2487. doi: 10.1002/ajmg.a.31487. [DOI] [PubMed] [Google Scholar]
- Friedman JM, Baross A, Delaney AD, et al. Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am J Hum Genet. 2006;79:500–513. doi: 10.1086/507471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont ML, Sanlaville D, Redon R, et al. Array-based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet. 2006;43:843–849. doi: 10.1136/jmg.2006.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopocki E, Schulze H, Strauss G, et al. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet. 2007;80:232–240. doi: 10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight LA, Yong MH, Tan M, Ng IS. Del(3) (p25.3) without phenotypic effect. J Med Genet. 1995;32:994–995. doi: 10.1136/jmg.32.12.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepischi-Santos AC, Vianna-Morgante AM, Jehee FS, et al. Whole-genome array-CGH screening in undiagnosed syndromic patients: old syndromes revisited and new alterations. Cytogenet Genome Res. 2006;115:254–261. doi: 10.1159/000095922. [DOI] [PubMed] [Google Scholar]
- Lapaire O, Lu XY, Johnson KL, et al. Array-CGH analysis of cell-free fetal DNA in 10 mL of amniotic fluid supernatant. Prenat Diagn. 2007;27:616–621. doi: 10.1002/pd.1752. [DOI] [PubMed] [Google Scholar]
- Larrabee PB, Johnson KL, Pestova E, et al. Microarray analysis of cell-free fetal DNA in amniotic fluid: a prenatal molecular karyotype. Am J Hum Genet. 2004;75:485–491. doi: 10.1086/423288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Shaw CA, Patel A, et al. Clinical implementation of chromosomal microarray analysis: summary of 2513 postnatal cases. PLoS ONE. 2007;2:e327. doi: 10.1371/journal.pone.0000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugtenberg D, de Brouwer AP, Kleefstra T, et al. Chromosomal copy number changes in patients with non-syndromic X-linked mental retardation detected by array CGH. J Med Genet. 2006;43:362–370. doi: 10.1136/jmg.2005.036178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmgren H, Sahlen S, Wide K, Lundvall M, Blennow E. Distal 3p deletion syndrome: detailed molecular cytogenetic and clinical characterization of three small distal deletions and review. Am J Med Genet A. 2007;143A:2143–2149. doi: 10.1002/ajmg.a.31902. [DOI] [PubMed] [Google Scholar]
- Malone FD, Canick JA, Ball RH, et al. First-trimester or second-trimester screening, or both, for Down’s syndrome. N Engl J Med. 2005;353:2001–2011. doi: 10.1056/NEJMoa043693. [DOI] [PubMed] [Google Scholar]
- Menten B, Maas N, Thienpont B, et al. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J Med Genet. 2006;43:625–633. doi: 10.1136/jmg.2005.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura S, Miura K, Masuzaki H, et al. Microarray comparative genomic hybridization (CGH)-based prenatal diagnosis for chromosome abnormalities using cell-free fetal DNA in amniotic fluid. J Hum Genet. 2006;51:412–417. doi: 10.1007/s10038-006-0376-7. [DOI] [PubMed] [Google Scholar]
- Mohammed NS, Bejjani BA, Shah S, Lupski JR, Shaffer LG. Development and validation of a high-resolution genomic microarray for identifying constitutional chromosome abnormalities. Am J Hum Genet. 2001;69:A5. [Google Scholar]
- Ou Z, Kang SH, Shaw CA, et al. Bacterial artificial chromosome-emulation oligonucleotide arrays for targeted clinical array-comparative genomic hybridization analyses. Genet Med. 2008;10:278–289. doi: 10.1097/GIM.0b013e31816b4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkel D, Segraves R, Sudar D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- Reddy UM. The evolving prenatal screening scene. Obstet Gynecol. 2007;110:2–4. doi: 10.1097/01.AOG.0000269044.84174.fc. [DOI] [PubMed] [Google Scholar]
- Rickman L, Fiegler H, Shaw-Smith C, et al. Prenatal detection of unbalanced chromosomal rearrangements by array-CGH. J Med Genet. 2005;43:353–361. doi: 10.1136/jmg.2005.037648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roa BB, Pulliam J, Eng CM, Cheung SW. Evolution of prenatal genetics: from point mutation testing to chromosomal microarray analysis. Expert Rev Mol Diagn. 2005;5:883–892. doi: 10.1586/14737159.5.6.883. [DOI] [PubMed] [Google Scholar]
- Rosenberg C, Knijnenburg J, Bakker E, et al. Array-CGH detection of micro rearrangements in mentally retarded individuals: clinical significance of imbalances present both in affected children and normal parents. J Med Genet. 2006;43:180–186. doi: 10.1136/jmg.2005.032268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo T, Cheung SW, Ward P, et al. Prenatal diagnosis of chromosomal abnormalities using array-based comparative genomic hybridization. Genet Med. 2006a;8:719–727. doi: 10.1097/01.gim.0000245576.47154.63. [DOI] [PubMed] [Google Scholar]
- Sahoo T, Peters SU, Madduri NS, et al. Microarray-based comparative genomic hybridization testing in deletion-bearing Angelman Syndrome patients: Genotype-phenotype correlations. J Med Genet. 2006b;43:512–516. doi: 10.1136/jmg.2005.036913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoumans J, Ruivenkamp C, Holmberg E, Kyllerman M, Anderlid BM, Nordenskjold M. Detection of chromosomal imbalances in children with idiopathic mental retardation by array based comparative genomic hybridisation (array-CGH) J Med Genet. 2005;42:699–705. doi: 10.1136/jmg.2004.029637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer LG, Kashork CD, Saleki R, et al. Targeted genomic microarray analysis for identification of chromosome abnormalities in 1500 consecutive clinical cases. J Pediatr. 2006;149:98–102. doi: 10.1016/j.jpeds.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Shaw-Smith C, Redon R, Rickman L, et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet. 2004;41:241–248. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CJ, Shaw CA, Yu W, et al. Comparative genomic hybridisation using a proximal 17p BAC/PAC array detects rearrangements responsible for four genomic disorders. J Med Genet. 2004;41:113–119. doi: 10.1136/jmg.2003.012831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simovich MJ, Yatsenko SA, Kang SH, et al. Prenatal diagnosis of a 9q34.3 microdeletion by array-CGH in a fetus with an apparently balanced translocation. Prenat Diagn. 2007;27:1112–1117. doi: 10.1002/pd.1841. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Beaudet AL. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr Opin Genet Dev. 2007;17:182–192. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Uhrig S, Schlembach D, Waldispuehl-Geigl J, et al. Impact of array comparative genomic hybridization-derived information on genetic counseling demonstrated by prenatal diagnosis of the TAR (thrombocytopenia-absent-radius) syndrome-associated microdeletion 1q21.1. Am J Hum Genet. 2007;81:866–868. doi: 10.1086/521338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, de Vries BB, Osoegawa K, et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet. 2003;73:1261–1270. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapner R, Thom E, Simpson JL, et al. First-trimester screening for trisomies 21 and 18. N Engl J Med. 2003;349:1405–1413. doi: 10.1056/NEJMoa025273. [DOI] [PubMed] [Google Scholar]
- Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet. 1991;49:995–1013. [PMC free article] [PubMed] [Google Scholar]
- Yatsenko SA, Cheung SW, Scott DA, et al. Deletion 9q34.3 syndrome: genotype-phenotype correlations and an extended deletion in a patient with features of Opitz C trigonocephaly. J Med Genet. 2005;42:328–335. doi: 10.1136/jmg.2004.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.