

Abstract
Ensuring sufficient drug solubility is a crucial problem in pharmaceutical-related research. For water-insoluble drugs, various formulation approaches are employed to enhance the solubility and bioavailability of lead compounds. The goal of this study was to enhance the dissolution and absorption of a new antitumor lead compound, T-OA. Early-stage preparation discovery concept was employed in this study. Based on this concept, a solid dispersion system was chosen as the method of improving drug solubility and bioavailability. Solid dispersions of T-OA in polyvinylpyrrolidone (PVP) K30 were prepared by the solvent evaporation method. Dissolution testing determined that the ideal drug-to-PVP ratio was 1:5. X-ray diffraction, Fourier transform infrared spectroscopy, and differential scanning calorimetry were employed to confirm the formation of solid dispersions. Scanning electron microscopy demonstrated that T-OA was converted into an amorphous form. Both in vitro dissolution testing and the in vivo studies demonstrated that the solubility and bioavailability of T-OA were significantly improved when formulated in a solid dispersion with PVP. The dissolution rate of the T-OA/PVP solid dispersion was greatly enhanced relative to the pure drug, and the relative bioavailability of T-OA solid dispersions was found to be 392.0%, which is 4-fold higher than the pure drug.
KEYWORDS: bioavailability; dissolution; early-stage preparation discovery concept (EPDC); solid dispersions, T-OA
INTRODUCTION
During drug discovery and development work, the permeability and solubility of drugs can be the limiting factors for oral absorption. Poor solubility causes drugs to dissolve very slowly in the gastrointestinal tract, thereby leading to a low bioavailability. Since permeability is an intrinsic drug property, various strategies have been developed with the aim of improving the dissolution rate. In addition to chemical modification, various formulation approaches are important and effective methods of enhancing solubility (1). In this study, we chose a new antitumor compound, T-OA, to serve as a model water-insoluble drug and investigated the effect of solid dispersion.
Early-stage preparation discovery concept (EPDC) is a promising method for screening new drugs. It is a specific novel approach in pharmaceutical research for drug discovery. In order to improve the disadvantageous physicochemical properties of lead compounds, the methods based on EPDC could be effective and might rescue some potential drugs in an early stage of their development.
T-OA is a newly discovered antitumor lead compound. The chemical name of this compound is 3β-hydroxyolea-12-en-28-oic acid-3,5,6-trimethylpyrazin-2-methyl ester (T-OA, C38H58O3N2, Fig. 1a). It is composed of one molecule each of trimethylpyrazine and oleanolic acid (OA), both of which are extracted from herbal medicines, conjugated via ester bonds. T-OA was reported to be a bioactive molecule. It has demonstrated toxicity in Bel-7402 and HCT-8 tumor cell lines (2). In our previous work carried out in the S180 mice model, T-OA showed tumor inhibition effects, with an inhibitory efficiency of up to 50%. Immunohistochemical analysis was employed to investigate T-OA’s antitumor properties, and it was concluded that T-OA could suppress nuclear transcription factor kappa B (NF-κB/p65) and COX-2 in mice. However, as a potential tumor therapeutic drug, a major problem remains to be resolved. T-OA is a water-insoluble drug; the water solubility of this drug is less than 0.1 μg/ml, which leads to a poor oral bioavailability. Therefore, it is important to establish effective methods of enhancing T-OA dissolution.
Fig. 1.
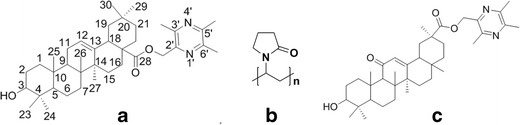
Structure of T-OA, PVPK30, and 43-PH
Many pharmaceutical methods can be employed to improve drug dissolution, including solid dispersions, microemulsion, and cyclodextrin inclusion complexes. In this investigation, we chose to prioritize the use of solid dispersions because the materials are readily available and the process of preparation is relatively rapid. There have been some reports in recent years on the use of binary or ternary solid dispersions for ameliorating the drug dissolution profile (3–5).
Solid dispersion is defined as the dispersion of active ingredients in carriers in a solid state; it can be prepared by fusion, solvent, or solvent–fusion methods (6–9). There are also various approaches regarding the method of preparing solid dispersions by the solvent method, including evaporation, spray, and freeze drying (10). A number of carriers can be employed for preparing solid dispersions, including polyethylene glycols (11,12), polyvinylpyrrolidone (13), hydroxypropyl methylcellulose (14,15), and poloxamer (16–18). The structure of PVP K30, a hydrophilic polymer, is shown in Fig. 1b. As it can form intermolecular hydrogen bonds with compounds of interest, PVP has been widely used to increase the solubility of hydrophobic drugs (19–22).
In this paper, to prepare solid dispersions, PVP K30 was selected as a polymeric carrier while T-OA was selected as a model water-insoluble drug. The solid dispersion was prepared through the solvent evaporation method and characterized by X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR), differential scanning calorimetry (DSC), and scanning electron microscopy (SEM). In addition, both the in vitro release and in vivo bioavailability of the solid dispersions was investigated.
MATERIALS AND METHODS
Materials
T-OA powder and 43-PH (internal standard, Fig. 1c) was obtained from Beijing University of Chinese Medicine (Beijing, China); their synthesis routes have both been reported previously (2). PVP K30 was obtained from Huzhou Pharmaceutical Co. Ltd (Zhejiang, China). Sodium dodecyl sulfate was obtained from Anhui Shanhe Pharmaceutical Excipients Co. Ltd (Anhui, China). HPLC-grade methanol and acetonitrile were obtained from Fisher Scientific (Shanghai, China). Sodium heparin (BR) was obtained from Sinopharm Chemical Reagent Co. Ltd (Beijing, China). All other reagents and solvents, such as dichloromethane, methanol, sodium acetate, acetic acid, hydrochloric acid, potassium dihydrogen phosphate, and sodium hydroxide, were of analytical grade. SPF male SD rats (250 ± 10 g) were provided by the Animal Center of Peking University Health Science Center (Animal production license SCXK (Beijing) 2011–0012, Quality certificate No. 0262466).
Preparation of Solid Dispersions
Briefly, T-OA was dissolved in dichloromethane with stirring. This was followed by the addition of PVP K30 and SLS, and stirring continued until a unique transparent solution formed. Under a ventilator, the organic solvent was removed by evaporation at 80°C in a bath. When it was almost dry, the solid was transferred to a vacuum drying apparatus (DZF-6050 Vacuum drying oven, Shanghai Boxun Industry & Commerce Co. Ltd, Medical Equipment Factory, Shanghai, China) to remove residual solvent. After about 2–3 h at 30°C, when the solvent was completely removed, the resultant solid dispersions were pulverized, sieved, and stored in a desiccator at room temperature.
Characterization of Solid Dispersions
X-ray Diffraction Study
Vacuum grease was applied over a glass slide to adhere the sample. About 100 mg of sample was sprinkled over it to make a layer with a thickness of 0.5 mm. All the experiments were performed on an XRD instrument (Japan Science D/max 2500) with a sensitivity of 0.001. The samples were exposed to CuKα radiation under 40 kV and 40 mA over the 2θ range from 5° to 90° in increments of 0.12°/s every 0.02°. T-OA and physical mixtures of T-OA and PVP K30 were also run as controls. The samples used for this study were freshly prepared (48 h prior) and preserved in a desiccator before using.
FT-IR Spectroscopy Study
FT-IR spectra of solid dispersions were obtained with the IR-21 Shimadzu Biorad FT-IR system (Kyoto, Japan). The sample was dispersed in dry potassium bromide (5 wt.% of sample). The disk was placed in the FT-IR sample holder and the IR spectra, in absorbance mode, was obtained in the spectral region 4,000 to 500 cm−1 with a resolution of 1 cm−1.
Differential Scanning Calorimetry Study
Finely powdered samples (10 ± 0.1 mg) were weighed and encapsulated in flat-bottomed aluminum pans with crimped-on lids. The scans were obtained in an air atmosphere with a Pyris 1 DSC instrument (Perkin Elmer, USA) by heating from 30°C to 300°C at a rate of 10°C/min. The apparatus, following calibration with indium, automatically calculated heats of fusion by integration of the area under the DSC peaks.
Scanning Electron Microscopy Study
Prior to imaging, samples were mounted onto aluminum stages using double-sided carbon tape and sputter-coated using an electron microscopy sputter coater equipped with an Au source. Samples were exposed to the Au for 2.5 min and then examined using a Hitachi S-4800 field emission scanning electron microscope (Hitachi High-Technologies Corp.; Tokyo, Japan).
In Vitro Dissolution Study
Dissolution tests were performed with a dissolution apparatus (RCZ-8M dissolution apparatus with RZQ-8D auto-sampling and Collection System; Tianda Tianfa Technology Co. Ltd; Tianjin, China) using the paddle method according to USP34. Samples of original T-OA, physical mixtures of T-OA and the polymeric carrier, and various solid dispersions equivalent to 100 mg of T-OA were added to 1,000 mL of pure water. Paddle rotation speed was set at 50 rpm, and the temperature was maintained at 37 ± 0.5°C. At predetermined intervals (5, 10, 15, 30, and 45 min), 5 mL of sample was withdrawn from each vessel and the same volume of fresh medium was replenished simultaneously. The samples were filtered with a 0.45-μm membrane filter. The absorbance of each sample was analyzed by spectrophotometer at 280 nm (UV-2550 UV-Visible Spectrophotometer, SHIMADZU). The percentage of T-OA dissolved in water was calculated using a regression equation generated from the standard curve. The regression equation employed was y = 0.0141x + 0.0051, the linear range was 0.25 μg∼50.4 μg/mL and the r2 value was 0.9998. Our previous tests have confirmed that there were no changes in the λmax of T-OA despite the presence of PVP K30 dissolved in the dissolution medium. All samples were diluted with methanol before analysis.
In Vivo Bioavailability Study
Animal Study
Ten male SD rats, weighing 250 ± 10 g, were randomly divided into two groups of five rats. All the animals were fasted for 12 h prior to initiation of the experiment. The drug was administrated by the intragastric method (300 mg/kg body weight). A total of 0.5 mL of blood was collected from the orbital venous plexus 0.5, 1, 2, 4, 6, 8, 10, and 12 h after the drug administration. Blood samples were placed into heparinized tubes. After centrifugation, the obtained plasma was stored at −20°C until further study. Two hundred microliters of plasma was used for quantification, 8 μL of internal standard, 43-PH, was precisely added, and this step was followed by the addition of 600 μL of acetonitrile. After being vortexed (VORTEX SHAKER, QL-861; Jiangsu, China) for 1 min, the mixture was centrifuged (refrigerated centrifuge, 1-15K, Sigma) at 12,000 rpm for 5 min at 4°C. The supernatant was then withdrawn and evaporated under ventilation with compressed air at room temperature (air oil-less compressor, YH-05; Shanghai Yuan Hui Mechanical and Electric Appliance Science and Technology Co., Ltd; Shanghai, China). The residue was redissolved in 100 μL of methanol by vortexing. The mixture was centrifuged at 12,000 rpm for 5 min at 4°C, and 10 μL of the supernatant was injected in the HPLC system for quantification.
HPLC Analysis of Plasma Samples
The concentration of T-OA in plasma was quantified by RP-HPLC (Agilent1100 HPLC including a set of quaternionic pump, online degassing unit, auto injector, DAD detector, chemstation; Agilent Co., Ltd; USA; Kromasil chromatographic column, 250*4.6 mm, 5 μm). The mobile phase used was methanol and water (98:2, v/v), and the signal was monitored at 278 nm. The flow rate was maintained at 1.0 mL/min, column temperature was kept at 25°C, and the sample volume was 10 μL.
The pharmacokinetic parameters associated with each animal were estimated by Kinetica v4.4 software. All the animal experiments were performed according to the Guidelines for Animal Experimentation, University of TCM, Beijing.
RESULTS
Dissolution Test
PVP Ratio
T-OA was found to be insoluble in water within 45 min of dissolution test, while the solid dispersions showed markedly improved dissolution characteristics in this study. As shown in Fig. 2, the dissolution rate of the solid dispersion was greatly enhanced relative to pure drug and physical mixture of drug and carrier; furthermore, the dissolution rate increased as the proportion of PVP increased. When the drug/PVP ratio was 1:8, the dissolution rate reached 80.8 ± 1.46%, which is 1.2-fold higher than that observed with the formulation with a ratio of 1:3. Solid dispersions prepared with drug/PVP ratios of 1:5, 1:6, and 1:8 exhibited similar dissolution profiles, as there were no significant differences between these three groups.
Fig. 2.
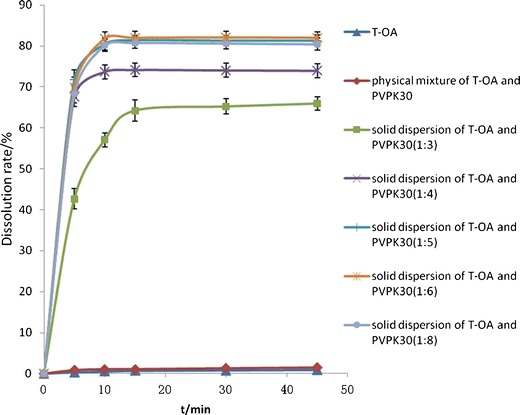
Dissolution profiles of T-OA, physical mixture with PVP, and T-OA solid dispersion
SLS Addition
Figure 3 shows the influence of SLS on the properties of the solid dispersion. The formulation composed of PVP and T-OA was unstable in dissolution test, and three batches of the same composition showed significant differences in the release profile. The dissolution ratio varied from 72.1 ± 1.46% to 93.4 ± 1.72%. This phenomenon was ameliorated by adding SLS to the solid dispersion. As depicted in Fig. 4, the internal addition of SLS enhanced the in vitro release profile of the drug compared with the physical mixtures. So it is not due to SLS surface activity and solubilization. SLS may act as crystal inhibitor as discussed in SEM paragraph. Although PVP is also a good crystal inhibitor, it could reduce the dissolution rate due to its viscosity, when the PVP amount is excessive. Henceforth, SLS was suitable for this study.
Fig. 3.

Dissolution profiles of different batches of solid dispersion and different ratio of SLS
Fig. 4.

Dissolution profiles of physical mixture of SLS and solid dispersion and solid dispersions with and without SLS
Figure 5 shows the dissolution profiles of solid dispersions in different release media. The dissolution rate was poor in a medium with a pH of 1.0. Further studies proved the sink condition was matched in water and pH 6.8 medium while not in pH 1.0, so the enteric dosage and corresponding dissolution test would be preferred in the future work, for formulations design.
Fig. 5.
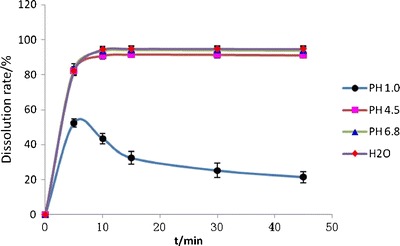
Dissolution profiles of solid dispersions in different dissolution mediums
Physical Characterization
X-ray Diffraction
To investigate the presence of crystallinity in the T-OA/PVP solid dispersion, the formulation underwent XRD analysis. The XRD patterns of the solid dispersion of T-OA and PVP K30 (1:5) are shown in Fig. 6a. The XRD patterns of pure T-OA and the solid dispersion of T-OA and PVP K30 of a different ratio are shown in Fig. 6b. The XRD patterns of T-OA and a physical mixture of T-OA and PVP K30 are shown in Fig. 6c. For T-OA, very sharp characteristic peaks at diffraction angles (2θ) of 14.12°, 14.38°, 15.82°, 17.24°, 19.22°, and 24.13° were observed, indicating the presence of crystallinity in the drug preparation; for the physical mixture of T-OA and PVP K30, these characteristic peaks were also observed. For the prepared solid dispersion, however, specific peaks indicative of crystallinity disappeared.
Fig. 6.
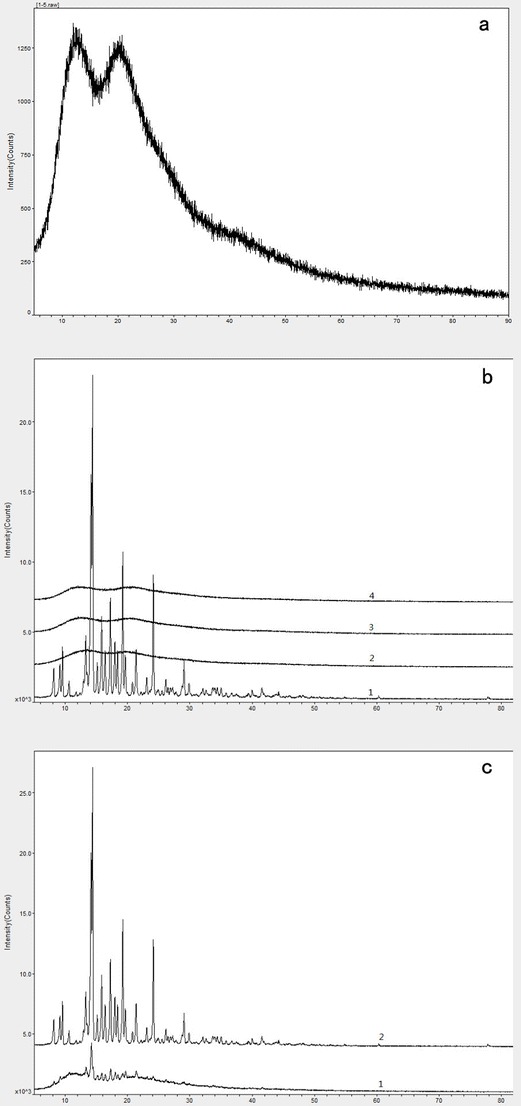
XRD spectra of solid dispersions and physical mixture of T-OA and PVP K30
FT-IR Spectroscopy
The T-OA/PVP solid dispersion was characterized by FT-IR spectroscopy. Pure T-OA (Fig. 7a), PVP K30 (Fig. 7b) and a physical mixture of T-OA and PVP K30 (Fig. 7c) were run as controls, the solid dispersion FT-IR spectrum was Fig. 7d. In the FT-IR spectrum, the strong absorption peaks at 1,726 and 1,149 cm−1 represent the specific stretching bands of C=O and C–O, respectively. In the IR spectrum of the solid dispersion, these two bands were both red shifted or absent.
Fig. 7.

FT-IR spectroscopy of T-OA (a), physical mixture of T-OA and PVP K30 (b), solid dispersion of T-OA and PVP K30 (c), and PVP K30 (d)
DSC Scans
Further analysis was carried out using DSC to study the thermal properties of the T-OA/PVP solid dispersions. Pure T-OA showed an endothermic peak at 178.467°C (peak temperature) with a fusion enthalpy of 68.495 J/g (Fig. 8a). In the T-OA/PVP solid dispersions, the endothermic peak indicative of T-OA was absent. Both types of solid dispersion preparations showed an endothermic peak at 66.8 ± 0.7°C (peak temperature) with a fusion enthalpy of 197.3 ± 5.2 J/g, as well as a weak inflection or exothermic change in their base line at about 152°C (Fig. 8b, c).
Fig. 8.

DSC traces of pure T-OA (a), solid dispersion of T-OA and PVP K30 (b), and solid dispersion of T-OA, PVP K30, and SLS (c) at a scanning rate of 10°C/min
Scanning Electron Microscopy
SEM analysis was performed to visualize the morphology of pure T-OA and T-OA/PVP solid dispersions. The T-OA samples exhibited rectangular crystal structures (Fig. 9a), while the T-OA/PVP solid dispersions presented an irregular bulk shape (Fig. 9b, d). T-OA/PVP K30 solid dispersions presented a new morphology that was different from pure T-OA. A very small number of T-OA crystals could be observed in the scanning electron micrograph of solid dispersion of T-OA and PVP K30 (Fig. 9b). In the scanning electron micrograph of a solid dispersion of T-OA, PVP K30, and SLS (Fig. 9d), the degree of crystallinity was obviously changed and the few crystalline areas remaining disappeared.
Fig. 9.

Morphology images of T-OA and solid dispersions. T-OA (a), solid dispersion of T-OA and PVP K30 (b, c), and solid dispersion of T-OA, PVP K30, and SLS (d)
In Vivo Bioavailability
In our in vivo rat tests, there was a good linearity between C and A (C means concentration and A refers to ratio of peak area. A = A sample/A internal standard). The regression equation was C = 1.5776A + 0.0488, the linear range was 0.1 μg∼12.8 μg/mL, and the r2 value was 0.9995. The validation parameters of precision (CV less than 5%) and accuracy (recovery of ±10%) were acceptable, and the lower limit of quantification was 0.732 μg/mL.
The pharmacokinetic parameters associated with each animal were estimated by Kinetica 4.4 software (Table I). The data were fitted with a noncompartment model and the results are displayed in Table I and Fig. 10. Our in vivo study found that the pharmacokinetic profile of T-OA was highly improved by solid dispersion formulation, as the Cmax, Tmax, T1/2, and MRT were all significantly enhanced or prolonged compared with pure T-OA drug. The relative bioavailability of the solid dispersion was 392.0%.
Table I.
Pharmacokinetic Results of T-OA and Its Solid Dispersion
| C max (μg/mL) | T max (h) | AUC (μg mL−1 h−1) | T 1/2 (h) | MRT (h) | |
|---|---|---|---|---|---|
| Solid dispersion(1:5) | 2.17 ± 0.67 | 1.80 ± 0.45 | 9.27 ± 2.50 | 4.17 ± 2.35 | 6.18 ± 1.90 |
| Pure drug | 0.32 ± 0.08 | 1.30 ± 0.67 | 2.36 ± 0.37 | 8.15 ± 2.86 | 13.01 ± 3.15 |
Fig. 10.

Bioavailability of T-OA solid dispersions and prototype drug in rats
DISCUSSION
During drug development research, EPDC is an effective method worth consideration. This method refers to carrying out pharmaceutical work normally performed later at the candidate compound discovery stage, in order to improve the physicochemical properties of leading compounds and the improve efficiency of drug discovery.
In general drug development process, the preformulation and formulation stages are always performed following early-stage drug discovery, while we intend to introduce these steps into the discovery stage. Some leading compounds often have to be abandoned at an early research stage because of undesirable physicochemical properties. A formulation approach based on EPDC could improve the unfavorable drug properties and influence drug biofate, such as by enhancing in vivo absorption, distribution, metabolism, and bioavailability, or by reducing specific accumulation and toxicity. This work aimed to improve the physicochemical properties and in vivo absorption of a lead compound, T-OA, through EPDC.
There are many approaches can be introduced into EPDC. In author’s opinion, at the rapid screening stage, the early preparation technology could not only save research time, but also is useful for the similar structured compounds. So designing compound delivery formulations with the minimal number of trials and the least time is very important for pharmaceutical scientists. The minimum types of experiments and the relatively unique process are needed, and the investigator should be able to reveal the role of experiments with single factor investigation. So solid dispersion technology was chosen by us firstly.
The in vitro studies demonstrated that formulation as a solid dispersion can greatly improve drug dissolution. Many hypotheses have been proposed to explain the mechanism by which formulation as a solid dispersion can ameliorate the disadvantageous pharmaceutical properties of drugs. Generally, the major factors that influence drug solubility and dissolution include degree of crystallinity, wettability, and particle size (23). The XRD results indicated that the drug was in an amorphous form within the crystalline polymer matrix. In an amorphous state, a drug possesses higher Gibbs energy than its stable crystalline state; therefore, in an amorphous state in the solid dispersion, T-OA had a higher dissolution rate. The FT-IR spectrum demonstrated that formulation as a solid dispersion notably weakened the C–O stretching band of T-OA, which is a reflection of the Van der Waals interactions and/or intermolecular hydrogen bonding between PVP K30 and T-OA; we posit this was responsible for accelerating the dissolution rate.
Formulation as a solid dispersion by the solvent method can greatly improve drug dissolution, but the issue of residual organic solvent needs to be considered. We compared three organic solvents: ethanol, methylene dichloride, and acetone. Methylene dichloride displayed the best drug dissolubility and the minimum required amount; thus, it was finally chosen. In the future industrial work, solvents of class III including acetone, propylene glycol, and isopropanol might be selected instead of methylene dichloride, on the purpose of avoiding the toxicity and environment protection.
By SEM analysis, T-OA and PVP K30 were observed to form an amorphous solid dispersion. There tiny crystals were observed in the SEM image of PVP K30/T-OA solid dispersions (Fig. 9b), while when SLS was included in the formulation, these structures disappeared. Thus, in this case, SLS may act as a crystal inhibitor, and this could explain the improved dissolution of the solid dispersion by the addition of SLS.
The in vivo experiments confirmed the high dissolution rate of PVP/T-OA solid dispersions. The pharmacokinetic parameters, including Cmax, Tmax, and MRT, were all significantly improved by formulation of the drug as a solid dispersion. Of course, this formulation can improve bioavailability, and this is consistent with the in vitro results.
In herbal extracts and chemical modification studies, there are many compounds that have similar structures and physicochemical properties. Solid dispersion systems may enhance the dissolution rate of novel compounds with similar structures, such as the C=O stretching band observed in T-OA. The employment of solid dispersion systems is a good direction in EPDC and may increase drug screening efficiency.
The results indicated that formulation as a solid dispersion can enhance the solubility of hydrophobic drug, and more importantly, it can also improve the bioavailability. There are still many approaches which can be introduced into EPDC to improve a new compound’s bioavailability, such as solid lipid nanoparticles (24,25), microemulsions, micropowders (26), micro drug coatings (27), liposomes (28), and engineered nanomaterials (29). During early-stage drug discovery research, EPDC cannot only save time but also simplify the process and increase efficiency.
CONCLUSIONS
The solid dispersion technique based on EPDC was successfully employed for the improvement of the dissolution and absorption of a water-insoluble compound. A solid dispersion of the hydrophobic compound T-OA in PVP was made, and both the drug’s solubility and bioavailability were highly improved by this solid dispersion system. Research based on EPDC can increase the efficiency of discovery of potential new drugs, and solid dispersion technology can be employed as one direction of the EPDC technical system.
ACKNOWLEDGMENTS
This study was financially supported by the National Natural Science Foundation of China (No. 81173519) and the Innovation Team Project Foundation of Beijing University of Chinese Medicine (Lead Compound Discovering and Developing Innovation Team Project Foundation).
REFERENCES
- 1.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. doi: 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- 2.Wang P, She G, Yang Y, Li Q, Zhang H, Liu J, et al. Synthesis and biological evaluation of new Ligustrazine derivatives as anti-tumor agents. Molecules. 2012;17:4972–4985. doi: 10.3390/molecules17054972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu L, Wang X. Improved dissolution of oleanolic acid with ternary solid dispersions. AAPS PharmSci Tech. 2007;8(4):E1–E5. doi: 10.1208/pt0804113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cirri M, Maestrelli F, Corti G, Mura P. Fast-dissolving tablets of glyburide based on ternary solid dispersions with PEG 6000 and surfactants. Drug Deliv. 2007;14:247–255. doi: 10.1080/10717540601067802. [DOI] [PubMed] [Google Scholar]
- 5.Kwon SH, Kim SY, Ha KW, Kang MJ, et al. Pharmaceutical evaluation of genistein-loaded pluronic micelles for oral delivery. Arch Pharm Res. 2007;30:1138–1143. doi: 10.1007/BF02980249. [DOI] [PubMed] [Google Scholar]
- 6.Damian F, Blaton N, Naesens L, et al. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur J Pharm Sci. 2000;10:311–322. doi: 10.1016/S0928-0987(00)00084-1. [DOI] [PubMed] [Google Scholar]
- 7.Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60:1281–1302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 8.Ford JL. The current status of solid dispersions. Pharm Acta Helv. 1986;61:69–88. [PubMed] [Google Scholar]
- 9.Passerini N, Albertini B, Gonzalez-Rodriguez ML, Cavallari C, Rodriguez L. Preparation and characterization of ibuprofen–poloxamer 188 granules obtained by melt granulation. Eur J Pharm Sci. 2002;15:71–78. doi: 10.1016/S0928-0987(01)00210-X. [DOI] [PubMed] [Google Scholar]
- 10.He X, Pei L, Tong HHY, Zheng Y. Comparison of spray freeze drying and the solvent evaporation method for preparing solid dispersions of baicalein with Pluronic F68 to improve dissolution and oral bioavailability. AAPS PharmSciTech. 2011;12(1):104–113. doi: 10.1208/s12249-010-9560-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janssens S, de Novoa AH, Roberts CJ, Van den Mooter G. Characterization of ternary solid dispersions of Itraconazole inpolyethylene glycol 6000/polyvidone-vinylacetate 64 blends. Eur J Pharm Biopharm. 2008;69(3):1114–1120. doi: 10.1016/j.ejpb.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Michoel A, Van den Mooter G. Study of the phase behavior of polyethylene glycol 6000–itraconazole solid dispersions using DSC. Int J Pharm. 2004;272(1–2):181–187. doi: 10.1016/j.ijpharm.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 13.Abdul-Fattah AM, Bhargava HN. Preparation and in vitro evaluation of solid dispersions of halofantrine. Int J Pharm. 2002;235:17–33. doi: 10.1016/S0378-5173(01)00941-3. [DOI] [PubMed] [Google Scholar]
- 14.Zheng X, Yang R, Tang X, Zheng L. Part I: characterization of solid dispersions of nimodipine prepared by hot-melt extrusion. Drug Dev Ind Pharm. 2007;33:791–802. doi: 10.1080/03639040601050213. [DOI] [PubMed] [Google Scholar]
- 15.LI DX, JANG K-Y, KANG W, BAE K, LEE MH, OH Y-K, et al. Enhanced solubility and bioavailability of sibutramine base by solid dispersion system with aqueous medium. Biol Pharm Bull. 2010;33(2):279–284. doi: 10.1248/bpb.33.279. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Zhang GGZ, Neilly J, Marsh K, Mawhinney D, Sanzgiri YD. Enhancing the bioavailability of ABT-963 using solid dispersion containing Pluronic F-68. Int J Pharm. 2004;286:69–80. doi: 10.1016/j.ijpharm.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 17.Yong CS, Yang CH, Rhee JD, et al. Enhanced rectal bioavailability of ibuprofen in rats by poloxamer 188 and menthol. Int J Pharm. 2004;269:169–176. doi: 10.1016/j.ijpharm.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 18.Castro SG, Bruni SS, Lanusse CE, Allemandi DA, Palma SD. Improved albendazole dissolution rate in Pluronic 188 solid dispersions. AAPS PharmSciTech. 2010;11(4):1518–1525. doi: 10.1208/s12249-010-9517-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sethia S, Squillante E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm. 2004;272:1–10. doi: 10.1016/j.ijpharm.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 20.Konno H, Handa T, Alonzo DE, Taylor LS. Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur J Pharm Biopharm. 2008;70(2):493–499. doi: 10.1016/j.ejpb.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 21.Marín MT, Margarit MV, Salcedo GE. Characterization and solubility study of solid dispersions of flunarizine and polyvinylpyrrolidone. Farmaco. 2002;57(9):723–727. doi: 10.1016/S0014-827X(02)01262-4. [DOI] [PubMed] [Google Scholar]
- 22.Kim M-S, Kim J-S, Park HJ, Cho WK, Cha K-H, Hwang S-J. Enhanced bioavailability of sirolimus via preparation of solid dispersion nanoparticles using a supercritical antisolvent process. Int J Nanomed. 2011;6:2997–3009. doi: 10.2147/IJN.S26546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guedes FL, de Oliveira BG, Hernandes MZ, De Simone CA, Veiga FJB, de Lima MdCA, et al. Solid dispersions of imidazolidinedione by PEG and PVP polymers with potential antischistosomal activities. AAPS PharmSciTech. 2011;12(1):401–410. doi: 10.1208/s12249-010-9556-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi F, Zhao J-H, Liu Y, Wang Z, Zhang Y-T, Feng N-P. Preparation and characterization of solid lipid nanoparticles loaded with frankincense and myrrh oil. Int J Nanomed. 2012;7:2033–2043. doi: 10.2147/IJN.S30085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.A.C. Silva, A. Kumar, W. Wild, D. Ferreira, D. Santos, B. Forbes, Long-term stability; biocompatibility and oral delivery potential of Risperidone-loaded solid lipid nanoparticles. Int J Nanomed. 2012. http://www.sciencedirect.com/science/article/pii/S037851731200779X. [DOI] [PubMed]
- 26.Chu KR, Lee E, Jeong SH, Park E-S. Effect of particle size on the dissolution behaviors of poorly water-soluble drugs. Arch Pharmacal Res. 2012;35(7):1187–1195. doi: 10.1007/s12272-012-0709-3. [DOI] [PubMed] [Google Scholar]
- 27.Zarie ES, Kaidas V, Gedamu D, Mishra YK, Adelung R, Furkert FH, et al. Solvent free fabrication of micro and nanostructured drug coatings by thermal evaporation for controlled release and increased effects. PLoS One. 2012;7(8):e40746. doi: 10.1371/journal.pone.0040746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spuch C, Hindawi CN. Liposomes for targeted delivery of active agents against neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease). J Drug Deliv. 2011. doi:10.1155/2011/469679. [DOI] [PMC free article] [PubMed]
- 29.Liang X-J, Chen C, Zhao Y, Lee J, Wang PC. Biopharmaceutics and therapeutic potential of engineered nanomaterials. Curr Drug Metab. 2008;9(8):697–709. doi: 10.2174/138920008786049230. [DOI] [PMC free article] [PubMed] [Google Scholar]